
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective generation of singlet oxygen from dioxygen with atomically dispersed catalysts†
Wenjie
Ma‡
ac,
Junjie
Mao‡
d,
Chun-Ting
He‡
 e,
Leihou
Shao
a,
Ji
Liu
a,
Ming
Wang
ac,
Ping
Yu
ac and
Lanqun
Mao
*ab
e,
Leihou
Shao
a,
Ji
Liu
a,
Ming
Wang
ac,
Ping
Yu
ac and
Lanqun
Mao
*ab
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry, The Chinese Academy of Sciences (CAS), Beijing 100190, China. E-mail: lqmao@bnu.edu.cn
bCollege of Chemistry, Beijing Normal University, Xinjiekouwai Street 19, Beijing 100875, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dKey Laboratory of Functional Molecular Solids, Ministry of Education, College of Chemistry and Materials Science, Anhui Normal University, Wuhu 241002, China
eMOE Key Laboratory of Functional Small Organic Molecule, College of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang 330022, China
First published on 19th April 2022
Abstract
Singlet oxygen (1O2) as an excited electronic state of O2 plays a significant role in ubiquitous oxidative processes from enzymatic oxidative metabolism to industrial catalytic oxidation. Generally, 1O2 can be produced through thermal reactions or the photosensitization process; however, highly selective generation of 1O2 from O2 without photosensitization has never been reported. Here, we find that single-atom catalysts (SACs) with atomically dispersed MN4 sites on hollow N-doped carbon (M1/HNC SACs, M = Fe, Co, Cu, Ni) can selectively activate O2 into 1O2 without photosensitization, of which the Fe1/HNC SAC shows an ultrahigh single-site kinetic value of 3.30 × 1010 min−1 mol−1, representing top-level catalytic activity among known catalysts. Theoretical calculations suggest that different charge transfer from MN4 sites to chemisorbed O2 leads to the spin-flip process and spin reduction of O2 with different degrees. The superior capacity for highly selective 1O2 generation enables the Fe1/HNC SAC as an efficient non-radiative therapeutic agent for in vivo inhibition of tumor cell proliferation.
Introduction
Dioxygen (O2) occupies a critical position in a great variety of oxidation reactions involved in both chemical reactions and biological processes.1–6 In aerobic biology, O2-related oxidation reactions are achieved by a series of evolutionary metalloenzymes such as cytochrome P450 through the interaction with O2, resulting in the formation of reactive oxygen species (ROS) such as singlet oxygen (1O2), hydroxyl radical (˙OH) and superoxide anion (O2−) or reactive metal-O2 intermediates including metal superoxo, (hydro)peroxo, and metal-oxo species.7–11 Among the active species, 1O2 with an unoccupied π* orbital has strong electrophilicity and unique reactivity and selectivity to serve as a synthetic reagent, attracting enormous interest from both fundamental studies and practical application fields.12–16 To this end, increasing attention has been drawn to the development of efficient approaches to highly selective generation of 1O2.17–22Conventional approaches to 1O2 production mainly include thermal processes using enzymatic or chemical reactions and photosensitization of O2 with rationally designed photosensitizers.23–26 However, the production of 1O2 through these approaches is always accompanied by other ROS generation. In addition, thermal processes often require harsh conditions such as rigorous pH and particular solvents.27,28 Despite the simplicity and controllability of the photosensitization route, it faces inherent limitations like poor selectivity of 1O2, photobleaching of sensitizers, and difficulty in large-scale production.29–31 Therefore, development of new approaches to highly selective production of 1O2 is of great significance not only in the investigation of 1O2-related biological and physical processes but also in the development of new materials and biological tools with 1O2.
Recently, single-atom catalysts (SACs) have shown considerable potential in industrial chemical processing and photo/electrochemical energy conversion with high catalytic activity and unique selectivity due to their excellent properties like maximum atom utilization efficiency, well-defined active centers, and tunable coordination environment.32–34 Among the developed SACs, metal-nitrogen (MNx) sites embedded in carbon skeletons represent a significant series of SACs with excellent catalytic activity toward oxygen-related reactions including the oxygen reduction reaction and advanced oxidation processes, benefitting from precise tuning of electronic structures of active sites.35–38 Thus, MNx SACs provide great possibilities and opportunities in the design and fabrication of selective catalysts for 1O2 production. However, despite some investigations on the capacity of MNx SACs toward oxygen activation with non-selective ROS production, the atomic engineering of MNx SACs is still needed to optimize the metal–O2 interaction to achieve selective 1O2 generation.
Here, we demonstrate a new approach to highly selective generation of 1O2 from O2 without photosensitization with single transition metal atoms anchored on hollow N-doped carbon as atomically dispersed catalysts (M1/HNC SACs, M = Fe, Co, Cu, Ni). The 1O2 generation efficiency is highly related to the metal centers, following the sequence of Fe1/HNC > Co1/HNC > Cu1/HNC > Ni1/HNC. Among the catalysts examined, the Fe1/HNC SAC with a single atomic motif of FeN4 coordination shows the best kinetic value of 0.140 min−1, exceeding those of Co1/HNC (0.033 min−1), Cu1/HNC (0.019 min−1) and Ni1/HNC (0.016 min−1), and the single-site kinetic value of the Fe1/HNC SAC reaches up to as high as 3.30 × 1010 min−1 mol−1, representing top-level catalytic activity among known catalysts. Density functional theory (DFT) calculations demonstrate that the selective 1O2 generation capacity originates from significant charge transfer from MN4 sites to chemisorbed O2, leading to the spin-flip process and spin reduction of O2 with the lowest value of 0.25 for O2 adsorbed on the FeN4 site. Based on the efficient production of 1O2 enabled by the Fe1/HNC SAC, we develop a non-radiative therapeutic platform for in vivo inhibition of tumor cell proliferation.
Results and discussion
To synthesize M1/HNC SACs, metal acetylacetonate encapsulated zeolitic imidazolate framework-8 (M/ZIF-8) was first synthesized as the raw material to form core–shell composites by tannic acid (TA) coating. The core–shell precursors were then pyrolyzed at 900 °C under an Ar atmosphere to obtain M1/HNC catalysts (Fig. 1a). Hollow N-doped carbon (HNC) was prepared with pure ZIF-8 without the encapsulation of metal acetylacetonate as the control catalyst. X-ray diffraction (XRD) patterns of the catalysts show peaks for (002) and (101) planes of graphitic carbon, located at around 24° and 43°, respectively, and no observable peaks for metal-related nanoparticles were found (Fig. S1†), indicating the absence of metal nanoparticles in M1/HNC catalysts.39 Raman spectra of the M1/HNC SACs and HNC reveal similar intensity ratios of the D band to the G band (Fig. S2†), suggesting that the catalysts possess a similar structure of the defective carbon skeleton. In addition, the nitrogen sorption isotherm analysis of M1/HNC SACs and HNC (Fig. S3†) gave a similar Brunner–Emmet–Teller (BET) surface area at around 700 cm2 g−1, further indicating the structural similarity of the prepared catalysts. Transmission electron microscopy (TEM) and high-angle annular dark-field scanning TEM (HAADF-STEM) images show that Fe1/HNC has a hollow polyhedral morphology with an average size of 200 nm and thickness of 10 nm and no metal nanoparticles were observed (Fig. 1b). The elemental mapping images show a homogeneous distribution of Fe, N, and C elements over the whole domain of the Fe1/HNC SAC (Fig. 1c). Furthermore, the aberration-corrected HAADF-STEM image was obtained to confirm single atomic Fe in the Fe1/HNC SAC. As shown in Fig. 1d, the bright dots highlighted with red circles clearly demonstrate that the atomically dispersed Fe atoms exist over N-doped carbon. Similarly, the morphology of other three M1/HNC SACs and the atomic dispersion were confirmed by using TEM and HAADF-STEM images, all showing a hollow polyhedral structure and atomically dispersed metal atoms (Fig. S4–S6†). | ||
| Fig. 1 Synthesis of M1/HNC SACs and structural characterization of the Fe1/HNC SAC. (a) Schematic depiction of the route to the fabrication of M1/HNC SACs. (b) TEM image of Fe1/HNC. (c) HAADF-STEM image and the corresponding elemental mapping images of Fe1/HNC (C, red; Fe, green; N, orange). (d) Aberration-corrected HAADF-STEM image of Fe1/HNC showing atomically dispersed Fe atoms as bright dots highlighted with red circles. | ||
The atomic electronic structure and coordination configuration of metal species in M1/HNC SACs were explored by X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) characterization experiments. Fig. 2a shows the XANES spectra of Fe1/HNC at the Fe K-edge in comparison with those of references including Fe foil and Fe2O3. The peak position of Fe1/HNC is situated between those of Fe2O3 and Fe foil, indicating that the valence state of the Fe atom is between 0 and +3. The Fourier-transform EXAFS spectrum of Fe1/HNC shows only one peak at about 1.5 Å, ascribed to the first coordination shell of Fe–N, and no Fe–Fe peak like that of Fe foil at around 2.2 Å was observed (Fig. 2b), suggesting the inexistence of metal-related nanoparticles or clusters in Fe1/HNC. In order to further investigate the atomic distribution of Fe in Fe1/HNC, wavelet transform (WT) analysis of Fe K-edge EXAFS was carried out. As illustrated in Fig. 2c, only one intensity maximum at approximately 4.2 Å−1 related to Fe–N coordination was observed from the WT contour plot of Fe1/HNC, which is different from the contour WT plots of Fe foil and Fe2O3 with the intensity maximum corresponding to Fe–Fe contribution, elucidating that no Fe–Fe bond is present in Fe1/HNC, but isolated Fe atoms exist. Moreover, the coordination configuration and structural parameters of Fe atoms were obtained from EXAFS fitting curves (Fig. 2d and S7†). As listed in Table S1,† the coordination number of Fe atoms and the bond length of Fe–N are 4.2 and 2.02 Å, respectively. From the above results, the atomic structure configuration of Fe1/HNC was constructed (Fig. 2e). The local structure of other three M1/HNC SACs was also confirmed with XANES and EXAFS spectra (Fig. S8–S10†), all possessing similar atomic configuration with Fe1/HNC.
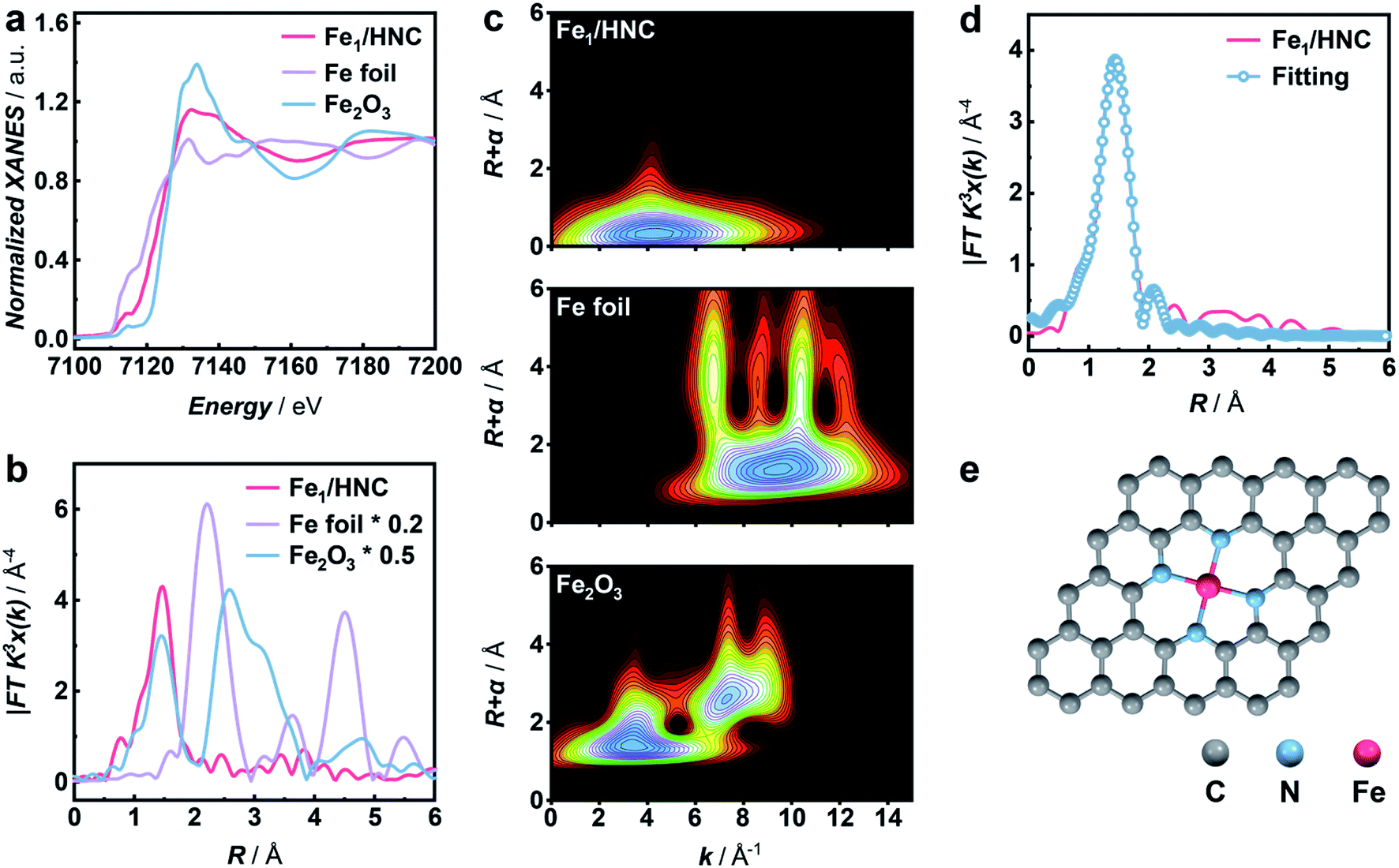 | ||
| Fig. 2 XAFS analysis of the Fe1/HNC SAC. (a) Normalized XANES spectra and (b) corresponding Fourier-transform EXAFS spectra at the Fe K-edge of Fe1/HNC (red line), Fe foil (purple line), and Fe2O3 (blue line). (c) Wavelet transforms of the Fe K-edge EXAFS spectra for Fe1/HNC, Fe foil, and Fe2O3. (d) Fourier-transform EXAFS spectrum and the corresponding fitting curve of Fe1/HNC in R-space. (e) Schematic structure of Fe1/HNC. | ||
In order to systematically evaluate the oxidizing capacity of M1/HNC SACs, the oxidation of colorless 3,3′,5,5′-tetramethylbenzidine (TMB) to blue oxidized TMB (ox-TMB) with the characteristic adsorption at ca. 652 nm was used as the catalytic model reaction.40–43 Considering that the pyrolysis temperature has significant effect on the coordination microenvironment of SACs,44,45 the TMB oxidation activities of the catalysts prepared at different temperatures were explored. As shown in Fig. 3a, the catalyst obtained at 900 °C (denoted as Fe1/HNC-900) exhibits the highest catalytic activity towards TMB oxidation, and 900 °C was thus chosen to prepare other SACs. We next compared the catalytic activity of M1/HNC SACs with different transition metal centers toward TMB oxidation. The results displayed in Fig. 3b and S11† show that the activity trends follow the sequence of Fe1/HNC > Co1/HNC > Cu1/HNC > Ni1/HNC > HNC, indicating that the single metal atom center plays a vital role in the oxidation reaction. Among the catalysts investigated, Cu1/HNC and Ni1/HNC SACs exhibit quite low catalytic activity towards TMB oxidation like HNC. Comparatively, both Fe1/HNC and Co1/HNC SACs show a high catalytic oxidation rate, far surpassing those of Cu1/HNC and Ni1/HNC SACs. The significant difference in catalytic performance of M1/HNC SACs may arise from the binding strength of different MN4 centers with O species.46–48
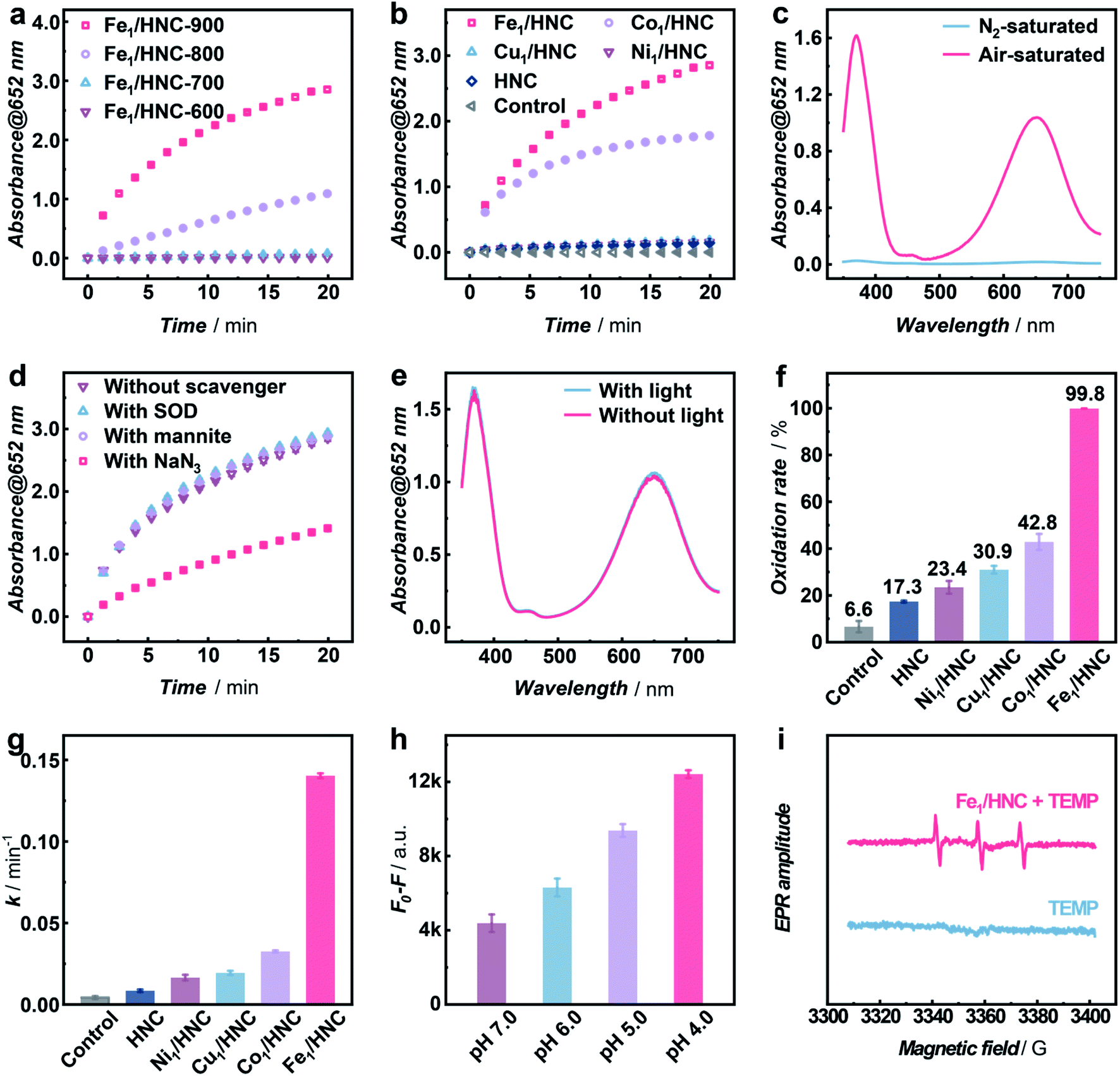 | ||
| Fig. 3 Selective generation of 1O2 with M1/HNC SACs. Time-dependent absorbance changes of ox-TMB monitored at 652 nm (a) with 5 μg mL−1 catalysts synthesized at different temperatures or (b) with 5 μg mL−1 M1/HNC and HNC in the Britton–Robinson (B–R) buffer (pH 4.0). (c) UV-vis absorption spectra of ox-TMB catalyzed by 5 μg mL−1 Fe1/HNC at 2 min in the N2-saturated (blue line) and air-saturated (red line) B–R buffer (pH 4.0). (d) Time-dependent absorbance changes of ox-TMB monitored at 652 nm catalyzed by 5 μg mL−1 Fe1/HNC in the B–R buffer (pH 4.0) without scavengers (brown line) or with SOD (blue line), mannite (purple line), and NaN3 (red line). (e) UV-vis absorption spectra of ox-TMB catalyzed by 5 μg mL−1 Fe1/HNC in 2 min without (red line) or with (blue line) light in the B–R buffer (pH 4.0). (f) Oxidation rate of ABDA with M1/HNC and HNC in the B–R buffer (pH 4.0). (g) k value of the ABDA oxidation reaction with 5 μg mL−1 M1/HNC and HNC in the B–R buffer (pH 4.0). (h) Decrease in fluorescence intensity (λex/λem = 380/433 nm) of ABDA with 5 μg mL−1 Fe1/HNC in 5 min in the B–R buffer at different pH values. (i) TEMPO ESR signals in the absence (blue line) and presence of 20 μg mL−1 Fe1/HNC in the B–R buffer (pH 4.0). | ||
To verify whether O2 is the only substrate for the oxidation reaction, Fe1/HNC was selected as the representative catalyst to evaluate the TMB oxidation reaction under a N2 atmosphere. As shown in Fig. 3c, the absorbance intensity of ox-TMB undergoes a sharp decrease in the N2-saturated B–R buffer compared with that under air-saturated conditions, indicating the indispensable role of O2 in the oxidation of TMB catalyzed by the Fe1/HNC SAC. In addition, as displayed in Fig. S12–S15,† the catalytic TMB oxidation reaction with M1/HNC (Fe1/HNC and Co1/HNC as examples) is dose- and pH-dependent.
It is generally known that the catalytic oxidation reactions with O2 are always accompanied with the generation of ROS like ˙OH, O2− and 1O2, thus, it is vital to identify the exact species produced during the process of the oxidation reaction by M1/HNC SACs. For this purpose, mannite, superoxide dismutase (SOD), and NaN3 were chosen as the ROS scavengers for ˙OH, O2−, and 1O2, respectively.49 Notably, only NaN3 exhibits efficient inhibitation of TMB oxidation catalyzed by the Fe1/HNC SAC, while the others display no obvious effects on the oxidation reaction (Fig. 3d), suggesting that the Fe1/HNC SAC selectively activates O2 into 1O2. In consideration of photosensitization usually implemented to produce 1O2, the comparison of the Fe1/HNC-catalyzed TMB oxidation reaction under normal light conditions and in a dark environment was performed. As shown in Fig. 3e, a negligible difference in characteristic absorbance of ox-TMB was observed, indicating the striking generation of 1O2 without photosensitization. Collectively, highly selective generation of 1O2 from O2 can be achieved by using the Fe1/HNC SAC without the assistance of the externally applied stimulus.
To further validate the selective production of 1O2 from O2 enabled by M1/HNC SACs, an 1O2-specific probe 9,10-anthracenediyl-bis(methylene) dimalonic acid (ABDA) was adopted to selectively recognize and react with 1O2.50,51 ABDA is a fluorescent molecule with λex and λem at ca. 380 nm and 433 nm, respectively, which turns to be a non-fluorescent endoperoxide product upon selective oxidation by 1O2via a [4 + 2]-cycloaddition. With ABDA as the probe, we evaluated the catalytic efficiency of M1/HNC SACs in generating 1O2 from O2. As can be seen in Fig. 3f and S16,† the activity towards ABDA oxidation follows the rate sequence of Fe1/HNC (99.8%) > Co1/HNC (42.8%) > Cu1/HNC (30.9%) > Ni1/HNC (23.4%) > HNC (17.3%), which is consistent with the trends in TMB oxidation. Notably, Fe1/HNC displays the highest catalytic activity toward ABDA oxidation with a rate of nearly 100%, indicative of more efficient 1O2 generation catalyzed by Fe1/HNC in comparison with other SACs. In addition, the kinetic profiles of ABDA oxidation were examined to assess 1O2 generation capacities of M1/HNC SACs by calculating the oxidation rate of ABDA with pseudo-first-order approximation. As shown in Fig. S17,† an exponential decrease in the fluorescence intensity of ABDA against the reaction time was observed and the pseudo-first-order kinetic constants (k values) were obtained from the linear relationship. The results shown in Fig. 3g reveal that Fe1/HNC catalyzes the generation of 1O2 for ABDA oxidation with a k value of 0.140 min−1, considerably higher than that of Co1/HNC (0.033 min−1), Cu1/HNC (0.019 min−1) and Ni1/HNC (0.016 min−1), further highlighting the critical role of different single metal atom coordination. The single-site kinetic constant (ksingle-site value) of the Fe1/HNC SAC was further evaluated to be as high as 3.30 × 1010 min−1 mol−1 due to the high catalytic efficiency with a low metal content of 0.17 wt% determined via inductively coupled plasma optical emission spectrometry (ICP-OES). Besides, the Fe1/HNC-enabled selective generation of 1O2 was investigated with different concentrations of the catalyst and in reaction media with different pH values (Fig. 3h and S18†). As shown in Fig. S19,† the ABDA oxidation reaction shows a dose-dependent behavior. In addition, the generation of 1O2 for ABDA oxidation is peculiarly prone to occur under acidic conditions.
To further confirm the selective generation of 1O2 enabled by Fe1/HNC, colorimetric and fluorescent probes for other ROSs were utilized, thereinto, ρ-phthalic acid (PTA) and iodonitrotetrazolium chloride (INT) were selected to detect ˙OH, and O2−, respectively. PTA, as a widely used ˙OH probe, can be selectively hydroxylated upon reaction with ˙OH to form 2-hydroxy terephthalic acid with strong fluorescence at λex/λem = 315/400 nm.52 INT is a specific probe for O2−, which can be reduced by O2− to generate the colored formanzan product with the maximum absorption at 510 nm. As shown in Fig. S20,† no obvious changes in the fluorescence intensity of PTA or absorbance of INT were observed in the oxidation reaction, demonstrating negligible generation of ˙OH and O2− but highly selective production of 1O2 during the O2 activation process. Furthermore, electron spin resonance (ESR) characterization using 2,2,6,6-tetramethylpiperidine (TEMP) as the 1O2 trapping agent and 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO) as the trapping agent for ˙OH and O2− were also conducted to give more direct evidence of the produced ROS.53,54Fig. 3i shows the typical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet ESR signal for 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) formed due to the 1O2 generation in the presence of the Fe1/HNC SAC. However, there are no observable signals when BMPO was used as the trapping agent (Fig. S21†), indicating that negligible ˙OH and O2− are produced in the process of Fe1/HNC-catalyzed O2 activation.
:1:1 triplet ESR signal for 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) formed due to the 1O2 generation in the presence of the Fe1/HNC SAC. However, there are no observable signals when BMPO was used as the trapping agent (Fig. S21†), indicating that negligible ˙OH and O2− are produced in the process of Fe1/HNC-catalyzed O2 activation.
To gain in-depth insight into the mechanism of selective 1O2 generation enabled by M1/HNC SACs, DFT calculations were performed to explore the electronic structure of the active sites and their interactions with O2.55–58 In accordance with the atomic structural analysis, the models of M1/HNC SACs adsorbed with O2 were constructed and optimized. The adsorption energy of O2 on the FeN4 site in the Fe1/HNC SAC was calculated to be −1.039 eV, which is less than those of other M1/HNC SACs (Fig. 4i), illustrating the strong interaction between the FeN4 site and O2. In order to further elucidate the electronic interactions between MN4 sites and absorbed O2, the projected density of states (PDOS) for M 3d and O 2p was calculated. As shown in Fig. 4a–d, more hybridization between the PDOS for Fe 3d orbitals and O 2p orbitals can be evidently observed in contrast with those for CuN4 or NiN4 centers. Meanwhile, the Fe 3d orbitals shift toward the Fermi level upon O2 adsorption (Fig. S22–S25†), further demonstrating the occurrence of O2 activation on FeN4.59,60 In addition, the Bader charge analysis and charge density difference analysis (Fig. 4e–h) reveal distinct charge transfer with 0.44 e from the FeN4 site to O2, leading to the elongation of the O–O bond with a length from 1.22 Å in free O2 to 1.284 Å in O2 adsorbed on the FeN4 site (Fig. 4i), which is longer than those on other MN4 sites. The longest O–O bond of O2 upon adsorption on the FeN4 site manifests the strongest interaction between the FeN4 site and O2. The transferred electrons will occupy the half-filled antibonding π* orbitals of O2 and give rise to the reduction of the Hirshfeld spin value of O2 from 1 in free O2 to 0.25, suggesting that a significant spin-flip process occurs for chemisorbed O2 on the FeN4 site. The spin value of O2 upon chemisorption on the FeN4 site (0.25) is close to that of 1O2 (0) and relatively lower than those on other MN4 sites, elucidating the superior capacity of the FeN4 site toward the generation of 1O2. Given the above, the charge transfer from the MN4 site to adsorbed O2 contributes to different spin-flip degrees of O2, thus bringing about a discrepancy in their capacities toward selective 1O2 generation.
 | ||
| Fig. 4 DFT calculations. PDOS of the M 3d (red line) and O 2p (blue line) orbitals of (a) FeN4, (b) CoN4, (c) CuN4 and (d) NiN4 sites adsorbed with O2. Calculated charge density differences of (e) FeN4, (f) CoN4, (g) CuN4 and (h) NiN4 centers adsorbed with O2. (i) Comparison of the DFT results for constructed MN4 structures. | ||
Having demonstrated the high efficiency of selective 1O2 generation without photosensitization from molecular O2 enabled by the Fe1/HNC SAC, we then employed the Fe1/HNC SAC as a therapeutic agent for suppressing tumor cell growth. To improve the dispersibility and biocompatibility of Fe1/HNC, an amphipathic molecule DSPE-PEG2000 was modified on the surface of Fe1/HNC to form PEGylated Fe1/HNC (donated as P-Fe1/HNC) through the hydrophobic interaction.61 The cellular uptake of P-Fe1/HNC was visualized by confocal laser scanning microscopy (CLSM) imaging with fluorescein isothiocyanate (FITC)-loaded P-Fe1/HNC. As displayed in Fig. 5a and S26,† the bright green fluorescence and the black dots in cytosol clearly indicate the efficient internalization of P-Fe1/HNC in HeLa cells. Next, the inhibition effect of P-Fe1/HNC on cell proliferation was systematically explored by the cell viability test with a standard MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium] assay. The results show that the treatment of HeLa cells with PEGylated HNC (P-HNC) without single Fe atomic centers exhibits a negligible effect on the proliferation of HeLa cells with cell viability exceeding 90% even at a concentration of P-HNC as high as 0.3 mg mL−1 (Fig. 5b), indicating the excellent biocompatibility and low cytotoxicity of P-HNC. In striking contrast, an obvious decrease in cell viability of HeLa cells to less than 50% upon the treatment of P-Fe1/HNC with a low concentration of 0.06 mg mL−1 was observed and the cell viability decreases with increasing P-Fe1/HNC concentration, providing direct evidence for the antiproliferation effect on HeLa cells.
 | ||
| Fig. 5 In vitro and in vivo inhibition of tumor growth with the Fe1/HNC SAC. (a) CLSM fluorescence image of HeLa cells treated with FITC-loaded P-Fe1/HNC for 6 h. (b) Cell viability of HeLa cells after 12 h-treatment of P-HNC or P-Fe/HNC at the concentrations of 0.06–0.3 mg mL−1. CLSM fluorescence images of HeLa cells stained with Calcein-AM and (c) PI or (d) DCFH-DA after the 12 h-treatment with PBS, P-HNC and P-Fe1/HNC. (e) Cell viability of HeLa cells pre-incubated with AA (0.25 mM, 0.5 mM, and 1 mM) after 12 h-treatment with 0.1 mg mL−1 P-Fe/HNC. In vivo (f) tumor proliferation and (g) body weight curves of mice treated with the intravenous injection of PBS (blue line), P-HNC (purple line), or P-Fe1/HNC (red line). | ||
In the meantime, the cytotoxicity of P-Fe1/HNC was further investigated by co-staining HeLa cells with calcein-AM (green fluorescent dye for viable cell staining) and propidium iodide (PI, red fluorescent dye for dead cell staining) and visualized with CLSM imaging.62 As exhibited in Fig. 5c and S27,† in comparison with the group treated with PBS or P-HNC showing bright green fluorescence, cancer cells treated with P-Fe1/HNC show strong red fluorescence, indicating the high cytotoxicity against HeLa cells of Fe1/HNC SACs. According to the results demonstrated above, such capacity of killing cancer cells was deemed to originate from the intracellular oxidative stress induced by Fe1/HNC SAC-enabled selective production of 1O2, which was distinctly confirmed with a cell-permeable ROS-sensitive probe (2′,7′-dichlorofluorescein diacetate, DCFH-DA).63,64 Obviously, cancer cells treated with P-Fe1/HNC show the brightest green fluorescence of DCF (Fig. 5d and S28†), indicative of most 1O2 generation enabled by Fe1/HNC. In order to further demonstrate the cytotoxicity against HeLa cells of the Fe1/HNC SAC from the oxidative damage induced by the generation of 1O2, ascorbic acid (AA) as an antioxidant was used to pre-incubate HeLa cells.61 As shown in Fig. 5e, the pre-incubation of AA can efficiently reduce the cytotoxicity of the Fe1/HNC SAC through the elimination of generated 1O2 with a concentration-dependent behavior.
The antitumor effect of the Fe1/HNC SAC in vivo was also investigated with HeLa tumor-bearing mice as a model. When the tumor volume reached about 100 mm3, the mice were randomly divided into three groups and intravenously injected with PBS, P-HNC, and P-Fe1/HNC, respectively. As shown in Fig. 5f, the mice treated with P-HNC exhibit a similar tumor growth tendency with that of PBS, indicating that HNC had no obvious therapeutic effect on suppressing tumor growth. In contrast, the group treated with P-Fe1/HNC displays a relatively higher tumor inhibition effect with the reduction of the tumor size to 46.5% in 12 days compared with the control group, which was attributed to the high capacity of the Fe1/HNC SAC for 1O2 generation within the tumor site. Moreover, photographs of the excised tumors further confirmed the good antitumor effect of Fe1/HNC (Fig. S29†). In addition, no obvious changes of the body weight of mice were observed during the experimental period upon administration of different catalysts, indicating the favorable biocompatibility of the Fe1/HNC SAC (Fig. 5g). Further blood biochemical analysis also implied the good biosafety of these catalysts (Fig. S30†).
Conclusions
In summary, we have demonstrated that atomically dispersed MN4 sites on hollow N-doped carbon can be used for the selective generation of strong oxidizing 1O2 from O2 without photosensitization. The as-prepared M1/HNC SACs show different 1O2 generation efficiencies in the sequence of Fe1/HNC > Co1/HNC > Cu1/HNC > Ni1/HNC. Among the developed catalysts, the Fe1/HNC SAC shows the best kinetic value of 0.14 min−1 and a single-site kinetic value of 3.30 × 1010 min−1 mol−1, originating from the spin-flip process and spin reduction of O2 to 0.25 induced via significant charge transfer with 0.44 e from the FeN4 site to O2. More importantly, the Fe1/HNC SAC with superior 1O2 generation capacity has been successfully utilized as an efficient non-radiative therapeutic agent for inhibiting tumor cell proliferation in vitro and in vivo. We believe that this finding would provide a facile method for the selective production of 1O2, opening up a new avenue for the development of functional SACs in biomedical applications.Data availability
All experimental and computational data is available in the ESI.†Author contributions
L. M., W. M. and J. M. conceived the idea for the project. W. M. and J. M. designed the experiments and conducted material synthesis, structural characterization, performance test and data analysis. C. H. carried out the DFT calculations. L. S. contributed to the animal experiments. J. L. and M. W. helped in the cell experiments. P. Y. contributed to the discussion of the catalytic part. W. M. drafted the manuscript, and L. M. finalized the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Key Research and Development Program (2018YFE0200800), the National Natural Science Foundation of China (Grant No. 22134002, 21790390 and 21790391 for L. M., 21705155 and 21790392 for W. M., 21971002 for J. M., and 21775151 and 21790053 for P. Y.), the National Basic Research Program of China (2016YFA0200104 and 2018YFA0703501), and the Strategic Priority Research Program of Chinese Academy of Sciences (XDB30000000). Beijing Academy of Science and Technology (BJAST) Scholar Programs (B), and Beijing Municipal Financial Project (PXM2021_178305_000004). All the animal experiments were conducted with the guidelines of the Animal Advisory Committee at the State Key Laboratory of Cognitive Neuroscience and Learning, and were approved by the Institutional Animal Care and Use Committee at Beijing Normal University.Notes and references
- Z. Li, Z. Wang, N. Chekshin, S. Qian, J. X. Qiao, P. T. Cheng, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, Science, 2021, 372, 1452–1457 CrossRef CAS PubMed.
- R. A. Copeland, S. Zhou, I. Schaperdoth, T. K. C. Shoda, J. M. Bollinger and C. Krebs, Science, 2021, 373, 1489–1493 CrossRef CAS PubMed.
- T. Richards, J. H. Harrhy, R. J. Lewis, A. G. R. Howe, G. M. Suldecki, A. Folli, D. J. Morgan, T. E. Davies, E. J. Loveridge, D. A. Crole, J. K. Edwards, P. Gaskin, C. J. Kiely, Q. He, D. M. Murphy, J.-Y. Maillard, S. J. Freakley and G. J. Hutchings, Nat. Catal., 2021, 4, 575–585 CrossRef CAS.
- Y. Wang, S. Liu, C. Pei, Q. Fu, Z.-J. Zhao, R. Mu and J. Gong, Chem. Sci., 2019, 10, 10531–10536 RSC.
- M. Ansari, D. Senthilnathan and G. Rajaraman, Chem. Sci., 2020, 11, 10669–10687 RSC.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. Jose, A. W. Schaefer, A. C. Roveda, W. J. Transue, S. K. Choi, Z. Ding, R. B. Gennis and E. I. Solomon, Science, 2021, 373, 1225–1229 CrossRef CAS PubMed.
- W. Peng, X. Qu, S. Shaik and B. Wang, Nat. Catal., 2021, 4, 266–273 CrossRef CAS.
- M. Wikström, K. Krab and V. Sharma, Chem. Rev., 2018, 118, 2469–2490 CrossRef PubMed.
- A. McEvoy, J. Creutzberg, R. K. Singh, M. J. Bjerrum and E. D. Hedegård, Chem. Sci., 2021, 12, 352–362 RSC.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS PubMed.
- C. P. Stanley, G. J. Maghzal, A. Ayer, J. Talib, A. M. Giltrap, S. Shengule, K. Wolhuter, Y. Wang, P. Chadha, C. Suarna, O. Prysyazhna, J. Scotcher, L. L. Dunn, F. M. Prado, N. Nguyen, J. O. Odiba, J. B. Baell, J.-P. Stasch, Y. Yamamoto, P. Di Mascio, P. Eaton, R. J. Payne and R. Stocker, Nature, 2019, 566, 548–552 CrossRef CAS PubMed.
- M. Agrachev, W. Fei, S. Antonello, S. Bonacchi, T. Dainese, A. Zoleo, M. Ruzzi and F. Maran, Chem. Sci., 2020, 11, 3427–3440 RSC.
- H.-B. Cheng, B. Qiao, H. Li, J. Cao, Y. Luo, K. M. Kotraiah Swamy, J. Zhao, Z. Wang, J. Y. Lee, X.-J. Liang and J. Yoon, J. Am. Chem. Soc., 2021, 143, 2413–2422 CrossRef CAS PubMed.
- P. Di Mascio, G. R. Martinez, S. Miyamoto, G. E. Ronsein, M. H. G. Medeiros and J. Cadet, Chem. Rev., 2019, 119, 2043–2086 CrossRef CAS PubMed.
- H. Zhang, D. Yu, S. Liu, C. Liu, Z. Liu, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2022, 61, e202109068 CAS.
- Z. Zheng, H. Liu, S. Zhai, H. Zhang, G. Shan, R. T. K. Kwok, C. Ma, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, K. S. Wong, X. Hu and B. Z. Tang, Chem. Sci., 2020, 11, 2494–2503 RSC.
- M. Huo, L. Wang, L. Zhang, C. Wei, Y. Chen and J. Shi, Angew. Chem., Int. Ed., 2020, 59, 1906–1913 CrossRef CAS PubMed.
- C. Wu, Z. Liu, Z. Chen, D. Xu, L. Chen, H. Lin and J. Shi, Sci. Adv., 2021, 7, eabj8833 CrossRef CAS PubMed.
- F. Gao, T. Shao, Y. Yu, Y. Xiong and L. Yang, Nat. Commun., 2021, 12, 745 CrossRef CAS PubMed.
- Y. Zhao, M. Sun, X. Wang, C. Wang, D. Lu, W. Ma, S. A. Kube, J. Ma and M. Elimelech, Nat. Commun., 2020, 11, 6228 CrossRef CAS PubMed.
- Y. Gao, T. Wu, C. Yang, C. Ma, Z. Zhao, Z. Wu, S. Cao, W. Geng, Y. Wang, Y. Yao, Y. Zhang and C. Cheng, Angew. Chem., Int. Ed., 2021, 60, 22513–22521 CrossRef CAS PubMed.
- H. Ma, S. Long, J. Cao, F. Xu, P. Zhou, G. Zeng, X. Zhou, C. Shi, W. Sun, J. Du, K. Han, J. Fan and X. Peng, Chem. Sci., 2021, 12, 13809–13816 RSC.
- X. Mi, P. Wang, S. Xu, L. Su, H. Zhong, H. Wang, Y. Li and S. Zhan, Angew. Chem., Int. Ed., 2021, 60, 4588–4593 CrossRef CAS PubMed.
- Z. Yang, J. Qian, A. Yu and B. Pan, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 6659–6664 CrossRef CAS PubMed.
- L.-S. Zhang, X.-H. Jiang, Z.-A. Zhong, L. Tian, Q. Sun, Y.-T. Cui, X. Lu, J.-P. Zou and S.-L. Luo, Angew. Chem., Int. Ed., 2021, 60, 21751–21755 CrossRef CAS PubMed.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2007, 129, 6916–6926 CrossRef CAS PubMed.
- T. Chen, P. Hou, Y. Zhang, R. Ao, L. Su, Y. Jiang, Y. Zhang, H. Cai, J. Wang, Q. Chen, J. Song, L. Lin, H. Yang and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 15006–15012 CrossRef CAS PubMed.
- X. Ye, Y. Li, P. Luo, B. He, X. Cao and T. Lu, Nano Res., 2021, 15, 1509–1516 CrossRef.
- T. Luo, G. T. Nash, Z. Xu, X. Jiang, J. Liu and W. Lin, J. Am. Chem. Soc., 2021, 143, 13519–13524 CrossRef CAS PubMed.
- D. Mao, F. Hu, Z. Yi, Kenry, S. Xu, S. Yan, Z. Luo, W. Wu, Z. Wang, D. Kong, X. Liu and B. Liu, Sci. Adv., 2020, 6, eabb2712 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- H. Jing, P. Zhu, X. Zheng, Z. Zhang, D. Wang and Y. Li, Adv. Powder Mater., 2022, 1, 100013 CrossRef.
- X. Zhang, H. Lin, J. Zhang, Y. Qiu, Z. Zhan, Q. Xu, G. Meng, W. Yan, L. Gu, L. Zheng, D. Wang and Y. Li, Chem. Sci., 2021, 12, 14599–14605 RSC.
- Y. Xiong, W. Sun, Y. Han, P. Xin, X. Zheng, W. Yan, J. Dong, J. Zhang, D. Wang and Y. Li, Nano Res., 2021, 14, 2418–2423 CrossRef CAS.
- X.-P. Qin, S.-Q. Zhu, L.-L. Zhang, S.-H. Sun and M.-H. Shao, J. Electrochem., 2021, 27, 185–194 CAS.
- W. Ma, F. Wu, P. Yu and L. Mao, Chem. Sci., 2021, 12, 7908–7917 RSC.
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C.-T. He, C. Peng, W.-C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X.-M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS PubMed.
- S. Ji, B. Jiang, H. Hao, Y. Chen, J. Dong, Y. Mao, Z. Zhang, R. Gao, W. Chen, R. Zhang, Q. Liang, H. Li, S. Liu, Y. Wang, Q. Zhang, L. Gu, D. Duan, M. Liang, D. Wang, X. Yan and Y. Li, Nat. Catal., 2021, 4, 407–417 CrossRef CAS.
- X. Chen, L. Zhao, K. Wu, H. Yang, Q. Zhou, Y. Xu, Y. Zheng, Y. Shen, S. Liu and Y. Zhang, Chem. Sci., 2021, 12, 8865–8871 RSC.
- B. Xu, H. Wang, W. Wang, L. Gao, S. Li, X. Pan, H. Wang, H. Yang, X. Meng, Q. Wu, L. Zheng, S. Chen, X. Shi, K. Fan, X. Yan and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 4911–4916 CrossRef CAS PubMed.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS PubMed.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS PubMed.
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS PubMed.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS PubMed.
- H. Wang, S. Jiang, S. Chen, D. Li, X. Zhang, W. Shao, X. Sun, J. Xie, Z. Zhao, Q. Zhang, Y. Tian and Y. Xie, Adv. Mater., 2016, 28, 6940–6945 CrossRef CAS PubMed.
- M. Wang, Y. Zhang, M. Ng, A. Skripka, T. Cheng, X. Li, K. K. Bhakoo, A. Y. Chang, F. Rosei and F. Vetrone, Chem. Sci., 2020, 11, 6653–6661 RSC.
- H. Chen, S. Li, M. Wu, Kenry, Z. Huang, C.-S. Lee and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 632–636 CrossRef CAS PubMed.
- F. Cao, L. Zhang, Y. You, L. Zheng, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2020, 59, 5108–5115 CrossRef CAS PubMed.
- Q. Liu, K. Wan, Y. Shang, Z.-G. Wang, Y. Zhang, L. Dai, C. Wang, H. Wang, X. Shi, D. Liu and B. Ding, Nat. Mater., 2021, 20, 395–402 CrossRef CAS PubMed.
- X. Hu, F. Li, F. Xia, X. Guo, N. Wang, L. Liang, B. Yang, K. Fan, X. Yan and D. Ling, J. Am. Chem. Soc., 2020, 142, 1636–1644 CrossRef CAS PubMed.
- H.-Y. Zhuo, X. Zhang, J.-X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- L. Li, X. Chang, X. Lin, Z.-J. Zhao and J. Gong, Chem. Soc. Rev., 2020, 49, 8156–8178 RSC.
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T.-S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS.
- Y. Pan, Y. Chen, K. Wu, Z. Chen, S. Liu, X. Cao, W.-C. Cheong, T. Meng, J. Luo, L. Zheng, C. Liu, D. Wang, Q. Peng, J. Li and C. Chen, Nat. Commun., 2019, 10, 4290 CrossRef PubMed.
- R. Gao, J. Wang, Z.-F. Huang, R. Zhang, W. Wang, L. Pan, J. Zhang, W. Zhu, X. Zhang, C. Shi, J. Lim and J.-J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- M. Huo, L. Wang, Y. Wang, Y. Chen and J. Shi, ACS Nano, 2019, 13, 2643–2653 CAS.
- R. Xiong, D. Hua, J. Van Hoeck, D. Berdecka, L. Léger, S. De Munter, J. C. Fraire, L. Raes, A. Harizaj, F. Sauvage, G. Goetgeluk, M. Pille, J. Aalders, J. Belza, T. Van Acker, E. Bolea-Fernandez, T. Si, F. Vanhaecke, W. H. De Vos, B. Vandekerckhove, J. van Hengel, K. Raemdonck, C. Huang, S. C. De Smedt and K. Braeckmans, Nat. Nanotechnol., 2021, 16, 1281–1291 CrossRef CAS PubMed.
- T. Xiong, M. Li, Y. Chen, J. Du, J. Fan and X. Peng, Chem. Sci., 2021, 12, 2515–2520 RSC.
- R. Cai, H. Xiang, D. Yang, K.-T. Lin, Y. Wu, R. Zhou, Z. Gu, L. Yan, Y. Zhao and W. Tan, J. Am. Chem. Soc., 2021, 143, 16113–16127 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01110g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |