
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBisulfite-free and single-nucleotide resolution sequencing of DNA epigenetic modification of 5-hydroxymethylcytosine using engineered deaminase†
Neng-Bin
Xie‡
ab,
Min
Wang‡
a,
Tong-Tong
Ji
a,
Xia
Guo
a,
Jiang-Hui
Ding
a,
Bi-Feng
Yuan
 *abc and
Yu-Qi
Feng
ab
*abc and
Yu-Qi
Feng
ab
aSauvage Center for Molecular Sciences, Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: bfyuan@whu.edu.cn
bSchool of Public Health, Wuhan University, Wuhan 430071, China
cCancer Precision Diagnosis and Treatment and Translational Medicine Hubei Engineering Research Center, Zhongnan Hospital of Wuhan University, Wuhan 430071, China
First published on 26th May 2022
Abstract
The discovery of 5-hydroxymethylcytosine (5hmC) in mammalian genomes is a landmark in epigenomics study. Similar to 5-methylcytosine (5mC), 5hmC is viewed as a critical epigenetic modification. Deciphering the functions of 5hmC necessitates the location analysis of 5hmC in genomes. Here, we proposed an engineered deaminase-mediated sequencing (EDM-seq) method for the quantitative detection of 5hmC in DNA at single-nucleotide resolution. This method capitalizes on the engineered human apolipoprotein B mRNA-editing catalytic polypeptide-like 3A (A3A) protein to produce differential deamination activity toward cytosine, 5mC, and 5hmC. In EDM-seq, the engineered A3A (eA3A) protein can deaminate C and 5mC but not 5hmC. The original C and 5mC in DNA are deaminated by eA3A to form U and T, both of which are read as T during sequencing, while 5hmC is resistant to deamination by eA3A and is still read as C during sequencing. Therefore, the remaining C in the sequence manifests the original 5hmC. By EDM-seq, we achieved the quantitative detection of 5hmC in genomic DNA of lung cancer tissue. The EDM-seq method is bisulfite-free and does not require DNA glycosylation or chemical treatment, which offers a valuable tool for the straightforward and quantitative detection of 5hmC in DNA at single-nucleotide resolution.
Introduction
DNA cytosine methylation (5-methylcytosine, 5mC) is the most critical epigenetic modification that plays sophisticated roles in regulating gene expression.1 5mC modification in DNA is dynamic and reversible.2 In 2009, the discovery of 5-hydroxymethylcytosine (5hmC) in mammalian genomes uncovered the mechanism of active DNA demethylation in mammals.3,4 It has been established that ten–eleven translocation (TET) proteins can oxidize 5mC to 5hmC, 5-formylcytosine (5fC), and 5-carboxycytosine (5caC).5 The resulting 5fC and 5caC undergo base excision repair or direct deformylation or decarboxylation to restore unmodified cytosines.5–9The discovery of 5hmC in mammalian genomes is a landmark in epigenomics study.10 Similar to 5mC in DNA, 5hmC is also viewed as a vital epigenetic marker that could modulate gene expression.11 A growing number of studies have demonstrated that 5hmC plays important roles in diverse biological processes, such as tumorigenesis and embryogenesis.12–14 Some methods have been developed to detect 5hmC in genomic DNA, including thin layer chromatography detection,3,4 liquid chromatography or capillary electrophoresis with mass spectrometry (LC-MS or CE-MS) analysis.15–25 In these methods, genomic DNA is normally digested to nucleosides or nucleotides followed by analysis with different platforms, which mainly provides the qualitative and quantitative detection of 5hmC.
Revealing the functions of 5hmC in DNA necessitates the location analysis of 5hmC in genomes.26–29 Several techniques have been developed to map 5hmC in DNA. Affinity enrichment-based purification followed by sequencing methods has been established to map 5hmC in genomes.30–32 However, these methods cannot precisely map 5hmC in DNA at single-nucleotide resolution.33 Afterwards, oxidative bisulfite sequencing (oxBS-seq)34 and TET-assisted bisulfite sequencing (TAB-seq)35 methods were established to detect 5hmC in DNA at single-nucleotide resolution. A limitation of these methods is the use of bisulfite because the harsh conditions for chemical treatment can lead to the degradation of input DNA as much as 99.9%.36 Detection of 5hmC through single-molecule, real-time (SMRT) sequencing is possible.37 However, SMRT sequencing currently has a relatively high rate of false-positive mapping of modified nucleobases.38
It has been reported that the A3A (apolipoprotein B mRNA-editing catalytic polypeptide-like 3A) protein can efficiently deaminate cytosine (C), 5mC and 5hmC in DNA but shows no deamination activity toward glycosylated 5hmC (β-glucosyl-5-hydroxymethyl-2′-deoxycytidine, 5gmC).39,40 Using this property of A3A, we and others reported A3A-mediated deamination sequencing (AMD-seq)40 and A3A-coupled epigenetic sequencing (ACE-seq)41 for mapping 5hmC in DNA at single-base resolution. In these approaches, 5hmC is first glycosylated to produce 5gmC by using β-glucosyltransferase (β-GT). Upon A3A treatment, C and 5mC in DNA are converted to U and T, respectively. Both U and T are read as T during sequencing, while 5gmC is resistant to the deamination by A3A and read as C during sequencing. These methods, however, require the pre-treatment of DNA with β-GT to glycosylate 5hmC.
In the current study, we generated a series of engineered A3A proteins from the wild-type A3A (wtA3A) protein. With the characterized unique deamination properties of engineered A3A proteins, we developed an engineered deaminase-mediated sequencing (EDM-seq) method for the quantitative detection of 5hmC at single-nucleotide resolution. The EDM-seq method is straightforward and does not require chemical treatment or glycosylation by β-GT. By the proposed EDM-seq, we achieved the direct and quantitative detection of 5hmC in genomic DNA of lung cancer tissue and the corresponding adjacent normal tissue.
Experimental methods
Materials and reagents
The 24-mer C-containing DNA (GC-C, AC-C, TC-C and CC-C), 24-mer 5mC-containing DNA (GC-5mC, AC-5mC, TC-5mC and CC-5mC), and 24-mer 5hmC-containing DNA (GC-5hmC, AC-5hmC, TC-5hmC and CC-5hmC) were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). The sequences of these oligonucleotides are listed in ESI Table S1.† 2′-Deoxycytidine (dC), 2′-deoxyguanosine (dG), 2′-deoxyadenosine (dA), thymidine (dT), 2′-deoxynucleoside 5′-triphosphates (dATP, dCTP, dGTP, and TTP) and phosphodiesterase I were purchased from Sigma-Aldrich (St. Louis, MO, USA). 5-Hydroxymethyl-2′-deoxycytidine-5′-triphosphate (5hmdCTP) and 5-methyl-2′-deoxycytidine-5′-triphosphate (5mdCTP) were purchased from TriLink BioTechnologies (San Diego, CA, USA). DNase I, S1 nuclease and alkaline phosphatase (CIAP) were purchased from Takara Biotechnology Co. Ltd. (Dalian, China). The lung cancer tissue and the corresponding adjacent normal tissue were collected from Hubei Cancer Hospital (Wuhan, China). All experiments were conducted in accordance with the guidelines and regulations of the Ethics Committee of Hubei Cancer Hospital.Preparation of double-stranded DNA with C, 5mC and 5hmC
Three kinds of 224-bp double-stranded DNA (dsDNA) substrates (DNA-C, DNA-5mC, and DNA-5hmC) and two kinds of 234-bp dsDNA substrates (DNA-C5mC and DNA-C5hmC) were prepared as the standards for the method development. The detailed sequence information can be found in ESI Table S2.† As for the preparation of DNA-C, 0.5 ng of synthetic DNA (Takara) was used as the template for PCR amplification. PCR amplification was carried out in a 50 μL reaction solution including 1 U of Q5 DNA polymerase (New England Biolabs), 5 μL of 10× reaction buffer, 4 μL of dATP, dGTP, TTP, and dCTP (2.5 mM for each), 2 μL of 10 μM forward primer (5′-GAGTGACGCTGAGCTTGACGTCGCGC-3′), and 2 μL of 10 μM reverse primer (5′-ATCCTCTCCAACATTCCACTAACAATTA-3′). DNA-5mC and DNA-5hmC were prepared by PCR amplification with dCTP being replaced by 5mdCTP or 5hmdCTP, respectively. As for the DNA-5mC and DNA-5hmC, all the cytosines were replaced by 5mC or 5hmC, respectively (except for the cytosines in PCR primers). The PCR reaction consisted of 95 °C for 5 min, 30 cycles of 95 °C for 1 min, 60 °C for 1 min, and 72 °C for 1 min, followed by 72 °C for 10 min. As for the preparation of DNA-C5mC and DNA-C5hmC, 0.5 ng of synthetic DNA (Takara) was used as the template for PCR amplification. PCR amplification was carried out under the same conditions as those for the preparation of DNA-C except that the forward primer was replaced by primer CX (5′-GAGTGACGCTGAGCTTGACGTCGCGCGTC-3′) and dCTP was replaced by 5mdCTP or 5hmdCTP, respectively. The PCR products were separated by agarose gel electrophoresis and recovered using a Gel Extraction kit (Omega Bio-Tek Inc., Norcross, GA, USA).Expression and purification of engineered A3A proteins
The full-length coding sequence of the wild-type A3A (wtA3A) protein or engineered A3A (eA3A) protein was cloned into the pET-41a(+) plasmid, which was transformed into the Escherichia coli (E. coli) BL21(DE3) pLysS strain. These recombinant proteins carried the human rhinovirus 3C protease (HRV 3C) site between the glutathione S-transferase (GST) tag and the wtA3A protein or eA3A protein. The transformed E. coli cells were grown in LB medium (tryptone 10 g L−1, yeast extract 5 g L−1, and NaCl 10 g L−1) at 37 °C supplemented with kanamycin (10 μg mL−1) and chloramphenicol (10 μg mL−1). The culture of bacteria was carried out under shaking at 180 rpm. Isopropyl-β-D-thiogalactoside (IPTG) was added to the medium with a final concentration of 0.5 mM when the OD600nm of E. coli cell suspension reached 0.6. The expression of recombinant proteins was induced at 25 °C for 20 h. Then the E. coli cells were harvested by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min. Cell pellets were lysed by sonication and then centrifuged at 12000 g at 4 °C for 10 min. The obtained supernatant was incubated with Glutathione Sepharose™ 4B beads (Sangon, Shanghai, China) according to the manufacturer's recommended procedure. After digestion with HRV 3C protease (Sangon, Shanghai, China), the wtA3A or eA3A protein was further purified with a size-exclusion column (Millipore, Darmstadt, Germany) and equilibrated with a storage solution containing 50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 0.01 mM EDTA, 0.5 mM dithiothreitol, and 0.01% Tween-20. The purified proteins were determined by SDS-PAGE and stored at −80 °C before use (the representative SDS-PAGE for wtA3A, eA3A-5 and eA3A-9 is shown in ESI Fig. S1†). The concentrations of purified proteins were quantified using the BCA protein assay kit (Beyotime, Shanghai, China).
000g for 5 min. Cell pellets were lysed by sonication and then centrifuged at 12000 g at 4 °C for 10 min. The obtained supernatant was incubated with Glutathione Sepharose™ 4B beads (Sangon, Shanghai, China) according to the manufacturer's recommended procedure. After digestion with HRV 3C protease (Sangon, Shanghai, China), the wtA3A or eA3A protein was further purified with a size-exclusion column (Millipore, Darmstadt, Germany) and equilibrated with a storage solution containing 50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 0.01 mM EDTA, 0.5 mM dithiothreitol, and 0.01% Tween-20. The purified proteins were determined by SDS-PAGE and stored at −80 °C before use (the representative SDS-PAGE for wtA3A, eA3A-5 and eA3A-9 is shown in ESI Fig. S1†). The concentrations of purified proteins were quantified using the BCA protein assay kit (Beyotime, Shanghai, China).
Deamination assay for single-stranded DNA
The 24-mer C-containing DNA (GC-C, AC-C, TC-C and CC-C), 5mC-containing DNA (GC-5mC, AC-5mC, TC-5mC and CC-5mC), and 5hmC-containing DNA (GC-5hmC, AC-5hmC, TC-5hmC and CC-5hmC) were used as the substrates to evaluate the deaminase activity of wtA3A and eA3A proteins toward C, 5mC and 5hmC. Typically, 60 ng of the oligonucleotide was incubated with different concentrations of the wtA3A or eA3A protein in a 20 μL solution of 20 mM MES (pH 6.5), 0.1% Triton X-100, and 2 μL of DMSO at 37 °C for 2 h. The deamination reaction was terminated by incubating at 95 °C for 10 min. The deamination rate of wtA3A and eA3A proteins was determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.Deamination assay for double-stranded DNA
Three kinds of 224-bp dsDNA (DNA-C, DNA-5mC, and DNA-5hmC) were used as the substrates to evaluate the deaminase activity of wtA3A and eA3A proteins. Typically, 60 ng of the dsDNA substrate was first denatured to single-stranded (ssDNA) by heating at 95 °C for 10 min and then chilling at 0 °C for 5 min. The denatured DNA was treated with 20 μM of the wtA3A or eA3A protein. The deamination reaction was carried out at 37 °C for 2 h in a 20 μL solution of 20 mM MES (pH 6.5), 0.1% Triton X-100, and 2 μL of DMSO. The deamination reaction was quenched by heating at 95 °C for 10 min. Deaminase-treated DNA was then amplified by PCR using EpiMark® Hot Start Taq DNA polymerase (New England Biolabs). PCR amplification was carried out with initial denaturation at 95 °C for 3 min and 25 cycles of 95 °C for 30 s, 55 °C for 1 min and 68 °C for 1 min, followed by 5 min of elongation at 68 °C. 20 pmol of forward primer (5′-GAGTGATGTTGAGTTTGATGTTGTGT-3′) and reverse primer (5′-CTCCAACATTCCACTAACAATTACTCTCT-3′) were used for PCR amplification. The resulting PCR products were subjected to Sanger sequencing and colony sequencing (TsingKe). Colony sequencing was carried out according to previous reports.41,42 The detailed procedure of colony sequencing can be found in the ESI.†Enzymatic digestion of DNA
The enzymatic digestion of DNA was carried out under neutral conditions according to a previous report.43,44 Briefly, a 50 μL mixture containing 1 μg of DNA, 2.5 U of DNase I, 0.125 U of phosphodiesterase I, 90 U of S1 nuclease, 7.5 U of CIAP and 5 μL of reaction buffer (50 mM Tris–HCl, pH 7.0, 10 mM NaCl, 1 mM MgCl2, and 1 mM ZnSO4) was incubated at 37 °C for 6 h. The digested samples were extracted with chloroform twice. The aqueous layer was lyophilized to dryness and reconstituted in 50 μL H2O followed by LC-MS/MS analysis.LC-MS/MS analysis
LC-MS/MS analysis of nucleosides was carried out on a LC-MS/MS system consisting of an AB 3200 QTRAP mass spectrometer (Applied Biosystems, Foster City, CA, USA) and a Shimadzu LC-20AD HPLC (Tokyo, Japan). LC separation was conducted on a Hisep C18-T column (150 mm × 2.1 mm i.d., 5 μm, Weltech Co., Ltd., Wuhan, China) with a flow rate of 0.2 mL min−1 at 35 °C. Water (solvent A) and methanol (solvent B) were employed as mobile phases with a gradient of 5–50% B for 25 min. Mass spectrometry detection was carried out in the positive electrospray ionization mode, and nucleosides were monitored in the multiple reaction monitoring (MRM) mode. Mass transitions (precursor ions → product ions) of dC (228.1 → 112.1), dT (243.1 → 127.1), dA (252.1 → 136.1), dG (268.1 → 152.1), 5mC (242.1 → 126.1), 5hmC (258.1 →142.1), and 5gmC (268.1 → 142.1) were utilized to determine these nucleosides.Quantitative and site-specific detection of 5hmC at individual sites in genomic DNA by EDM-seq
Genomic DNA of lung cancer tissue and the corresponding normal tissue was extracted using the tissue DNA kit (Omega Bio-Tek Inc., Norcross, GA, USA) according to the manufacturer's recommended procedure. The genomic DNA was fragmented into an average size of 500 bp using a JY92-II N Ultrasonic Homogenizer (Scientz) with the following settings: 130 W peak incident power for 30 cycles (1 cycle = 5 s on and 5 s off). As for the EDM-seq, the fragment DNA (60 ng) was denatured and treated with 20 μM of the eA3A protein. As for the AMD-seq method, 1 μg of the fragment DNA was treated with β-GT (New England Biolabs) at 37 °C for 2 h followed by purification using 0.9 × KAPA pure beads (Roche). The resulting DNA (60 ng) was denatured and treated with 20 μM of wtA3A. The deaminase-treated DNA was amplified using site-specific primers (ESI Table S3†). The PCR products were subjected to Sanger sequencing and colony sequencing. As for colony sequencing, 50 clones for each sample were randomly picked up and subjected to sequencing.Results and discussion
Principle of the engineered deaminase-mediated sequencing (EDM-seq)
In the current study, we aimed to establish a straightforward method for quantitative detection of 5hmC at single-nucleotide resolution. We and others previously demonstrated that the wtA3A protein could deaminate C, 5mC, and 5hmC in DNA to generate U, T, and 5-hydroxymethyluracil, respectively (ESI Fig. S2†).40,41 However, wtA3A shows weaker deamination activity toward 5hmC than toward C and 5mC.39It has been reported that the neighbouring 5′ nucleobase of cytosine may affect the deamination activity of wtA3A toward cytosine.45 Here we further evaluated the readouts of C, 5mC, and 5hmC in different sequence contexts of GC, AC, TC and CC sites after wtA3A treatment. In this respect, three 224-bp dsDNA (DNA-C, DNA-5mC, and DNA-5hmC) were utilized for the evaluation (ESI Table S2†). The dsDNA was denatured to single-stranded DNA (ssDNA) and treated with wtA3A. The resulting DNA was amplified and subjected to Sanger sequencing. It can be seen that all the C in DNA-C and all the 5mC in DNA-5mC were read as T after wtA3A treatment (ESI Fig. S3†). However, 5hmC in TC and CC sites was read as T; while 5hmC in GC and AC sites was partially read as T and partially read as C (ESI Fig. S3†), indicating that wtA3A could fully deaminate 5hmC in TC and CC sites and partially deaminate 5hmC in GC and AC sites.
The property of the wtA3A protein that exhibits weaker deamination activity toward 5hmC than toward C and 5mC inspired us to engineer wtA3A to further increase the deamination selectivity of wtA3A toward 5hmC and C/5mC. Along this line, we expect that an engineered A3A (eA3A) protein can deaminate C and 5mC but not 5hmC. In this respect, the original C and 5mC in DNA will be deaminated by eA3A to form U and T, both of which are read as T during sequencing, while 5hmC is resistant to the deamination by eA3A and is still read as C during sequencing (Fig. 1). Therefore, the remaining C in the sequence manifests the original 5hmC, which offers the single-nucleotide resolution detection of 5hmC in DNA (Fig. 1).
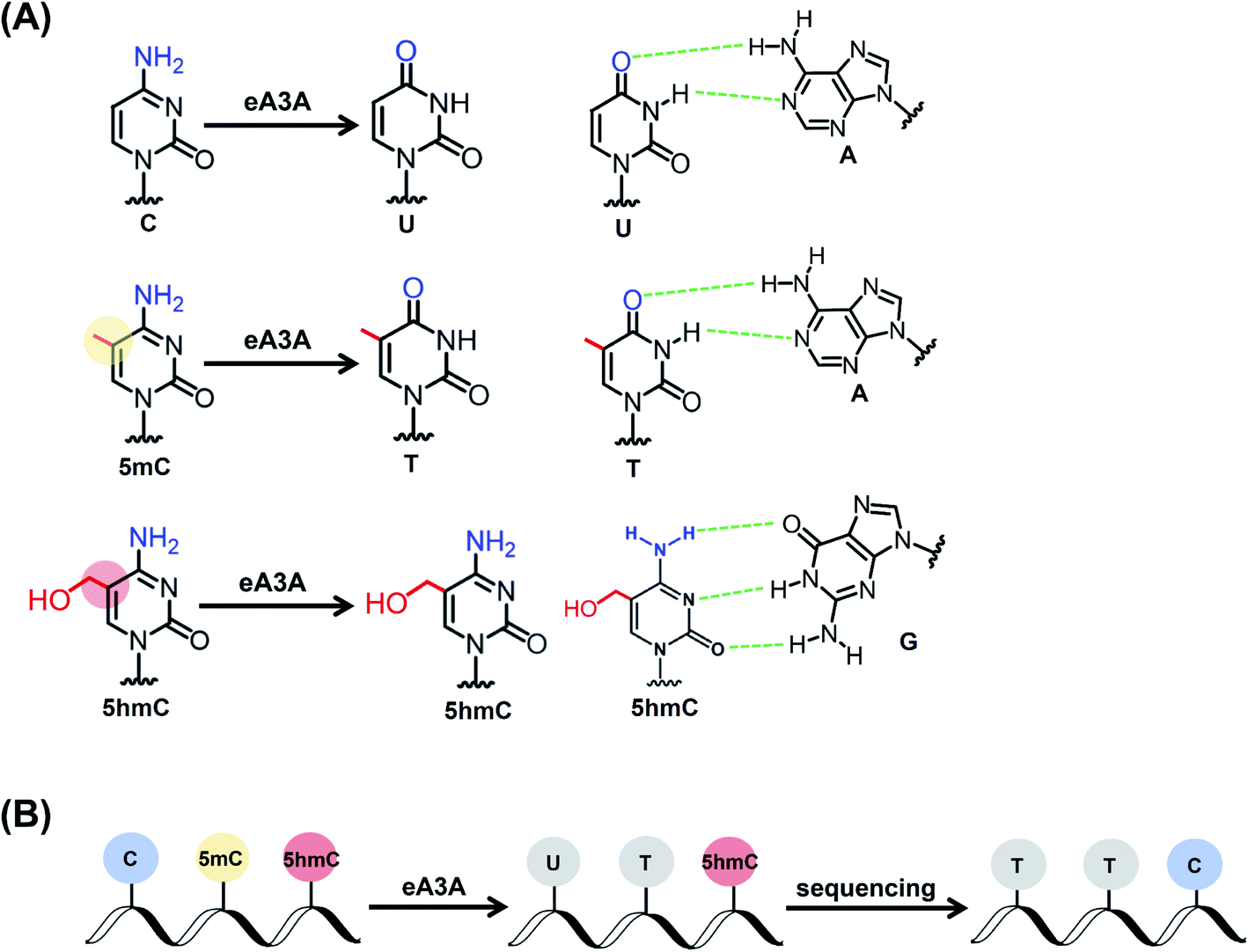 | ||
| Fig. 1 Principle of the EDM-seq method. (A) C and 5mC can be deaminated by the eA3A protein to form U and T, respectively. Both U and T base pair with A. 5hmC is resistant to the deamination by the eA3A protein and still base pairs with G. (B) In EDM-seq, C and 5mC are deaminated to form U and T, both of which will be read as T during sequencing. However, 5hmC is not deaminated by the eA3A protein and still read as C during sequencing. Thus, the remaining C manifests the original 5hmC in DNA. | ||
Engineered A3A proteins
According to the crystal structure of wtA3A, the amino acid residues 31 (threonine, T) and 130 (tyrosine, Y) contribute to the cytosine positioning by directly interacting with the pyrimidine ring.46 We speculate that the residues T31 and Y130 should be critical for the deamination capability of the wtA3A protein toward C, 5mC and 5hmC. Indeed, alteration of the residue T31 or Y130 reduces the deamination activity of the wtA3A protein toward C.47 Previous reports demonstrated that loop 1 and loop 7 of wtA3A played crucial roles in the intrinsic substrate preference of wtA3A.45,47 We therefore engineered a subset of residues around loop 1 and loop 7 of wtA3A and generated a series of engineered A3A proteins (eA3A-1 to eA3A-9, Fig. 2A). The deamination activities toward C, 5mC, and 5hmC by these eA3A proteins were evaluated using three 224-bp dsDNA (DNA-C, DNA-5mC, and DNA-5hmC).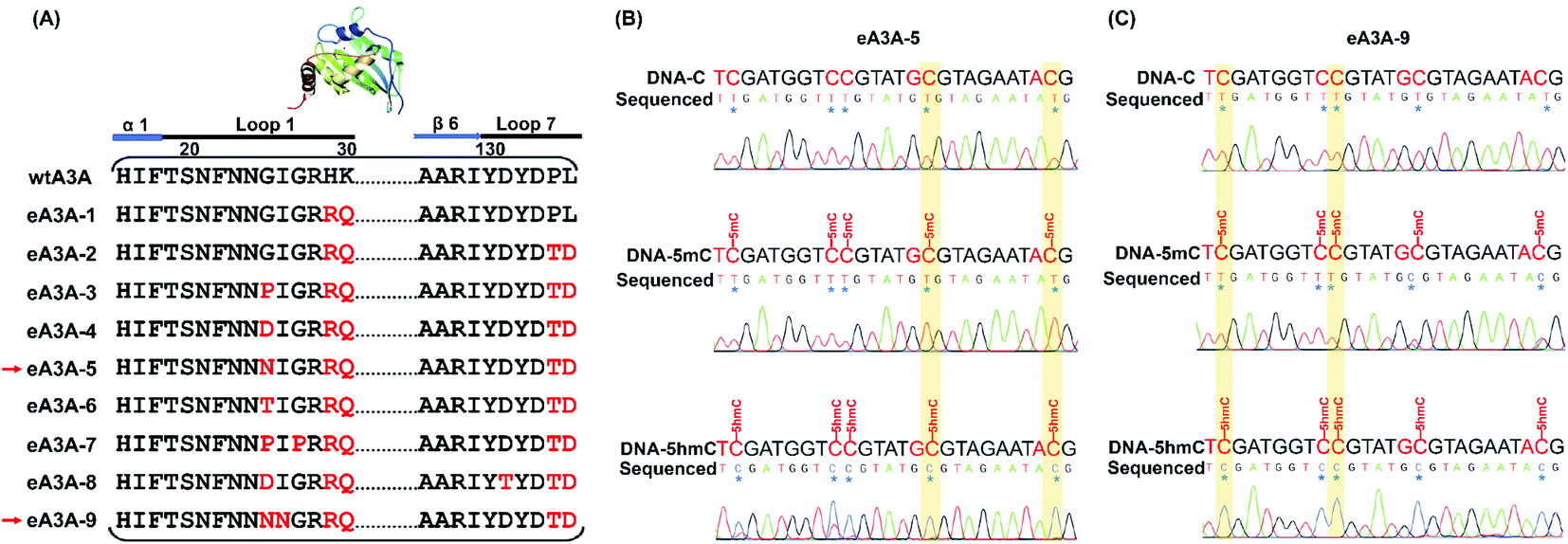 | ||
| Fig. 2 Characterization of the deaminase selectivity of eA3A-5 and eA3A-9 toward C, 5mC and 5hmC in different sequence contexts by Sanger sequencing. (A) The amino acid composition of wtA3A and eA3A proteins in the loop 1 and loop 7 regions. (B) The sequencing results of three 224-bp dsDNA (DNA-C, DNA-5mC, and DNA-5hmC) after eA3A-5 treatment. In GC and AC sites, all the C and 5mC were deaminated by eA3A-5 and then read as T; but all the 5hmC were resistant to deamination by eA3A-5 and still read as C. (C) The sequencing results of three 224-bp dsDNA (DNA-C, DNA-5mC, and DNA-5hmC) after eA3A-9 treatment. In TC and CC sites, all the C and 5mC were deaminated by eA3A-9 and then read as T; but all the 5hmC was resistant to deamination by eA3A-9 and was still read as C. | ||
We initially obtained eA3A protein 1 (eA3A-1) with arginine (R) and glutamine (Q) replacing residues 29 (histidine, H) and 30 (lysine, K) of wtA3A (Fig. 2A). The Sanger sequencing results showed that eA3A-1 could readily deaminate C but partially deaminate 5mC and 5hmC (ESI Fig. S4†). eA3A protein 2 (eA3A-2) was generated with P134T and L135D mutations in loop 7 of eA3A-1 (Fig. 2A). eA3A-2 showed excellent deamination activity toward both C and 5mC (ESI Fig. S5†). However, eA3A-2 only moderately deaminated 5hmC in TC and CC sites and could not deaminate 5hmC in GC and AC sites (ESI Fig. S5†). Since eA3A-2 is capable of deaminating C and 5mC but not 5hmC in GC and AC sites, the differential deamination performance of eA3A-2 toward C/5mC and 5hmC can be utilized to detect 5hmC in GC and AC sites.
It has been observed that alteration of residue 25 (glycine, G) in loop 1 of wtA3A could affect the deamination activity of wtA3A.45,47,48 On the basis of eA3A-2, we further generated eA3A proteins 3, 4, 5, and 6 (eA3A-3, eA3A-4, eA3A-5, and eA3A-6) (Fig. 2A). These four eA3A proteins all exhibited adequate deamination activity toward C (Fig. 2B and ESI Fig. S6–S8†). However, eA3A-3 and eA3A-4 could not efficiently deaminate 5mC and showed no deamination activity toward 5hmC (ESI Fig. S6 and S7†) and therefore are not suitable to be utilized to discriminate C/5mC and 5hmC. eA3A-5 and eA3A-6 could effectively deaminate 5mC and showed a moderate activity toward 5hmC in TC and CC sites (Fig. 2B and ESI Fig. S8†). On the contrary, eA3A-5 and eA3A-6 could not deaminate 5hmC in GC and AC sites (Fig. 2B and ESI Fig. S8†). Consequently, eA3A-5 and eA3A-6 could be used to discriminate 5hmC from C/5mC in GC and AC sites.
Next, we further generated eA3A-7 (with G27P mutation in loop 1 of eA3A-3), eA3A-8 (with D131T mutation in loop 7 of eA3A-4), and eA3A-9 (with I26N mutation in loop 1 of eA3A-5) (Fig. 2A). All these three eA3A proteins of eA3A-7, eA3A-8 and eA3A-9 could efficiently deaminate C and 5mC in TC and CC sites but showed attenuated deamination activity toward 5mC in GC and AC sites (Fig. 2C and ESI Fig. S9 and S10†). Conversely, eA3A-7, eA3A-8 and eA3A-9 couldn't deaminate 5hmC in all the cytosine sites (Fig. 2C and ESI Fig. S9 and S10†). Therefore, eA3A-7, eA3A-8 and eA3A-9 all could be used to discriminate 5hmC from C/5mC in TC and CC sites.
Collectively, based on the reported crystal structure of wtA3A and the characterized region that is critical to the substrate preference and enzyme activity of wtA3A, we engineered and generated a series of A3A mutant proteins (eA3A-1 to eA3A-9). Three eA3A proteins (eA3A-2, eA3A-5 and eA3A-6) exhibit effective deamination activity toward C/5mC but show no deamination activity toward 5hmC in GC and AC sites. Likewise, the other three eA3A proteins (eA3A-7, eA3A-8 and eA3A-9) exhibit effective deamination activity toward C/5mC but show no deamination activity toward 5hmC in TC and CC sites. As for the underlying mechanism of the changed substrate preference of eA3A-5 and eA3A-9, the crystal structures of eA3A-5 and eA3A-9 should be obtained, which can be carried out in future studies.
Quantitative and site-specific detection of 5hmC in DNA by EDM-seq
After characterization of the deamination properties of these eA3A proteins toward C, 5mC and 5hmC, we proposed an engineered deaminase-mediated sequencing (EDM-seq) method for the quantitative detection of 5hmC at single-nucleotide resolution (Fig. 3). In this method, two eA3A proteins, eA3A-5 and eA3A-9, were employed as the deaminases for the deamination reaction. According the aforementioned properties of eA3A proteins, eA3A-5 is able to totally deaminate C/5mC but shows no deamination activity toward 5hmC in GC and AC sites, while eA3A-9 is able to totally deaminate C/5mC but shows no deamination activity toward 5hmC in TC and CC sites. In addition, a similar deamination of 5mC in both C5mC and 5mC5mC sites (or resistance to deamination of 5hmC in C5hmC and 5hmC5hmC sites) can be obtained by eA3A-9 (ESI Fig. S11†). Therefore, the combined use of eA3A-5 and eA3A-9 could offer the direct detection of 5hmC in various sequence contexts, including GC, AC, TC and CC sites (Fig. 3). | ||
| Fig. 3 Schematic illustration of the EMD-seq method. (A) After eA3A-5 treatment, C and 5mC in GC and AC sites were deaminated and read as T, while 5hmC was not deaminated and read as C. (B) After eA3A-9 treatment, C and 5mC in TC and CC sites were deaminated and read as T, while 5hmC was not deaminated and read as C. The combined use of eA3A-5 and eA3A-9 enables the quantitative and site-specific detection of 5hmC in DNA within various sequence contexts. | ||
In EDM-seq, eA3A-5 is used to detect 5hmC at GC and AC sites, while eA3A-9 is used to detect 5hmC at TC and CC sites. The 24-mer C-containing DNA (GC-C, AC-C, TC-C and CC-C), 5mC-containing DNA (GC-5mC, AC-5mC, TC-5mC and CC-5mC), and 5hmC-containing DNA (GC-5hmC, AC-5hmC, TC-5hmC and CC-5hmC) were used as the substrates to evaluate the deamination efficiency of eA3A-5 and eA3A-9 toward C, 5mC and 5hmC by LC-MS/MS analysis. Three different mixtures of DNA (GC-C and AC-C; GC-5mC and AC-5mC; GC-5hmC and AC-5hmC) were separately treated with eA3A-5 followed by LC-MS/MS analysis. The results showed that neither the dC nor 5mC signal was observed after eA3A-5 treatment; however, the signal intensity of 5hmC was almost intact after eA3A-5 treatment (Fig. 4A). Similarly, another three different mixtures of DNA (TC-C and CC-C; TC-5mC and CC-5mC; TC-5hmC and CC-5hmC) were separately treated with eA3A-9 followed by LC-MS/MS analysis. The results demonstrated that both dC and 5mC were undetectable after eA3A-9 treatment, whereas there is no obvious change of the signal intensity of 5hmC after eA3A-9 treatment (Fig. 4B). In addition, other canonical nucleosides of dA, dG and dT were not affected by either eA3A-5 or eA3A-9 treatment (ESI Fig. S12†). The LC-MS/MS results demonstrated that eA3A-5 could efficiently deaminate C/5mC but couldn't deaminate 5hmC in GC and AC sites, while eA3A-9 totally deaminated C/5mC but showed no deamination activity toward 5hmC in TC and CC sites.
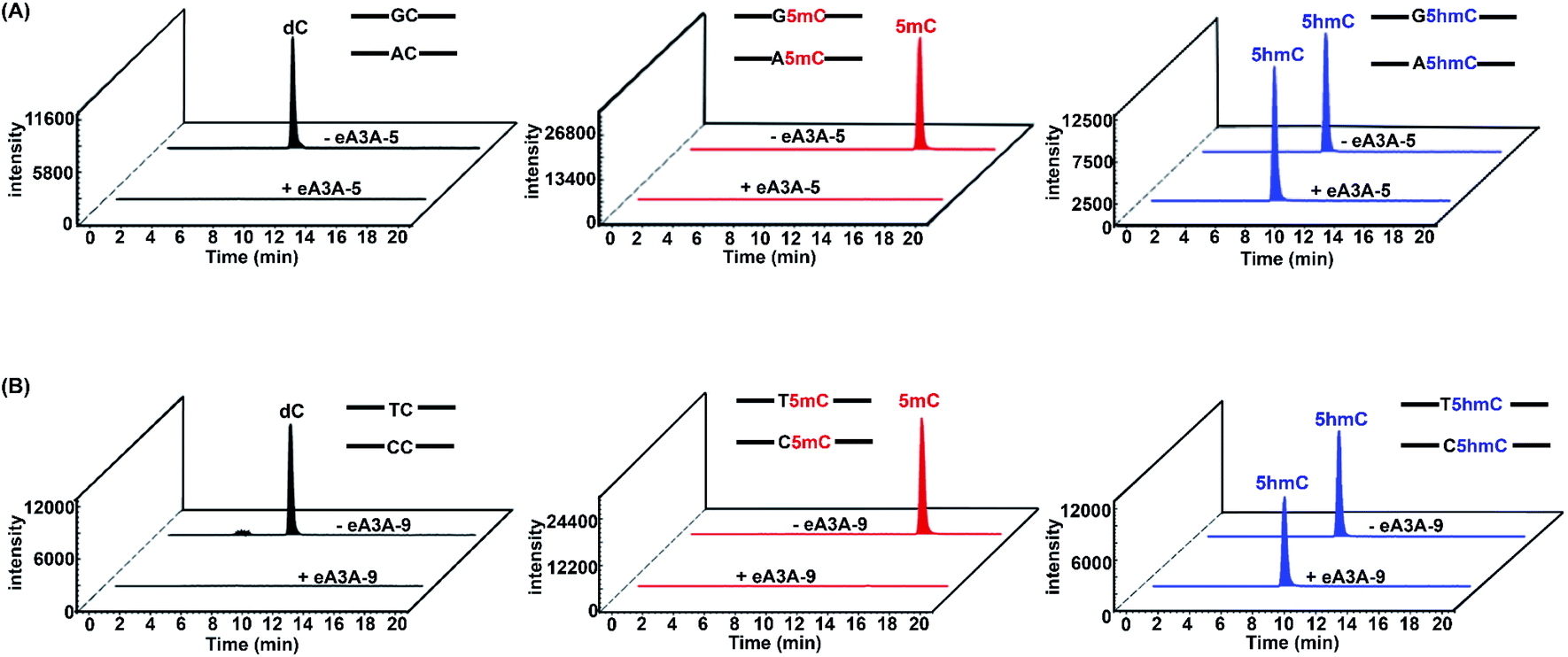 | ||
| Fig. 4 Extracted-ion chromatograms of dC, 5mC and 5hmC from eA3A-5 or eA3A-9 treated DNA by LC-MS/MS analysis. Three different mixtures of DNA (GC-C and AC-C; GC-5mC and AC-5mC; GC-5hmC and AC-5hmC) were separately treated with eA3A-5. Another three different mixtures of DNA (TC-C and CC-C; TC-5mC and CC-5mC; TC-5hmC and CC-5hmC) were separately treated with eA3A-9. (A) Extracted-ion chromatograms of dC (from the GC-C and AC-C mixture), 5mC (from the GC-5mC and AC-5mC mixture) and 5hmC (from the GC-5hmC and AC-5hmC mixture) without or with eA3A-5 treatment. (B) Extracted-ion chromatograms of dC (from the TC-C and CC-C mixture), 5mC (from the TC-5mC and CC-5mC mixture) and 5hmC (from the TC-5hmC and CC-5hmC mixture) without or with eA3A-9 treatment. | ||
We further treated these oligonucleotide mixtures with different concentrations of eA3A-5 or eA3A-9. The results revealed that the deamination rates of C and 5mC in GC and AC sites continuously increased and eventually reached up to almost 100% with the concentration of eA3A-5 being 1 μM, while 5hmC showed no obvious deamination with the increased concentration of eA3A-5 (Fig. 5A). Similarly, the deamination rates of C and 5mC in TC and CC sites gradually increased and eventually reached up to almost 100% with the concentration of eA3A-9 being 1 μM, but 5hmC still remained almost intact without deamination (Fig. 5B). On the other hand, it can be observed that wtA3A deaminates all the C, 5mC and 5hmC without selectivity, even though the deamination activity of wtA3A was weaker toward 5hmC (Fig. 5C and D). These LC-MS/MS results are in line with the Sanger sequencing results (ESI Fig. S3†).
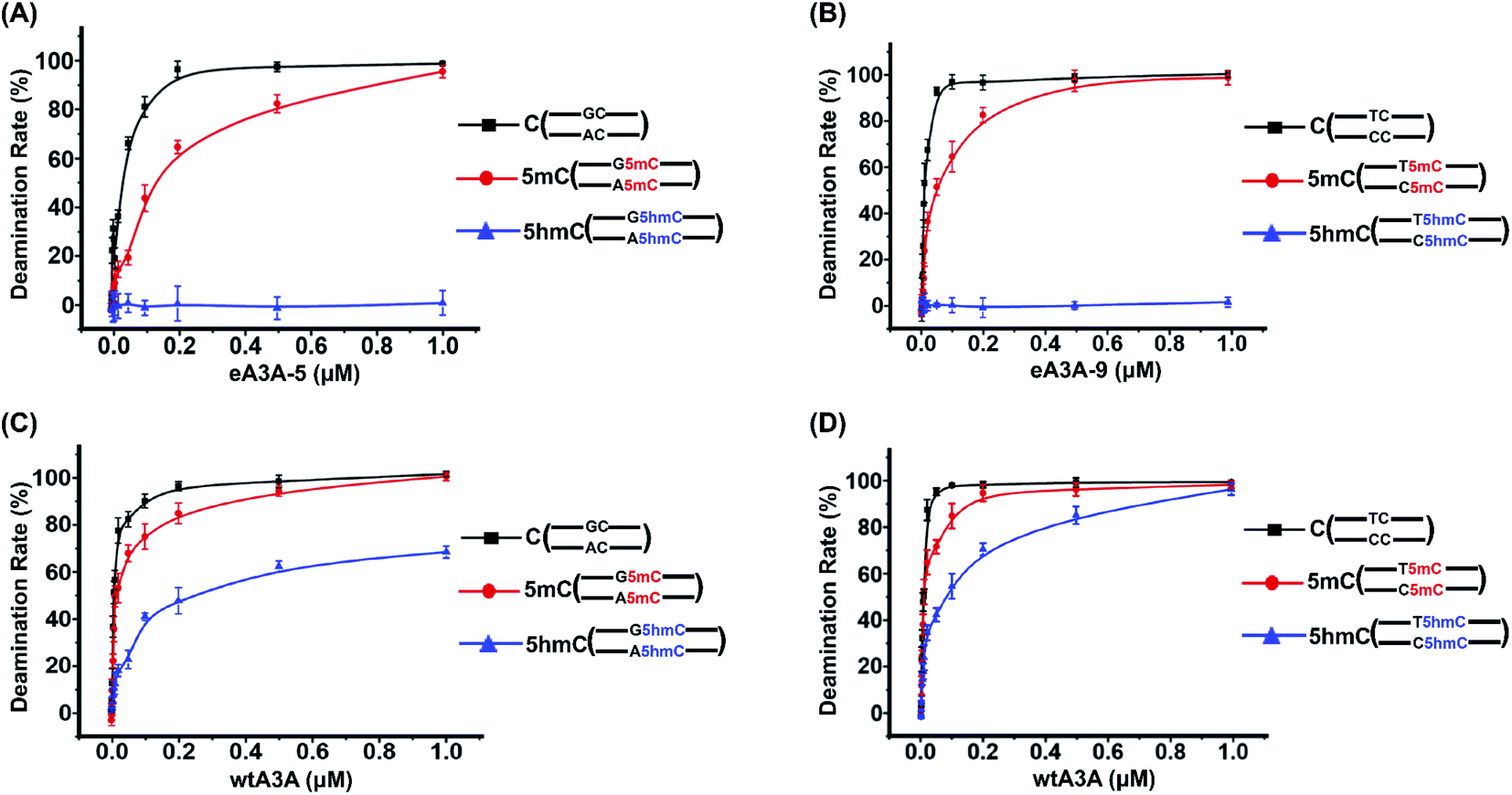 | ||
| Fig. 5 Evaluation of the deaminase activity of eA3A-5, eA3A-9 and wtA3A toward C, 5mC and 5hmC in different sequence contexts by LC-MS/MS analysis. Three different mixtures of DNA (GC-C and AC-C; GC-5mC and AC-5mC; GC-5hmC and AC-5hmC) were separately treated with different concentrations of eA3A-5 or wtA3A followed by LC-MS/MS analysis. Another three different mixtures of DNA (TC-C and CC-C; TC-5mC and CC-5mC; TC-5hmC and CC-5hmC) were separately treated with different concentrations of eA3A-9 or wtA3A followed by LC-MS/MS analysis. (A) The deamination rate of C, 5mC, and 5hmC in GC and AC sites using different concentrations of eA3A-5. (B) The deamination rate of C, 5mC, and 5hmC in TC and CC sites using different concentrations of eA3A-9. (C) The deamination rate of C, 5mC, and 5hmC in GC and AC sites using different concentrations of wtA3A. (D) The deamination rate of C, 5mC, and 5hmC in TC and CC sites using different concentrations of wtA3A. | ||
We next employed colony sequencing to quantitatively evaluate the 5hmC level by the EDM-seq method. There 224-bp dsDNA (DNA-C, DNA-5mC, and DNA-5hmC) were treated with eA3A-5, eA3A-9, or wtA3A followed by colony sequencing (Fig. 6A). The results suggested that all the C and 5mC in GC and AC sites were fully deaminated and read as T, while 5hmC in GC and AC sites were resistant to deamination and read as C after eA3A-5 treatment (Fig. 6B and ESI Fig. S13†). Likewise, after eA3A-9 treatment, all the C and 5mC in TC and CC sites were fully deaminated and read as T, while 5hmC in TC and CC sites were resistant to deamination and read as C (Fig. 6C and ESI Fig. S14†). However, wtA3A couldn't achieve the differential deamination between C/5mC and 5hmC (Fig. 6D and E, ESI Fig. S15 and S16†). The colony sequencing results are also consistent with the LC-MS/MS analysis (Fig. 5 and 6). Taken together, the combined use of eA3A-5 and eA3A-9 could achieve the quantitative detection of 5hmC in all the different cytosine sites, including GC, AC, TC and CC sites.
 | ||
| Fig. 6 Evaluation of the deaminase activity of eA3A-5, eA3A-9 and wtA3A toward C, 5mC and 5hmC in different sequence contexts by colony sequencing. (A) The 224-bp DNA-C, DNA-5mC and DNA-5hmC were first denatured to ssDNA and then separately treated with eA3A-5, eA3A-9, or wtA3A followed by colony sequencing. Fifty clones for each sample were randomly picked up and sequenced. (B) After eA3A-5 treatment, C and 5mC in GC and AC sites were all deaminated and read as T, while all the 5hmC in GC and AC sites was resistant to deamination and read as C. (C) After eA3A-9 treatment, C and 5mC in TC and CC sites were all read as T, while all the 5hmC in TC and CC sites was resistant to deamination and read as C. (D) After wtA3A treatment, C and 5mC in GC and AC sites were all deaminated and read as T, while 5hmC in GC and AC sites was partially deaminated and partially read as C and partially read as T. (E) After wtA3A treatment, C, 5mC and 5hmC in TC and CC sites were all deaminated and read as T. | ||
We next further evaluated the quantitative capability of the EDM-seq method in measuring the stoichiometry of 5hmC at given sites in DNA. In this respect, DNA-C and DNA-5hmC were mixed at different ratios with DNA-5hmC ranging from 0% to 100%. The prepared mixtures were subjected to EDM-seq with Sanger sequencing. The results showed that the measured percentages of C/(C + T) at given sites increased linearly with the increased ratios of 5hmC in the mixture of DNA-C and DNA-5hmC (ESI Fig. S17†), suggesting that the EDM-seq method is capable of site-specific and quantitative measurement of 5hmC with different stoichiometries. EDM-seq with Sanger sequencing could offer the site-specific quantification of 5hmC with a stoichiometry as low as 10%. Since the engineered A3A proteins could fully deaminate C and 5mC but show no deamination activity toward 5hmC, accurate quantification of 5hmC with lower modification stoichiometry at given sites could be achieved by the EDM-seq method while being coupled with high-throughput sequencing or colony sequencing.
In the previous AMD-seq method, β-GT is used to selectively add a glucosyl group to 5hmC to form 5gmC, thus obtaining the resistance to wtA3A deamination. Compared to AMD-seq, EDM-seq is simpler and more straightforward without the need of glycosylation of 5hmC by β-GT. Chemical treatment or chemical labelling strategies have been employed to map 5hmC, such as TET assisted pyridine borane sequencing (TAPSβ) assay.49 However, these methods require relatively harsh reaction conditions. In addition, the selectivity of chemical reactions may not be sufficient, which would affect the subsequent mapping analysis. On the contrary, the EDM-seq method is not involved in chemical reactions or labelling, thus allowing the whole procedure to be carried out under mild conditions, and DNA is not prone to degradation. Collectively, the EDM-seq proves to be a straightforward and easy to handle method in quantitative and site-specific detection of 5hmC.
Detection of 5hmC in genomic DNA of lung cancer tissue
It has been reported that 5hmC is dysregulated in a wide variety of cancers.50,51 Specifically, the global reduction of the 5hmC level in genomic DNA is considered to be a hallmark of cancers.52 Here, we employed the EDM-seq method to examine individual 5hmC sites in lung cancer tissue as well as in adjacent normal tissue.In the development of the EDM-seq method, two engineered A3A proteins (eA3A-5 and eA3A-9) were screened and employed for mapping 5hmC in different sequence contexts since the neighbouring 5′ nucleobase of cytosine (5′XC3′) could affect the deamination activity of eA3A. We then further applied this EDM-seq method to analyse and quantify 5hmC in genomic DNA. Similar to the method development, different neighbouring 5′ nucleobases of cytosine in genomic DNA (GC, AC, TC, and CC sites) were selected to demonstrate the applicability of the EDM-seq method. Four cytosine sites that have been previously identified to be hydroxylmethylated were selected for the examination (ESI Table S3†).53 These four sites are separately located in the GC, AC, TC, and CC sites (ESI Table S3†). The isolated genomic DNA was fragmented and then directly treated with eA3A-5 (for examining GC and AC sites) or eA3A-9 (for examining TC and CC sites). The subsequent Sanger sequencing showed that all these four sites were partially read as C and partially read as T in normal tissue (Fig. 7A and ESI Fig. S18†), indicating that all these four sites are partially hydroxymethylated. However, all these four sites were read as T in lung cancer tissue (Fig. 7B and ESI Fig. S18†), suggesting that there is no detectable 5hmC in these sites in lung cancer tissue. Consistent with a previous study, the results demonstrated that the 5hmC level was significantly decreased in lung cancer tissue compared to that in normal tissue.
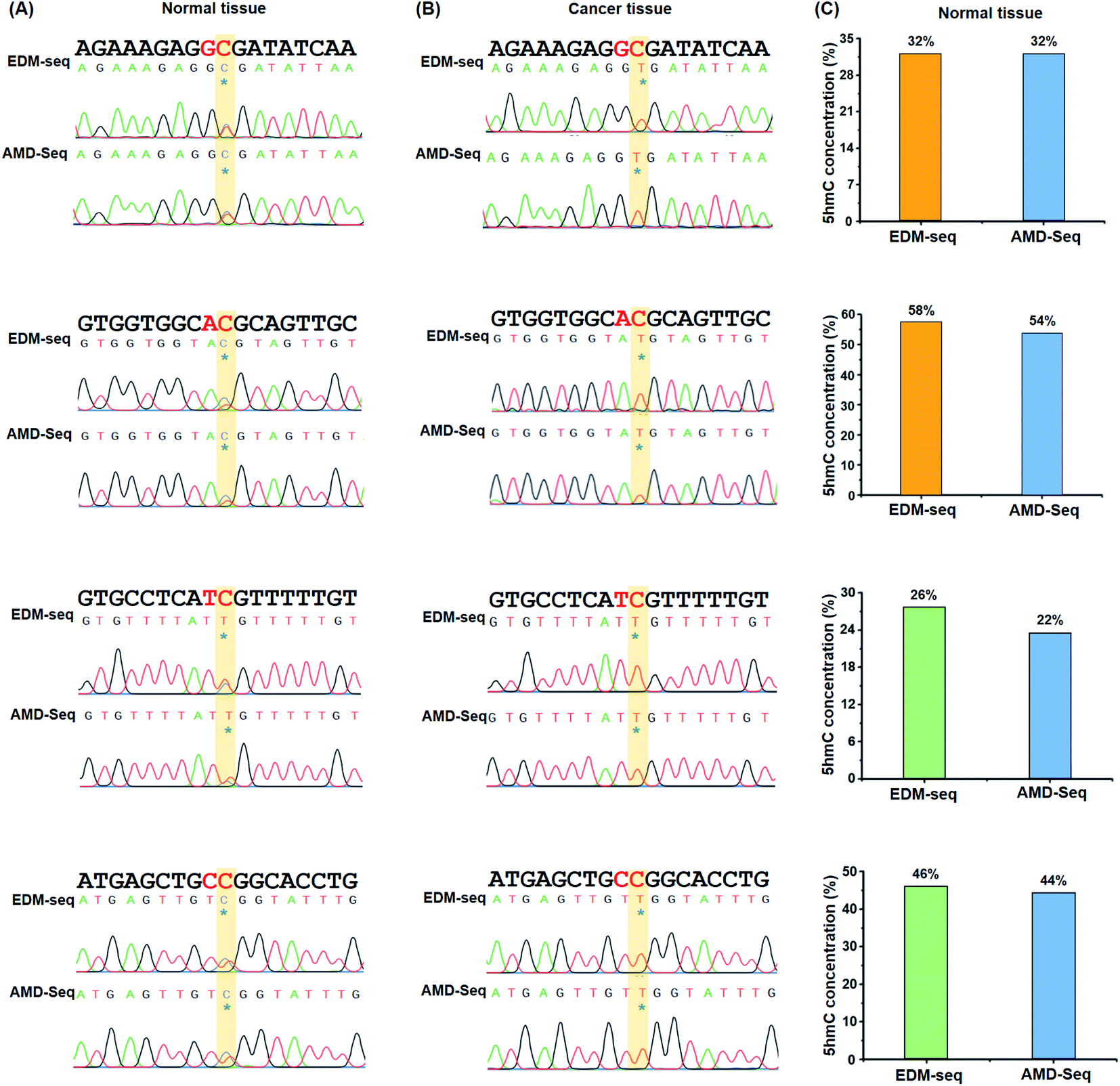 | ||
| Fig. 7 Quantitative and site-specific detection of 5hmC in genomic DNA of lung cancer tissue and adjacent normal tissue by EDM-seq and AMD-seq. (A) 5hmC at chr5:14180809 (CRCh37, GC site), chr3:29918016 (CRCh37, AC site), chr4:169198493 (CRCh37, TC site) and chr1:68672111 (CRCh37, CC site) in genomic DNA of normal tissue was determined by EDM-seq and AMD-seq methods. (B) 5hmC at chr5:14180809 (CRCh37, GC site), chr3:29918016 (CRCh37, AC site), chr4:169198493 (CRCh37, TC site) and chr1:68672111 (CRCh37, CC site) in genomic DNA of lung cancer tissue was determined by EDM-seq and AMD-seq methods. (C) Quantitative 5hmC level in different sites in genomic DNA of normal tissue by EDM-seq and AMD-seq methods though colony sequencing. | ||
In addition, we utilized the previous AMD-seq method to examine 5hmC in these four sites. It can be seen that the similar sequencing results were obtained by both EDM-seq and AMD-seq methods (Fig. 7A and B and ESI Fig. S18†). Moreover, the quantitative results from colony sequencing by EDM-seq showed that 5hmC levels at these four sites in genomic DNA of normal tissue were 32%, 58%, 26%, and 46%, respectively (Fig. 7C). Similarly, the quantitative results from colony sequencing by AMD-seq revealed that 5hmC levels at these four sites were 32%, 54%, 22%, and 44%, respectively (Fig. 7C). It's clear that the results obtained by both EDM-seq and AMD-seq are highly consistent (Fig. 7 and ESI Fig. S18†). Collectively, the results demonstrated that the EDM-seq method is capable of the quantitative detection of 5hmC at single-nucleotide resolution. This approach, when coupled with high throughput sequencing, can enable the quantitative and genome-wide mapping of 5hmC at single-nucleotide resolution in future studies. In the EDM-seq method, two eA3A proteins are needed to achieve the mapping analysis of 5hmC in different sequence contexts. It can be envisaged that a single engineered A3A protein can meet the differential deamination toward C/5mC and 5hmC with further evolution of the A3A protein in future studies.
Conclusions
In summary, we proposed an EDM-seq method for the quantitative detection of 5hmC in DNA at single-nucleotide resolution. We generated a series of engineered A3A proteins. The deamination activities of these engineered A3A proteins toward C, 5mC and 5hmC were evaluated by using Sanger sequencing, colony sequencing and LC-MS/MS analysis. We found that eA3A-5 could fully deaminate C and 5mC but showed no deamination activity toward 5hmC in GC and AC sites, while eA3A-9 showed an adequate deamination activity toward C and 5mC but couldn't deaminate 5hmC in TC and CC sites. Gratifyingly, the combined use of eA3A-5 and eA3A-9 enabled the quantitative and site-specific detection of 5hmC in DNA within various sequence contexts. By the EDM-seq method, we achieved the quantitative analysis of individual 5hmC sites in genomic DNA of lung cancer tissue and adjacent normal tissue. Compared to previous 5hmC mapping methods, the EDM-seq method is bisulfite-free, chemical labelling-free, and does not require DNA glycosylation or chemical treatment. Taken together, the EDM-seq method offers a valuable tool for the straightforward, quantitative and accurate detection of 5hmC in DNA at single-nucleotide resolution.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work was supported by the National Natural Science Foundation of China (22074110 and 21721005). We thank Dr Chu-Bo Qi (Hubei Cancer Hospital) for providing the lung cancer tissue and the adjacent normal tissue.References
- A. Parry, S. Rulands and W. Reik, Nat. Rev. Genet., 2021, 22, 59–66 CrossRef CAS PubMed.
- C. Luo, P. Hajkova and J. R. Ecker, Science, 2018, 361, 1336–1340 CrossRef CAS PubMed.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef CAS PubMed.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- X. Wu and Y. Zhang, Nat. Rev. Genet., 2017, 18, 517–534 CrossRef CAS PubMed.
- K. Iwan, R. Rahimoff, A. Kirchner, F. Spada, A. S. Schroder, O. Kosmatchev, S. Ferizaj, J. Steinbacher, E. Parsa, M. Muller and T. Carell, Nat. Chem. Biol., 2018, 14, 72–78 CrossRef CAS PubMed.
- Y. Feng, N. B. Xie, W. B. Tao, J. H. Ding, X. J. You, C. J. Ma, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, CCS Chem., 2020, 2, 994–1008 Search PubMed.
- Y. Feng, J. J. Chen, N. B. Xie, J. H. Ding, X. J. You, W. B. Tao, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 11322–11329 RSC.
- E. Kaminska, E. Korytiakova, A. Reichl, M. Muller and T. Carell, Angew. Chem., Int. Ed. Engl., 2021, 60, 23207–23211 CrossRef CAS PubMed.
- M. Munzel, D. Globisch and T. Carell, Angew. Chem., Int. Ed. Engl., 2011, 50, 6460–6468 CrossRef PubMed.
- H. Wu and Y. Zhang, Cell, 2014, 156, 45–68 CrossRef CAS PubMed.
- H. Stroud, S. Feng, S. Morey Kinney, S. Pradhan and S. E. Jacobsen, Genome Biol., 2011, 12, R54 CrossRef CAS PubMed.
- L. Scourzic, E. Mouly and O. A. Bernard, Genome Med., 2015, 7, 9 CrossRef PubMed.
- M. L. Chen, F. Shen, W. Huang, J. H. Qi, Y. Wang, Y. Q. Feng, S. M. Liu and B. F. Yuan, Clin. Chem. (N. Y.), 2013, 59, 824–832 CrossRef CAS PubMed.
- R. Yin, J. Mo, M. Lu and H. Wang, Anal. Chem., 2015, 87, 1846–1852 CrossRef CAS PubMed.
- S. Liu, J. Wang, Y. Su, C. Guerrero, Y. Zeng, D. Mitra, P. J. Brooks, D. E. Fisher, H. Song and Y. Wang, Nucleic Acids Res., 2013, 41, 6421–6429 CrossRef CAS PubMed.
- W. Y. Lai, J. Z. Mo, J. F. Yin, C. Lyu and H. L. Wang, TrAC, Trends Anal. Chem., 2019, 110, 173–182 CrossRef CAS.
- F. Yuan, X. H. Zhang, J. Nie, H. X. Chen, Y. L. Zhou and X. X. Zhang, Chem. Commun., 2016, 52, 2698–2700 RSC.
- Y. Yu, S. H. Zhu, F. Yuan, X. H. Zhang, Y. Y. Lu, Y. L. Zhou and X. X. Zhang, Chem. Commun., 2019, 55, 7595–7598 RSC.
- Y. Dai, B. F. Yuan and Y. Q. Feng, RSC Chem. Biol., 2021, 2, 1096–1114 RSC.
- B. F. Yuan, Chem. Res. Toxicol., 2020, 33, 695–708 Search PubMed.
- S. Liu and Y. Wang, Chem. Soc. Rev., 2015, 44, 7829–7854 RSC.
- R. Zhang, W. Lai and H. Wang, Anal. Chem., 2021, 93, 15567–15572 CrossRef CAS PubMed.
- M. Y. Chen, Z. Gui, K. K. Chen, J. H. Ding, J. G. He, J. Xiong, J. L. Li, J. Wang, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2022, 33, 2086–2090 CrossRef CAS.
- Q. Wang, J. H. Ding, J. Xiong, Y. Feng, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2021, 32, 3426–3430 CrossRef CAS.
- M. Berney and J. F. McGouran, Nat. Rev. Chem., 2018, 2, 332–348 CrossRef CAS.
- L. Y. Zhao, J. Song, Y. Liu, C. X. Song and C. Yi, Protein Cell, 2020, 11, 792–808 CrossRef CAS PubMed.
- H. Zeng, B. He, B. Xia, D. Bai, X. Lu, J. Cai, L. Chen, A. Zhou, C. Zhu, H. Meng, Y. Gao, H. Guo, C. He, Q. Dai and C. Yi, J. Am. Chem. Soc., 2018, 140, 13190–13194 CrossRef CAS PubMed.
- C. B. Qi, J. H. Ding, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2019, 30, 1618–1626 CrossRef CAS.
- G. Ficz, M. R. Branco, S. Seisenberger, F. Santos, F. Krueger, T. A. Hore, C. J. Marques, S. Andrews and W. Reik, Nature, 2011, 473, 398–402 CrossRef CAS PubMed.
- W. A. Pastor, U. J. Pape, Y. Huang, H. R. Henderson, R. Lister, M. Ko, E. M. McLoughlin, Y. Brudno, S. Mahapatra, P. Kapranov, M. Tahiliani, G. Q. Daley, X. S. Liu, J. R. Ecker, P. M. Milos, S. Agarwal and A. Rao, Nature, 2011, 473, 394–397 CrossRef CAS PubMed.
- C. X. Song, K. E. Szulwach, Y. Fu, Q. Dai, C. Yi, X. Li, Y. Li, C. H. Chen, W. Zhang, X. Jian, J. Wang, L. Zhang, T. J. Looney, B. Zhang, L. A. Godley, L. M. Hicks, B. T. Lahn, P. Jin and C. He, Nat. Biotechnol., 2011, 29, 68–72 CrossRef CAS PubMed.
- J. Peng, B. Xia and C. Yi, Sci. China: Life Sci., 2016, 59, 219–226 CrossRef CAS PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 2012, 336, 934–937 CrossRef CAS PubMed.
- M. Yu, G. C. Hon, K. E. Szulwach, C. X. Song, L. Zhang, A. Kim, X. Li, Q. Dai, Y. Shen, B. Park, J. H. Min, P. Jin, B. Ren and C. He, Cell, 2012, 149, 1368–1380 CrossRef CAS PubMed.
- K. Tanaka and A. Okamoto, Bioorg. Med. Chem. Lett., 2007, 17, 1912–1915 CrossRef CAS PubMed.
- C. X. Song, T. A. Clark, X. Y. Lu, A. Kislyuk, Q. Dai, S. W. Turner, C. He and J. Korlach, Nat. Methods, 2012, 9, 75–77 CrossRef CAS PubMed.
- Z. K. O'Brown, K. Boulias, J. Wang, S. Y. Wang, N. M. O'Brown, Z. Hao, H. Shibuya, P. E. Fady, Y. Shi, C. He, S. G. Megason, T. Liu and E. L. Greer, BMC Genomics, 2019, 20, 445 CrossRef PubMed.
- E. K. Schutsky, C. S. Nabel, A. K. F. Davis, J. E. DeNizio and R. M. Kohli, Nucleic Acids Res., 2017, 45, 7655–7665 CrossRef CAS PubMed.
- Q. Y. Li, N. B. Xie, J. Xiong, B. F. Yuan and Y. Q. Feng, Anal. Chem., 2018, 90, 14622–14628 CrossRef CAS PubMed.
- E. K. Schutsky, J. E. DeNizio, P. Hu, M. Y. Liu, C. S. Nabel, E. B. Fabyanic, Y. Hwang, F. D. Bushman, H. Wu and R. M. Kohli, Nat. Biotechnol., 2018, 36, 1083–1090 CrossRef CAS PubMed.
- F. Tang, S. Liu, Q. Y. Li, J. Yuan, L. Li, Y. Wang, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2019, 10, 4272–4281 RSC.
- M. Y. Cheng, X. J. You, J. H. Ding, Y. Dai, M. Y. Chen, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 8149–8156 RSC.
- C. J. Ma, L. Li, W. X. Shao, J. H. Ding, X. L. Cai, Z. R. Lun, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 14126–14132 RSC.
- F. Ito, Y. Fu, S. A. Kao, H. Yang and X. S. Chen, J. Mol. Biol., 2017, 429, 1787–1799 CrossRef CAS PubMed.
- T. Kouno, T. V. Silvas, B. J. Hilbert, S. M. D. Shandilya, M. F. Bohn, B. A. Kelch, W. E. Royer, M. Somasundaran, N. Kurt Yilmaz, H. Matsuo and C. A. Schiffer, Nat. Commun., 2017, 8, 15024 CrossRef PubMed.
- K. Shi, M. A. Carpenter, S. Banerjee, N. M. Shaban, K. Kurahashi, D. J. Salamango, J. L. McCann, G. J. Starrett, J. V. Duffy, O. Demir, R. E. Amaro, D. A. Harki, R. S. Harris and H. Aihara, Nat. Struct. Mol. Biol., 2017, 24, 131–139 CrossRef CAS PubMed.
- Y. Fu, F. Ito, G. Zhang, B. Fernandez, H. Yang and X. S. Chen, Biochem. J., 2015, 471, 25–35 CrossRef CAS PubMed.
- Y. Liu, P. Siejka-Zielinska, G. Velikova, Y. Bi, F. Yuan, M. Tomkova, C. Bai, L. Chen, B. Schuster-Bockler and C. X. Song, Nat. Biotechnol., 2019, 37, 424–429 CrossRef CAS PubMed.
- J. P. Thomson and R. R. Meehan, Epigenomics, 2017, 9, 77–91 CrossRef CAS PubMed.
- X. Tian, B. Sun, C. Chen, C. Gao, J. Zhang, X. Lu, L. Wang, X. Li, Y. Xing, R. Liu, X. Han, Z. Qi, X. Zhang, C. He, D. Han, Y. G. Yang and Q. Kan, Cell Res., 2018, 28, 597–600 CrossRef CAS PubMed.
- G. Ficz and J. G. Gribben, Genomics, 2014, 104, 352–357 CrossRef CAS PubMed.
- X. Li, Y. Liu, T. Salz, K. D. Hansen and A. Feinberg, Genome Res., 2016, 26, 1730–1741 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1–S3 and Fig. S1–S18. See https://doi.org/10.1039/d2sc01052f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |