
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymeric frustrated Lewis pairs in CO2/cyclic ether coupling catalysis†
Thomas A. R.
Horton
 ab,
Meng
Wang
ab and
Michael P.
Shaver
*ab
ab,
Meng
Wang
ab and
Michael P.
Shaver
*ab
aDepartment of Materials, School of Natural Sciences, The University of Manchester, Manchester, UK. E-mail: michael.shaver@manchester.ac.uk
bSustainable Materials Innovation Hub, Royce Hub Building, The University of Manchester, Oxford Road, Manchester, UK
First published on 8th March 2022
Abstract
Frustrated Lewis pairs (FLPs) are now ubiquitous as metal-free catalysts in an array of different chemical transformations. In this paper we show that this reactivity can be transferred to a polymeric system, offering advantageous opportunities at the interface between catalysis and stimuli-responsive materials. Formation of cyclic carbonates from cyclic ethers using CO2 as a C1 feedstock continues to be dominated by metal-based systems. When paired with a suitable nucleophile, discrete aryl or alkyl boranes have shown significant promise as metal-free Lewis acidic alternatives, although catalyst reuse remains illusive. Herein, we leverage the reactivity of FLPs in a polymeric system to promote CO2/cyclic ether coupling catalysis that can be tuned for the desired epoxide or oxetane substrate. Moreover, these macromolecular FLPs can be reused across multiple reaction cycles, further increasing their appeal over analogous small molecule systems.
Introduction
Valorisation of CO2 as a renewable carbon feedstock is desirable in the pursuit of a sustainable, carbon-neutral society. Current efforts to use this typically unreactive substrate rely either on designer gas-capture systems and subsequent reactions, or combining CO2 directly with suitably reactive substrates.1 Epoxides are one such reactive substrate that has been extensively explored, where insertion of CO2 yields the corresponding cyclic or polycarbonate products.2 While the majority of early research efforts in this area have employed expensive or non-abundant metal-based systems, the use of metal-free catalysts to facilitate these transformations has expanded in recent years.3Frustrated Lewis pairs (FLPs) are ubiquitous as metal-free catalysts for a myriad of chemical transformations.4–7 The frustration induced by sterically hindered Lewis acid (LA) and base (LB) centres permits cooperative action of, and catalysis with, a multitude of substrates including carbon dioxide, dihydrogen and cyclic ethers.8–15 In 2017, our group demonstrated that FLP reactivity is maintained when incorporated into a polymeric system, revealing a new class of stimuli-responsive materials that exploit FLP-mediated small molecule activation.16,17 Inspired by these systems, Yan et al. later reported similar macromolecular FLPs capable of activating CO2 and catalysing amine formylation.18 Polymeric FLPs have also been reported in C–H functionalisation, amination and hydrogenation catalysis with demonstrated potential for catalyst recovery and reuse.19–22
Recently, we reported the successful crosslinking of highly Lewis acidic styrenic copolymers with a corresponding Lewis basic copolymer, via ring-opening of cyclic ether substrates.23,24 Given these results, we wondered whether our systems would enable effective catalytic insertion of CO2 into the ring-opened cyclic ether substrates. Aryl and alkyl boranes have previously been applied successfully in the formation of both cyclic and polycarbonates when paired with a phosphonium or ammonium salt partner.25–31 More recently, superbasic phosphazenes have also proved active under mild conditions.32 The use of a phosphine LB in these reactions is however, to our knowledge, previously unreported. Herein, we report the first use of both conventional small molecule and polymeric FLPs to catalyse the insertion of CO2 into cyclic ether substrates with high selectivity towards the cyclic product (Fig. 1).
 | ||
| Fig. 1 (A) Previously reported borane-mediated CO2 insertion catalysts. (B) Cyclic ether triggered poly(FLP) networks. (C) Poly(FLP) catalysed cyclic carbonate formation. | ||
Results and discussion
Screening of LA/LB copolymer catalysts
Having previously reported three boron-containing copolymers of varying Lewis acidity (B1–3) we first sought to optimise the LA component of our proposed system (Fig. 2). Propylene oxide (PO) was selected as a model substrate. A readily synthesised triphenylphosphine functionalised copolymer, P1, was selected as the Lewis basic co-catalyst. For the PO/P1/B2 system, conversion to propylene carbonate was observed but remained low, as the catalysis promoted reverse hydroboration to decouple the catalytic site from the polymer backbone (Fig. S5 and S6†). We know that the addition of epoxides affords stable networks, suggesting that the addition of CO2 promotes decomposition after the first turnover. Decomposition occurs at both moderate and elevated temperatures and mimics reversible reactions commonly associated with polymeric alkyl boranes,33 therefore preventing further catalytic testing using B2. | ||
| Fig. 2 Performance of polymeric LAs for CO2 insertion to PO when paired with P1. Structure of P2 to be used in later reaction screening. AN = acceptor number calculated using the Gutmann–Beckett method. | ||
Aryl-boron-containing polymers have a higher thermal stability.21 This was proven when pairing a polymeric LB with B1 and B3. While the capacity of B3 to ring-open cyclic ethers was already established,24 we anticipated B1 would also facilitate this reaction based on previous small-molecule systems.15,27 Previous studies into small molecule borane catalysed formation of cyclic carbonates reported a reactivity decrease when using stronger LAs as a result of adduct stability.27 Indeed, the same trend can be observed within our polymeric systems when considering estimations of Lewis acidity by the Gutmann–Beckett method.34,35 Reactions employing B3 (acceptor number, AN = 84.4) were notably slower than those with the less Lewis acidic B1 (AN = 67.6) under the same optimised conditions (Fig. 2 and S5†). Given its superior performance, we selected B1 as the Lewis acidic component for the rest of this study.
With catalytic conversions using B1 remaining low, optimisation of the system was essential. We hypothesised that catalyst decomposition was occurring, as extended reaction times did not increase product formation. Analysis of the reaction mixture by 31P NMR spectroscopy revealed the presence of a new oxidised phosphine species, a commonly encountered off-cycle product during phosphine-promoted catalysis.36 As direct CO2 activation is not observed with this FLP system under the mild conditions used in catalysis, the mechanism likely proceeds via ring-opening of the epoxide substrate, followed by CO2 insertion as reported with other aryl borane-containing systems.27 Triphenylphosphine oxidation was not observed in the absence of epoxide, meaning that an on-cycle decomposition process involving PO must be occurring. Deoxygenation could potentially form propene, however analysis of the crude reaction mixture by 1H NMR spectroscopy revealed no obvious side products, potentially due to their gaseous nature.
As we previously noted that ring-opening of styrene oxide (SO) by poly(FLP) systems was rapid,23 we hoped to use this substrate to better understand this catalytic system. Under the same conditions, only 19% conversion from SO to styrene carbonate was observed. However, the system showed increased phosphine oxidation and the concomitant formation of styrene as confirmed by the presence of vinyl peaks at 5.3 ppm and 5.8 ppm in 1H NMR spectra (Fig. S7 and S9†). Inspired by this finding, we re-attempted the reaction with PO under milder conditions and preventing gaseous product release, and indeed observed propene in situ (Fig. S8†).
Evidence of alkene formation prompted us to consider the possible routes to epoxide deoxygenation that would result in phosphine oxidation. Previous work had shown ring-opening of epoxides by tertiary phosphines results in Wittig-like reactivity,37 while heating an FLP activated N2O complex releases N2 gas, forming a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–B linked species.38 Ring-opening of episulfides, the sulfuric analogue of epoxides, also leads to formation of similar linkages.15 It is thus hypothesised that, at elevated temperatures, CO2 insertion is competitive with alkene elimination and phosphine oxidation, gradually leading to catalyst decomposition (Fig. 3).
O–B linked species.38 Ring-opening of episulfides, the sulfuric analogue of epoxides, also leads to formation of similar linkages.15 It is thus hypothesised that, at elevated temperatures, CO2 insertion is competitive with alkene elimination and phosphine oxidation, gradually leading to catalyst decomposition (Fig. 3).
 | ||
| Fig. 3 Proposed route of catalyst deactivation for P1, represented using small-molecule model compounds. | ||
Catalyst activity could thus be renewed through the addition of disiloxane reducing agents36,39–42 used to regenerate the phosphine. Indeed, we found that addition of 1,3-diphenyldisiloxane (DPDS) to our PO/B1/P1/CO2 reaction mixture allowed for >99% conversion of PO (Fig. S10†). Although DPDS addition increased catalytic performance, the mix of desired carbonate products and alkene side products obtained when using SO both rendered the reaction less green and made product separation more difficult.
From this understanding, we sought to design a system to preclude this decomposition pathway. In N2O systems, elimination of N2 occurred when the phosphorus and boron components were oriented in the less energetically favourable cis conformation.38 If the same conformational requirements are present, as previous small molecule studies suggested the cis configuration is preferred for PO activation by a BPh3/PtBu2Me FLP pair,15 a bulkier polyphosphine may be preferred, especially as PPh3 has a lower cone angle than PtBu2Me.43 This doesn't preclude decomposition from the trans configurations as attack of the phosphine copolymer on SO occurs at the more hindered carbon of the epoxide ring (i.e. adjacent to the phenyl ring) as assigned by 31P–13C coupling constants (44.5 Hz, Fig. 4A). With this mode of attack, rotation from conformations I to II (Fig. 3) affords the undesired conformation, with decomposition encouraged by the enthalpic driving force of conjugated product formation (styrene).
 | ||
| Fig. 4 HSQC analysis demonstrating the influence of phosphine steric bulk on mode of SO ring-opening. Red = CH/CH3, blue = CH2. (A) P1/B1 system, attack of phosphine LB observed exclusively at more hindered carbon. (B) P2/B1 catalytic system, attack of phosphine LB at least hindered carbon is preferred by a factor of 7. Solvent peaks omitted for clarity. | ||
The switch to our previously reported mesityl-substitute P2 (ref. 16) would increase both steric bulk (favouring the trans attack and precluding rotation) and increase Lewis basicity to enhance its reactivity. The impact of this increased steric bulk from the mesityl groups was readily observable using small molecule model compounds, with a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 preference for ring-opening at the less hindered carbon observed for SO (Fig. 4B). Under the same reaction conditions, the P2/B1 catalyst combination led to >99% conversion of SO to styrene carbonate. Notably, no vinyl peaks were observed in the reaction mixture by 1H NMR spectroscopy (Fig. S13†), with minimal phosphine oxidation observed by 31P NMR spectroscopy. Taken together, the mesityl groups and polymer backbone bulk mitigate unproductive alkene elimination.
:1 preference for ring-opening at the less hindered carbon observed for SO (Fig. 4B). Under the same reaction conditions, the P2/B1 catalyst combination led to >99% conversion of SO to styrene carbonate. Notably, no vinyl peaks were observed in the reaction mixture by 1H NMR spectroscopy (Fig. S13†), with minimal phosphine oxidation observed by 31P NMR spectroscopy. Taken together, the mesityl groups and polymer backbone bulk mitigate unproductive alkene elimination.
Substrate scope
With an optimised system in hand, we investigated the substrate scope and overall reactivity of the P2/B1 system. Conversion of PO to propylene carbonate reaches >99% conversion in just 7 hours, dramatically outperforming the less Lewis basic P1. Realising that CO2/epoxide catalysis with small molecule FLPs is also unreported, we compared our macromolecular catalysts to their small molecule equivalents, BPh3 and PMes2Ph. These conventional FLPs, under the same reaction conditions, gave >99% conversion of PO within just 6 hours (Fig. S27†), the first example of conventional FLP use in CO2 insertion to epoxides. Use of a B/P system therefore significantly outperforms previously reported organocatalytic systems which typically require higher catalyst loadings.3,44 Use of a phosphine LB also allows for milder conditions than ammonium salt systems,29 while maintaining the same selectivity, although phosphonium systems are substantially more active.27A variety of substrates were screened for P2/B1 using similar reaction conditions (Fig. 5). All terminal epoxide substrates were quantitatively converted in under 24 hours, although their rates differed markedly depending on sterics and electronics. 3-Chloropropylene oxide (CPO) outperformed all other tested substrates, consistent with trends reported for other systems.27,45 While the electron withdrawing chlorine promotes rapid turnover, the sterically similar methyl group (butylene oxide, BO) shows decreased conversion. The role of steric bulk was also apparent for internal epoxides. While ring-opening of cyclohexene oxide (CHO) is possible using both P2/B1 and our previously reported poly(FLP) systems,23,24 productive conversion into product is not observed. Previous attempts to use conventional FLPs in the copolymerisation of CHO and carbonyl sulfide were also unsuccessful,26 although borane/phosphonium salts do form polycarbonates.27,28 To check if this was due to the strain induced during CO2 insertion preventing cyclohexyl rearrangements, cis-2,3-butylene oxide, CBO, was trialled. With no conversion for this derivative, it is likely down to steric clashes during ring-closure that prevent product formation.
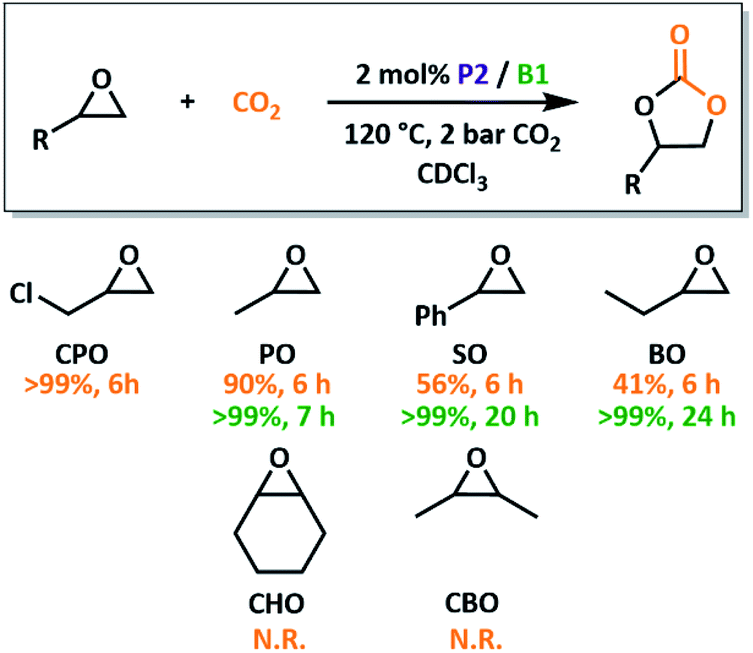 | ||
| Fig. 5 Screening of cyclic ether substrates for CO2 insertion to obtain cyclic carbonate products. Conversions calculated by 1H NMR spectroscopy after 6 h are shown in orange (N.R. = no reaction) and the times required for >99% conversion are shown in green. Reactions were carried out in a sealed ampoule on a 0.8 mmol scale using 2 bar CO2 pressure and catalyst P2/B1 (2 mol% as calculated from the number of expected active functional groups). | ||
Reactivity with oxetane substrates
Notably, oxetane (OX) can also be successfully ring-opened by P2/B1. Conversion to trimethylene carbonate (TMC) was slower due to the lower ring strain of OX relative to epoxide substrates,46,47 as well as the relatively high strength of the intermediate crosslinked networks formed as reported in our previous work.23 Attempts to expand the OX substrate scope with both 3-bromooxetane and 3,3-dimethyloxetane were unsuccessful. Although a change in chemical shift was observed in 11B NMR spectra, indicating LA coordination, no change was observed by 31P NMR spectroscopy for the LB counterpart, suggesting steric clashes prevent nucleophilic attack. Selectivity for cyclic 6-membered carbonates over their polymeric counterparts is often cited as problematic when using a CO2 insertion synthetic route.48 With epoxide substrates, high selectivity for the cyclic product is achieved at all temperatures screened. However, if the high temperatures optimised for epoxide ring opening are used (120 °C) with oxetane, only 61% preference for the cyclic product TMC (Table 1, entry 1) is observed. Lowering the temperature to 100 °C gives a significant improvement in selectivity, although longer reaction times are required to achieve high conversions (Table 1, entry 2 and 3). This additionally supports the formation of the polycarbonate product directly, rather than viaTMC polymerisation. Interestingly, while better leaving groups have been proposed as routes to improve cyclic product selectivity in small molecule catalysts,29 the opposite trend is observed herein where the better P1 leaving group gave less selectivity (Table 1, entry 4).
Catalyst recyclability
Polymer-supported catalysts have also shown potential for facile reuse or incorporation into flow reactors as an enabler for green manufacturing.49,50 Previous polymeric Lewis bases have been synthesised in pursuit of multiple use cycles but require more forcing conditions and suffer from limited substrate scopes.51,52 We thus decided to explore the catalytic performance of P2/B1 across multiple reaction cycles (Fig. 6) using a precipitation-recovery strategy (Section 6, ESI†), comparing it to the spiking of the reaction mixture with fresh substrate. This more rigorous recovery procedure is a true stress test of catalyst reuse using P2/B1. | ||
| Fig. 6 Conversions obtained in consecutive reaction cycles for CO2 insertion to PO, as calculated by 1H NMR spectroscopy, using 1,3,5-trimethoxybenzene as an internal standard. Reaction conditions as stated in Fig. 5. The data shown in green was obtained by adding fresh substrate at the end of each cycle (i.e. without purification and catalyst recovery). The recovered P2/B1 catalysts remain active after 4 cycles, achieving >99% conversion with extended reaction times (cycle 5). | ||
As can be seen in Fig. 6, and for either reuse methodology, catalytic performance decreases significantly for P2/B1 after the first reaction cycle. Smaller rate decreases are observed over subsequent cycles. Gradual phosphine oxidation can be observed (Fig. S40†), although attempted regeneration using DPDS did not substantially increase reaction rates, as the additional steric bulk from the mesityl groups could prohibit silane attack.39 It is important to note that this is a reduction in rate only, as extending the reaction time gives quantitative conversion (Fig. 6, cycle 5 and S39†).
It is important to highlight that these macromolecular catalysts are generated in situ, with the addition of substrate triggering gelation. This means that the accessibility of the catalyst active site is a key factor in reuse efficiency. Post-reaction, we observed that the catalyst resting state was present in its gelated, cross-linked form, supported by 31P and 11B NMR spectroscopy, and GPC measurements of hydrodynamic volume (Fig. S43 and S44†). Precipitation thus forms a superstructure that does not dissolve, but instead swells (Fig. S42†), creating a physical constraint on substrate access to catalytic sites.20 In our case, crosslinking occurs at the active components, limiting polymer chain segmental motions in the vicinity of the FLP and likely suppressing the rate of epoxide capture and CO2 insertion. This hypothesis is consistent with the observation that a significant decrease in activity is only observed after the first cycle. Future work in the team directed at incorporating these metal-free catalysts into flow reactors is an important step in overcoming these practical constraints.
Conclusions
In summary, we report the first use of both conventional and polymeric FLPs in the formation of carbonates from cyclic ethers using CO2, with high selectivity for the cyclic carbonate product. The synthesised macromolecular catalysts displayed good reactivity for a variety of terminal epoxide substrates and are also active catalysts for the formation of trimethylene carbonate from oxetane and CO2. Post-reaction, the poly(FLPs) used can be easily recovered and reused, with gradual decreases in catalyst efficiency attributed to partial phosphine oxidation as well as increased crosslinking. Further tuning of both the catalytic system and reaction conditions is expected to offer a more diverse substrate scope with higher selectivity for oxetane substrates. Further development of intermolecular poly(FLPs) for catalysis is expected to instead utilise the effects seen from crosslinking to enhance reactivity with other substrates via compartmentalisation.Author contributions
Thomas A. R. Horton and Meng Wang: investigation, methodology and manuscript writing. Michael P. Shaver: conceptualisation, manuscript writing – review and editing, supervision and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly thank the Leverhulme Trust (81420) and the University of Manchester for financial support. This work was also supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1, and EP/P025498/1 and the Sustainable Materials Innovation Hub, funded through the European Regional Development Fund OC15R19P.Notes and references
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- F. G. Fontaine and D. W. Stephan, Curr. Opin. Green Sustain. Chem., 2017, 3, 28–32 CrossRef.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed.
- S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476–3477 CrossRef CAS PubMed.
- B. Birkmann, T. Voss, S. J. Geier, M. Ullrich, G. Kehr, G. Erker and D. W. Stephan, Organometallics, 2010, 29, 5310–5319 CrossRef CAS.
- G. C. Welch, R. Prieto, M. A. Dureen, A. J. Lough, O. A. Labeodan, T. Höltrichter-Rössmann and D. W. Stephan, Dalton Trans., 2009, 1559–1570 RSC.
- S. Kronig, E. Theuergarten, D. Holschumacher, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2011, 50, 7344–7359 CrossRef CAS PubMed.
- T. Krachko, E. Nicolas, A. W. Ehlers, M. Nieger and J. C. Slootweg, Chem.–Eur. J., 2018, 24, 12669–12677 CrossRef CAS PubMed.
- M. Wang, F. Nudelman, R. R. Matthes and M. P. Shaver, J. Am. Chem. Soc., 2017, 139, 14232–14236 CrossRef CAS PubMed.
- U. Yolsal, M. Wang, J. R. Royer and M. P. Shaver, Macromolecules, 2019, 52, 3417–3425 CrossRef CAS.
- L. Chen, R. Liu and Q. Yan, Angew. Chem., Int. Ed., 2018, 57, 9336–9340 CrossRef CAS PubMed.
- A. Willms, H. Schumacher, T. Tabassum, L. Qi, S. L. Scott, P. J. C. Hausoul and M. Rose, ChemCatChem, 2018, 10, 1835–1843 CrossRef CAS.
- F. Vidal, J. McQuade, R. Lalancette and F. Jäkle, J. Am. Chem. Soc., 2020, 142, 14427–14431 CrossRef CAS PubMed.
- Q. Liu, L. Yang, C. Yao, J. Geng, Y. Wu and X. Hu, Org. Lett., 2021, 23, 3690 Search PubMed.
- N. Bouchard and F. G. Fontaine, Dalton Trans., 2019, 48, 4846–4856 RSC.
- U. Yolsal, T. A. R. Horton, M. Wang and M. P. Shaver, J. Am. Chem. Soc., 2021, 143, 12980–12984 CrossRef PubMed.
- M. Wang, J. Holland, T. A. R. Horton, U. Yolsal and M. P. Shaver, Polymer, 2022, 124576 CrossRef CAS.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- J. L. Yang, H. L. Wu, Y. Li, X. H. Zhang and D. J. Darensbourg, Angew. Chem., Int. Ed., 2017, 56, 5774–5779 CrossRef CAS PubMed.
- K. A. Andrea and F. M. Kerton, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS.
- K. A. Andrea and F. M. Kerton, RSC Adv., 2019, 9, 26542–26546 RSC.
- C. J. Zhang, S. Q. Wu, S. Boopathi, X. H. Zhang, X. Hong, Y. Gnanou and X. S. Feng, ACS Sustainable Chem. Eng., 2020, 8, 13056–13063 CrossRef CAS.
- Y. Y. Zhang, G. W. Yang, R. Xie, L. Yang, B. Li and G. P. Wu, Angew. Chem., Int. Ed., 2020, 132, 2–10 CrossRef.
- G. G. Gu, L. Y. Wang, R. Zhang, T. J. Yue, B. H. Ren and W. M. Ren, Polym. Chem., 2021, 12, 6436–6443 RSC.
- J. Zhang, L. Wang, S. Liu and Z. Li, Angew. Chem., Int. Ed., 2022, 61, e202111197 CAS.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106(6), 1235–1257 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- D. E. Bergbreiter, Y. C. Yang and C. E. Hobbs, J. Org. Chem., 2011, 76, 6912–6917 CrossRef CAS PubMed.
- D. E. Bissing and A. J. Speziale, J. Am. Chem. Soc., 1965, 87, 2683–2690 CrossRef CAS.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS PubMed.
- J. A. Buonomo, C. G. Eiden and C. C. Aldrich, Chem.–Eur. J., 2017, 23, 14434–14438 CrossRef CAS PubMed.
- C. J. O'Brien, J. L. Tellez, Z. S. Nixon, L. J. Kang, A. L. Carter, S. R. Kunkel, K. C. Przeworski and G. A. Chass, Angew. Chem., Int. Ed., 2009, 48, 6836–6839 CrossRef PubMed.
- E. Nicolas, A. Guerriero, V. Lyaskovskyy, M. Peruzzini, K. Lammertsma, L. Gonsalvi and J. C. Slootweg, Inorganics, 2016, 4, 34 CrossRef.
- M. Mehta, I. Garcia De La Arada, M. Perez, D. Porwal, M. Oestreich and D. W. Stephan, Organometallics, 2016, 35, 1030–1035 CrossRef CAS.
- J. A. Bilbrey, A. H. Kazez, J. Locklin and W. D. Allen, J. Comput. Chem., 2013, 34, 1189–1197 CrossRef CAS PubMed.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- E. Fazekas, G. S. Nichol, M. P. Shaver and J. A. Garden, Dalton Trans., 2018, 47, 13106 RSC.
- J. L. Wolk, M. Sprecher, H. Basch and S. Hoz, Org. Biomol. Chem., 2004, 2, 1065–1069 RSC.
- J. L. Wolk, T. Hoz, H. Basch and S. Hoz, J. Org. Chem., 2001, 66, 915–918 CrossRef CAS PubMed.
- J. Huang, C. Jehanno, J. C. Worch, F. Ruipérez, H. Sardon, A. P. Dove and O. Coulembier, ACS Catal., 2020, 10, 5399–5404 CrossRef CAS.
- U. Yolsal, T. A. R. Horton, M. Wang and M. P. Shaver, Prog. Polym. Sci., 2020, 111, 101313 CrossRef CAS.
- D. E. Bergbreiter, J. Tian and C. Hongfa, Chem. Rev., 2009, 109, 530–582 CrossRef CAS PubMed.
- S. N. Talapaneni, O. Buyukcakir, S. H. Je, S. Srinivasan, Y. Seo, K. Polychronopoulou and A. Coskun, Chem. Mater., 2015, 27, 6818–6826 CrossRef CAS.
- J. Lu and P. H. Toy, Synlett, 2011, 2011, 659–662 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00894g |
| This journal is © The Royal Society of Chemistry 2022 |