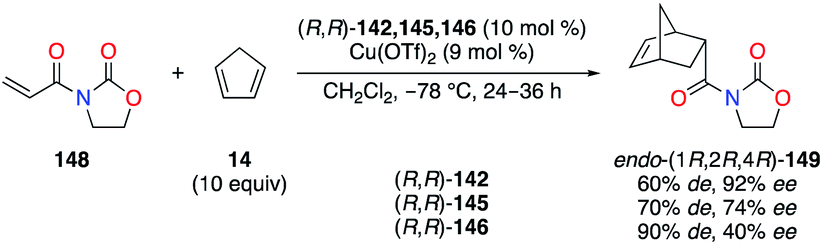DOI:
10.1039/D2SC01096H
(Review Article)
Chem. Sci., 2022,
13, 10985-11008
Fluorine in metal-catalyzed asymmetric transformations: the lightest halogen causing a massive effect
Received
21st February 2022
, Accepted 6th August 2022
First published on 8th August 2022
Abstract
This review aims at providing an overview of the most significant applications of fluorine-containing ligands reported in the literature starting from 2001 until mid-2021. The ligands are classified according to the nature of the donor atoms involved. This review highlights both metal–ligand interactions and the structure–reactivity relationships resulting from the presence of the fluorine atom or fluorine-containing substituents on chiral catalysts.
 From left to right: Samuel Lauzon and Thierry Ollevier | Samuel Lauzon was born in Québec, Canada. He joined the research group of Professor Thierry Ollevier as an undergraduate intern student in 2015. He obtained his MSc in 2018 working on asymmetric iron catalysis. He received his PhD in organic chemistry at Université Laval (Québec, Canada) under the supervision of Professor Thierry Ollevier in 2022. His work involved the development of designer 2,2′-bipyridinediol ligands for applications in metal-catalyzed asymmetric transformations. He is currently interested in ligand design, crystallization of chiral catalysts, transition state models, and environmentally benign catalysis. Thierry Ollevier was born in Brussels and obtained his Licence (1991) and PhD (1997) at the University of Namur (Belgium) and was a post doctorate fellow at the Université catholique de Louvain (Belgium), under István E. Markó (1997), a NATO and BAEF postdoctorate fellow at Stanford University under Barry M. Trost (1998–2000), and then a post doctorate fellow at the Université de Montréal under André B. Charette (2000–2001). After an Assistant Professor appointment (2001) at Université Laval (Québec, Canada), he became Associate (2006) and is currently Full Professor. Current research in his group aims at designing novel catalysts, developing catalytic reactions and applying these methods to chemical synthesis. He is active in the areas of asymmetric catalysis, iron catalysis, diazo chemistry, fluorine chemistry, and flow chemistry. He has served as a member of the Advisory Board of SynOpen since 2019, as an Associate Editor of RSC Advances since 2015 and was admitted as a Fellow of the Royal Society of Chemistry (2016). |
1. Introduction
Fluoroorganic molecules have received widespread interest in recent research in view of the fact that their synthesis is virtually missing from any biological processes. Being the 13th most abundant element in the lithosphere, fluorine is mostly present in water-insoluble minerals, i.e., fluorspar (CaF2), cryolite (Na3AlF6), and fluoroapatite (Ca5(PO4)3F), which limits its uptake into living organisms.1 Also, the nucleophilicity of fluoride is diminished by its high hydration energy, and therefore, this anion is inadequate for any nucleophilic substitution reactions in aqueous media. As a result, only a dozen of naturally occurring fluorometabolites, i.e., fluoroacetic acid, ω-fluorinated fatty acids, (2R,3R)-2-fluorocitric acid, (2S,3S,4R,5R)-nucleocidin, and (2S,3S)-4-fluorothreonine, have been identified so far.2 Since then, such scarcity has been compensated for by man-made fluorine-containing pharmaceuticals, of which many have been welcomed as blockbuster drugs on the market.3 Molecular conformation, membrane permeability, and metabolic stability are all properties affected by fluorine substitution, but the impact on the pharmacodynamics and pharmacokinetics of a lead remains quite unpredictable.4 The specific behaviour of fluorinated compounds arises from the short, strong, and highly polarized C–F bond that electrostatically pairs with the neighbouring atoms, bonds, and lone pairs.5 Instead of being targeted only for biological activity purposes, various chiral organofluorine compounds were employed as catalysts in asymmetric transformations.6 Among the known strategies for designing enhanced stereodiscriminating catalysts,7 the conversion of known catalytic systems into F-containing ones unlocked new interesting features ensuing from the fluorine effect. Chiral fluorinated catalysts benefit from (i) electronically and (ii) sterically customized properties and (iii) being used in fluorous biphasic systems (FBS). Since an increased reactivity in acid catalysts is often a synonym of electronic deficiency, Pauling (χP), empirical (χe), Huheey (χH), and Jaffé (χJ) electronegativities values are provided for a selection of atoms and groups (Table 1).8 Fluorine is the most electronegative element on the Pauling scale (χP = 3.98) and is most frequently used as an electronically impoverishing substituent in ligands. The CF3 group is also electronically deficient, much more than other C-based substituents, and its χ value is similar to the ones of the CN and NO2 groups, known as strongly electron withdrawing groups (EWG). A set of steric parameters, e.g., A values (−ΔG),9 Taft (−Es),10 Charton (ν),11 and biphenyl rotational interference values (IX–H),12 and Boltzmann-weighted Sterimol parameters (wB1, wL, and wB5),13 eases the comparison of bulkiness between selected atoms and functional groups (Table 2). Fluorine is a small element (van der Waals radius = 1.47 Å vs. 1.20 Å for H vs. 1.52 Å for O)14 and generally causes minimal steric perturbation upon H to F exchange, whereas the substitution of C–H by C–F in a methyl group successively leads to an increase in size (Me < CH2F < CHF2 < CF3). Thus, fluorine is a powerful tool to increase the steric hindrance – even more in polyfluoroalkyl groups – rendering the CF3 group as a bulky substituent in the general order Me < iPr ∼ CF3 ≪ Ph ∼ tBu. The CF3 group is a key component for fine-tuning simultaneously the steric and electronic properties, which is undeniably of great interest for ligand design. Following the “like dissolves like” principle, a chiral ligand bearing sufficiently long perfluoroalkyl chains, i.e., ponytails (RF), acquires a strong affinity for the fluorous phase, also called “fluorophilicity”.15 This engineered technology involves the temperature-dependent miscibility of fluorous-organic solvents leading to partition of two phases at lower temperature. The fluorous ligand (or catalyst) can be selectively separated from the reactant/product mixture and recovered using various experimental methods.
Table 1 Electronegativities of selected atoms and functional groups
| Atom or group |
χ
P
|
χ
e
|
χ
H
|
χ
J
|
| H |
2.20 |
2.28 |
— |
— |
| F |
3.98 |
3.95 |
— |
— |
| Cl |
3.16 |
3.03 |
— |
— |
| Br |
2.96 |
2.80 |
— |
— |
| I |
2.66 |
2.47 |
— |
— |
| NO2 |
— |
3.40 |
— |
— |
| CN |
— |
3.30 |
— |
— |
| CF3 |
— |
3.35 |
3.46 |
3.29 |
| CHF2 |
— |
— |
3.00 |
2.94 |
| CH2F |
— |
— |
2.61 |
2.61 |
| CH3 |
— |
2.30 |
2.27 |
2.30 |
Table 2 Steric parameters of selected atoms and functional groups
| Atom or group |
−ΔG (kcal mol−1) |
−Es |
ν
|
I
X–H (kJ mol−1) |
wSterimol |
|
wB1 (Å) |
wL (Å) |
wB5 (Å) |
|
Thickness/width values.
|
| H |
— |
0.00 |
0.00 |
∼4.0 |
— |
— |
— |
| F |
0.15 |
0.46 |
0.27 |
19.2 |
1.52 |
3.12 |
2.03 |
| Cl |
0.43 |
0.97 |
0.55 |
38.1 |
1.80 |
3.99 |
2.03 |
| Br |
0.38 |
1.16 |
0.65 |
42.5 |
1.95 |
4.29 |
2.03 |
| CN |
0.17 |
0.51 |
— |
25.6 |
— |
— |
— |
| NO2 |
1.10 |
1.01/2.52a |
— |
32.4 |
— |
— |
— |
| Me |
1.70 |
1.24 |
0.52 |
40.4 |
1.88 |
3.43 |
2.02 |
| CH2F |
1.59 |
1.48 |
0.62 |
— |
— |
— |
— |
| CHF2 |
1.85 |
1.91 |
0.68 |
— |
— |
— |
— |
| CF3 |
2.10 |
2.40 |
0.91 |
50.6 |
1.87 |
3.75 |
2.60 |
|
i
Pr |
2.15 |
1.71 |
0.76 |
52.6 |
1.91 |
4.56 |
3.17 |
| SiMe3 |
2.50 |
3.36 |
1.40 |
47.2 |
— |
— |
— |
| Ph |
3.00 |
1.01/3.82a |
1.66 |
33.1 |
1.70 |
6.80 |
3.15 |
|
t
Bu |
∼4.20 |
2.78 |
1.24 |
76.6 |
2.76 |
4.54 |
3.17 |
The synergistic participation of the steric, electronic, and physical properties brought up by the chiral fluorinated ligand may modulate the stereoselective event of a reaction positively vs. “fluorine-free transition states (TS)”. This review highlights both metal–ligand interactions and structure–reactivity relationship related to the presence of the fluorine atom or fluorine-containing substituents on chiral catalysts. The article focusses on the molecular architecture of the ligands rather than on the type of reaction they were employed in. The selection of chiral fluoroorganic ligands are classified according to O-, N,O-, N-, P, N-, P- and C-binding modes with metals. The advantages of using catalytic systems involving fluorine for both the reactivity and the stereochemical outcomes are highlighted, compared, and discussed. Furthermore, the effects on the stereochemical outcome arising from using fluorinated ligands vs. non-fluorinated ones are often provided when both results were available in the literature. The set of examples for promoting the massive effect of fluorine in metal-catalyzed asymmetric catalysis is not exhaustive; hence fluorinated reactants, solvents, additives, and Brønsted acids-based catalysts are not reviewed herein.
2. Chiral fluoroorganometallic complexes
2.1
O-Based binding modes
2.1.1 Phosphoric acids.
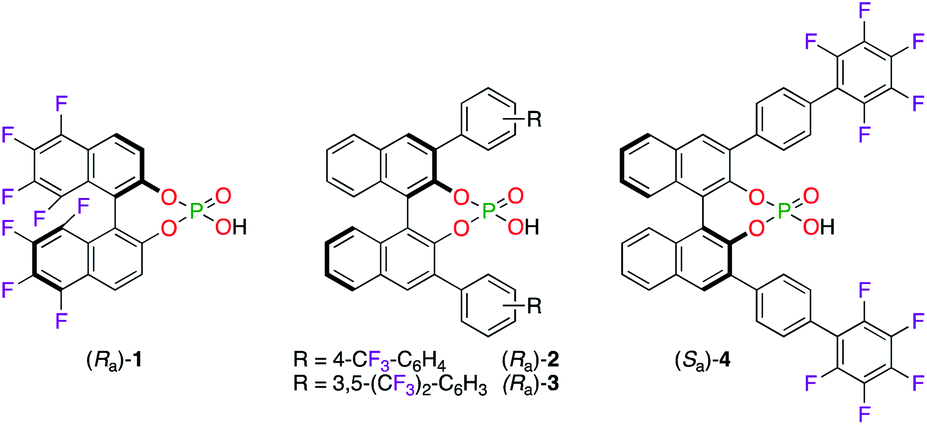
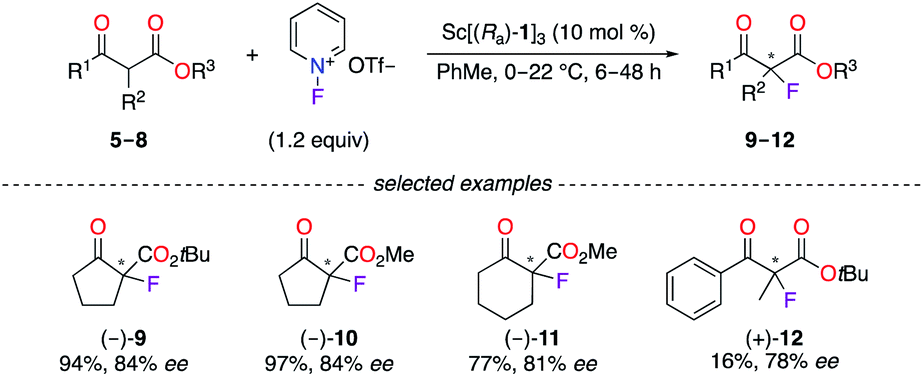
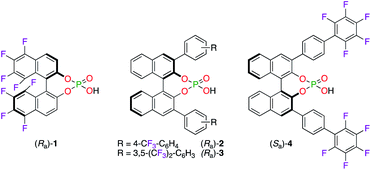
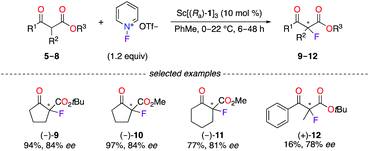
Chiral phosphoric acid (CPA) ligands (Ra)-1–3 and (Sa)-4 were obtained by the phosphorylation of fluorinated 1,1′-bi-2-naphthol (BINOL)-type ligands and were described as promising candidates for enantioselective applications (Fig. 1). As an example, the fluorination reaction of β-ketoesters 5–8 using the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Ra)-1/ScCl3 complex afforded the corresponding α-fluorinated products 9–12 with 78–84% ee (Scheme 1).16 The importance of the eight fluorine atoms was demonstrated based on the low 11% ee obtained for (+)-9 using the ScIII salt of the non-fluorinated CPA. On the other hand, (Ra)-217 and (Ra)-318 were respectively employed with CuI salts in the synthesis of chiral polyfluoro N-containing compounds, albeit with modest ees.
:1 (Ra)-1/ScCl3 complex afforded the corresponding α-fluorinated products 9–12 with 78–84% ee (Scheme 1).16 The importance of the eight fluorine atoms was demonstrated based on the low 11% ee obtained for (+)-9 using the ScIII salt of the non-fluorinated CPA. On the other hand, (Ra)-217 and (Ra)-318 were respectively employed with CuI salts in the synthesis of chiral polyfluoro N-containing compounds, albeit with modest ees.
 |
| | Fig. 1 Chiral fluorinated CPA ligands 1–4 in metal-catalyzed reactions. | |
 |
| | Scheme 1 [(Ra)-1]3/ScIII-catalyzed fluorination reaction. | |
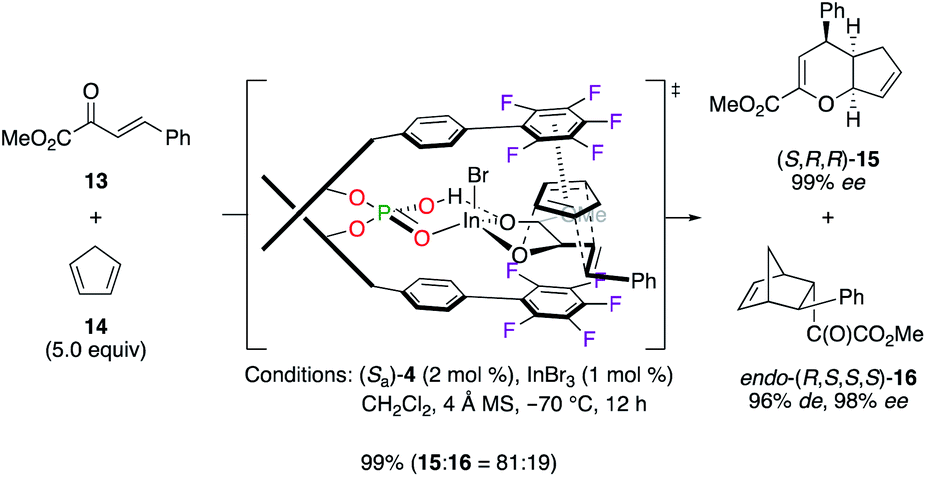
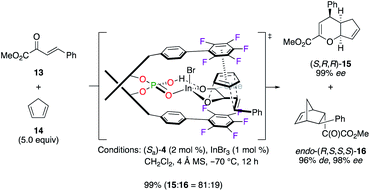
In another application, cycloadducts (S,R,R)-15 and endo-(R,S,S,S)-16, respectively obtained from the hetero-Diels–Alder (HDA) and the Diels–Alder (DA) reactions between dienophile 13 and cyclopentadiene 14, were synthesized with excellent yields and stereoselectivities using the binary (Sa)-4/InIII catalytic system (Scheme 2).19 In most cases, a higher chemoselectivity was noted for the HDA cycloadduct vs. the DA one. The stereoselective induction was explained by the important π, π interaction, arising from an electrostatic pairing between one electronically deficient pentafluorophenyl ring and diene 14, together with highly sensitive ortho positions occupied by F atoms.
 |
| | Scheme 2 (Sa)-4/InIII-catalyzed HDA and DA reactions. | |
2.1.2 Carboxylates and alkoxides.
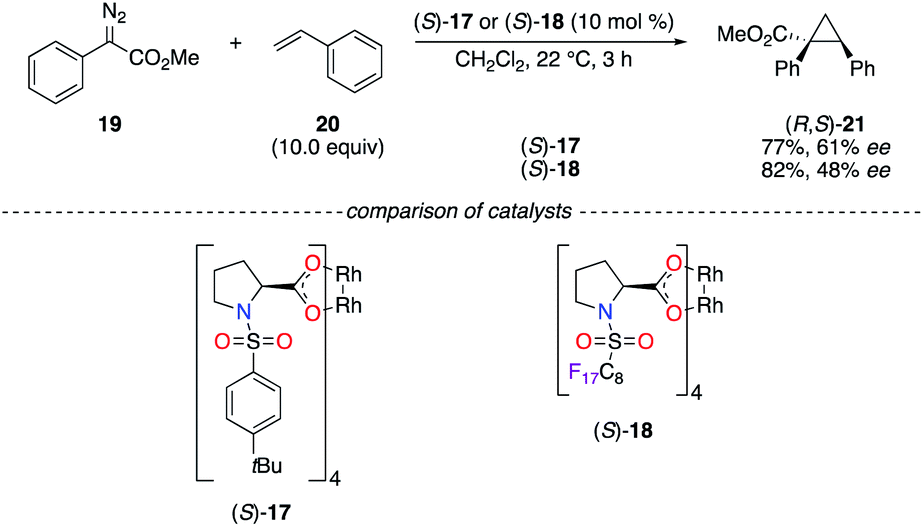
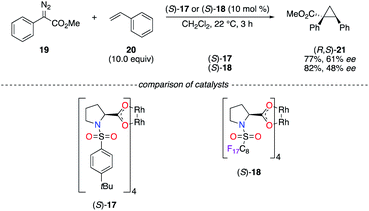
Known as valuable catalysts in the chemistry of diazo compounds, chiral RhII tetrakis(carboxylates) are rather expensive to be easily used in industrial applications. The recyclable chiral fluorous RhII complex (S)-18 was then targeted in the cyclopropanation reaction of diazoester 19 and styrene 20 (Scheme 3).20 Whereas it was less selective in terms of ee than (S)-17, the perfluoroalkyl chain allowed an efficient recovery of the catalyst using both liquid and solid fluorous phase extraction strategies. Slight improvements of the chiral induction were obtained in the insertion reaction of 19 into the C–H bond of cyclohexane using (S)-18 under either homogeneous or heterogeneous conditions.
 |
| | Scheme 3 Cyclopropanation reaction using chiral dirhodium catalysts. | |
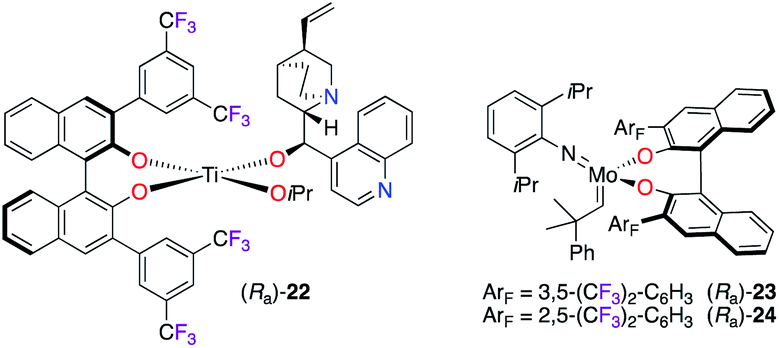
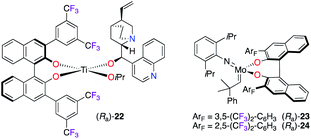
Various enantioselective organic transformations were performed using more sophisticated CF3-containing BINOL-based chiral catalysts (Fig. 2). Indeed, the synergetic activation using self-assembled bifunctional chiral catalysis was shown as an efficient strategy in the hydrophosphonylation reaction of aldehydes.21 The multiple reactive sites of catalyst (Ra)-22, generated in situ in the presence of Ti(OiPr)4, CF3-aryl-substituted BINOL, and (–)-cinchonidine, allowed cooperative interactions between steric and electronic properties, as postulated in the transition state model of the reaction. Also, trifluoromethylated BINOLates showed excellent chiral induction in the desymmetrization of Cs-symmetric phosphaferrocenes via ring-closing metathesis using MoVI-catalysts (Ra)-23 and (Ra)-24.22
 |
| | Fig. 2 TiIV– and MoVI–BINOLate complexes. | |
2.1.3 Diols.
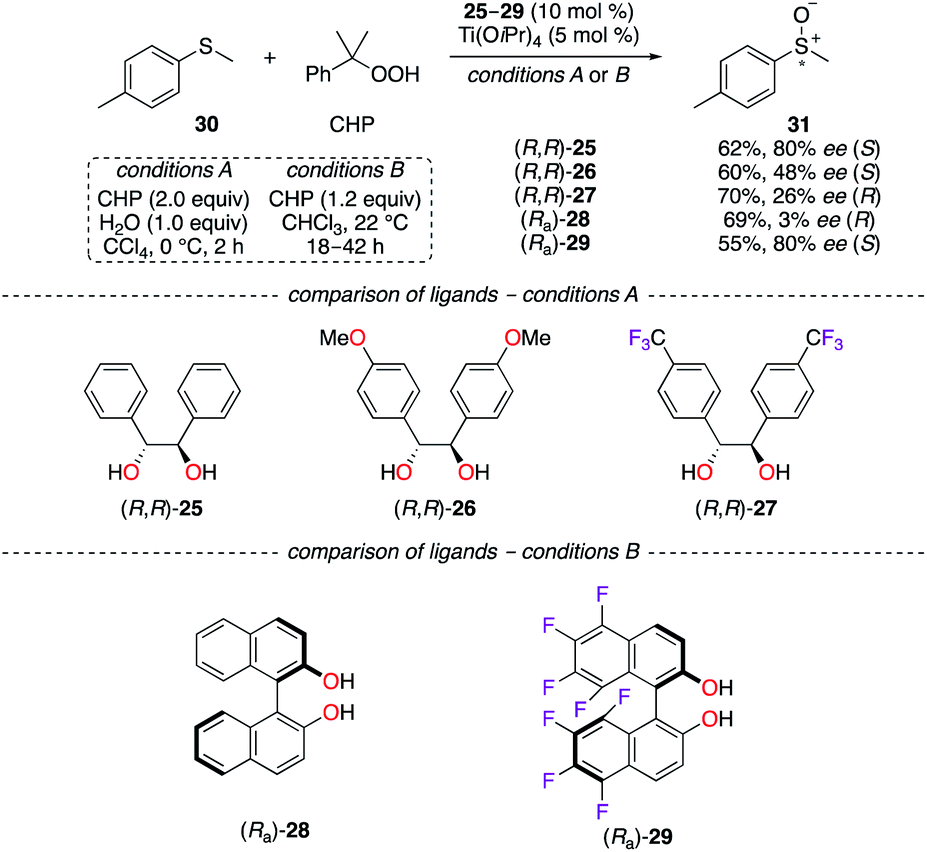
Since hydrobenzoin 25 is cheap and readily available in its R,R and S,S enantiomeric forms, chiral TiIV complexes have employed the 1,2-diphenylethane-1,2-diol scaffold in the enantioselective oxidation reaction of 4-methyl-thioanisole 30 using cumyl hydrogen peroxide (CHP) as the oxidant (Scheme 4, conditions A).23 The incorporation of OMe and CF3 groups onto the chiral backbone of (R,R)-26 and (R,R)-27 led to electronically modified TiIV catalysts of lower capacity for chiral induction. As observed experimentally, a reversal of the asymmetric induction was observed using the 4-CF3-C6H4 group (26% ee for (R)-36) vs. the unsubstituted one (80% ee for (S)-36), resulting in optically active sulfoxides of opposite signs. As postulated, competing mechanisms, arising from divergent binding modes with the TiIV centre, led to a decreased purity of 31 as the S enantiomer and to a complete reversal of the sense of chiral induction in favour of (R)-31. The TiIV-catalyzed asymmetric oxidation of aromatic sulfides was also demonstrated using a BINOL ligand comprising fluorine atoms at the 5, 5′, 6, 6′, 7, 7′, 8, and 8′ positions (Scheme 4, conditions B).24 The oxidation of 30 by the (Ra)-28/TiIV system afforded (R)-31 with virtually no enantioselectivity. Again, a reversal of the chiral induction was observed when (Ra)-29 was employed and sulfoxide 31 was obtained as the S enantiomer in 80% ee. In the case of F8BINOL (Ra)-29, the tenfold increase in acidity of the hydroxyl groups (pKa = 9.28 vs. 10.28 for (Ra)-28) and the more positive oxidation potential (E2 = 2.07 V vs. 1.47 V for (Ra)-28) are believed to result in a more configurationally stable chiral environment of the TiIV-based catalyst.
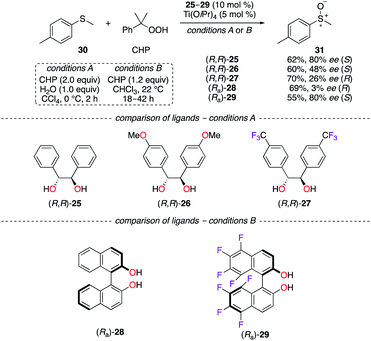 |
| | Scheme 4 TiIV-Catalyzed sulfoxidation reaction – (R,R)-hydrobenzoin and (Ra)-BINOL derivatives. | |
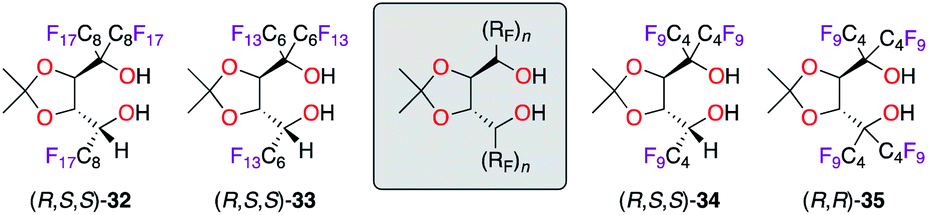
Being recognized along with BINOL as “privileged ligands”,25 α,α,α′,α′-tetraaryl-1,3-dioxolane-4,5-dimethanol (TADDOL) ligands have inspired the design of ligands 32–35, bearing α,α,α′-triperfluoroalkyl and α,α,α′,α′-tetraperfluorobutyl chains (Fig. 3).26 Being successfully employed in the Ti(iPrO)4-catalyzed methylation of various aldehydes, ligands (R,S,S)-32–34 afforded similar levels of chiral induction (ca. 95% ee) in the benchmark reaction using benzaldehyde. Notably, shortening the fluorinated ponytails had no effect on the efficiency of the catalyst (C4F9vs. C8F17), but the recyclability of the ligand was reduced; hence the use of an expensive fluorous solvent was therefore needed for extraction. Interestingly, only the tetrakis(perfluoroalkyl) analogue (R,R)-35 was obtained from (R,S,S)-34, whereas the incorporation of a fourth perfluoroalkyl chain failed for diol (R,S,S)-32 and (R,S,S)-33 due to higher steric congestion. However, the (R,R)-35/TiIV catalyst was noticed to be inactive in the tested catalytic application.
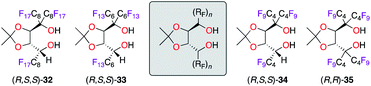 |
| | Fig. 3 TADDOL-like fluorous ligands. | |
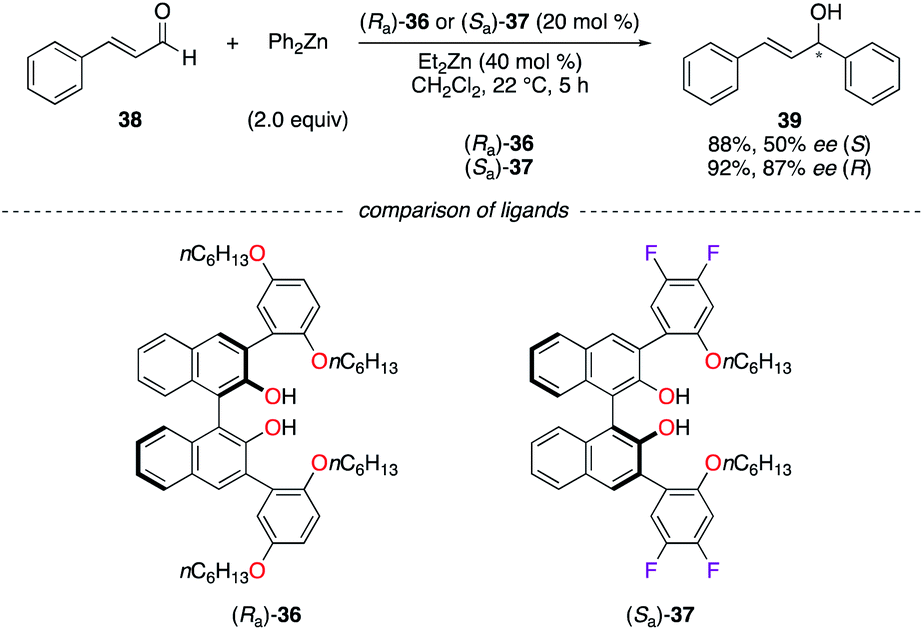
BINOLs (Ra)-36 and (Sa)-37 were subjected to comparison studies in the addition of diphenylzinc to (E)-cinnamaldehyde 38 (Scheme 5).27 As can be seen in (Sa)-37, the fluorine atoms increase the Lewis acidity of the ZnII chelated metal centre. The higher enantioselectivity observed for (R)-39 was then attributed to a better activation of the aldehyde by a more active catalyst, which favours the ligand-controlled pathway vs. the uncatalyzed background reaction.
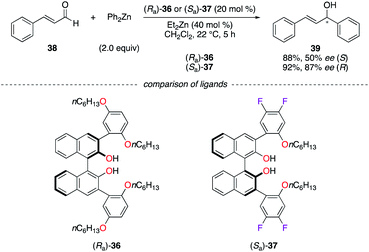 |
| | Scheme 5 Alkylation addition reaction of Ph2Zn to cinnamaldehyde – (Ra)-36vs. (Sa)-37. | |
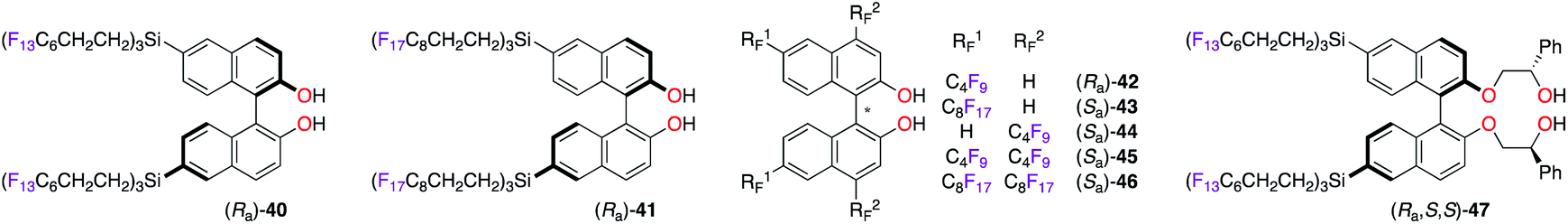
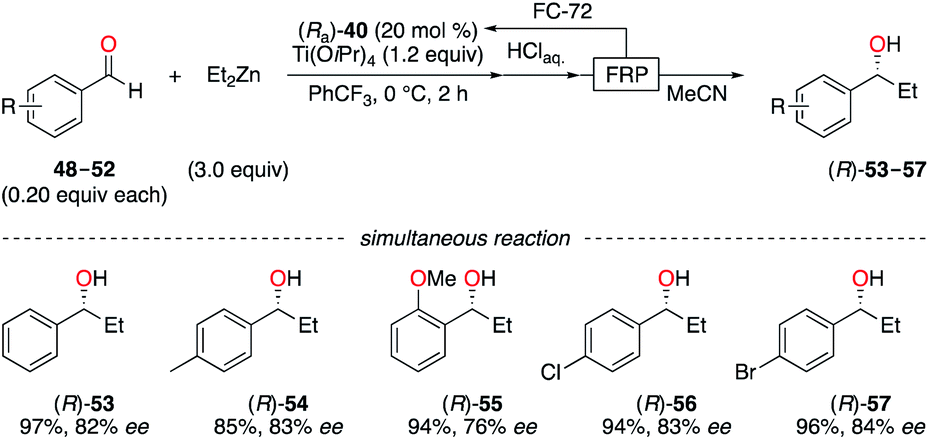
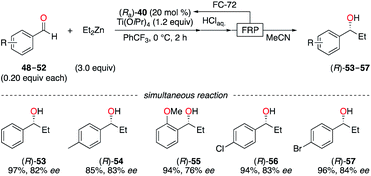
Enantioselective applications in fluorous biphasic systems (FBS) were described for perfluorobutyl, -hexyl, and -octyl BINOL derivatives, i.e., (Ra)-40–42,28 (Sa)-43–46,28c,29 and (Ra,S,S)-47 (Fig. 4).30 As an example, the addition of diethylzinc to a solution containing aldehydes 48–52 (0.20 equiv. each, total = 1.0 equiv.) in α,α,α-trifluorotoluene was performed using (Ra)-40 (Scheme 6).28a All five aldehydes simultaneously reacted with the nucleophile under the optimal reaction conditions. Elution of the reaction mixture with acetonitrile, using fluorous reverse phase (FRP) silica gel chromatography, afforded alcohols (R)-53–57 with ees up to 84%. The recovery of (Ra)-40 from the fluorous phase was achieved using tetradecafluorohexane (FC-72) as the eluent. Also, a recycling protocol using a fluorous biphasic system based on (perfluoro)methyldecalin was suitable to recover (Sa)-46, which was reused in up to nine subsequent runs and afforded (S)-1-phenylpropane-1-ol without loss of chiral induction.28c The protonation of the SmIII-enolate, obtained from 2-methoxy-2-phenylcyclohexanone, afforded the corresponding enantioenriched ketone with similar yields and ees by using both (Ra,S,S)-47 and its non-fluorinated analogue.30 The main advantage of (Ra,S,S)-47 is its easy recyclability via a simple filtration. The enantioselective fluorescent recognition of trans-1,2-diaminocyclohexane was performed using a fluorous BINOL in an FC-72/MeOH biphasic system.31
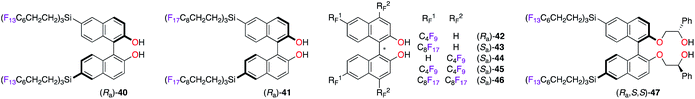 |
| | Fig. 4 Chiral RF–BINOL ligands for TiIV-catalyzed reactions in FBS. | |
 |
| | Scheme 6 Simultaneous ethylation reaction of aldehydes in FBS. | |
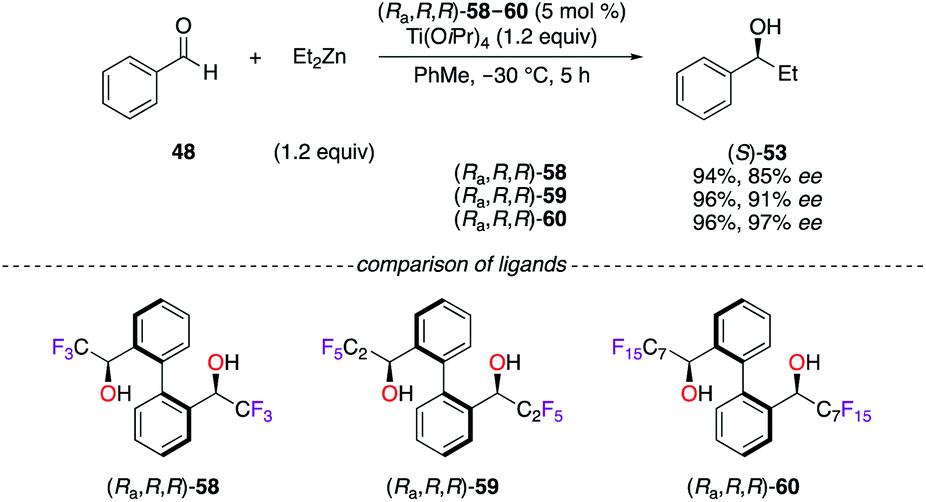
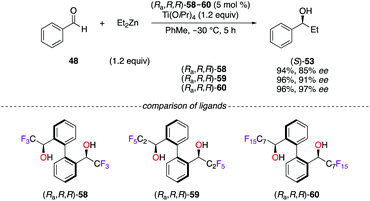
The success encountered with the axially chiral BINOL-type ligand fostered the development of designer diol ligands, i.e., (Ra,R,R)-58,32 (Ra,R,R)-59,32a–c and (Ra,R,R)-60,32a,b,33 bearing benzylic α-CnFn+1-alcohols, whose acidities are closely similar to the ones of phenolic compounds. The TiIV-catalyzed addition of Et2Zn to benzaldehyde 48 was tackled to evaluate the increase of the steric bulk arising from the various perfluoroalkyl chains (Scheme 7).32a The highest enantiomeric excess of (S)-53 was obtained using (Ra,R,R)-60, where the C7F15 chains were inducing maximum steric hindrance. Moreover, the (Ra,R,R)-60/TiIV catalytic system was profitably applied in fluorous catalysis, and the chiral ligand was recovered quantitatively over seven cycles using an optimized binary solvent system of FC-72/CH2Cl2 (2:1).33b
 |
| | Scheme 7 TiIV-Catalyzed ethylation reaction using α,α′-CnFn+1-diol ligands. | |
Overall, the increase in acidity of hydroxylic groups is a direct effect from the incorporation of fluorine atoms onto chiral ligands. Similarly, electronically deficient O-binding sites allow better interactions with the metal involved giving a more compact TS, resulting in an increased asymmetric induction and even in a complete reversal of it. Perfluoroalkyl chains have provided chiral ligands the ability to be recycled through various fluorous phase extraction strategies.
2.2
N,O-Based binding modes
2.2.1 Diols.
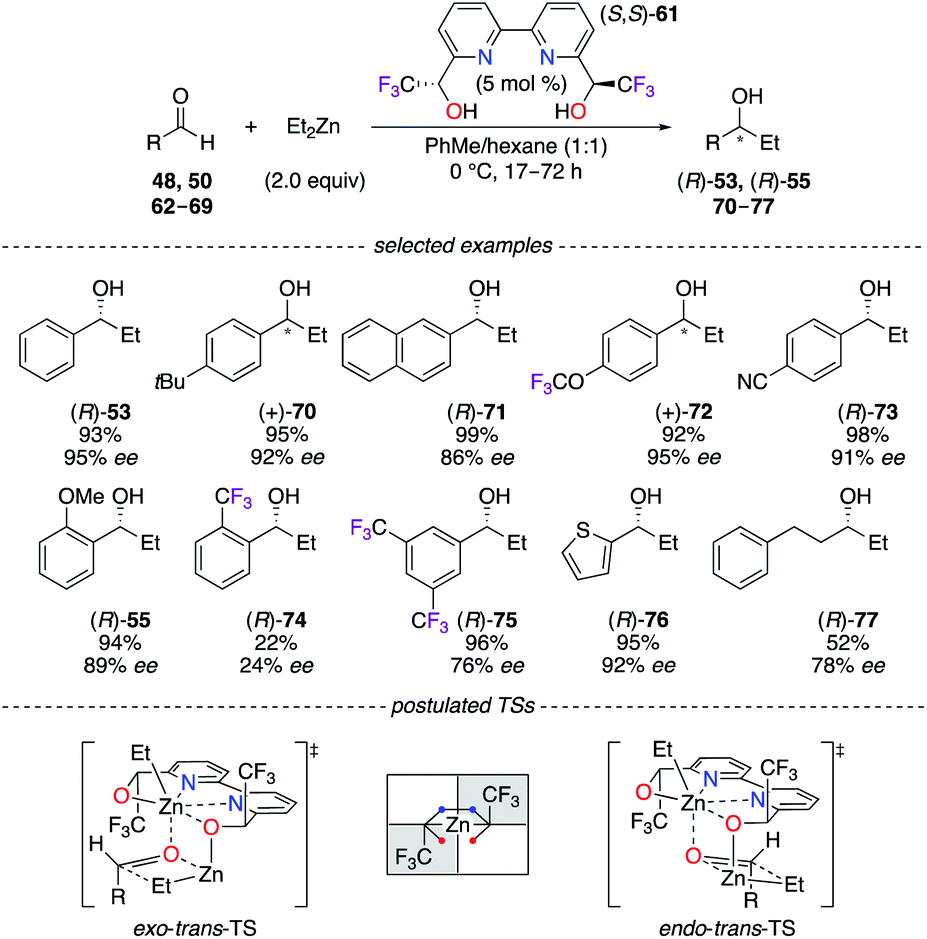
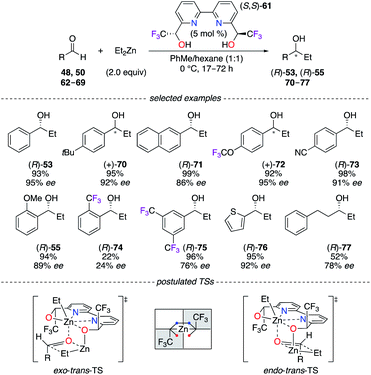
A sterically hindered catalytic site was built from 2,2′-bipyridinediol (S,S)-61 to take advantage of the stereoelectronic properties of CF3 groups at the α,α′-positions of the OH moieties. A selection of aromatic, heteroaromatic, and aliphatic alcohols (R)-53, (R)-55, and 70–77 were obtained in good to excellent yields (up to 99%) and enantioselectivities (up to 95% ee) using a ZnII-mediated reaction (Scheme 8).34 As observed from X-ray diffraction (XRD) analysis, the hexacoordinated (R,R)-61/Zn(OTf)2 complex led to the hypothesis of coherent transition state models of distinct configurations – either as exo–trans or endo–trans – giving the major and minor enantiomers, respectively. Interestingly, (S)-ibuprofen was employed for the resolution of the α-CF3-alcohols, and 2,2′-bipydiol61 was synthesized in both enantiomeric forms with excellent stereoselectivities (97% de and >99% ee for R,R; >99.5% de and >99.5% ee for S,S).
 |
| | Scheme 8 2,2′-Bipyridine-α,α′-CF3-diol/ZnII-mediated ethylation reaction of aldehydes. | |
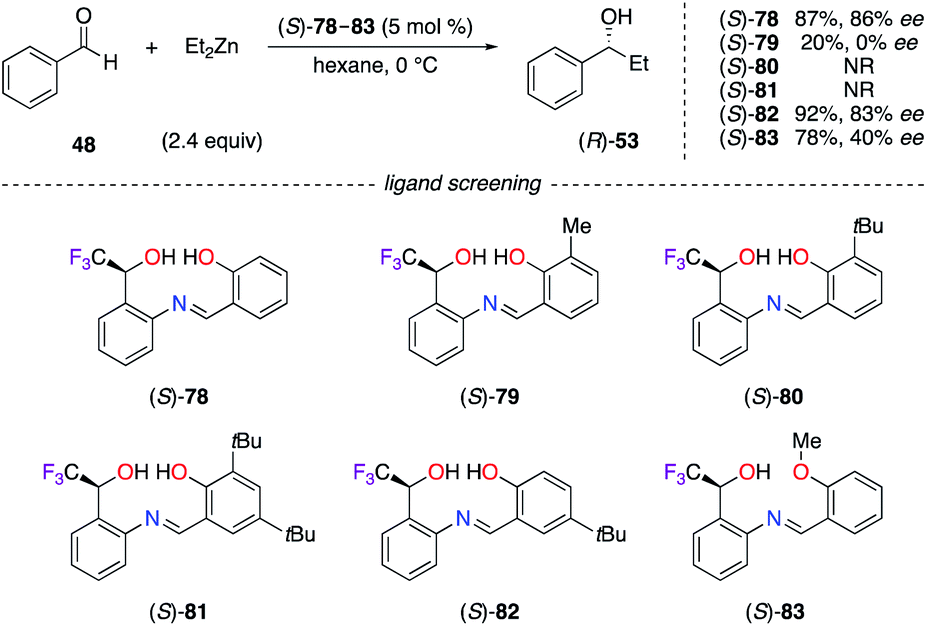
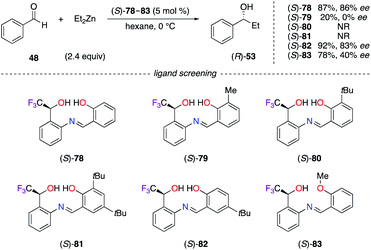
Chiral Schiff's base ligands (S)-78–83, containing an α-CF3-alcohol at a C-stereocentre, were used in the addition of diethylzinc to benzaldehyde 48 (Scheme 9).35 Ligands (S)-79–81, substituted at the ortho position with a Me or a tBu group, failed to increase the chiral induction. Only para-substituted (S)-82 afforded (R)-53 with similar ee to (S)-78. A non-linear relationship with a minimum enantiomeric amplification, together with high resolution mass spectrometry (HRMS) analysis, suggested that the dimeric [(S)-78]2/ZnII complex is the active catalyst. The C-stereocentre of Schiff's base ligands (S)-78–83 was constructed by the enantioselective reduction of the o-nitrophenyl-α-CF3-ketone using the (R)-CBS oxazaborolidine reagent. Surprisingly, the non-fluorinated analogues of these Schiff's base ligands have not been described in the literature so far.
 |
| | Scheme 9 ZnII-Mediated ethylation reaction of 48 using Schiff's bases bearing an α-CF3-alcohol. | |
2.2.2 β-Amino alcohols.
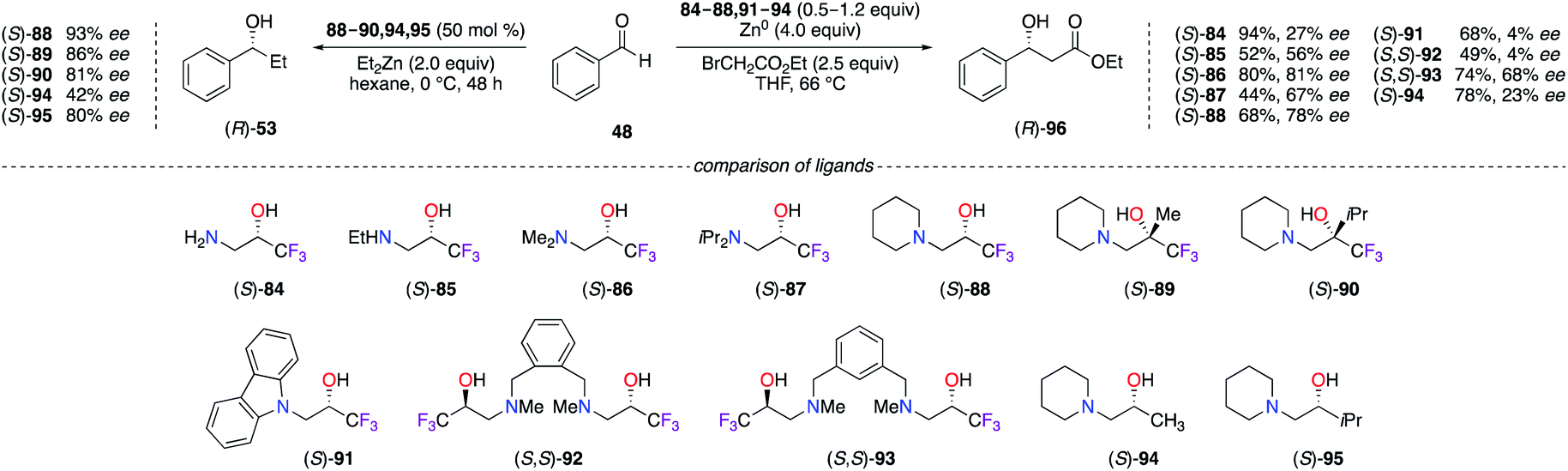
Fluorinated β-amino alcohol ligands 84–95 have been used in various asymmetric transformations involving the alkylation of aldehydes (Scheme 10). In the Et2Zn alkylation reaction of benzaldehyde,36 only the organozinc catalyst prepared from (S)-88 led to a maximum enantioselectivity, notwithstanding the presence of the Me or iPr substituents on the ligand at the α-position of the hydroxyl group giving (S)-89 and (S)-90 with bulkier quaternary carbon stereocentres (Scheme 10, left). Correlation studies on the catalyst loading (2–50 mol%) have demonstrated a strong dependence between the increased amount of (S)-88–90 and the ee observed on (R)-53, whereas no dependency was determined for the non-fluorinated analogues (S)-94 and (S)-95; the superior degree of aggregation of CF3-containing β-amino alcohol ligands, particularly for the (S)-88/ZnII catalyst, strongly participated in the mechanism to reach higher chiral induction of alcohol (R)-53.36a A wider library of β-amino α-CF3-alcohol ligands were screened in the Reformatsky reaction of PhCHO (Scheme 10, right).37 The ees obtained with ligands (S)-84 and (S)-85, containing a primary or a secondary amine, were much lower than the ones provided using tertiary amine-based ligands. Furthermore, (S)-86 possessing a N,N-dimethyl-amino group led to the highest 81% ee of (R)-96 in comparison with the other ligands having diisopropylamine ((S)-87), piperidine ((S)-88), and carbazole ((S)-91) motifs at the β-position. As shown on (S,S)-92 and (S,S)-93, the benzene ring bearing β-amino α-CF3-alcohols tethered at the 1,2 and 1,3 positions were considerably less efficient than (S)-86. The aggregation effect of such trifluoromethylated ligands with ZnII species was also found beneficial for the enantioselectivity ((S)-88vs. (S)-94).
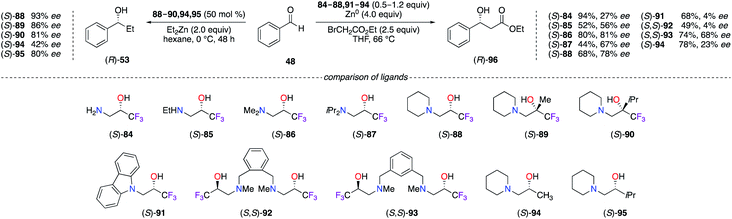 |
| | Scheme 10 Et2Zn alkylation (left) and Reformatsky (right) reactions of benzaldehyde using amino-alcohol derivatives. | |
ZnII-Catalyzed alkylation of aldehydes was also performed using substituted N-methyl-L-prolinol bearing α,α-aryl groups substituted by a F atom38 or a C8F17 chain39 at the para position. Through AgI catalysis, the 1,3-dipolar cycloaddition reaction was disclosed using 2,3-dihydroimidazo[1,2-a]pyridine-based DHIPOH ligands, substituted by a CF3 group at the C6 position of the quinoline backbone, but they only showed modest chiral inductions among the tested ligands.40
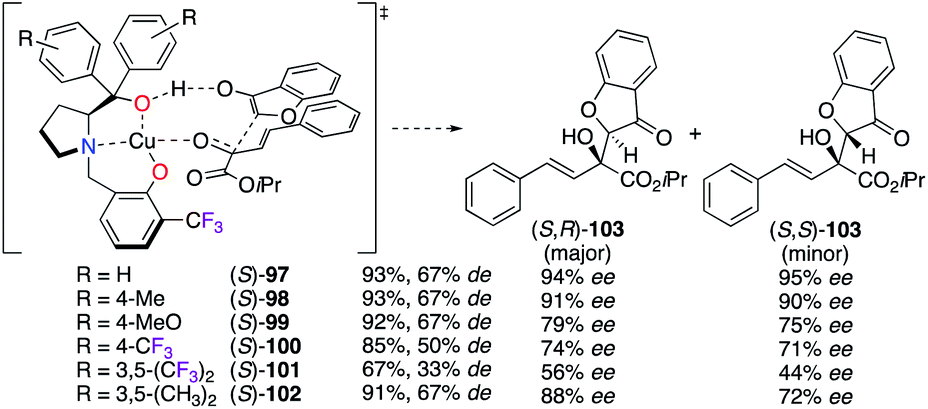
The CuII-catalyzed aldol reaction of β,γ-unsaturated α-ketoesters with coumarin-3-ones was investigated using various prolinol derivatives (S)-97–102(Scheme 11).41 Supported by density-functional theory (DFT) calculations, the nucleophilic attack of coumarin-3-one was hypothesized to occur from the Si-face of the α-ketoester in order to prepare predominantly the S,R diastereoisomer of 103 with good yields (67–93%) and low to excellent enantioselectivities (56–94% ee). According to the experimental results, the values of the observed ees were decreased when electron withdrawing groups were present on the aromatic rings.
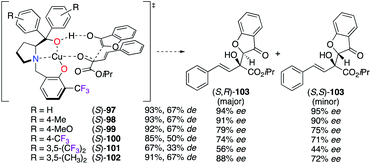 |
| | Scheme 11 Postulated TS for the CuII-catalyzed aldol reaction. | |
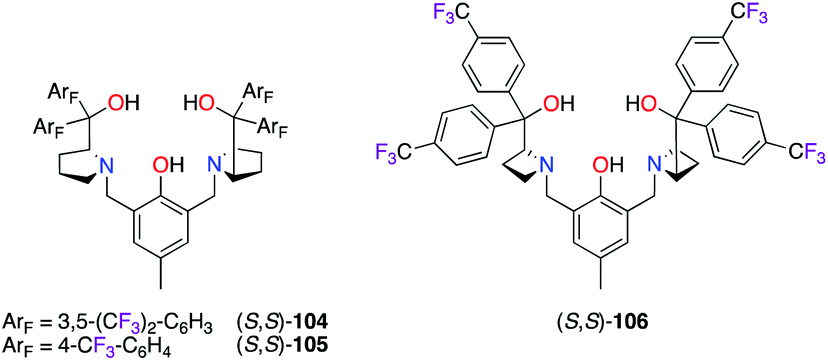
The Brønsted base/Lewis acid cooperative catalysis was highlighted by the formation of dinuclear ZnII catalysts in the presence of bis(prolinol)phenols (S,S)-104–106 (Fig. 5).42 Unfortunately, lower stereoselectivities were obtained using (S,S)-104–106vs. the non-fluorinated ligands in both the alkynylation43 and the domino Michael/Mannich [3 + 2] cycloaddition reactions.44
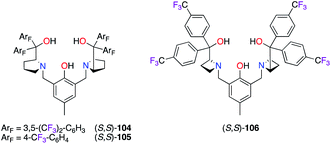 |
| | Fig. 5 Fluorinated bis(prolinol)phenol-type of ligands. | |
2.2.3 β-Amido chalcogens.
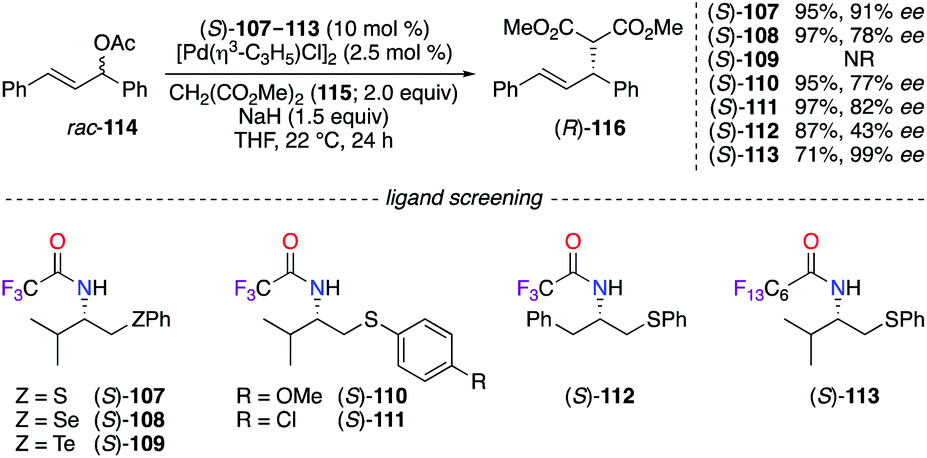
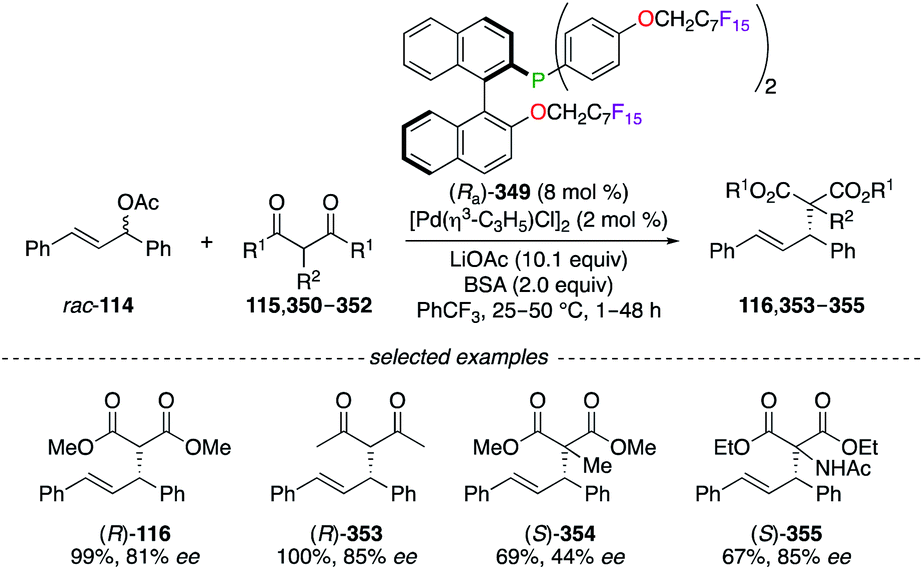
N-Polyfluoroacyl β-chalcogeno amide ligands (S)-107–113 were employed conjointly with a PdII salt in the asymmetric allylic alkylation reaction of rac-114 using dimethyl malonate 115 (Scheme 12).45 Ligand (S)-107 containing a sulfur atom provided (R)-116 with the best enantioselectivity among the chalcogen series (S > Se > Te). Additionally, modifications of the electronic nature of the thioether group ((S)-110, (S)-111) and the side chain attached to the stereocentre ((S)-112) failed to improve the level of chiral induction. Surprisingly, an excellent 99% ee was observed on (R)-116 when a perfluorinated amide group was introduced on the optimal skeleton of ligand (S)-113, which was recovered by a liquid–liquid extraction and was further employed in a subsequent allylic alkylation reaction with no loss of chiral induction.
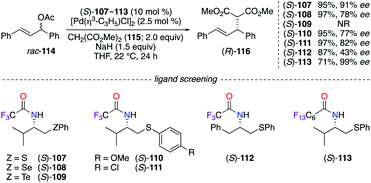 |
| | Scheme 12 β-Amido chalcogen/PdII-catalyzed asymmetric allylic alkylation reaction. | |
2.2.4 Salens.
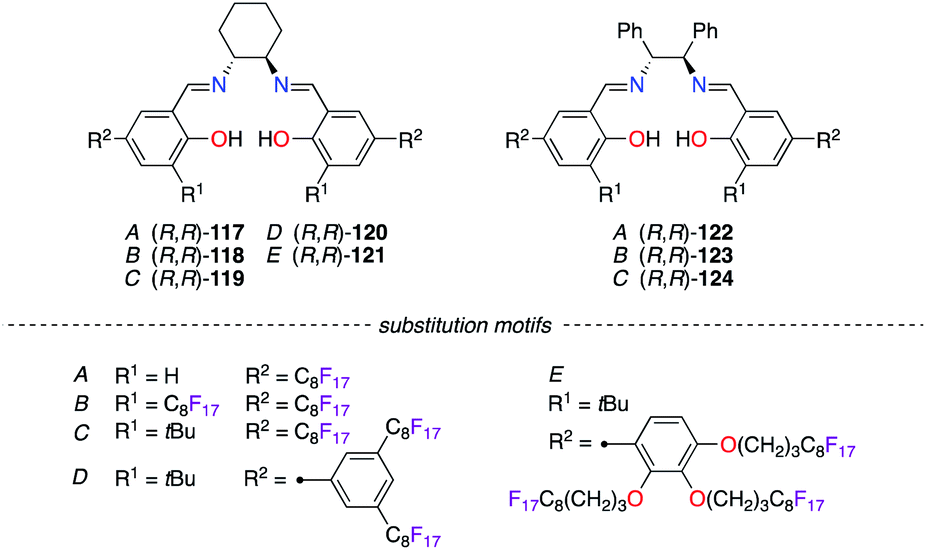
Condensation of numerous fluorous salicylaldehydes (A–E) with (R,R)-1,2-diaminocyclohexane (DACH) and (R,R)-1,2-diphenylethylenediamine (DPEN) generated salen ligands (R,R)-117–121 and (R,R)-122–124 (Fig. 6). Both types of ligands were mixed with MnIII,46 CoII,47 or IrII salts,48 and these salen/metal catalysts showed high levels of stereoinduction in various catalytic applications using FBS.
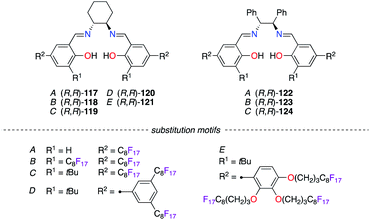 |
| | Fig. 6 Salen ligands bearing fluorinated motifs. | |
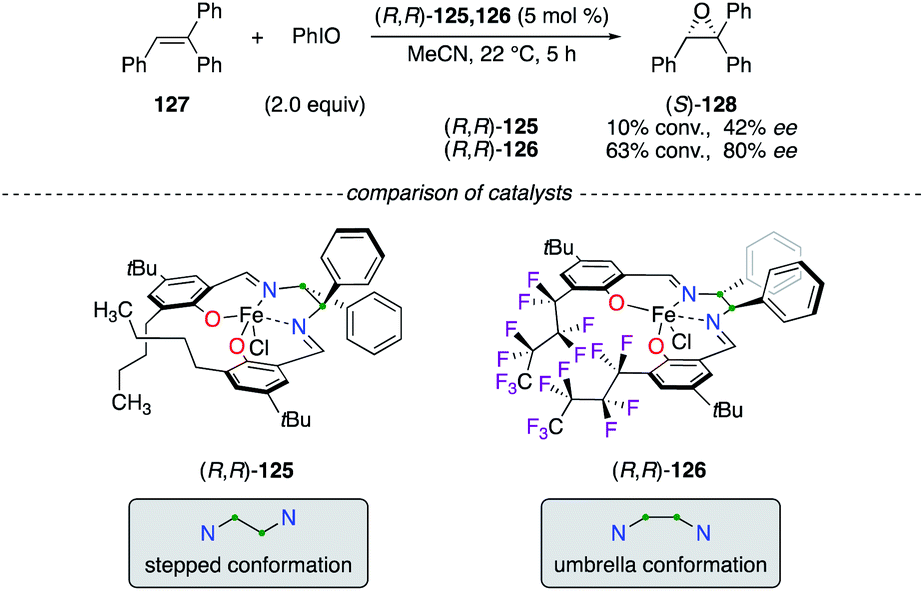
The efficiency of FeIII(salen) catalysts was compared in the asymmetric epoxidation reaction of 127, from which a higher level of chiral induction was mainly attributed to the fluorophilic effect (Scheme 13).49 According to crystallographic experiments, two distinct structures of the catalyst (R,R)-125 and (R,R)-126, bearing respectively C4H9 and C4F9 chains, were presented. Catalyst (R,R)-126, because of intramolecular stacking of the C4F9 chains arising in it, was described to adopt a unique umbrella conformation, which was more efficient to afford enantiomerically enriched (S)-128 than the usual C2-symmetrical stepped conformation of metal(salen) complexes.
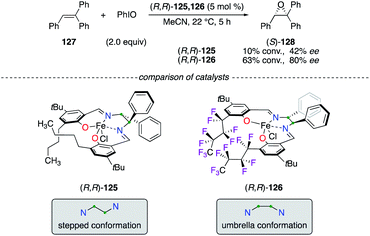 |
| | Scheme 13 PhIO-assisted epoxidation reaction using chiral FeIII complexes. | |
Structural insights into the C2-symmetric complexes prepared from the combination of fluorous diamino–dialkoxy ligands and TiIV and ZrIV cations were obtained from various spectroscopic and crystallographic analyses.50
2.2.5 Carboxamidates.
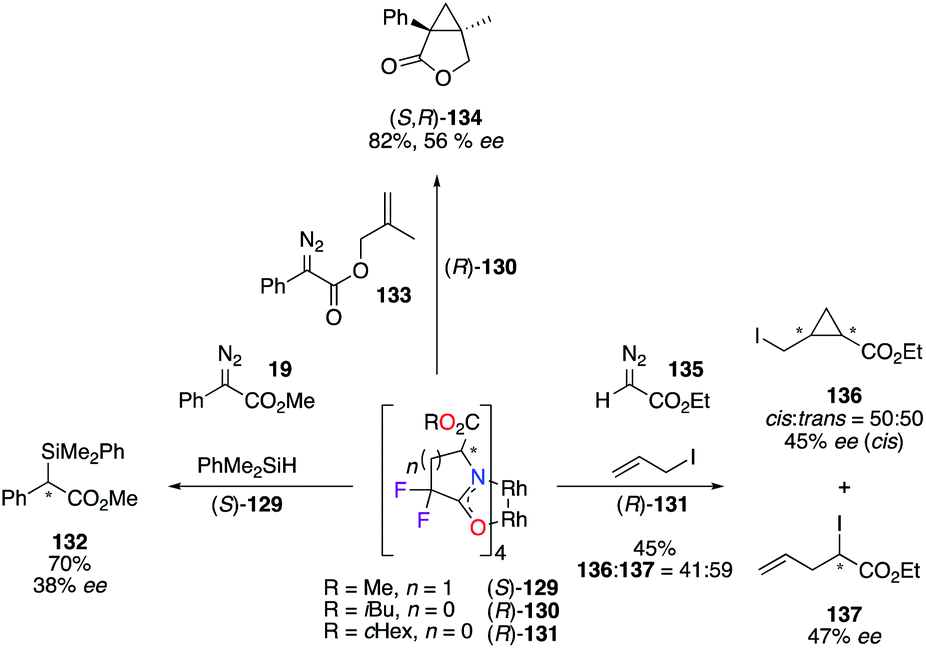
Tetrakis(carboxamidates) derivatives of chiral dirhodium catalysts 129–131, bearing α,α-fluorine atoms, were developed by Doyle to increase the catalytic activity, while maintaining high levels of asymmetric induction for selected transformations: (i) insertion reaction into the Si–H bond (left), (ii) intramolecular cyclopropanation (up), and (iii) ylide formation/[2,3]-sigmatropic rearrangement (right) reactions (Scheme 14).51 All tested catalysts, (S)-129, (R)-130, and (R)-131, showed increased reactivity towards diazo compounds 19, 133, and 135, although the stereoselectivities of products 132, (S,R)-134, 136, and 137 were lower than the ones obtained with their corresponding non-fluorinated analogues.
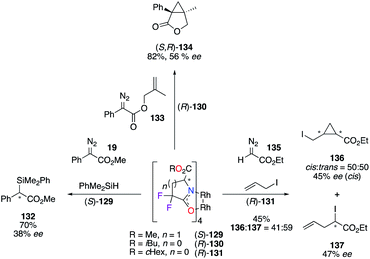 |
| | Scheme 14 Various applications of chiral RhII2 catalysts in diazo chemistry. | |
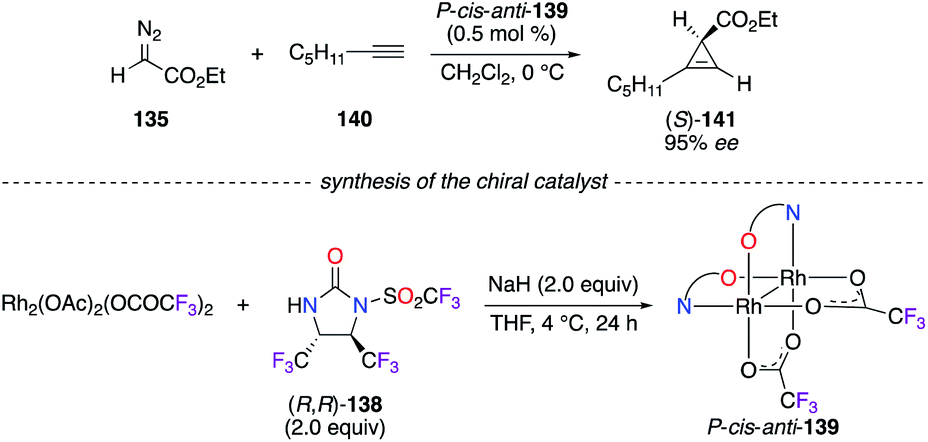
Chiral dirhodium catalyst P-cis–anti-139 was employed in the [2 + 1]-cycloaddition reaction of ethyl α-diazoacetate 135 and hept-1-yne 140 (Scheme 15).52 Other mono- and tris(amidate)-bridged RhII2 complexes were prepared from the N-triflylimidazolidinone precursor (R,R)-138, but all led to (S)-141 with lower enantioselectivities (<95% ee). The trans-vicinal CF3 groups on the imidazolidinone backbone brought up interesting features to (R,R)-138, i.e., high sterically hindered and electron deficient chelating N atoms.
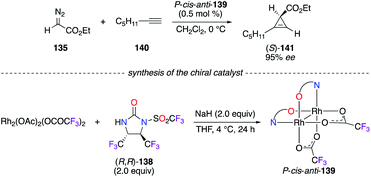 |
| | Scheme 15 [2 + 1]-Cycloaddition reaction catalyzed by the [(R,R)-138]2/RhII2 complex. | |
To sum up, ligands bearing a trifluoromethyl substituent for inducing chirality are scarce, but the potential of using the CF3 group to provide an important steric hindrance at the α position of alcohols and amines has been disclosed in various studies. Furthermore, the beneficial synergy of both the steric and the electronic properties for optimal enantioselectivity was demonstrated in the comparison study performed using CF3-containing ligand 88 and its Me- and iPr-analogs in the ethylation reaction of benzaldehyde. The fluorophilic effect is also responsible for significant conformational changes to classical salen complexes leading to a new conformation adopted by the F-tagged ligand–metal complex.
2.3
N-Based binding modes
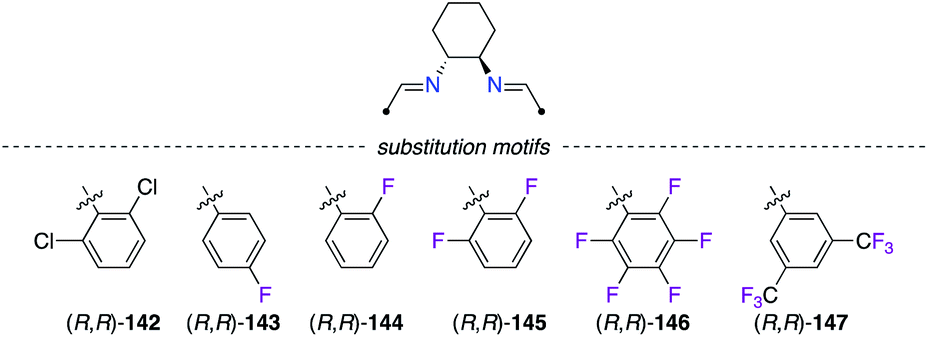
2.3.1 Diamines and diimines.
Chiral 1,2-diaminocyclohexane (DACH)-derived diimine ligands (R,R)-142–147 have been promoted in asymmetric catalysis in the pioneering work by Jacobsen in both the olefin aziridination and the carbene insertion into the Si–H bond (Fig. 7).53 In CuII catalysis, Evans described the Diels–Alder reaction of 3-acryloyl-2-oxazolidinone 148 with cyclopentadiene 14 as being highly enantioselective when (R,R)-142, bearing two 2,6-dichlorophenyl arms, was used (Scheme 16).54 Interestingly, fluorine substitution at the 2,6- and 2,3,4,5,6-positions of the aryl group led to lower enantioselectivities of (1R,2R,4R)-149, but higher endo/exo diastereoselectivities were observed (142 < 145 < 146).
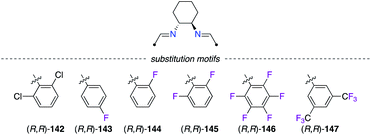 |
| | Fig. 7 Fluorinated motifs incorporated into DACH-type diimine ligands. | |
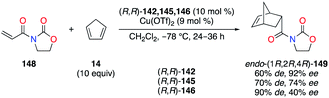 |
| | Scheme 16 CuII-Catalyzed Diels–Alder reaction – diimine ligand screening. | |
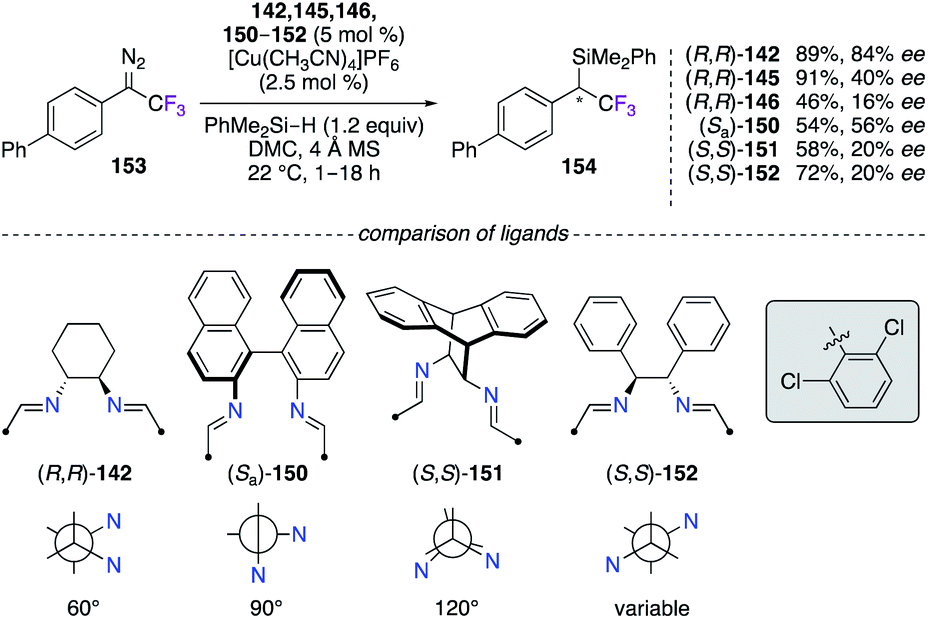
The insertion reaction of 1-(1,1′-biphenyl)-2,2,2-trifluoro-1-diazo-ethane 153 into PhMe2Si–H was performed using chiral diimine CuI complexes (Scheme 17).55 In comparison with the best 84% ee obtained with (R,R)-142, the fluorinated diimine analogues (R,R)-145 and (R,R)-146 led to unsatisfactory 40% ee and 16% ee, respectively. Being very interested towards a ligand design approach, 2,6-dichlorobenzaldehyde and other chiral diamine scaffolds, i.e., (Sa)-1,1′-binaphthyl-2,2′-diamine, (S,S)-11,12-diamino-9,10-dihydro-9,10-ethanoanthracene, and (S,S)-DPEN, were condensed together to generate new diimine-type ligands with distinct bite angles.56 However, the 150–152/CuI catalysts failed to give the insertion product with higher enantioselectivities than (R,R)-142. As shown in this study, any mix-and-match combinations involving a chiral diamine precursor and a desired aldehyde could lead to potential candidates being used for chiral induction.
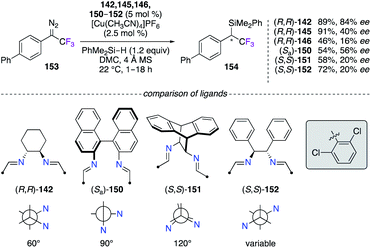 |
| | Scheme 17 CuI-Catalyzed carbene insertion reaction into the Si–H bond – chiral diimine possessing various bite angles. | |
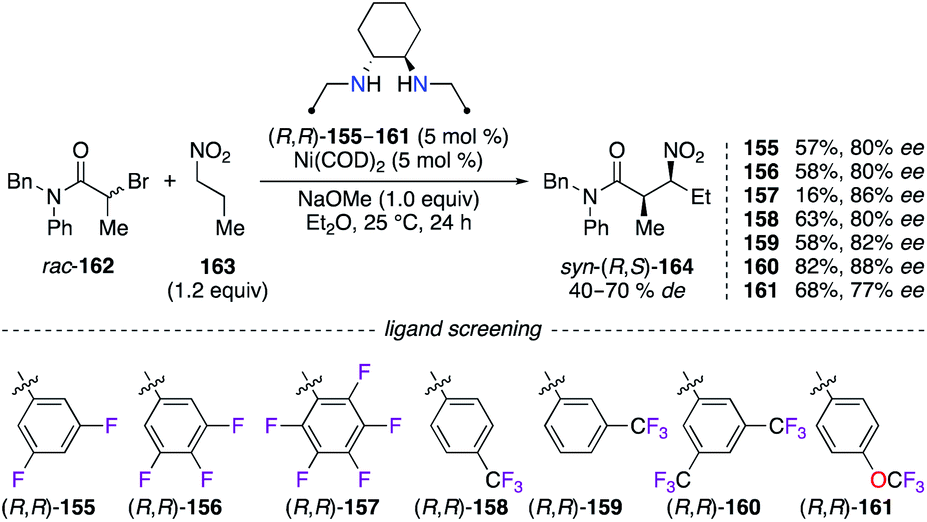
Chiral diamine ligands are prepared from the simple reductive amination of diimine precursors, e.g., using sodium borohydride.57 Numerous aldehydes containing different F-patterns were first treated with (R,R)-DACH and then reduced with NaBH4 or NaBH(OAc)3, to give chiral 1,2-diamine ligands (R,R)-155–161. The ability for chiral induction of these electronically deficient ligands was studied in the Ni0-catalyzed C-alkylation reaction of 1-nitropropane 163 by α-bromoamide rac-162 (Scheme 18).58 Independent of the nature of the substituent (F, CF3, or OCF3), fluorine substitution at the 3-, 4-, 2,6-, and 3,4,5-positions of the aromatic ring afforded syn-(R,S)-164 with up to 82% ee, whereas a good 86% ee, together with a low 16% yield, was observed when using the C6F5 analogue. Optimal results were obtained with the (R,R)-160/Ni0 system, and this catalyst was proven highly efficient to prepare 26 β-nitroamides with excellent stereoselectivities (up to 90% desyn and 99% eesyn).
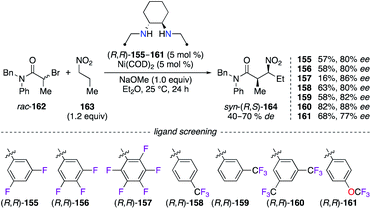 |
| | Scheme 18 Ni0-Catalyzed C-alkylation reaction – optimization of the chiral diamine ligand. | |
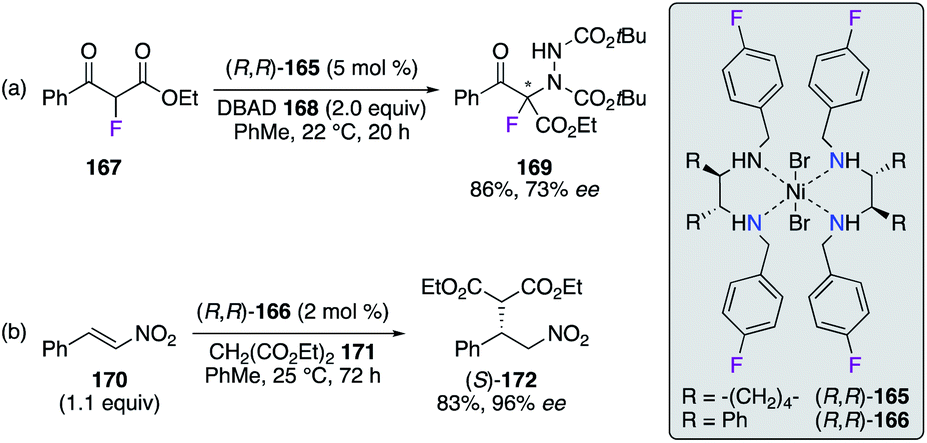
Chiral DACH- and DPEN-based NiII complexes (R,R)-165 and (R,R)-166 with N,N′-4-fluorobenzylamine arms were employed as moisture- and air-stable pre-catalysts in enantioselective transformations (Scheme 19). The α-hydrazination reaction of α-fluoro-β-ketoester 167 with di-tert-butyl azodicarboxylate (DBAD) 168 was successfully performed using (R,R)-165, where α-amino-β-ketoester 169 was obtained in 86% yield and 73% ee (Scheme 19, eqn (a)).59 Similarly, the (R,R)-166-catalyzed conjugate addition reaction of diethyl malonate 171 to β-nitrostyrene 170 was reported to give an excellent enantioselectivity on Michael adduct (S)-172 (Scheme 19, eqn (b)).60 When using 1-nitropent-1-ene as the substrate, (R,R)-166 afforded promising 89% ee of (R)-diethyl 2-(1-nitropenthan-2-yl)malonate, which is a key intermediate in the synthesis of brivaracetam having antiepileptic properties.
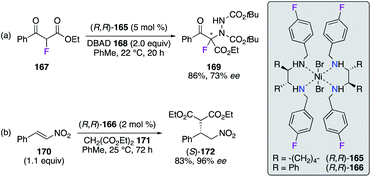 |
| | Scheme 19 NiII complexes employed in addition-type reactions. | |
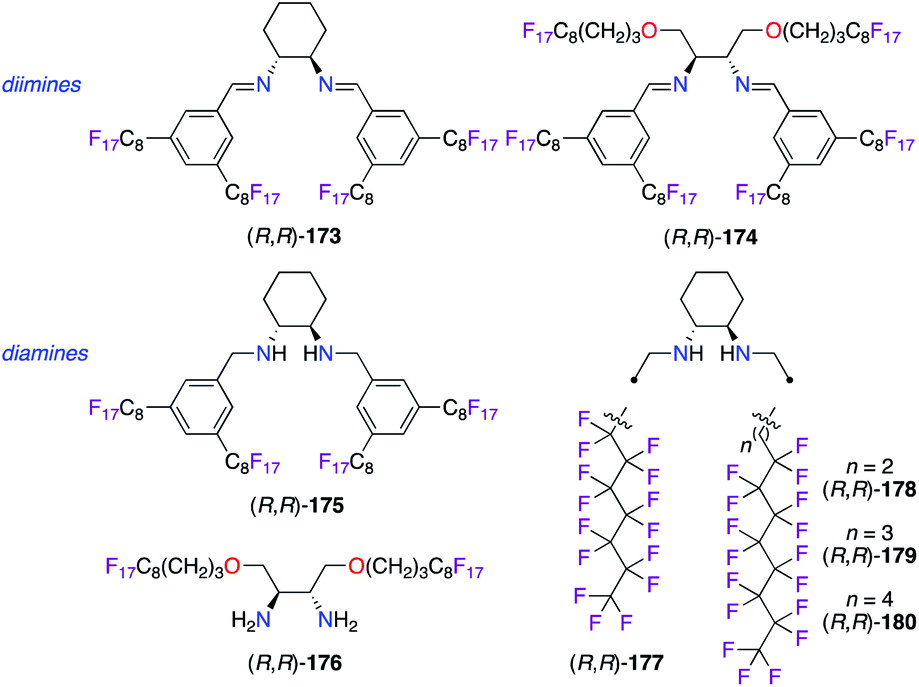
Polyfluorinated diamine and diimine types of ligands were obtained by the incorporation of perfluoalkyl chains to be used in various synthetic applications involving fluorous biphasic systems (Fig. 8). Chiral perfluorinated DACH-type diamine ligand (R,R)-175 was more tolerant towards decomposition vs. diimine (R,R)-173 over the recycling of both IrI and CuI catalysts.48,61 In terms of chiral induction, the (R,R)-175/CuI catalyst afforded the trans-(1R,2R)-cyclopropane, obtained from styrene 20 and ethyl α-diazoacetate 135, in 62% ee, whereas a low 6% ee of the opposite 1S,2S enantiomer was observed when using the diimine analogue. Furthermore, non-cyclic C2-symmetric tertiary diamines also led to poor levels of enantioselectivity (ca. 15% ee) probably due to a too flexible environment around the CuI centre. Starting from (4R,5R)-2,2-dimethyl-1,3-dioxolane-4,5-dimethanol, the incorporation of C8F17 ponytails tethered to the stereogenic carbons of the 1,2-diamine core allowed the preparation of unique fluorous diimine and diamine ligands. Unfortunately, poor levels of stereoinduction were obtained in the ATH reaction of acetophenone in a fluorous solvent using either IrI or RuII catalysts made from (R,R)-174 or (R,R)-176.62 Alternatively, the introduction of perfluoro-heptyl and -octyl chains directly on both N atoms led to aliphatic diaminocyclohexane (R,R)-177–180, which were found promising only in the CuI-catalyzed cyclopropanation of styrene 20 among the tested reactions.63
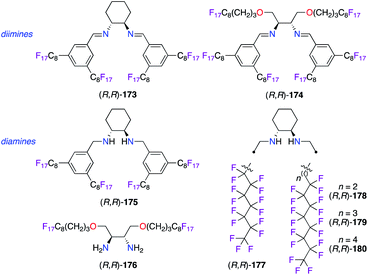 |
| | Fig. 8 Perfluorinated motifs in chiral diamine- and diimine-based ligands. | |
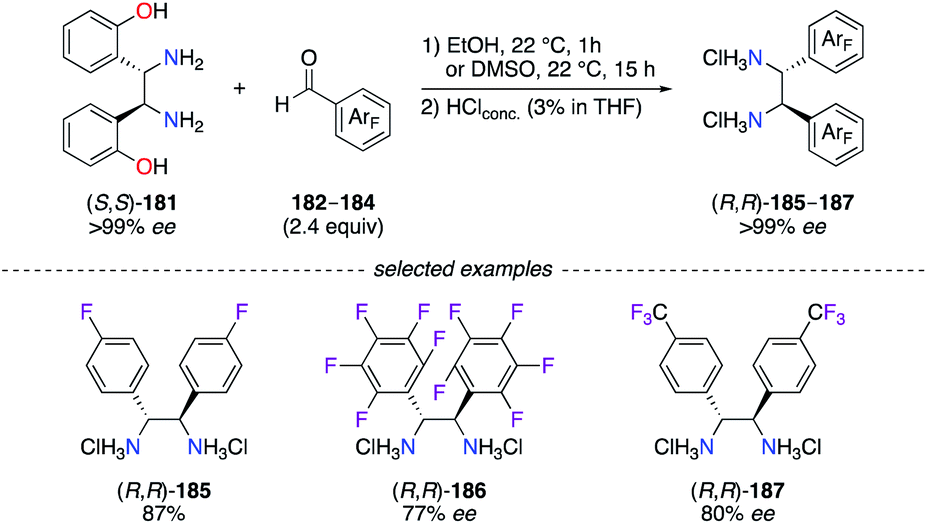
Overall, the 1,2-diphenylethylenediamine scaffold is scarcely described in the literature for the synthesis of fluorinated diimine and diamine ligands. To overcome this situation, an efficient method was described for the synthesis of DPEN-type ligands with diaryl-substituted backbones via a diaza-Cope rearrangement of (S,S)-1,2-bis-(2-hydroxyphenyl)-ethylenediamine 181 (or (R,R)-181) in the presence of the desired aldehyde.64 Following the described protocol, fluorinated DPEN (R,R)-185–187 were synthesized in high yields (77–87%) via the condensation of (S,S)-181 with aldehyde 182–184, i.e., 4-F-C6H4-CHO, C6F5-CHO, and 4-CF3-C6H4-CHO (Scheme 20). The diaza-Cope rearrangement was postulated to proceed through a six-membered chair transition state giving the desired diimines with chiral inversion of the C-stereocentres. This method is highly useful for the preparation of F-containing DPENs, which could be used as enantiopure building blocks – either as the R,R, or the S,S – to give diimine- and diamine-based designer ligands.
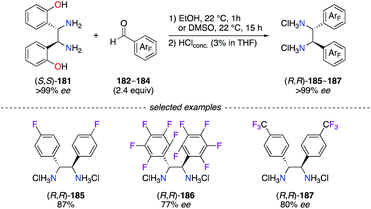 |
| | Scheme 20 Stereospecific preparation of C2-symmetric fluorinated DPEN ligands. | |
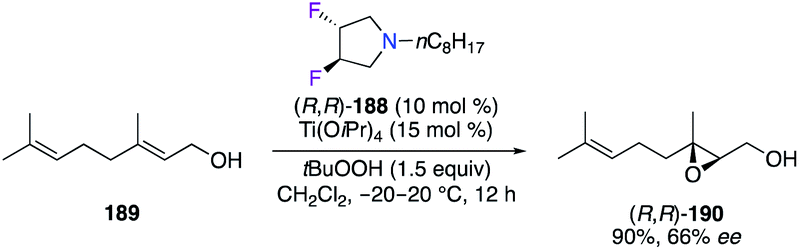
Trans-(R,R)-3,4-difluoropyrrolidine (R,R)-188, used as monodentate amine, was combined with Ti(OiPr)4 to catalyze the epoxidation of geraniol 189, giving the corresponding epoxide (R,R)-190 with a moderate 66% ee (Scheme 21).65
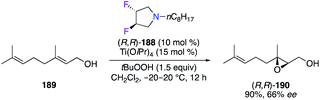 |
| | Scheme 21 TiIV-Catalyzed epoxidation reaction of geraniol. | |
2.3.2 Sulfonamides.
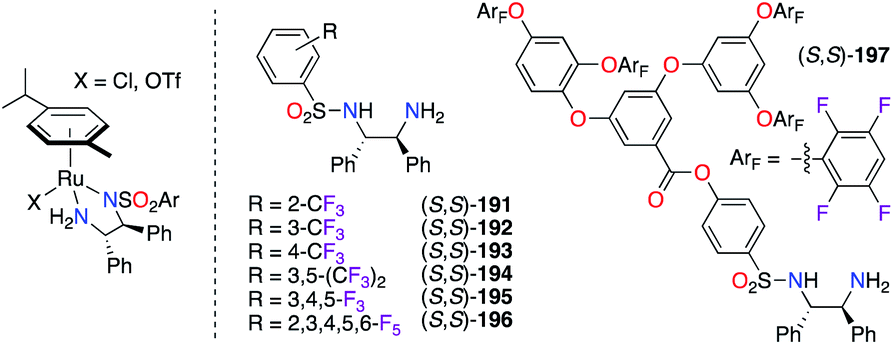
The sulfonylation of simple DPEN ligands with various aromatic sulfonyl precursors where fluorine substitution appears on all carbons of the aryl paved the way to a wide library of N-(tosyl)-1,2-phenylethylenediamine (TsDPEN) ligands (S,S)-191–197 (Fig. 9). Their complexation with RuII salts was proven valuable for both the tandem hydration/asymmetric transfer hydrogenation (ATH) of alkynes66 and the asymmetric hydrogenation (AH) of bisquinoline,67 where (S,S)-191–196 and (R,R)-193 were respectively used. Fluorous dendritic (S,S)-197 showed an enhanced activity and recyclability of the RuII catalyst towards the ATH reaction of aromatic ketones into secondary alcohols, which were obtained with excellent ees.68 During the optimization of the reaction conditions, (R,R)-193 and (R,R)-194 were screened, but the latter was chosen as the optimal chiral ligand in the IrIII-catalyzed ATH reaction of 2,9-dimethyl-1,10-phenanthroline.69
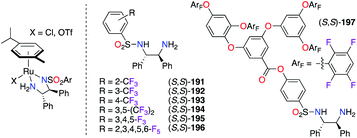 |
| | Fig. 9 F-containing tosylated-(S,S)-DPEN ligands in RuII complexes. | |
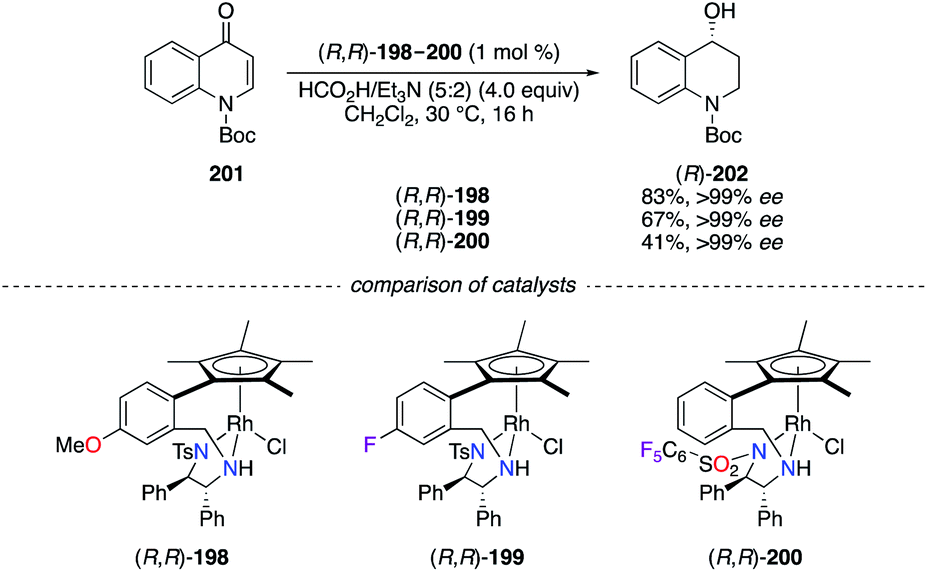
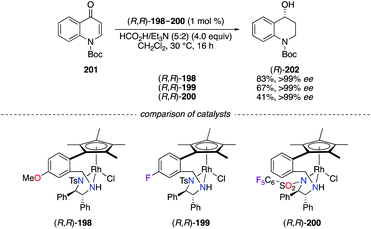
The ATH of N-protected 4-quinolone 201 using the formic acid/triethylamine mixture as the hydrogen source was performed using chiral RhIII complexes (R,R)-198–200 (Scheme 22).70 All three catalysts afforded excellent chiral inductions, but the partial cleavage of the tert-butyloxycarbonyl (Boc) group occurred when using a stronger Lewis acidic catalyst; hence lower yields of (R)-202 were obtained using (R,R)-199 and (R,R)-200.
 |
| | Scheme 22 RhIII-Catalyzed ATH reaction of 201 into (R)-202. | |
A 3,5-ditrifluoromethylated phenyl ring was incorporated into a Cinchona-alkaloid-based sulfonamide ligand, which was employed as the chiral source in the CuII-catalyzed radical oxytrifluoromethylation of alkenyl oximes.71 The Simmons-Smith cyclopropanation of allylic alcohols was performed using in situ generated ZnII complexes from fluorous disulfonamide ligands, where both ligands were easily recovered by fluorous solid phase extraction.72
2.3.3 Imidazolines.
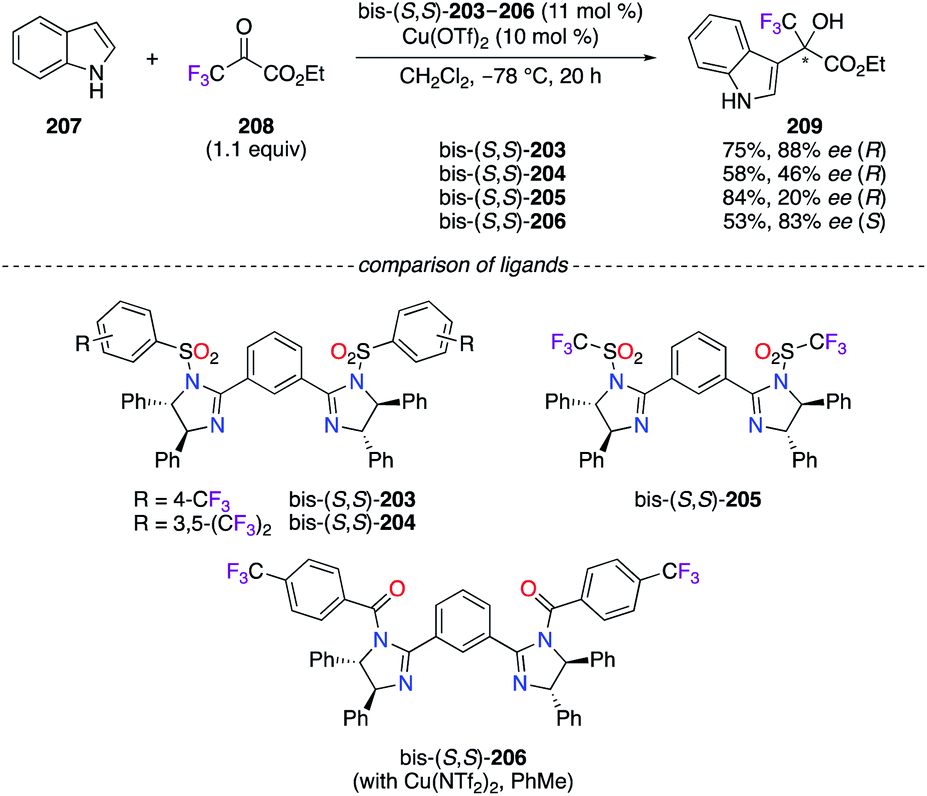
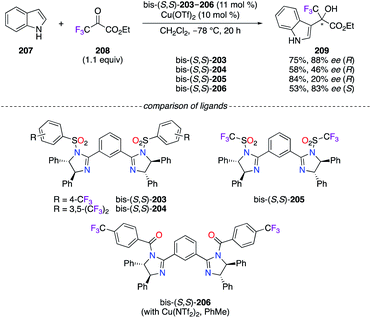
The (S,S)-DPEN scaffold was further employed as the chiral synthon for the synthesis of imidazoline-based ligands in the highly enantioselective Friedel–Crafts alkylation reaction of indoles with ethyl 3,3,3-trifluoropyruvate 208 (Scheme 23).73 Among them, fluorous phenylene bis(imidazoline) (PheBIM) bis-(S,S)-203–206 – particularly ligands bis-(S,S)-203 and bis-(S,S)-206 possessing a CF3 group at the para position of the phenyl ring – afforded 209 with good enantioselectivities when indole 207 was used as the nucleophile.
 |
| | Scheme 23 Friedel–Crafts alkylation reaction using chiral PHEBIM ligands. | |
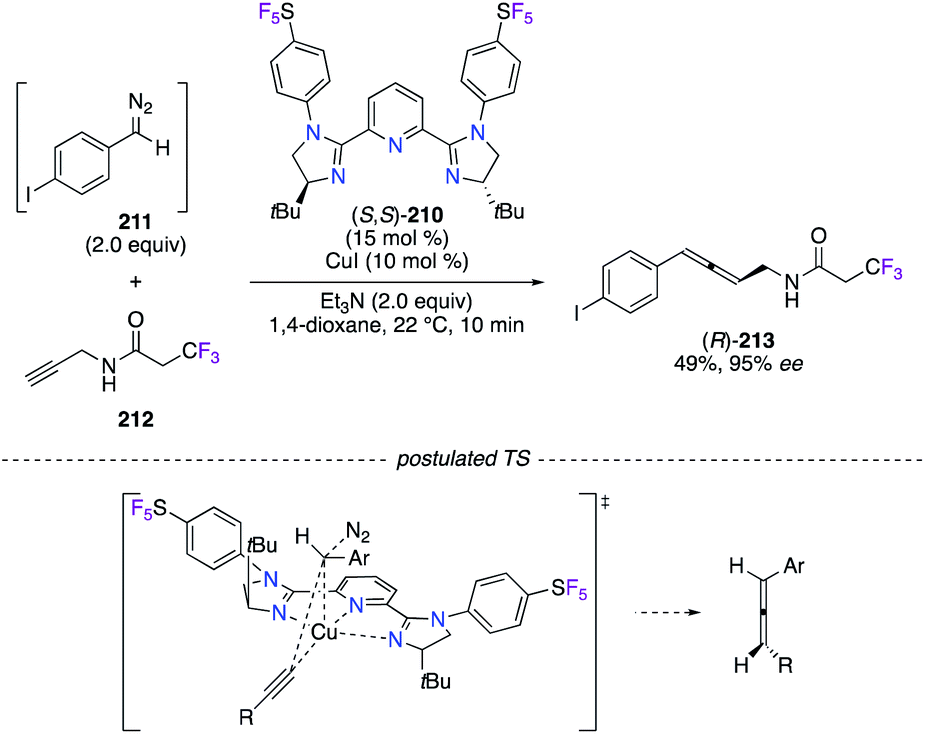
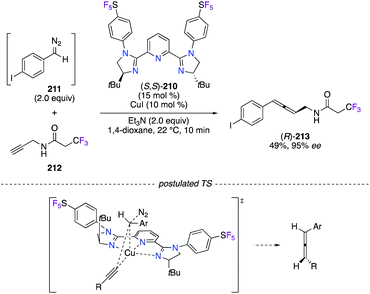
The allenylation of terminal alkynes and diazo compounds, generated in situ by MnO2-assisted oxidation of their corresponding hydrazone in a continuous flow system, was disclosed as being highly enantioselective (89–97% ee) under CuI catalysis (Scheme 24).74 A library of pyridine bis(imidazoline) (PyBIM) ligands bearing N-aryl substituents, i.e., 4-CF3-C6H4, 3,5-(CF3)2-C6H3, and 4-CF3-3,5-F2-C6H2, was screened, but only (S,S)-210, comprising 4-SF5-C6H4 groups, showed an optimal chiral induction. Noteworthily, these chiral ligands were synthesized from the treatment of a pyridine-2,6-diimidoyl chloride precursor, derived from (S)-tert-leucinol, with the corresponding fluorinated aniline, such as 4-(pentafluorothio)aniline in the case of (S,S)-210. As postulated, enantioenriched allene (R)-213 was synthesized via the concerted Cu–C bond insertion of the (S,S)-210/CuI acetylide intermediate into the diazo compound 211, which approaches with the H near the tBu group of the ligand to induce minimal steric strains. The scope was further extended to propargylamides derived from (S)-ibuprofen, penicillin G, (R,R)-atorvastatin, and others, and excellent stereoselectivities were highlighted in the disclosed method.
 |
| | Scheme 24 CuI-Catalyzed allenylation reaction and the postulated TS. | |
2.3.4 Oxazolines.
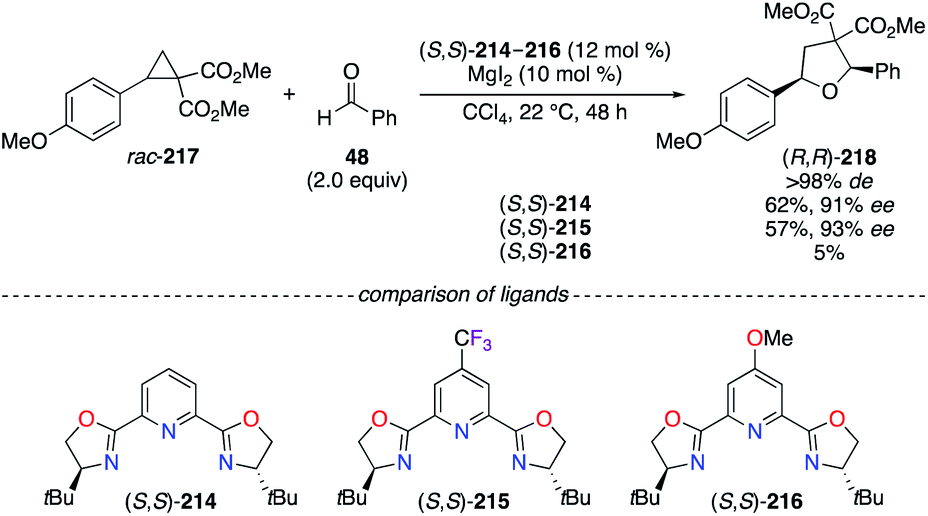
Tetrahydrofuran derivatives were synthesized with excellent stereoselectivities via the [3 + 2] cycloaddition of racemic cyclopropanes with various aldehydes using a tBu–pyridine bis(oxazoline) (PyBOX) ligand.75 The selection of electronically modified para-substituted tBu-PyBOX ligands (S,S)-214–216 has induced significant alteration of the yields rather than the ees, even though (S,S)-215 afforded 2,5-diaryl-THF (R,R)-218 with the best enantiocontrol (Scheme 25). However, the p-Cl-tBu-PyBOX showed a slightly better efficiency (74% yield, >98% de, 92% ee) and, therefore, was chosen to evaluate the scope of the reaction. An opposite trend was observed in the CuI-catalyzed allenylation reaction presented above, where the optimization studies performed on model substrates, using p-R-iPr-PyBOX ligands instead of tBu-PyBOX, led to the corresponding allene with 47% ee (R = CF3), 68% ee (R = H), and 70% ee (R = OMe).74
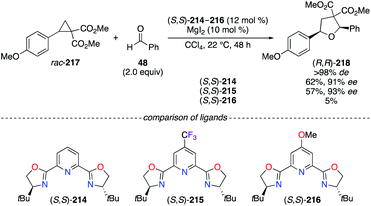 |
| | Scheme 25 MgII-Catalyzed dynamic kinetic asymmetric [3 + 2] cycloaddition reaction. | |
Chiral pyridine(oxazoline) (PyOX) ligands bearing a CF3-substituted C5 position were tested, but virtually no conversion was noted in the PdII-catalyzed dihydroxylation reaction of catechol and trans-1-phenyl-1,3-butadiene.76 Other examples of mono-oxazoline tethered CF3-based ligands were presented as amido-oxazolinate/ZnII and sulfoxide-oxazoline/PdII complexes.77
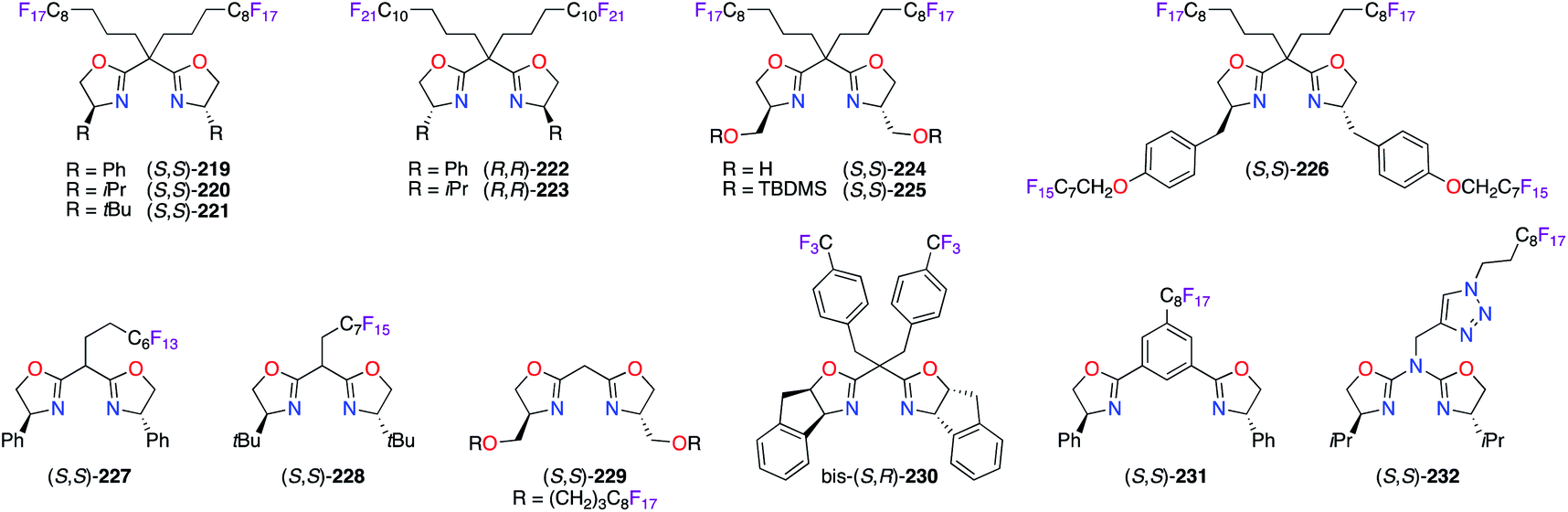
Similarly, fluorous bis(oxazoline) (BOX) ligands were synthesized and used with various metals to attain solubilization of the obtained chiral catalysts in fluorous solvents (Fig. 10). Starting from simple BOX derivatives, the incorporation of perfluoroalkyl substituents, comprising C8F17 and C10F21 chains, on the methylene bridge afforded (S,S)-219–221 and (R,R)-222 and (R,R)-223. High ees were obtained using these chiral ligands in the PdII-catalyzed allylic alkylation,78 CuI-catalyzed allylic oxidation78b and cyclopropanation,79 and CuII-catalyzed ene80 reactions. Used in these reactions, (S,S)-224 and (S,S)-225 both showed a favoured complexation with [Pd(η3-C3H5)Cl]2 to reach high chiral inductions, whereas low reactivity was obtained with Cu(OTf)·0.5C6H6.78b To facilitate the recycling of fluorinated BOX ligands, up to four perfluorooctyl chains were introduced on (S,S)-226, which was synthesized from (S)-tyrosine, to increase its fluorine content up to 59.3%.81 Mono-perfluoroalkyl bridged BOX ligands (S,S)-22782 and (S,S)-22878b were essentially designed to be used in common organic solvents for optimum catalytic activity, but to be recovered via a fluorous solid-phase extraction. Moving the perfluoroalkyl chains closer to the centre of chirality had no tremendous influence on the obtained ees using catalysts prepared from (S,S)-229 together with PdII or CuI salts.81 As part of the ligand screening, the indane-based bis-(S,R)-230/CuII catalyst afforded tert-butyl α-fluoroester with a modest 61% ee via the fluorination of diazoester 19.83 The synthetic utility of (S,S)-231 was demonstrated in the synthesis of enantioenriched compounds bearing 3-hydroxy-2-oxindole and quinazolinone scaffolds using CuII or ScIII catalysis.82c,d Importantly, the great ability of the triazole ring for coordination with copper salts was offset by the pefluoroalkyl group effect on azabis(oxazoline) ligand (S,S)-232 or its F51N9-tripodal analogue. As a result, high enantioenrichments were afforded in the benzoylation, Friedel–Crafts alkylation, and Henry reactions.84
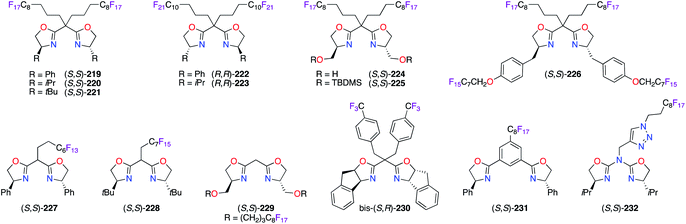 |
| | Fig. 10 Fluorous chiral bis(oxazoline)-derived ligands. | |
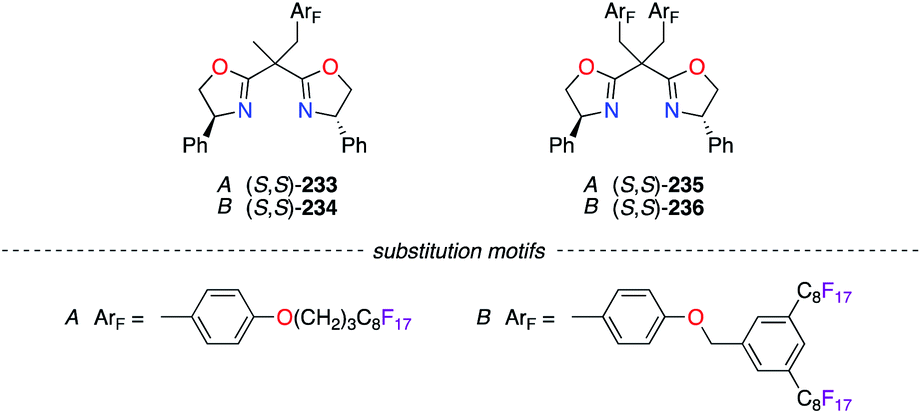
The strategy of preventing the immobilization of BOX chiral ligands onto poly(ethyleneglycol) (PEG) materials was further explored using (S,S)-233–236 (Fig. 11). Two substitution motifs comprising perfluoroalkyl chains, A85 or B,86 were included in the structure of the BOX ligands. The fluorinated moieties, which were separated from the coordination sites by an appropriate spacer to reduce any undesired interactions, allowed excellent recovery and recycling of the ligands via practical procedures.
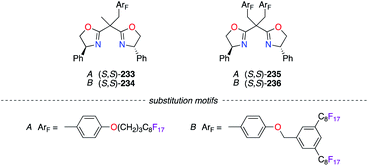 |
| | Fig. 11 Perfluoroalkyl motifs for chiral BOX ligands. | |
All things considered, the availability of many fluorinated aldehydes gives the access to a larger diversity of C2-symmetric chiral diamine and diimine ligands. The variety of functionalization patterns brought by introducing fluorine at every carbon of the aromatic ring has facilitated the fine-tuning of the Lewis acidity of a catalyst through electronically modified properties. Chiral ligands bearing the SF5 group have been limited due to the scarcity of the availability of the building blocks, and therefore, the pyridine bis(imidazoline) ligand is considered as a major breakthrough. Importantly, not only the recycling of chiral fluorous catalyts remains a key objective using various strategies, but the incorporation of diverse fluorous chains also led to ligand design in fluorous biphasic systems.
2.4
N,P-Based binding modes
2.4.1 Monophosphines.
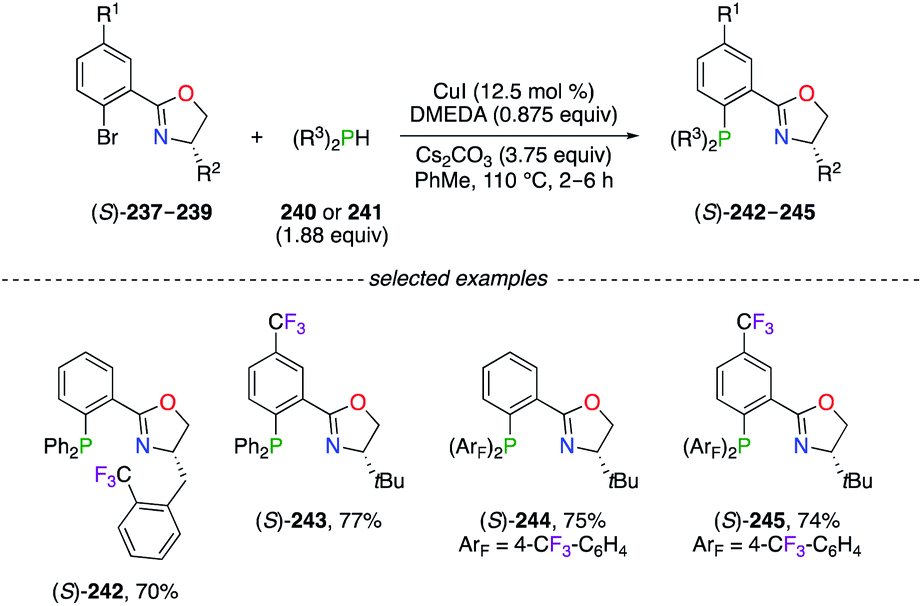
A wide variety of F-containing phosphino(oxazoline) (PHOX) chiral ligands were synthesized via the Ullmann-like coupling reaction of 2-bromo aryl derivatives (S)-237–239 with the corresponding diarylphosphine (Scheme 26).87 The simple reaction conditions, involving copper iodide and 1,2-dimethylethylenediamine (DMEDA), afforded (S)-242–245 with good yields, notwithstanding the steric and the electronic properties of the substrate. Noteworthily, a considerably increased catalytic activity of the PdII catalysts was observed using (S)-243 and (S)-245, which was not observed with the CF3-substitution pattern only on the phenyl rings of (S)-244. The great utility of (S)-245 was highlighted in the PdII-catalyzed intramolecular decarboxylative allylic alkylation reaction, where highly enantioenriched cyclohexanone derivatives bearing all-carbon quaternary stereocentres at the α-position were obtained with excellent yields.87,88 Furthermore, the (S)-245/PdII-catalyzed asymmetric alkylation reaction was described as an important key step in the synthetic route of (+)-elatol, a spirobicyclic natural product belonging to the family of chamigrene.88c Favourable complexation with palladium salts was achieved using strong π-acceptor bis(perfluoroalkyl)phosphino-oxazoline (FOX) ligands for the alkylation of monosubstituted allyl esters.89
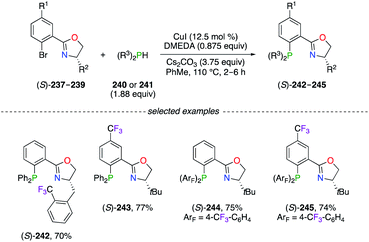 |
| | Scheme 26 Ullman-like coupling reaction for the synthesis of PHOX ligands. | |
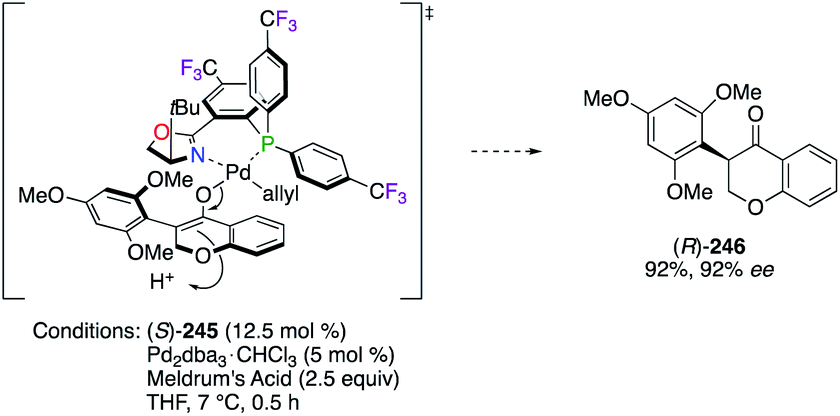
The synthesis of naturally occurring and non-natural isoflavones, members of the flavonoid class, was also targeted using (S)-245 through a Pd0-catalyzed decarboxylative protonation reaction.90 According to the postulated TS, both tBu and MeO groups pointing upwards – from the ligand and the substrate, respectively – induce a high sterically hindered environment and lead the protonation to occur preferentially via the Si-face (Scheme 27).90a As a result, isoflavone (R)-246 was obtained with excellent yield and enantioselectivity.
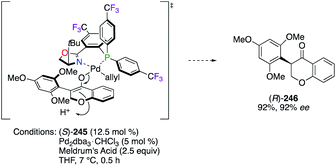 |
| | Scheme 27 Postulated TS for the synthesis of isoflavone (R)-246. | |
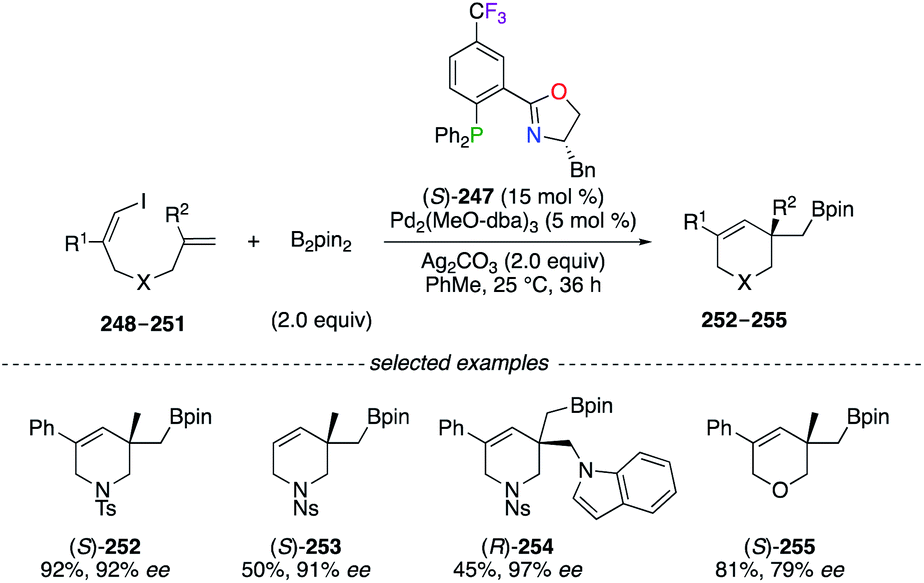
A highly enantioselective Pd0-catalyzed vinylborylation of (Z)-1-iodine-dienes 248–251 with B2pin2 was disclosed using (S)-247 for the construction of quaternary C-stereocentres on 252–255 (Scheme 28).91 Interestingly, ligand screenings of non-halogenated analogues of (S)-247 led to only moderate enantioselectivities (52–80% ee). Fluorinated PHOX ligands were employed in structure–reactivity correlation studies using chiral (π-allyl)palladium complexes and suggested that the electronic tuning of the ligand for maximizing both reactivity and selectivity mainly depends on the nature of the substituent rather than its position on the ligand.92
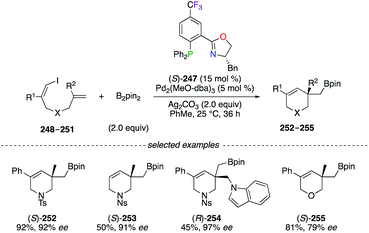 |
| | Scheme 28 (S)-247/Pd0-Catalyzed vinylborylation of alkenes. | |
In the presence of CuClO4, a chiral sulfinamide monophosphine (MingPhos) ligand, bearing a pentafluorophenyl substituent at the C-stereocentre, led to the [3 + 2]-cycloaddition product with lower enantioselectivity than its non-fluorinated analogue (66% ee vs. 82% ee).93
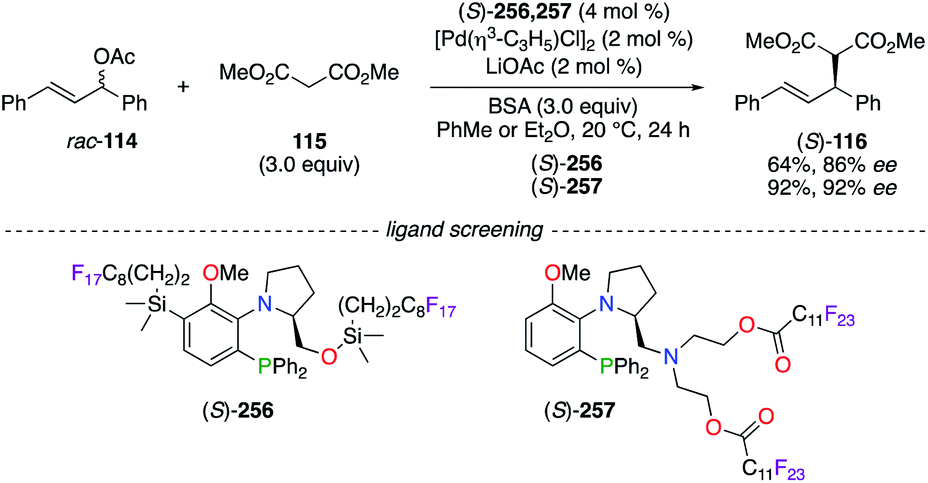
The PdII-catalyzed allylic alkylation reaction was performed using (S)-prolinol-based fluorous aminophosphine (S)-256 and (S)-257 (Scheme 29).94 High enantioselectivities were obtained from the addition of dimethyl malonate 115, at first deprotonated by LiOAc–N,O-bis(trimethylsilyl)acetamide (BSA), to 1,3-diphenyl-2-propenyl acetate rac-114 by using both chiral catalysts ((S)-116: 86% ee using (S)-256; 92% ee using (S)-257). The temperature-dependent solubility of the (S)-257/PdII catalyst was investigated, and these studies revealed the possibility to recover the fluorous catalyst via its precipitation in cold hexane.
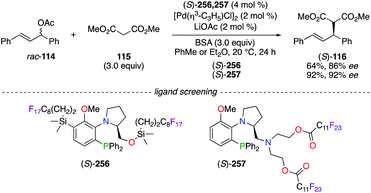 |
| | Scheme 29 PdII-Catalyzed allylic alkylation reaction using (S)-256 and (S)-257. | |
2.4.2 Diphosphines.
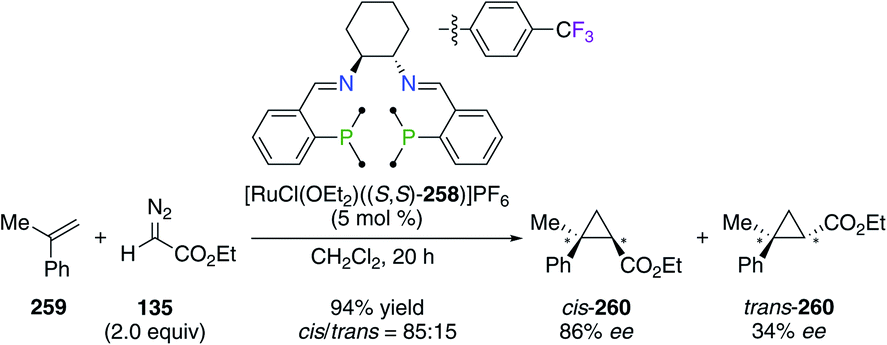
Fluorinated (S,S)-DACH-derived P2N2-tetradentate ligand (S,S)-258 was employed in the RuII-catalyzed cyclopropanation reaction of α-methyl styrene 259 with ethyl α-diazoacetate 135 (Scheme 30).95 The presence of 4-CF3-C6H4 groups was beneficial for the synthesis of cyclopropane 260 with good 70% decis, 86% eecis, and 34% eetrans, whereas the non-fluorinated analogue afforded 260 with low 52% decis, 23% eecis, and 18% eetrans. When using styrene 20 and 1-octene as substrates, the [RuCl(OEt2)((S,S)-258)]PF6 catalyst was also highly diastereoselective for the corresponding cis cyclopropanes. Again, CF3-substituted aryl groups were incorporated into a (S,S)-DPEN-based PNNP ligand, of which chiral FeII complexes were found to be highly electronically deficient, but inactive in the asymmetric transfer hydrogenation of acetophenone.96
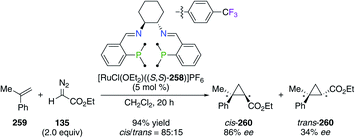 |
| | Scheme 30 Asymmetric cyclopropanation reaction catalyzed by a CF3-containing PNNP/RuII complex. | |
2.4.3 Axially chiral monophosphines.
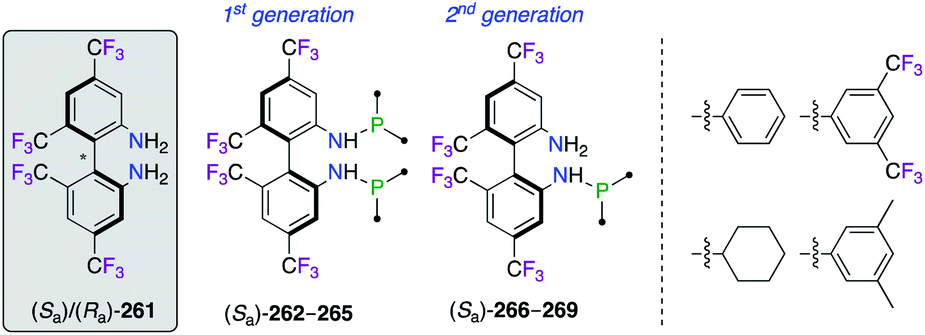
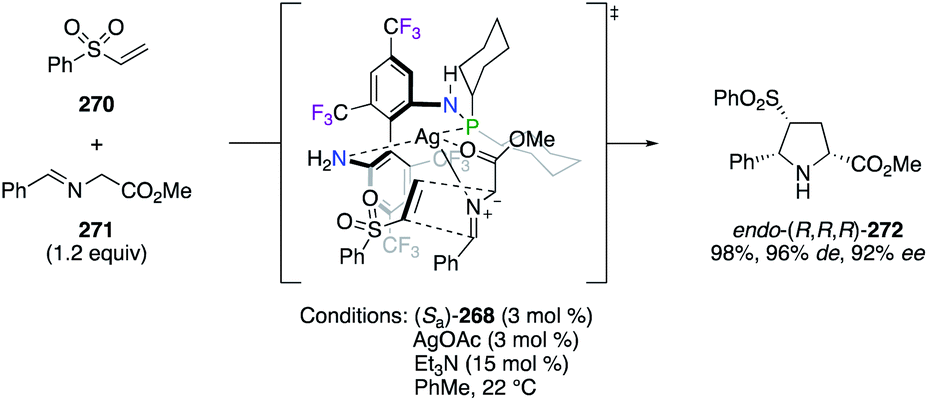
The important F-containing atropisomeric 1,1′-biphenyl architecture, i.e., 4,4′,6,6′-tetrakis-trifluoromethyl-biphenyl-2,2′-diamine (TF-BIPHAM) (Sa)/(Ra)-261, was obtained as an enantiopure material via the resolution of (Sa,S)- and (Ra,S)-10-camphornyl-based disulfonamide diastereoisomers and was employed in the synthesis of chiral amine–phosphine ligands (Fig. 12). The first generation of C2-symmetric N,N′-PR2-TF-BIPHAM ligands (Sa)-262–265, comprising diaryl- and dialkylphosphinyl groups, exhibited excellent asymmetric induction in the RhI-catalyzed hydrogenation of enamides.97 Catalytic applications towards the synthesis of enantioenriched saturated heterocycles via a highly efficient 1,3-dipolar cycloaddition reaction, using CuI (ref. 98) or AgI (ref. 99) catalysis, were developed using the second generation of ligands. Indeed, the scope of these ligands was extended to include mono-N-phospanyl (TF-BIPHAMPhos) derivatives (Sa)-266–269. When using the (Sa)-268/AgI catalytic system, pyrrolidine endo-(R,R,R)-272 was afforded in excellent yields and stereoselectivities via the 1,3-dipolar cycloaddition of in situ generated azomethine ylides, as shown using imine 271, to vinyl sulfone 270 through the more accessible Si face of the imine (Scheme 31).99b
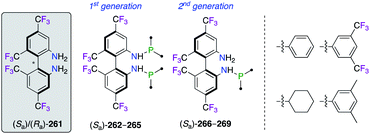 |
| | Fig. 12 Library of amine–phosphine ligands comprising the TF-BIPHAM scaffold. | |
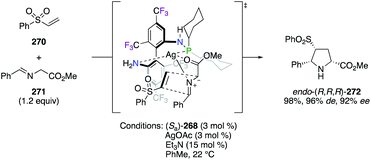 |
| | Scheme 31 Postulated TS for the attack of the azomethine ylide intermediate. | |
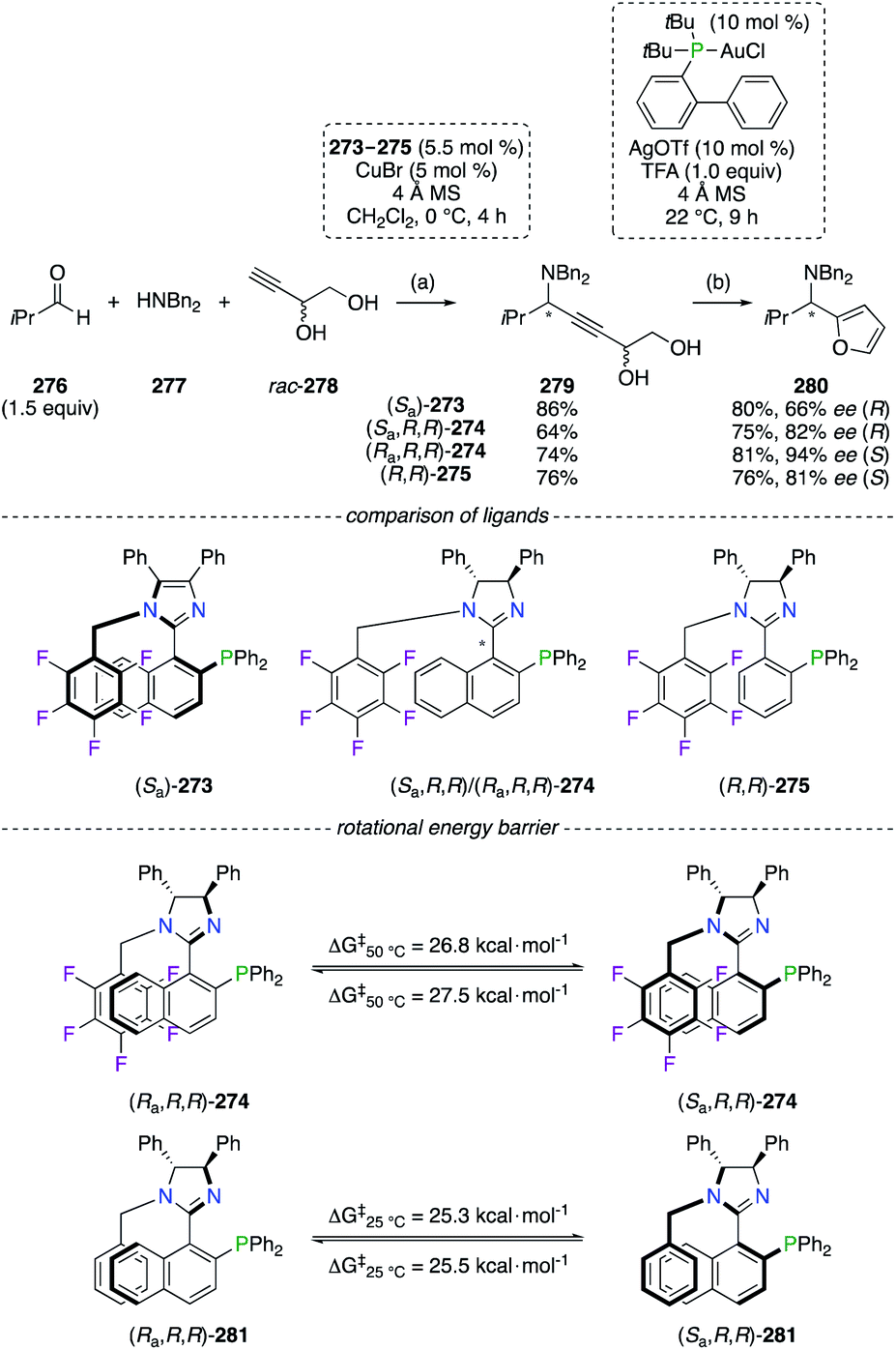
The CuI-catalyzed three-component alkynylation reaction from 276, 277, and rac-278, followed by the AuI-catalyzed dehydrative cyclization reaction of alkynediol 279 into 280, was described as highly enantioselective using phosphino(imidazoline) (StackPHIM) (Ra,R,R)-274 (Scheme 32).100 Since the imidazole analogue (Sa)-273 (StackPhos ligands)101 and (R,R)-275 (having no axial chirality) both led to 2-aminoalkyl furan 280 (R or S) with lower enantiomeric enrichments, the complementary between the stereocentres and the chiral axis to reach higher ee was demonstrated. Further fine-tuning of the 274/CuI catalyst was highlighted by the combination of the (R,R)-DPEN scaffold with the Ra or the Sa atropisomer, resulting in an increased chiral induction of 280 from 82% ee (R) to 94% ee (S). Noteworthily, atropisomerism of configurationally stable P,N-ligands 273 and 274 arises from π,π-stacking interactions between the naphthyl and C6F5 moieties. Being determined experimentally at 50 °C, rotational energy barriers (ΔG‡) of 26.8 kcal mol−1 (Ra into Sa) and 27.5 kcal mol−1 (Sa into Ra) have proven that both atropisomers of 274 could be synthesized, separated and successfully employed as chiral ligands in metal catalysis. However, the absence of fluorine atoms considerably lowered the ΔG‡ values of Ra–Sa (or Sa–Ra) interconversion, and epimerization of 281 occurred easily even at room temperature.
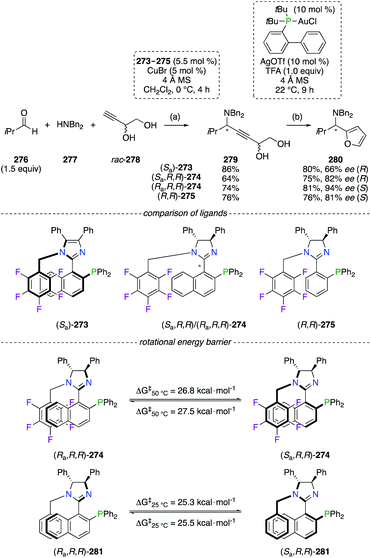 |
| | Scheme 32 Two-step synthesis of 2-aminoalkyl furan via the alkynylation/cyclization reaction sequence. | |
2.4.4 Ferrocenes.
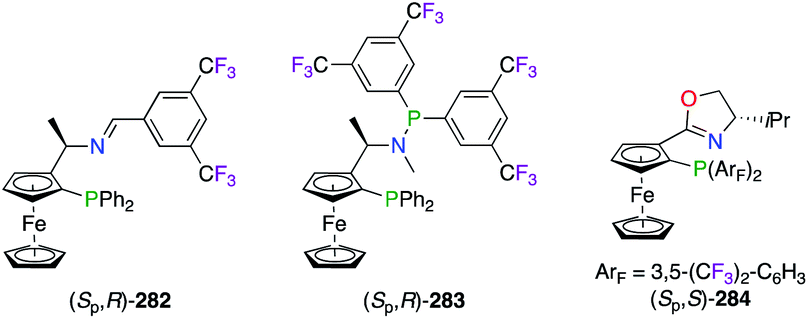
The popular 3,5-bis(trifluoromethyl)phenyl scaffold was incorporated into chiral ferrocenyl-derived P,N-containing ligands (Fig. 13). Indeed, imine-, amine-, and oxazoline-based phosphine ligands 282–284 were successfully used in the PdII-catalyzed allylic alkylation,102 the RhII-catalyzed hydrogenation,103 and the CuI-catalyzed 1,3-dipolar cycloaddition reactions.104 In the last case, the divergent exo/endo selectivities observed from the experimental results were rationalized through computational studies. Interestingly, the postulated TS models suggested that two different chelation modes of the substrate were arising from the electron-deficient ArF substituents of (Sp,S)-284vs. the electron-rich phenyl rings of its non-fluorinated analogue. Moreover, closely related ferrocenyl-based bis(perfluoroalkyl)phosphino(oxazoline) ligands were designed in a series of bulky tBu, Ph and Bn substituents at the C-stereocentre.89,105
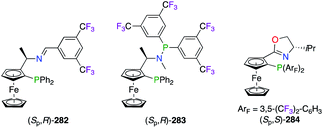 |
| | Fig. 13 Chiral ferrocenyl-derived ligands containing the 3,5-(CF3)2-C6H3 group. | |
In brief, the widespread use of 4-CF3 and 3,5-(CF3)2 motifs on the aromatic scaffold of the ligands was highlighted in this section. Importantly, the C6F5 group has been demonstrated as highly efficient when used in axially chiral phosphines, whereas its electrostatic pairing with the π system of the naphthyl moiety has induced a rotational energy barrier allowing atropisomerism. Also, the use of o,o′-CF3 within the TF-BIPHAM architecture was proved valuable in this strategy.
2.5
P-Based binding modes
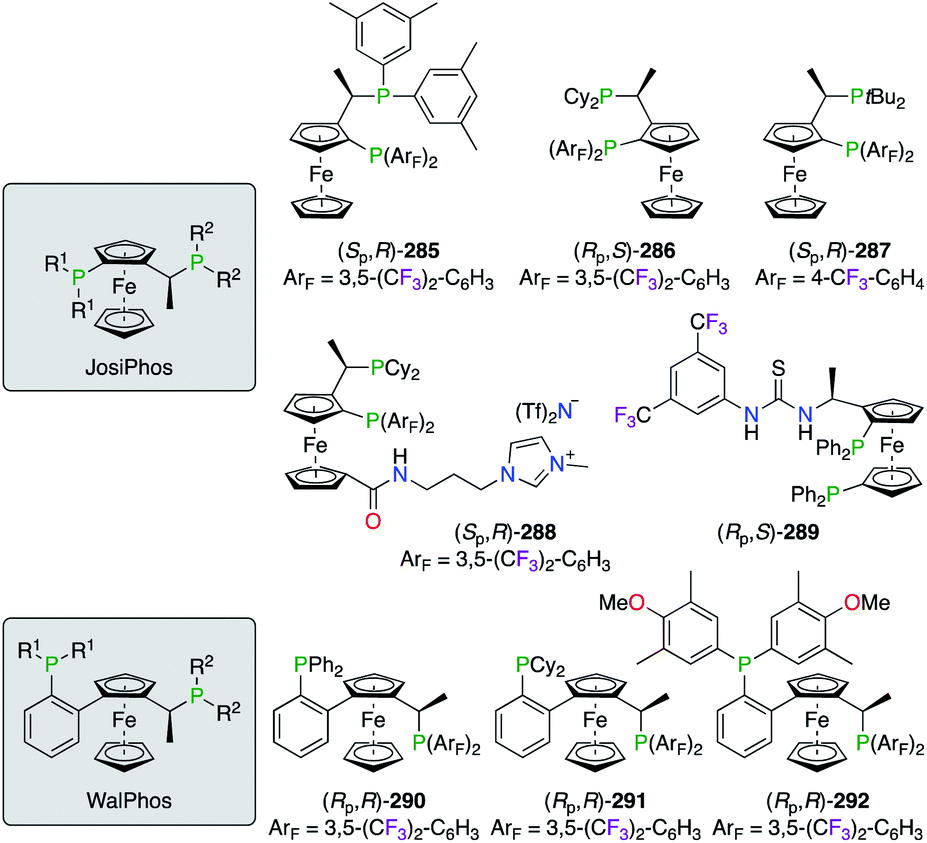
2.5.1 Planar chiral diphosphines.
Electron-poor diphosphine ligands were described for JosiPhos- and WalPhos-type ferrocenes 285–289 and (Rp,R)-290–292, respectively (Fig. 14). Chiral ligands (Sp,R)-285,106 (Rp,S)-286,106a,107 and (Sp,R)-287106b were tested in the IrI- or RhIII-catalyzed hydrogenation and the PdII/CuI-co-catalyzed Heck/Sonogashira asymmetric reactions, but only high chiral inductions were obtained by using the (Rp,S)-286/PdII catalyst. An application of fluorinated JosiPhos ligands in ionic liquids was demonstrated using (Sp,R)-288, i.e., the imidazolium-based analogue of (Sp,R)-286, in combination with [Rh(norbornadiene)2]BF4.107b The asymmetric hydrogenation reaction of methyl acetamidoacrylate, run under biphasic tert-butyl methyl ether/[bmim]BF4 conditions, afforded the corresponding product with 99% ee using either (Sp,R)-286 or (Sp,R)-288. More importantly, the ionic tag on (Sp,R)-288 allows better recyclability of the fluorinated catalyst in the chosen co-solvent system. Chiral thiourea-derived diphosphine ligand (Rp,S)-289 was used in the RhI-catalyzed hydrogenation reactions,108 where synergetic dual catalysis was induced by both the P2 ligated Lewis acid and the Brønsted acid. Control experiments revealed an important gain of ee when a stronger hydrogen bonding was provided by the thiourea moiety. WalPhos-type diphosphines (Rp,R)-290–292 were also described as a promising class of ligands in RhI, RuII, and CuI catalysis.109
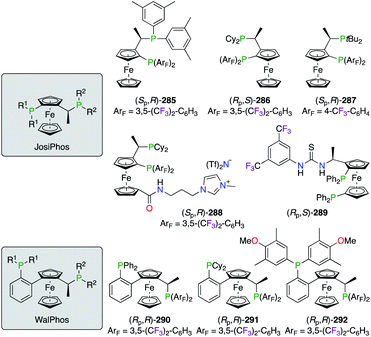 |
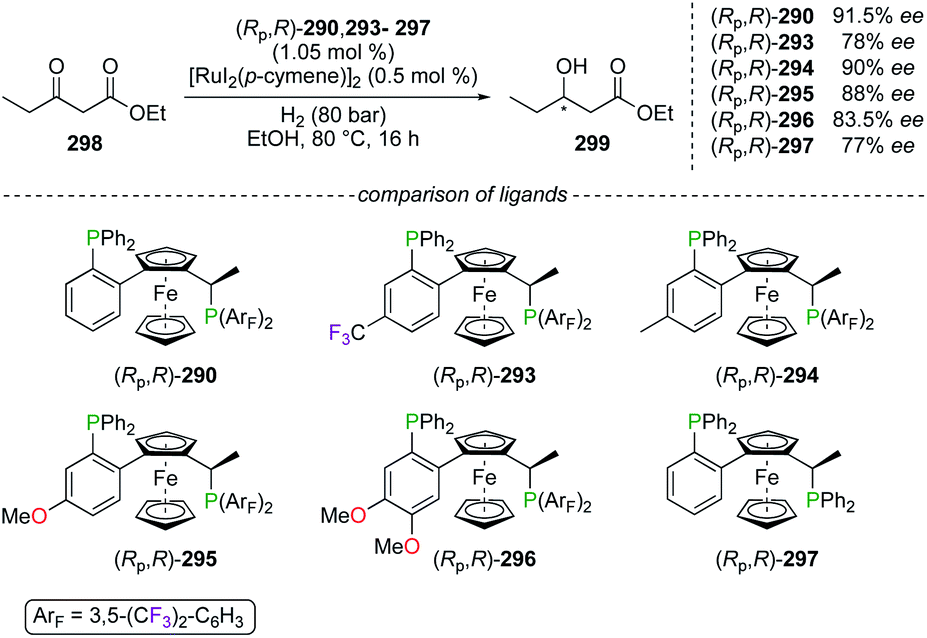
| | Fig. 14 Fluorinated JosiPhos- and WalPhos-type of ligands. | |
Other fluorinated WalPhos-type diphosphines ligands (Rp,R)-293–296 were designed to incorporate various electronic substituents on the aromatic backbone, not directly at the P atom. Such fine-tuning of the chiral ligand was studied in the RuII-catalyzed hydrogenation of ethyl 3-oxopentanoate 298 into 299 (Scheme 33).109b Unfortunately, no significant improvement of the chiral induction, arising from the electronic modifications of (Rp,R)-293–296, was demonstrated in comparison with the results obtained using ligands (Rp,R)-290 and (Rp,R)-297 as references. Noteworthily, conformational structures of chiral RuII complexes, obtained from (Rp,R)-293, (Rp,R)-296, and (Rp,R)-297, were investigated through XRD analysis.
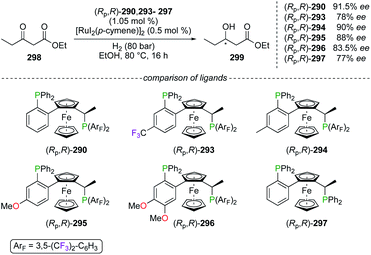 |
| | Scheme 33 Hydrogenation reaction using electronically modified WalPhos ligands. | |
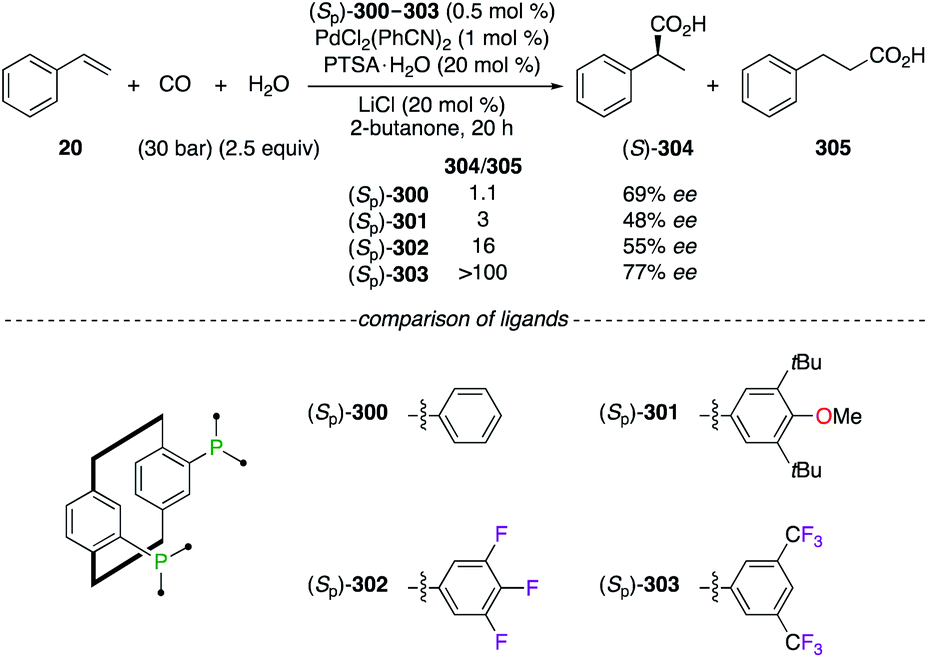
Planar chirality was further exploited in the hydroxycarbonylation reaction of styrene 20 using PhanePhos ligands (Sp)-300–303, all being available either in their Sp or Rp enantiomeric forms (Scheme 34). Chiral dinuclear PdII catalysts made from fluorinated ligands (Sp)-302 and (Sp)-303 afforded an increased selectivity for the branched regioisomer (S)-304vs. the selectivity obtained using the electron-rich PhanePhos ligands (Sp)-300 and (Sp)-301. Moreover, the highest ee was achieved using the (Sp)-303/PdII2 catalytic system.110
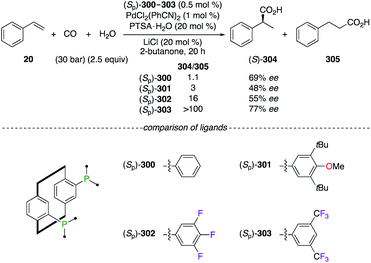 |
| | Scheme 34 PdII-Catalyzed carbonylation reaction of styrene 20. | |
2.5.2 Axially chiral diphosphines.
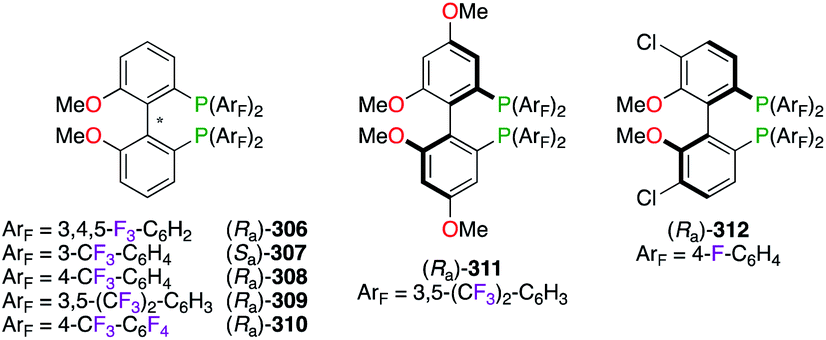
A PdII-catalyzed P–C coupling reaction,111 between enantiomerically enriched phosphanes and aryl iodides, inspired the synthesis of a wide library of F-containing atropisomeric 2,2′-bis(diphenylphosphino)-6,6′-dimethoxy-1,1′-biphenyl (MeO-BIPHEP) ligands 306–312 (Fig. 15). The development of polyfluorinated MeO-BIPHEP derivatives, comprising F12-((Ra)-306,112 (Sa)-307,113 (Ra)-308),113,114 F24-((Ra)-309)114,115 and F28-analogues ((Ra)-310),112a was undertaken, and these ligands were used as the chiral source for a variety of enantioselective transformations. Electronic modifications to the diaryl-core were explored for methoxy- or chlorine-substitutions at the 4,4′- and 5,5′-positions giving (Ra)-311116 – commercialized as (R)-BTFM-GarPhos™ – and (Ra)-312,117 respectively.
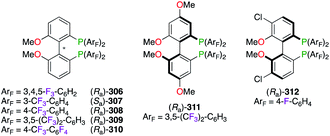 |
| | Fig. 15 F-Containing MeO-BIPHEP derivatives. | |
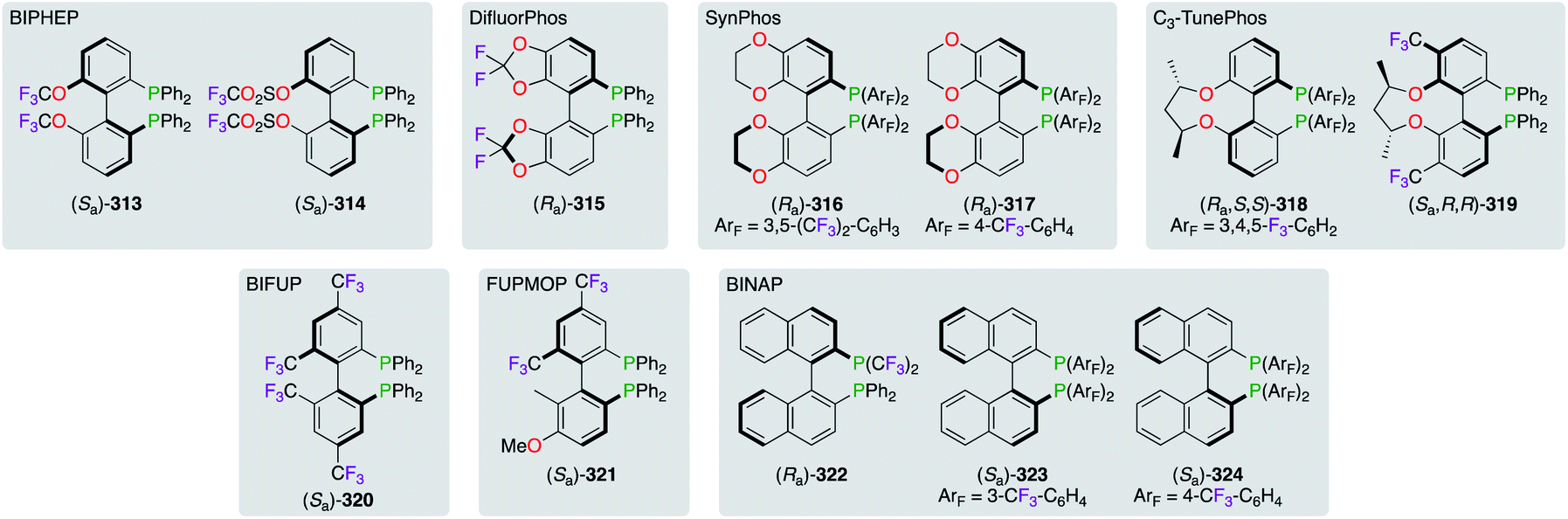
The family of axially chiral F-containing bisphosphine ligands was extended to achieve a priority objective, where the fine-tuning of the dihedral angle between the two aromatic rings was targeted (Fig. 16). Ligand design targeting structural variations arising from steric or electronic interactions, for getting optimum stereoselectivities, were extensively explored. Accordingly, all ligands were classified based on the 1,1′-biphenyl backbone: (i) BIPHEP-((Sa)-313 and (Sa)-314),118 (ii) DifluorPhos-((Ra)-315),106a,119 (iii) SynPhos-((Ra)-316 and (Ra)-317),120 (iv) C3-TunePhos-((Ra,S,S)-318 and (Sa,R,R)-319),121 (v) BIFUP- or FUPMOP-((Sa)-320 and (Sa)-321),122 and (vi) 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP)-type of ligands (322–324).119b,123 Based on comparative studies toward five selected classes of ligands, the narrowest dihedral angle was attributed to DifluorPhos-type ligands (θ ∼ 67°), whereas BINAP derivatives (θ ∼ 86°) were located at the other end of the steric scale.119a,b Overall, all electronically impoverished ligands, with specific steric profiles, showed excellent chiral inductions when complexed with the appropriate Lewis acid in various asymmetric reactions.
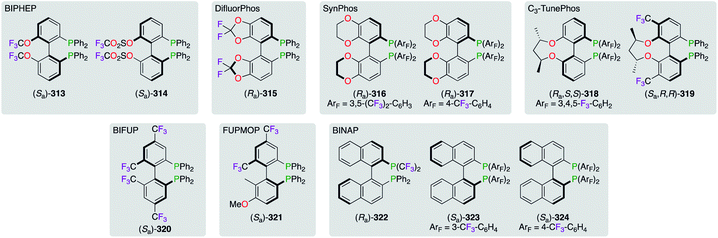 |
| | Fig. 16 Classes of axially chiral bisphosphine ligands comprising the 1,1′-biphenyl system. | |
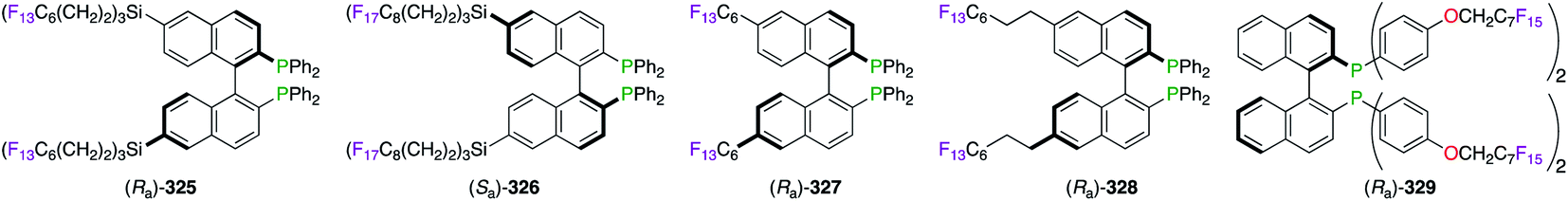
Strategies towards the recycling of the chiral ligand have encouraged the derivatization of BINAPs to incorporate perfluoroalkyl chains within their skeleton (Fig. 17). Fluorine substitution upon the naphthyl-backbone was then chosen for the synthesis of (Ra)-325. The asymmetric Heck reaction of 2,3-dihydrofuran was successfully performed in the benzene/FC-72 system using (Ra)-325/PdII.28b,124 The RuII-catalyzed hydrogenation reaction of dimethyl itaconate was described using heavier fluorinated BINAP (Sa)-326, which was immobilized on fluorous silica gel.125 Excellent retention of the fluorous ligand within the silica pores, via noncovalent interactions with the C8F17 chains, permitted its recycling without the use of conventional biphasic extraction methods. Similar to (Ra)-325, F-containing BINAPs (Ra)-327 and (Ra)-328 were developed for their great ability as “light” fluorous ligands to be extracted from the other organic compounds via simple FRP column chromatography.126 Introducing the perfluoroalkyl chain at the P atom, as highlighted by (Ra)-329, afforded poor ees in three metal-catalyzed reactions potentially due to the proximity of the fluorous tails to the activating site.127 The relatively low fluorine content of (Ra)-329 (51.5%) failed to give satisfactory chiral inductions in FBS. However, (Ra)-329 was separated from the reaction mixture quickly using liquid–liquid extraction by perfluorocarbons.
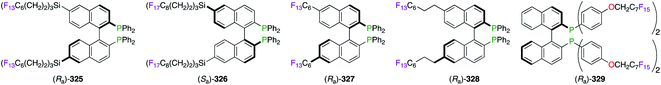 |
| | Fig. 17 Fluorous BINAPs employed in FBS. | |
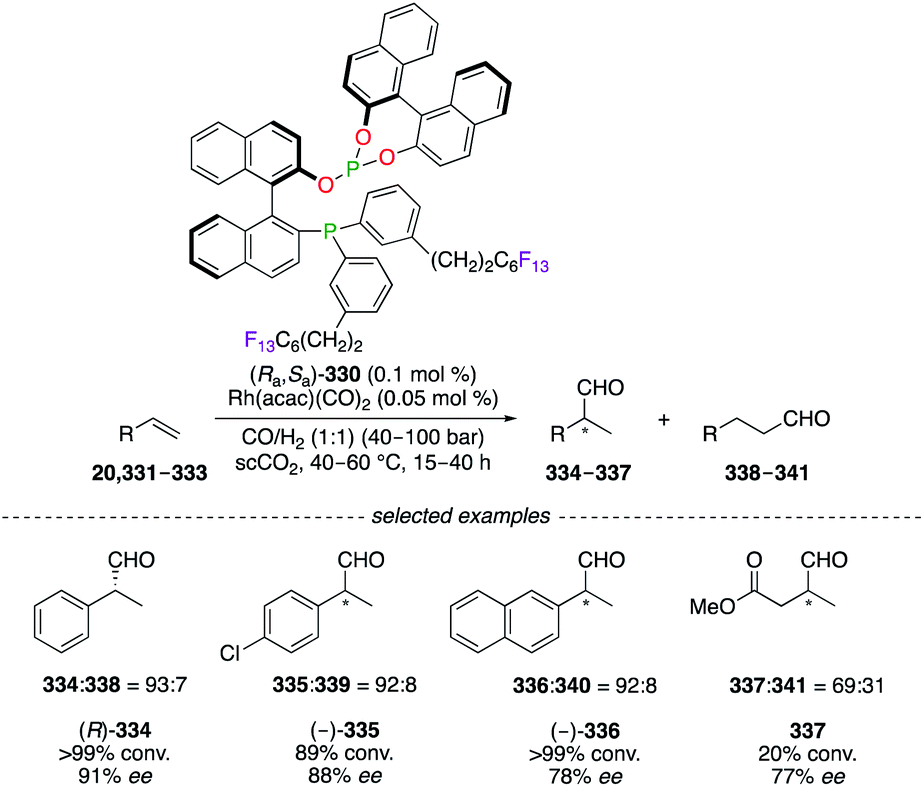
Research towards the development of greener synthetic methods focused on the replacement of commonly used organic solvents by less hazardous and more environmentally benign alternatives, e.g., supercritical carbon dioxide (scCO2). The RhI-catalyzed hydroformylation reaction of monosubstituted alkenes 20, 331–333 was performed in supercritical fluids using phosphine–phosphite BINAPHOS (Ra,Sa)-330 (Scheme 35).128 The aldehydes 334–337 were obtained with good regioselectivities and enantioselectivities using the (Ra,Sa)-330/RhI catalytic system. Considered as CO2-philic, the perfluoralkyl chains permitted sufficient solubility of the ligand to be used under homogeneous reaction conditions. Moreover, C4F9, C6F13, and C8F17 ponytails were incorporated into the 1,1′-binaphthyl core to generate [RF(CH2)3]–BINAPHOS analogues.129
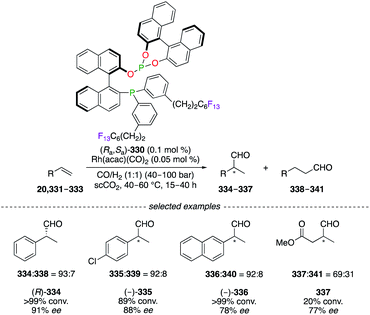 |
| | Scheme 35 (Ra,Sa)-330/RhI-Catalyzed hydroformylation reaction of alkenes. | |
2.5.3 Phosphoramidites.
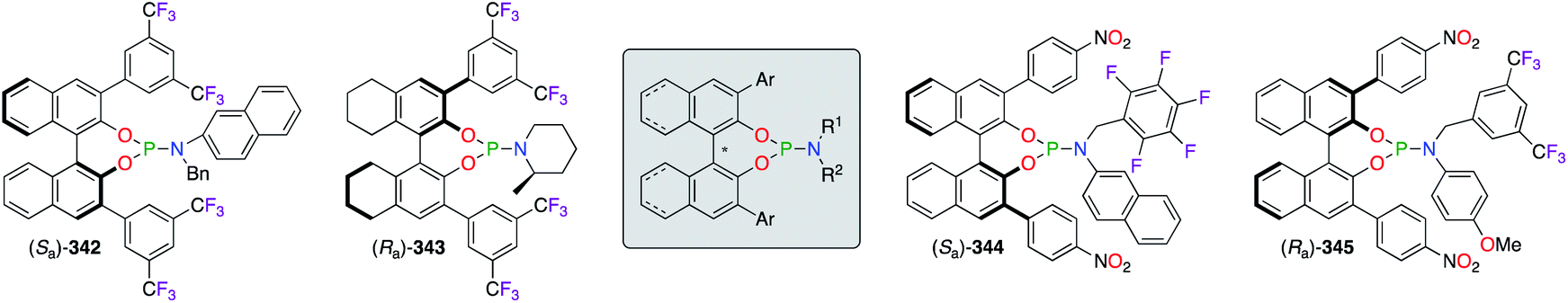
A wide library of monodentate phosphoramidite ligands was designed for numerous metal-catalyzed asymmetric reactions (Fig. 18).130 Following the long-arm approach, substituted phenyl rings were incorporated at the 3,3′-positions of the binaphthol skeleton in order to enhance chiral inductions. Indeed, the space surrounding the ligated metal centre was considerably restricted by bulky 3,5-(CF3)2 and 4-NO2 aromatic rings, as observed in (Sa)-342/(Ra)-343 and (Sa)-344/(Ra)-345, respectively. An additional fine-tuning of the chiral ligands was possible at the amine moiety, where electron-deficient benzyl substituents ((Sa)-344/(Ra)-345) or hindered piperidine ((Ra)-343) were generally more beneficial as observed from the obtained ees. A set of (R,R)-TADDOL-derived phosphoramidite ligands were screened in the PdII/CuI-catalyzed alkynylation reaction, but only the aryl substituents bearing bulky TMS groups and one electron withdrawing F atom afforded the optimal chiral induction.131
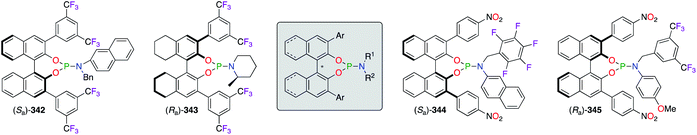 |
| | Fig. 18 Selected examples of chiral phosphoramidite ligands. | |
(R)-BINOL-based phosphite ligands, generated from 3,5-bis(trifluoromethyl)phenol and 2,2,2-trifluoroethanol, were considered as promising ligands in the synthesis of RhI complexes and their use in enantioselective catalysis in ionic liquids.132
2.5.4 Other P ligands.
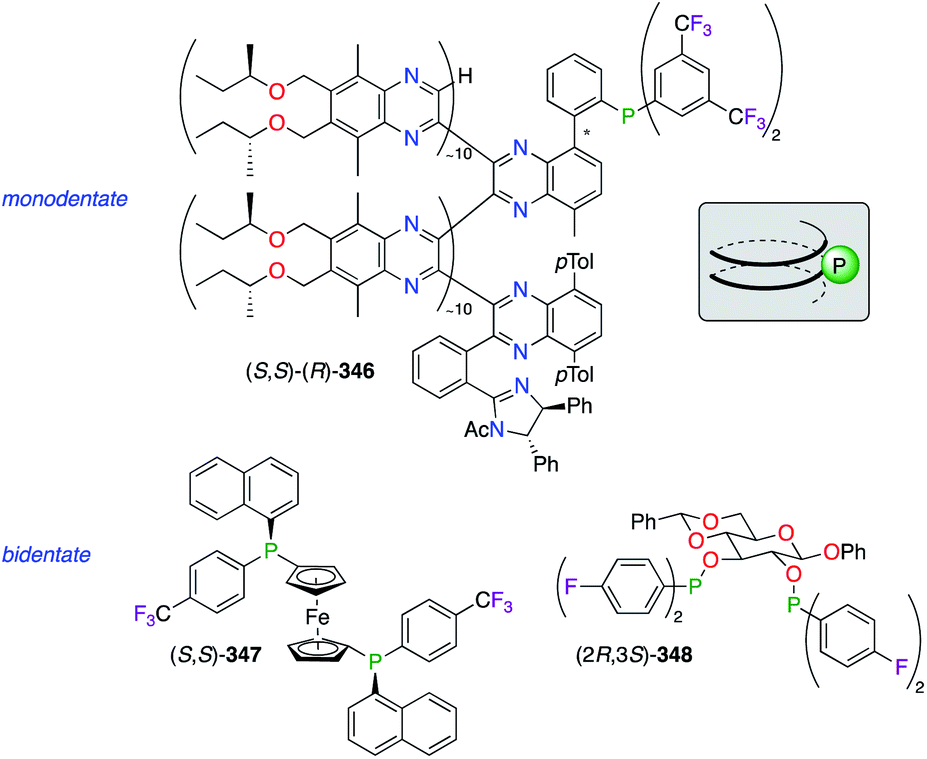
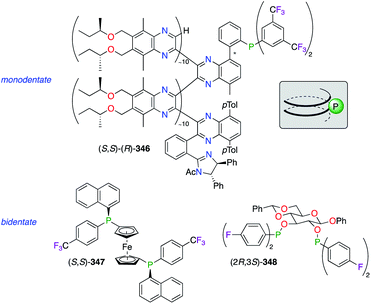
Fluorinated monophosphine, stereogenic phosphine, and sugar-derived phophinite ligands have been complexed with various noble metals (Fig. 19). The (10–1–10) copolymer poly(quinoxaline-2,3-diyl)phosphine (PQXphos) (S,S)–(R)-346, having P helicity, was employed as a screened ligand in the asymmetric Pd0-catalyzed Suzuki–Miyaura coupling reaction, giving only a moderate chiral induction.133 Interestingly, the sense of the chirality (Ra or Sa) at the active site of 346 would be induced, according to the structure models,133b by the helicity adopted by the polymer either in the right-handed (P) or the left-handed (M) helix geometry, respectively. Another monodentate (1R,3R,4S)-menthyl-based phosphine ligand, bearing two C6F13 and C8F17 ponytails, was reported to give fluorous chiral catalysts when mixed with IrI and RhI salts.134 Chiral at P atoms, diphosphine ligand (S,S)-347, belonging to the class of 1,1′-bis(diphenylphosphino)ferrocenes (dppf), was designed to modulate the stereoselective event in the PdII-catalyzed nucleophilic substitution of allylic acetates.135 When rac-114 and 115 were used as substrates, the combination of the bulky 1-naphthyl substituent with the electronically deficient 4-F-C6H4 unit afforded a slightly lower enantioselectivity (61% ee of (S)-116) than the unfluorinated one (68% ee of (R)-116) and its electron donating analogue (4-OMe-C6H4; 69% ee of (S)-116). A PtII-catalyzed alkylation of linked secondary phosphines (HRP–PRH) in the presence of various benzyl bromides, comprising F- and CF3-containing ones, led to P-stereogenic diphosphines with low stereoselectivities.136 The hydrogenation of a variety dehydroamino acids was reported using chiral phosphinite/RhI catalysts made from various carbohydrate scaffolds.137 Being highly dependent on the P-aryl substituent, the level of chiral induction was demonstrated to be considerably higher when using electron-rich bis(3,5-dimethylphenyl) groups vs. electron-poor ones, such as C2,C3-bis-(di-4-fluorophenyl)phosphinite ligand (2R,3S)-348, derived from phenyl β-D-glucopyranoside. Furthermore, a (R,R)-DIOP-like 4-(trifluoromethyl)phenylboronate diphosphine ligand was synthesized and mixed with RhI, PdII, and PtII salts to generate heterobimetallic complexes.138
 |
| | Fig. 19 Miscellaneous mono- and bidentate P-ligands containing fluorine atoms. | |
In general, the 3,5-(CF3)2-C6H3 substituent has been widely used in P-based chiral ligands. Various fluorous ponytails were incorporated into BINAP ligands to give “heavy” and “light” fluorous analogues to be used in distinct synthetic applications. Major advancements were demonstrated using CO2-philic BINAPHOS ligands bearing perfluoroalkyl chains, in the enantioselective hydroformylation reaction performed in supercritical carbon dioxide. Noteworthily, this section has detailed numerous mono- and diphosphines incorporating a large range of structurally diverse fluorine containing groups.
2.6
P,O-Based binding modes
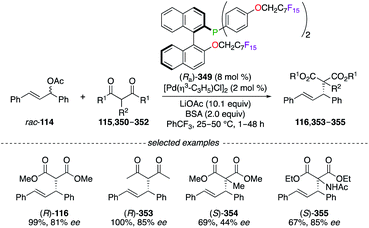
Being O-alkylated with perfluoroalkyl chains at the very last step of the synthesis, 2-(diphenylphosphino)-2′-alkoxy-1,1′-binaphthyl (MOP) (Ra)-349 was used, together with [Pd(η3-C3H5)Cl]2, in the asymmetric alkylation reaction of β-dicarbonyl derivatives with 1,3-diphenyl-2-propenyl rac-114 (Scheme 36).127,139 The corresponding alkylated products (R)-116 and 353–355 were obtained in moderate to excellent yields, and good ees were obtained. Noteworthily, (Ra)-349 was completely extracted from the reaction mixture using n-perfluorooctane, whereas the catalytic activity of the recycled PdII complex was lost when used in subsequent reactions.
 |
| | Scheme 36 (Ra)-349/PdII-catalyzed alkylation reaction of rac-114. | |
2.7
C-Based binding modes
2.7.1 Diaminocarbenes.
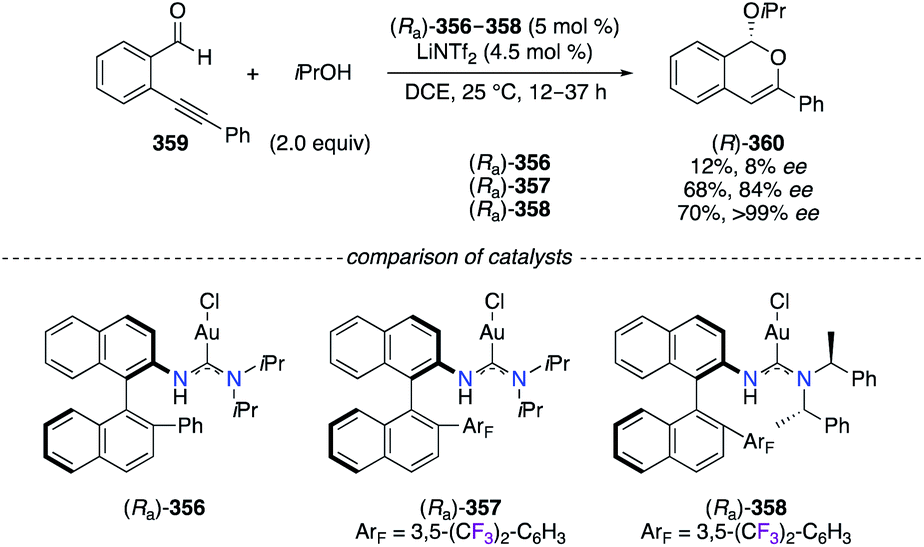
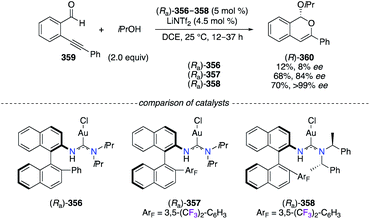
Chiral AuI complexes (Ra)-356–358 involving acyclic diaminocarbene ligands were described as efficient catalysts in the cyclization reaction of alkynylbenzaldehyde 359 and iPrOH (Scheme 37).140 Cycloadduct (R)-360 was obtained in various levels of enantioselectivity depending on the presence of the 3,5-(CF3)2-C6H3 group on the adjacent naphthyl ring ((Ra)-357 and (Ra)-358vs. (Ra)-356). As revealed by XRD and DFT studies, strong AuI–π interactions with the electron-deficient F-arene give an increased stability to the catalyst, by limiting the number of possible rotamers, therefore locating the chiral environment in the optimal orientation. Bulkier substituents at the amine moiety ((S)-PhMeCH vs.iPr) further enhanced steric interactions around the AuI centre and thus the obtained enantioselectivity.
 |
| | Scheme 37 AuI-Catalyzed cyclization of 359 into (R)-360. | |
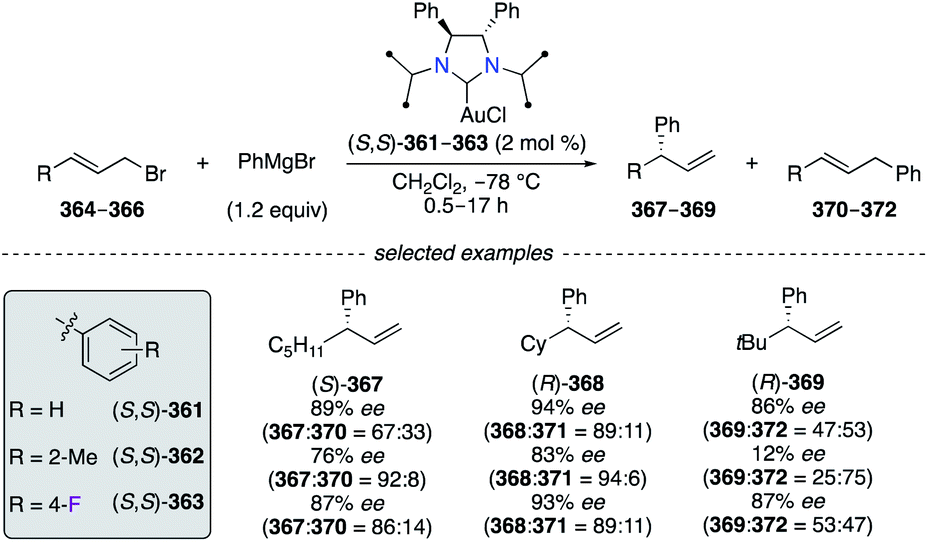
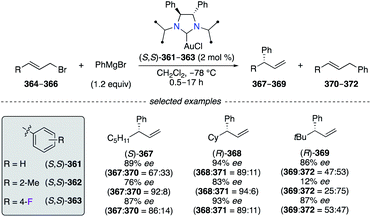
N-heterocyclic carbene (NHC)–CuI catalysts (S,S)-361–363 were employed in the asymmetric allylic arylation (AAAr) of aliphatic allylic bromides 364–366 with PhMgBr (Scheme 38).141 Overall, the substitution by sterically hindered and electron-deficient aryl groups onto chiral NHC–CuI complexes, e.g., (S,S)-362 and (S,S)-363, was found beneficial to obtain a higher regioselectivity towards the γ-regioisomers 367–369 than (S,S)-361. When using the allylic bromide 364, the best enantioselectivity was afforded by using (S,S)-361, but sterically and electronically fine-tuned catalysts considerably improved the γ-selectivity (367:370 up to 92:8). Excellent regioselectivity for (R)-368 was obtained particularly with (S,S)-362, whereas excellent enantioselectivities were rather observed when using (S,S)-361 and (S,S)-363. The NHC–CuI-catalyzed AAAr of 366 led to α-product 372 preferentially, and only the F-containing catalyst (S,S)-363 afforded (R)-369 with the best regio- and enantioselectivity when using a sterically hindered tBu-substrate.
 |
| | Scheme 38 NHC–CuI-Catalyzed AAAr reaction of aliphatic allylic bromides with phenyl magnesium bromide. | |
2.7.2 Dienes.
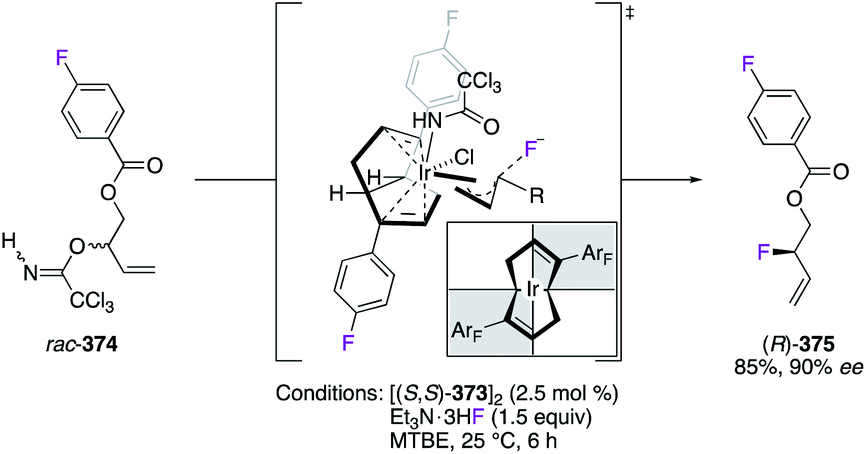
The fluorination reaction of allylic trichloroacetimidates rac-374 was performed via a dynamic kinetic asymmetric transformation (DKAT) mechanism using the IrI pre-catalyst [(S,S)-373]2 (Scheme 39).142 Chelated by the fluorinated (S,S)-bicyclo[3.3.0]octadiene ligand, the IrI centre generated, via ionization of the substrate, the lowest energetically π-allyl intermediate of the computed diastereomeric TSs. Accordingly, the most substituted carbon undergoes nucleophilic attack of the fluoride from the outer-sphere, and (R)-375 was obtained with excellent regio- and enantioselectivities.
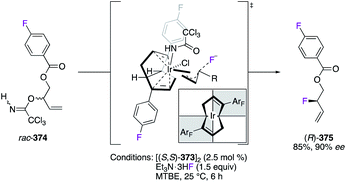 |
| | Scheme 39 Fluorination reaction via the optimized TS. | |
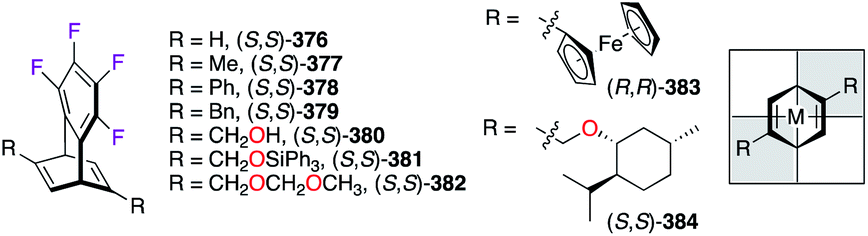
RhI- or IrI-catalyzed highly enantioselective organic transformations, i.e., arylation,143 conjugate addition,144 and cyclization reactions,145 were demonstrated using C2-symmetric tetrafluorobenzobarrelene (tfb) ligands 376–384 (Fig. 20). The bicyclic [2.2.2]octatriene skeleton was obtained in both R,R and S,S enantiomeric forms via the resolution of the racemic mixture by high pressure chiral liquid chromatography. Once the desired arms attached, the electronically deficient diene ligands 376–384 strongly chelate metal cations, and high sterically hindered environments are then created in the upper right and lower left quadrants.146
 |
| | Fig. 20 Chiral tfb-substituted ligands and their complexation modes with metals. | |
3. Conclusions
The introduction of the fluorine atom has revealed a positive modulation of stereoselective events using metal catalysis. An optimal balance between an ideal substrate activation and an inherent stability of the catalyst can be established by using these electron-deficient ligands. The examples presented herein demonstrate that the fluorine atom fine-tunes both electronic and steric properties of the chiral catalyst. The high availability of F-containing aromatic compounds, particularly bearing the 3,5-(CF3)2-C6H3 and the 4-CF3-C6H4 motifs, favours their incorporation into desired specific positions. Generally, their high electron withdrawing ability was exploited for reaching increased acidity of the chelated Lewis acid. Also, the chiral environments were often better defined by the participation of the fluorinated aromatic ring through electrostatic pairing, both with the metal centre in the inner sphere and the substrate orientation in the outer sphere. The use of the fluorine atom and polyfluorinated groups as a steric bulk remains underdeveloped in asymmetric catalysis. Unexpected high chiral inductions, which are scarcely reported in the literature, are likely to arise from the intrinsic steric properties of bulky polyfluorinated substituents. Besides stereoelectronic reasons, the presence of fluorine in chiral ligands is also valuable from a green chemistry perspective. The fluorophilic effect, which is observed in the presence of perfluoroalkyl chains, has indeed opened the field of asymmetric catalysis into fluorous biphasic systems. Due to easy extraction procedures, many advantages were highlighted by the recycling of the chiral catalyst over multiple runs without the loss of chiral induction over time. Not only is the fluorine atom considered as a highly multifunctional tool for ligand design, but also its judicious employment is of paramount importance for metal-catalyzed asymmetric transformations.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant RGPIN-2017-04272, the FRQNT Centre in Green Chemistry and Catalysis (CGCC) Strategic Cluster FRQNT-2020-RS4-265155-CCVC, and Université Laval (UL). S. L. thanks NSERC and FRQNT for graduate scholarships.
Notes and references
-
(a)
K. Seppelt, The Curious World of Fluorinated Molecules: Molecules Containing Fluorine, Elsevier, Amsterdam, 2021 Search PubMed;
(b)
A. Tressaud, Fluorine: A Paradoxal Element, Elsevier, Amsterdam, 2019 Search PubMed;
(c)
P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Weinheim, 2013 CrossRef.
- D. O'Hagan and D. B. Harper, J. Fluorine Chem., 1999, 100, 127–133 CrossRef.
-
(a) H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem.–Eur. J., 2019, 25, 11797–11819 CrossRef CAS PubMed;
(b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed;
(c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
-
(a) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed;
(b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed;
(c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed;
(d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC;
(e) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
-
(a) D. Cahard and V. Bizet, Chem. Soc. Rev., 2014, 43, 135–147 RSC;
(b) F. Fache, New J. Chem., 2004, 28, 1277–1283 RSC;
(c) G. Pozzi and I. Shepperson, Coord. Chem. Rev., 2003, 242, 115–124 CrossRef CAS;
(d) L. E. Zimmer, C. Sparr and R. Gilmour, Angew. Chem. Int. Ed., 2011, 50, 11860–11871 CrossRef CAS PubMed.
-
M. Stradiotto and R. J. Lundgren, Ligand Design in Metal Chemistry: Reactivity and Catalysis, Chichester, 2016 Search PubMed.
-
(a) A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS;
(b)
P. R. Wells, in Progress in Physical Organic Chemistry, ed. A. Streitwieser Jr and R. W. Taft, John Wiley & Sons, New York, 1968, vol. 6, pp. 111–145 Search PubMed;
(c) J. E. Huheey, J. Phys. Chem., 1965, 69, 3284–3291 CrossRef CAS;
(d) J. Hinze, M. A. Whitehead and H. H. Jaffé, J. Am. Chem. Soc., 1963, 85, 148–154 CrossRef CAS.
-
(a)
J. A. Hirsch, in Topics in Stereochemistry, ed. N. Allinger and E. L. Eliel, John Wiley & Sons, New York, 1967, vol. 1, pp. 199−222 Search PubMed;
(b)
E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of Organic Compounds, John Wiley & Sons, New York, 1994 Search PubMed;
(c) Y. Carcenac, M. Tordeux, C. Wakselman and P. Diter, New J. Chem., 2006, 30, 447–457 RSC.
-
S. H. Unger and C. Hansch, in Progress in Physical Organic Chemistry, ed. R. W. Taft, John Wiley & Sons, New York, 1976, vol. 12, pp. 91–118 Search PubMed.
-
(a) M. Charton, J. Am. Chem. Soc., 1975, 97, 1552–1556 CrossRef CAS;
(b) M. Charton, J. Org. Chem., 1976, 41, 2217–2220 CrossRef CAS.
- G. Bott, L. D. Field and S. Sternhell, J. Am. Chem. Soc., 1980, 102, 5618–5626 CrossRef CAS.
- A. V. Brethomé, S. P. Fletcher and R. S. Paton, ACS Catal., 2019, 9, 2313–2323 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
-
(a)
J. A. Gladysz, D. P. Curran and I. T. Horváth, Handbook of Fluorous Chemistry, Wiley-VCH, Weinheim, 2004 CrossRef;
(b)
K. Mikami, Green Reaction Media in Organic Synthesis, Blackwell Publishing, Oxford, 2005 CrossRef.
- S. Suzuki, H. Furuno, Y. Yokoyama and J. Inanaga, Tetrahedron: Asymmetry, 2006, 17, 504–507 CrossRef CAS.
- J.-S. Lin, F.-L. Wang, X.-Y. Dong, W.-W. He, Y. Yuan, S. Chen and X.-Y. Liu, Nat. Commun., 2017, 8, 1–11 CrossRef CAS PubMed.
- T. Li, P. Yu, Y.-M. Du, J.-S. Lin, Y. Zhi and X.-Y. Liu, J. Fluorine Chem., 2017, 203, 210–214 CrossRef CAS.
- J. Lv, L. Zhang, S. Hu, J.-P. Cheng and S. Luo, Chem.–Eur. J., 2012, 18, 799–803 CrossRef CAS PubMed.
- A. Biffis, M. Braga, S. Cadamuro, C. Tubaro and M. Basato, Org. Lett., 2005, 7, 1841–1844 CrossRef CAS PubMed.
- F. Yang, D. Zhao, J. Lan, P. Xi, L. Yang, S. Xiang and J. You, Angew. Chem. Int. Ed., 2008, 47, 5646–5649 CrossRef CAS PubMed.
- M. Ogasawara, S. Watanabe, K. Nakajima and T. Takahashi, J. Am. Chem. Soc., 2010, 132, 2136–2137 CrossRef CAS PubMed.
- S. Superchi, M. I. Donnoli and C. Rosini, Tetrahedron Lett., 1998, 39, 8541–8544 CrossRef CAS.
- L. J. P. Martyn, S. Pandiaraju and A. K. Yudin, J. Organomet. Chem., 2000, 603, 98–104 CrossRef CAS.
-
(a)
Q.-L. Zhou, Privileged Chiral Ligands and Catalysts, Wiley-VCH, Weinheim, 2011 CrossRef;
(b) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
-
(a) Y. S. Sokeirik, A. Hoshina, M. Omote, K. Sato, A. Tarui, I. Kumadaki and A. Ando, Chem.–Asian J., 2008, 3, 1850–1856 CrossRef CAS PubMed;
(b) Y. S. Sokeirik, H. Mori, M. Omote, K. Sato, A. Tarui, I. Kumadaki and A. Ando, Org. Lett., 2007, 9, 1927–1929 CrossRef CAS PubMed.
- W.-S. Huang and L. Pu, Tetrahedron Lett., 2000, 41, 145–149 CrossRef CAS.
-
(a) Y. Nakamura, S. Takeuchi, K. Okumura, Y. Ohgo and D. P. Curran, Tetrahedron, 2002, 58, 3963–3969 CrossRef CAS;
(b) Y. Nakamura, S. Takeuchi and Y. Ohgo, J. Fluorine Chem., 2003, 120, 121–129 CrossRef CAS;
(c) Y. Tian, Q. C. Yang, T. C. W. Mak and K. S. Chan, Tetrahedron, 2002, 58, 3951–3961 CrossRef CAS;
(d) Y. Nakamura, S. Takeuchi, Y. Ohgo and D. P. Curran, Tetrahedron Lett., 2000, 41, 57–60 CrossRef CAS.
- Y. Tian and K. S. Chan, Tetrahedron Lett., 2000, 41, 8813–8816 CrossRef CAS.
- Y. Nakamura, S. Takeuchi, Y. Ohgo and D. P. Curran, Tetrahedron, 2000, 56, 351–356 CrossRef CAS.
- C. Wang, X. Wu and L. Pu, Chem.–Eur. J., 2017, 23, 10749–10752 CrossRef CAS PubMed.
-
(a) M. Omote, Y. Nishimura, K. Sato, A. Ando and I. Kumadaki, Tetrahedron, 2006, 62, 1886–1894 CrossRef CAS;
(b) M. Omote, Y. Nishimura, K. Sato, A. Ando and I. Kumadaki, Tetrahedron Lett., 2005, 46, 319–322 CrossRef CAS;
(c) M. Omote, T. Hasegawa, K. Sato, A. Ando and I. Kumadaki, Heterocycles, 2003, 59, 501–504 CrossRef CAS;
(d) M. Omote, A. Kominato, M. Sugawara, K. Sato, A. Ando and I. Kumadaki, Tetrahedron Lett., 1999, 40, 5583–5585 CrossRef CAS.
-
(a) M. Omote, N. Tanaka, A. Tarui, K. Sato, I. Kumadaki and A. Ando, Tetrahedron Lett., 2007, 48, 2989–2991 CrossRef CAS;
(b) M. Omote, Y. Nishimura, K. Sato, A. Ando and I. Kumadaki, J. Fluorine Chem., 2005, 126, 407–409 CrossRef CAS;
(c) M. Omote, Y. Nishimura, K. Sato, A. Ando and I. Kumadaki, J. Fluorine Chem., 2006, 127, 74–78 CrossRef CAS.
- S. Lauzon and T. Ollevier, Chem. Commun., 2021, 57, 11025–11028 RSC.
- Y. S. Sokeirik, M. Omote, K. Sato, I. Kumadaki and A. Ando, Tetrahedron: Asymmetry, 2006, 17, 2654–2658 CrossRef CAS.
-
(a) T. Katagiri, Y. Fujiwara, S. Takahashi, N. Ozaki and K. Uneyama, Chem. Commun., 2002, 986–987 RSC;
(b) A. Harada, Y. Fujiwara and T. Katagiri, Tetrahedron: Asymmetry, 2008, 19, 1210–1214 CrossRef CAS.
- Y. Fujiwara, T. Katagiri and K. Uneyama, Tetrahedron Lett., 2003, 44, 6161–6163 CrossRef CAS.
- G. Zhao, X.-G. Li and X.-R. Wang, Tetrahedron: Asymmetry, 2001, 12, 399–403 CrossRef CAS.
- J. K. Park, H. G. Lee, C. Bolm and B. M. Kim, Chem.–Eur. J., 2005, 11, 945–950 CrossRef CAS PubMed.
- Y.-Z. Liu, S.-J. Shang, W.-L. Yang, X. Luo and W.-P. Deng, J. Org. Chem., 2017, 82, 11141–11149 CrossRef CAS PubMed.
- K. Li, X. Sun, L. Li, Z. Zha, F.-L. Zhang and Z. Wang, Chem.–Eur. J., 2021, 27, 581–584 CrossRef CAS PubMed.
- B. M. Trost, V. S. C. Yeh, H. Ito and N. Bremeyer, Org. Lett., 2002, 4, 2621–2623 CrossRef CAS PubMed.
- A. M. Cook and C. Wolf, Angew. Chem. Int. Ed., 2016, 55, 2929–2933 CrossRef CAS PubMed.
- Y. Yi, Y.-Z. Hua, H.-J. Lu, L.-T. Liu and M.-C. Wang, Org. Lett., 2020, 22, 2527–2531 CrossRef CAS PubMed.
- J. A. Sehnem, P. Milani, V. Nascimento, L. H. Andrade, L. Dorneles and A. L. Braga, Tetrahedron: Asymmetry, 2010, 21, 997–1003 CrossRef CAS.
-
(a) M. Cavazzini, A. Manfredi, F. Montanari, S. Quici and G. Pozzi, Eur. J. Org. Chem., 2001, 4639–4649 CrossRef CAS;
(b) M. Cavazzini, A. Manfredi, F. Montanari, S. Quici and G. Pozzi, Chem. Commun., 2000, 2171–2172 RSC;
(c) G. Pozzi, M. Cavazzini, F. Cinato, F. Montanari and S. Quici, Eur. J. Org. Chem., 1999, 1947–1955 CrossRef CAS;
(d) G. Pozzi, F. Cinato, F. Montanari and S. Quici, Chem. Commun., 1998, 877–878 RSC.
-
(a) M. Cavazzini, S. Quici and G. Pozzi, Tetrahedron, 2002, 58, 3943–3949 CrossRef CAS;
(b) I. Shepperson, M. Cavazzini, G. Pozzi and S. Quici, J. Fluorine Chem., 2004, 125, 175–180 CrossRef CAS.
- D. Maillard, G. Pozzi, S. Quici and D. Sinou, Tetrahedron, 2002, 58, 3971–3976 CrossRef CAS.
- Y. Kobayashi, R. Obayashi, Y. Watanabe, H. Miyazaki, I. Miyata, Y. Suzuki, Y. Yoshida, T. Shioiri and M. Matsugi, Eur. J. Org. Chem., 2019, 2401–2408 CrossRef CAS.
-
(a) E. Kirillov, L. Lavanant, C. Thomas, T. Roisnel, Y. Chi and J.-F. Carpentier, Chem.–Eur. J., 2007, 13, 923–935 CrossRef CAS PubMed;
(b) L. Lavanant, T.-Y. Chou, Y. Chi, C. W. Lehmann, L. Toupet and J.-F. Carpentier, Organometallics, 2004, 23, 5450–5458 CrossRef CAS;
(c) T. Tsukahara, D. C. Swenson and R. F. Jordan, Organometallics, 1997, 16, 3303–3313 CrossRef CAS.
- M. P. Doyle, W. Hu, I. M. Phillips, C. J. Moody, A. G. Pepper and A. M. Z. Slawin, Adv. Synth. Catal., 2001, 343, 112–117 CrossRef CAS.
- T. Remarchuk and E. J. Corey, Tetrahedron Lett., 2018, 59, 2256–2259 CrossRef CAS.
-
(a) Z. Li, K. R. Conser and E. N. Jacobsen, J. Am. Chem. Soc., 1993, 115, 5326–5327 CrossRef CAS;
(b) Z. Li, R. W. Quan and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5889–5890 CrossRef CAS;
(c) L. A. Dakin, S. E. Schaus, E. N. Jacobsen and J. S. Panek, Tetrahedron Lett., 1998, 39, 8947–8950 CrossRef CAS.
- D. A. Evans, T. Lectka and S. J. Miller, Tetrahedron Lett., 1993, 34, 7027–7030 CrossRef CAS.
- V. Carreras, C. Besnard, V. Gandon and T. Ollevier, Org. Lett., 2019, 21, 9094–9098 CrossRef CAS PubMed.
-
(a) B. M. Trost, D. L. Van Vranken and C. Bingel, J. Am. Chem. Soc., 1992, 114, 9327–9343 CrossRef CAS;
(b) R. C. Verboom, B. J. Plietker and J.-E. Bäckvall, J. Organomet. Chem., 2003, 687, 508–517 CrossRef CAS.
- H. Tye, C. Eldred and M. Wills, Tetrahedron Lett., 2002, 43, 155–158 CrossRef CAS.
- V. Devannah, R. Sharma and D. A. Watson, J. Am. Chem. Soc., 2019, 141, 8436–8440 CrossRef CAS PubMed.
- J. Y. Mang, D. G. Kwon and D. Y. Kim, J. Fluorine Chem., 2009, 130, 259–262 CrossRef CAS.
- A. N. Reznikov, L. E. Kapranov, V. V. Ivankina, A. E. Sibiryakova, V. B. Rybakov and Y. N. Klimochkina, Helv. Chim. Acta, 2018, 101, e1800170 CrossRef.
- I. Shepperson, S. Quici, G. Pozzi, M. Nicoletti and D. O'Hagan, Eur. J. Org. Chem., 2004, 4545–4551 CrossRef CAS.
- J. Bayardon, D. Maillard, G. Pozzi and D. Sinou, Tetrahedron: Asymmetry, 2004, 15, 2633–2640 CrossRef CAS.
- J. Bayardon, D. Sinou, O. Holczknecht, L. Mercs and G. Pozzi, Tetrahedron: Asymmetry, 2005, 16, 2319–2327 CrossRef CAS.
- H. Kim, Y. Nguyen, C. P.-H. Yen, L. Chagal, A. J. Lough, B. M. Kim and J. Chin, J. Am. Chem. Soc., 2008, 130, 12184–12191 CrossRef CAS PubMed.
-
(a) C. M. Marson and R. C. Melling, J. Org. Chem., 2005, 70, 9771–9779 CrossRef CAS PubMed;
(b) C. M. Marson and R. C. Melling, Chem. Commun., 1998, 1223–1224 RSC.
- H. Liu, S. Liu, H. Zhou, Q. Liu and C. Wang, RSC Adv., 2018, 8, 14829–14832 RSC.
- W. Ma, J. Zhang, C. Xu, F. Chen, Y.-M. He and Q.-H. Fan, Angew. Chem. Int. Ed., 2016, 55, 12891–12894 CrossRef CAS PubMed.
-
(a) W.-W. Wang and Q.-R. Wang, Chem. Commun., 2010, 46, 4616–4618 RSC;
(b) W.-W. Wang, Z.-M. Li, L. Su, Q.-R. Wang and Y.-L. Wu, J. Mol. Catal. A: Chem., 2014, 387, 92–102 CrossRef CAS.
- C. Xu, L. Zhang, C. Dong, J. Xu, Y. Pan, Y. Li, H. Zhang, H. Li, Z. Yu and L. Xu, Adv. Synth. Catal., 2016, 358, 567–572 CrossRef CAS.
-
(a) B. He, P. Phansavath and V. Ratovelomanana-Vidal, Org. Chem. Front., 2020, 7, 975–979 RSC;
(b) B. He, L.-S. Zheng, P. Phansavath and V. Ratovelomanana-Vidal, ChemSusChem, 2019, 12, 3032–3036 CrossRef CAS PubMed;
(c) L.-S. Zheng, Q. Llopis, P.-G. Echeverria, C. Férard, G. Guillamot, P. Phansavath and V. Ratovelomanana-Vidal, J. Org. Chem., 2017, 82, 5607–5615 CrossRef CAS PubMed;
(d) L.-S. Zheng, C. Férard, P. Phansavath and V. Ratovelomanana-Vidal, Chem. Commun., 2018, 54, 283–286 RSC.
- X.-T. Li, Q.-S. Gu, X.-Y. Dong, X. Meng and X.-Y. Liu, Angew. Chem. Int. Ed., 2018, 57, 7668–7672 CrossRef CAS PubMed.
-
(a) Y. Kawashima, T. Ezawa, M. Yamamura, T. Harada, T. Noguchi, T. Miura and N. Imai, Tetrahedron, 2015, 71, 8585–8592 CrossRef CAS;
(b) T. Miura, K. Itoh, Y. Yasaku, N. Koyata, Y. Murakami and N. Imai, Tetrahedron Lett., 2008, 49, 5813–5815 CrossRef CAS;
(c) N. Imai, K. Sakamoto, M. Maeda, K. Kouge, K. Yoshizane and J. Nokami, Tetrahedron Lett., 1997, 38, 1423–1426 CrossRef CAS.
- S. Nakamura, K. Hyodo, Y. Nakamura, N. Shibata and T. Toru, Adv. Synth. Catal., 2008, 350, 1443–1448 CrossRef CAS.
- J.-S. Poh, S. Makai, T. von Keutz, D. N. Tran, C. Battilocchio, P. Pasau and S. V. Ley, Angew. Chem. Int. Ed., 2017, 56, 1864–1868 CrossRef CAS PubMed.
- A. T. Parsons and J. S. Johnson, J. Am. Chem. Soc., 2009, 131, 3122–3123 CrossRef CAS PubMed.
- T. Fan, H.-C. Shen, Z.-Y. Han and L.-Z. Gong, Chin. J. Chem., 2019, 37, 226–232 CrossRef CAS.
-
(a) S. Abbina and G. Du, Organometallics, 2012, 31, 7394–7403 CrossRef CAS;
(b) R. Ma, J. Young, R. Promontorio, F. M. Dannheim, C. C. Pattillo and M. C. White, J. Am. Chem. Soc., 2019, 141, 9468–9473 CrossRef CAS PubMed.
-
(a) J. Bayardon and D. Sinou, Tetrahedron Lett., 2003, 44, 1449–1451 CrossRef CAS;
(b) J. Bayardon and D. Sinou, J. Org. Chem., 2004, 69, 3121–3128 CrossRef CAS PubMed.
- J. Bayardon, O. Holczknecht, G. Pozzi and D. Sinou, Tetrahedron: Asymmetry, 2006, 17, 1568–1572 CrossRef CAS.
- R. Kolodziuk, C. Goux-Henry and D. Sinou, Tetrahedron: Asymmetry, 2007, 18, 2782–2786 CrossRef CAS.
- J. Bayardon and D. Sinou, Tetrahedron: Asymmetry, 2005, 16, 2965–2972 CrossRef CAS.
-
(a) T. Deng and C. Cai, J. Fluorine Chem., 2013, 156, 183–186 CrossRef CAS;
(b) T. Deng, H. Wang and C. Cai, Eur. J. Org. Chem., 2014, 2014, 7259–7264 CrossRef CAS;
(c) T. Deng, H. Wang and C. Cai, J. Fluorine Chem., 2015, 169, 72–77 CrossRef CAS;
(d) T. Deng, H. Wang and C. Cai, Org. Biomol. Chem., 2014, 12, 5843–5846 RSC.
- M. Buchsteiner, L. Martinez-Rodriguez, P. Jerabek, I. Pozo, M. Patzer, N. Nöthling, C. W. Lehmann and A. Fürstner, Chem.–Eur. J., 2020, 26, 2509–2515 CrossRef CAS PubMed.
- R. Rasappan, T. Olbrich and O. Reiser, Adv. Synth. Catal., 2009, 351, 1961–1967 CrossRef CAS.
- B. Simonelli, S. Orlandi, M. Benaglia and G. Pozzi, Eur. J. Org. Chem., 2004, 2669–2673 CrossRef CAS.
- R. Annunziata, M. Benaglia, M. Cinquini, F. Cozzi and G. Pozzi, Eur. J. Org. Chem., 2003, 1191–1197 CrossRef CAS.
- K. Tani, D. C. Behenna, R. M. McFadden and B. M. Stoltz, Org. Lett., 2007, 9, 2529–2531 CrossRef CAS PubMed.
-
(a) Y. Lu, E. L. Goldstein and B. M. Stoltz, Org. Lett., 2018, 20, 5657–5660 CrossRef CAS PubMed;
(b) S. E. Shockley, J. C. Hethcox and B. M. Stoltz, Tetrahedron Lett., 2017, 58, 3341–3343 CrossRef CAS PubMed;
(c) D. E. White, I. C. Stewart, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 810–811 CrossRef CAS PubMed;
(d) N. T. McDougal, J. Streuff, H. Mukherjee, S. C. Virgil and B. M. Stoltz, Tetrahedron Lett., 2010, 51, 5550–5554 CrossRef CAS PubMed.
- Z. Hu, Y. Li, K. Liu and Q. Shen, J. Org. Chem., 2012, 77, 7957–7967 CrossRef CAS PubMed.
-
(a) M. P. Carroll, H. Müller-Bunz and P. J. Guiry, Chem. Commun., 2012, 48, 11142–11144 RSC;
(b) R. Doran, M. P. Carroll, R. Akula, B. F. Hogan, M. Martins, S. Fanning and P. J. Guiry, Chem.–Eur. J., 2014, 20, 15354–15359 CrossRef CAS PubMed.
- Z. Jiang, L. Hou, C. Ni, J. Chen, D. Wang and X. Tong, Chem. Commun., 2017, 53, 4270–4273 RSC.
- P. B. Armstrong, E. A. Dembicer, A. J. DesBois, J. T. Fitzgerald, J. K. Gehrmann, N. C. Nelson, A. L. Noble and R. C. Bunt, Organometallics, 2012, 31, 6933–6946 CrossRef CAS.
- B. Liu, Z.-M. Zhang, B. Xu, S. Xu, H.-H. Wu and J. Zhang, Adv. Synth. Catal., 2018, 360, 2144–2150 CrossRef CAS.
- T. Mino, Y. Sato, A. Saito, Y. Tanaka, H. Saotome, M. Sakamoto and T. Fujita, J. Org. Chem., 2005, 70, 7979–7984 CrossRef CAS PubMed.
- C. Bonaccorsi, S. Bachmann and A. Mezzetti, Tetrahedron: Asymmetry, 2003, 14, 845–854 CrossRef CAS.
- P. E. Sues, A. J. Lough and R. H. Morris, Organometallics, 2011, 30, 4418–4431 CrossRef CAS.
- C.-J. Wang, F. Gao and G. Liang, Org. Lett., 2008, 10, 4711–4714 CrossRef CAS PubMed.
-
(a) C.-J. Wang, G. Liang, Z.-Y. Xue and F. Gao, J. Am. Chem. Soc., 2008, 130, 17250–17251 CrossRef CAS PubMed;
(b) Q.-H. Li, M.-C. Tong, J. Li, H.-Y. Taoa and C.-J. Wang, Chem. Commun., 2011, 47, 11110–11112 RSC;
(c) T.-L. Liu, Z.-L. He, H.-Y. Tao, Y.-P. Cai and C.-J. Wang, Chem. Commun., 2011, 47, 2616–2618 RSC;
(d) Q.-H. Li, L. Wei, X. Chen and C.-J. Wang, Chem. Commun., 2013, 49, 6277–6279 RSC;
(e) J. Li, H.-Y. Tao and C.-J. Wang, Chem. Commun., 2017, 53, 1657–1659 RSC.
-
(a) C.-J. Wang, Z.-Y. Xue, G. Liang and Z. Lu, Chem. Commun., 2009, 2905–2907 RSC;
(b) G. Liang, M.-C. Tong and C.-J. Wang, Adv. Synth. Catal., 2009, 351, 3101–3106 CrossRef CAS;
(c) Z.-Y. Xue, T.-L. Liu, Z. Lu, H. Huang, H.-Y. Tao and C.-J. Wang, Chem. Commun., 2010, 46, 1727–1729 RSC.
- P. H. S. Paioti, K. A. Abboud and A. Aponick, ACS Catal., 2017, 7, 2133–2138 CrossRef CAS.
-
(a) F. S. P. Cardoso, K. A. Abboud and A. Aponick, J. Am. Chem. Soc., 2013, 135, 14548–14551 CrossRef CAS PubMed;
(b) S. Mishra, J. Liu and A. Aponick, J. Am. Chem. Soc., 2017, 139, 3352–3355 CrossRef CAS PubMed.
- X. Hu, C. Bai, H. Dai, H. Chen and Z. Zheng, J. Mol. Catal. A: Chem., 2004, 218, 107–112 CrossRef CAS.
- X. Li, X. Jia, L. Xu, S. H. L. Kok, C. W. Yip and A. S. C. Chan, Adv. Synth. Catal., 2005, 347, 1904–1908 CrossRef CAS.
- X.-X. Yan, Q. Peng, Y. Zhang, K. Zhang, W. Hong, X.-L. Hou and Y.-D. Wu, Angew. Chem. Int. Ed., 2006, 45, 1979–1983 CrossRef CAS PubMed.
- Z.-W. Lai, R.-F. Yang, K.-Y. Ye, H. S. Sun and S.-L. You, Beilstein J. Org. Chem., 2014, 10, 1261–1266 CrossRef PubMed.
-
(a) X. Bai, C. Wu, S. Ge and Y. Lu, Angew. Chem. Int. Ed., 2020, 59, 2764–2768 CrossRef CAS PubMed;
(b) D. J. Cowan, J. L. Collins, M. B. Mitchell, J. A. Ray, P. W. Sutton, A. A. Sarjeant and E. E. Boros, J. Org. Chem., 2013, 78, 12726–12734 CrossRef CAS PubMed.
-
(a) F. Brüning, H. Nagae, D. Käch, K. Mashima and A. Togni, Chem.–Eur. J., 2019, 25, 10818–10822 CrossRef PubMed;
(b) X. Feng, B. Pugin, E. Kusters, G. Sedelmeier and H.-U. Blaser, Adv. Synth. Catal., 2007, 349, 1803–1807 CrossRef CAS.
-
(a) Q. Zhao, S. Li, K. Huang, R. Wang and X. Zhang, Org. Lett., 2013, 15, 4014–4017 CrossRef CAS PubMed;
(b) Q. Zhao, J. Wen, R. Tan, K. Huang, P. Metola, R. Wang, E. V. Anslyn and X. Zhang, Angew. Chem. Int. Ed., 2014, 126, 8607–8610 CrossRef;
(c) J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Chem. Sci., 2016, 7, 3047–3051 RSC.
-
(a) T. Sturm, W. Weissensteiner and F. Spindler, Adv. Synth. Catal., 2003, 345, 160–164 CrossRef CAS;
(b) Y. Wang, T. Sturm, M. Steurer, V. B. Arion, K. Mereiter, F. Spindler and W. Weissensteiner, Organometallics, 2008, 27, 1119–1127 CrossRef CAS;
(c) A. Saito, N. Kumagai and M. Shibasaki, Angew. Chem. Int. Ed., 2017, 56, 5551–5555 CrossRef CAS PubMed.
-
(a) T. M. Konrad, J. T. Durrani, C. J. Cobley and M. L. Clarke, Chem. Commun., 2013, 49, 3306–3308 RSC;
(b) S. H. Gilbert, S. Tin, J. A. Fuentes, T. Fanjul and M. L. Clarke, Tetrahedron, 2021, 80, 131863 CrossRef CAS.
- L. Leseurre, F. Le Boucher d'Herouville, K. Püntener, M. Scalone, J.-P. Genêt and V. Michelet, Org. Lett., 2011, 13, 3250–3253 CrossRef CAS PubMed.
-
(a) T. Korenaga, K. Osaki, R. Maenishi and T. Sakai, Org. Lett., 2009, 11, 2325–2328 CrossRef CAS PubMed;
(b) T. Korenaga, R. Maenishi, K. Hayashi and T. Sakai, Adv. Synth. Catal., 2010, 352, 3247–3254 CrossRef CAS.
- J. H. Koh, A. O. Larsen and M. R. Gagné, Org. Lett., 2001, 3, 1233–1236 CrossRef CAS PubMed.
- F. Le Boucher d'Herouville, A. Millet, M. Scalone and V. Michelet, J. Org. Chem., 2011, 76, 6925–6930 CrossRef PubMed.
- M. Scalone and P. Waldmeier, Org. Process Res. Dev., 2003, 7, 418–425 CrossRef CAS.
- K. Aikawa, K. Yabuuchi, K. Torii and K. Mikami, Beilstein J. Org. Chem., 2018, 14, 576–582 CrossRef CAS PubMed.
- B. Drießen-Höscher, J. Kralik, F. Agel, C. Steffens and C. Hu, Adv. Synth. Catal., 2004, 346, 979–982 CrossRef.
-
(a) D.-Y. Zhang, C.-B. Yu, M.-C. Wang, K. Gao and Y.-G. Zhou, Tetrahedron Lett., 2012, 53, 2556–2559 CrossRef CAS;
(b) D.-Y. Zhang, D.-S. Wang, M.-C. Wang, C.-B. Yu, K. Gao and Y.-G. Zhou, Synthesis, 2011, 2796–2802 CAS.
-
(a) S. Jeulin, S. Duprat de Paule, V. Ratovelomanana-Vidal, J.-P. Genêt, N. Champion and P. Dellis, Angew. Chem. Int. Ed., 2004, 43, 320–325 CrossRef CAS PubMed;
(b) D. E. Kim, C. Choi, I. S. Kim, S. Jeulin, V. Ratovelomanana-Vidal, J.-P. Genêt and N. Jeong, Adv. Synth. Catal., 2007, 349, 1999–2006 CrossRef CAS;
(c) Z. Yan, B. Wu, X. Gao, M.-W. Chen and Y.-G. Zhou, Org. Lett., 2016, 18, 692–695 CrossRef CAS PubMed;
(d) L. Yin, L. Brewitz, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2014, 136, 17958–17961 CrossRef CAS PubMed.
- F. Berhal, O. Esseiva, C.-H. Martin, H. Tone, J.-P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Org. Lett., 2011, 13, 2806–2809 CrossRef CAS PubMed.
-
(a) H.-P. Xie, Z. Yan, B. Wu, S.-B. Hu and Y.-G. Zhou, Tetrahedron Lett., 2018, 59, 2960–2964 CrossRef CAS;
(b) S.-B. Hu, Z.-P. Chen, J. Zhou and Y.-G. Zhou, Tetrahedron Lett., 2016, 57, 1925–1929 CrossRef CAS.
- M. Murata, T. Morimoto and K. Achiwa, Synlett, 1991, 827–828 CrossRef CAS.
-
(a) N. Armanino, R. Koller and A. Togni, Organometallics, 2010, 29, 1771–1777 CrossRef CAS;
(b) D. E. Kim, C. Choi, I. S. Kim, S. Jeulin, V. Ratovelomanana-Vidal, J.-P. Genêt and N. Jeong, Synthesis, 2006, 4053–4059 CAS;
(c) N. Jeong, D. H. Kim and J. H. Choi, Chem. Commun., 2004, 1134–1135 RSC.
- Y. Nakamura, S. Takeuchi, S. Zhang, K. Okumura and Y. Ohgo, Tetrahedron Lett., 2002, 43, 3053–3056 CrossRef CAS.
-
(a) J. Horn and W. Bannwarth, Eur. J. Org. Chem., 2007, 2058–2063 CrossRef CAS;
(b) V. Andrushko, D. Schwinn, C. C. Tzschucke, F. Michalek, J. Horn, C. Mössner and W. Bannwarth, Helv. Chim. Acta, 2005, 88, 936–949 CrossRef CAS.
-
(a) E. G. Hope, A. M. Stuart and A. J. West, Green Chem., 2004, 6, 345–350 RSC;
(b) D. J. Birdsall, E. G. Hope, A. M. Stuart, W. Chen, Y. Hu and J. Xiao, Tetrahedron Lett., 2001, 42, 8551–8553 CrossRef CAS.
- J. Bayardon, M. Cavazzini, D. Maillard, G. Pozzi, S. Quici and D. Sinou, Tetrahedron: Asymmetry, 2003, 14, 2215–2224 CrossRef CAS.
-
(a) G. Franciò, K. Wittmann and W. Leitner, J. Organomet. Chem., 2001, 621, 130–142 CrossRef;
(b) G. Franciò and W. Leitner, Chem. Commun., 1999, 1663–1664 RSC.
- D. Bonafoux, Z. Hua, B. Wang and I. Ojima, J. Fluorine Chem., 2001, 112, 101–108 CrossRef CAS.
-
(a) L. Wang, W. Meng, C.-L. Zhu, Y. Zheng, J. Nie and J.-A. Ma, Angew. Chem. Int. Ed., 2011, 50, 9442–9446 CrossRef CAS PubMed;
(b) P.-S. Wang, P. Liu, Y.-J. Zhai, H.-C. Lin, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 12732–12735 CrossRef CAS PubMed;
(c) P.-S. Wang, M.-L. Shen, T.-C. Wang, H.-C. Lin and L.-Z. Gong, Angew. Chem. Int. Ed., 2017, 56, 16032–16036 CrossRef CAS PubMed;
(d) H.-C. Lin, P.-S. Wang, Z.-L. Tao, Y.-G. Chen, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2016, 138, 14354–14361 CrossRef CAS PubMed.
- F.-N. Sun, W.-C. Yang, X.-B. Chen, Y.-L. Sun, J. Cao, Z. Xu and L.-W. Xu, Chem. Sci., 2019, 10, 7579–7583 RSC.
- M. V. Escárcega-Bobadilla, L. Rodríguez-Pérez, E. Teuma, P. Serp, A. M. Masdeu-Bultó and M. Gómez, Catal. Lett., 2011, 141, 808–816 CrossRef.
-
(a) T. Yamamoto, Y. Akai, Y.-U. Nagata and M. Suginome, Angew. Chem. Int. Ed., 2011, 50, 8844–8847 CrossRef CAS PubMed;
(b) H. Yamamoto and M. Suginome, Angew. Chem. Int. Ed., 2009, 48, 539–542 CrossRef PubMed.
- A. Klose and J. A. Gladysz, Tetrahedron: Asymmetry, 1999, 10, 2665–2674 CrossRef CAS.
- U. Nettekoven, M. Widhalm, H. Kalchhauser, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, J. Org. Chem., 2001, 66, 759–770 CrossRef CAS PubMed.
- B. J. Anderson, D. S. Glueck, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2008, 27, 4992–5001 CrossRef CAS.
- T. V. RajanBabu, T. A. Ayers, G. A. Halliday, K. K. You and J. C. Calabrese, J. Org. Chem., 1997, 62, 6012–6028 CrossRef CAS.
- L. B. Fields and E. N. Jacobsen, Tetrahedron: Asymmetry, 1993, 4, 2229–2240 CrossRef CAS.
- M. Cavazzini, G. Pozzi, S. Quici, D. Maillard and D. Sinou, Chem. Commun., 2001, 1220–1221 RSC.
- S. Handa and L. M. Slaughter, Angew. Chem. Int. Ed., 2012, 51, 2912–2915 CrossRef CAS PubMed.
- K. B. Selim, H. Nakanishi, Y. Matsumoto, Y. Yamamoto, K.-i. Yamada and K. Tomioka, J. Org. Chem., 2011, 76, 1398–1408 CrossRef CAS PubMed.
-
(a) A. M. Sorlin, J. C. Mixdorf, M. E. Rotella, R. T. Martin, O. Gutierrez and H. M. Nguyen, J. Am. Chem. Soc., 2019, 141, 14843–14852 CrossRef CAS PubMed;
(b) J. C. Mixdorf, A. M. Sorlin, Q. Zhang and H. M. Nguyen, ACS Catal., 2018, 8, 790–801 CrossRef CAS;
(c) Q. Zhang, D. P. Stockdale, J. C. Mixdorf, J. J. Topczewski and H. M. Nguyen, J. Am. Chem. Soc., 2015, 137, 11912–11915 CrossRef CAS PubMed.
-
(a) R. Takeshi and T. Nishimura, Org. Biomol. Chem., 2015, 13, 4918–4924 RSC;
(b) T. Nishimura, H. Kumamoto, M. Nagaosa and T. Hayashi, Chem. Commun., 2009, 5713–5715 RSC.
-
(a) T. Nishimura, J. Wang, M. Nagaosa, K. Okamoto, R. Shintani, F.-y. Kwong, W.-y. Yu, A. S. C. Chan and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 464–465 CrossRef CAS PubMed;
(b) T. Nishimura, M. Nagaosa and T. Hayashi, Chem. Lett., 2008, 37, 860–861 CrossRef CAS;
(c) T. Nishimura, Y. Yasuhara, T. Sawano and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 7872–7873 CrossRef CAS PubMed.
-
(a) M. Nagamoto, T. Yanagi, T. Nishimura and H. Yorimitsu, Org. Lett., 2016, 18, 4474–4477 CrossRef CAS PubMed;
(b) T. Nishimura, Y. Yasuhara, M. Nagaosa and T. Hayashi, Tetrahedron: Asymmetry, 2008, 19, 1778–1783 CrossRef CAS.
- T. Nishimura, Y. Ichikawa, T. Hayashi, N. Onishi, M. Shiotsuki and T. Masuda, Organometallics, 2009, 28, 4890–4893 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Thierry
Ollevier
and
Thierry
Ollevier