
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of double perovskite and quadruple perovskite nanocrystals through post-synthetic transformation reactions†
Hanjun
Yang
 ,
Tong
Cai
,
Lacie
Dube
and
Ou
Chen
*
,
Tong
Cai
,
Lacie
Dube
and
Ou
Chen
*
Department of Chemistry, Brown University, 324 Brook St., Providence, Rhode Island 02912, USA. E-mail: ouchen@brown.edu
First published on 30th March 2022
Abstract
Lead-free halide perovskite nanocrystals (NCs) represent a group of emerging materials which hold promise for various optical and optoelectronic applications. Exploring facile synthetic methods for such materials has been of great interest to not only fundamental research but also technological implementations. Herein, we report a fundamentally new method to access lead-free Bi-based double perovskite (DP) and quadruple perovskite (or layered double perovskite, LDP) NCs based on a post-synthetic transformation reaction of Cs3BiX6 (X = Cl, Br) zero-dimensional (0D) perovskite NCs under mild conditions. The produced NCs show good particle uniformity, high crystallinity, and comparable optical properties to the directly synthesized NCs. The relatively slow kinetics and stop-on-demand feature of the transformation reaction allow real-time composition–structure–property investigations of the reaction, thus elucidating a cation-alloyed intermediate-assisted transformation mechanism. Our study presented here demonstrates for the first time that post-synthetic transformation of 0D perovskite NCs can serve as a new route towards the synthesis of high-quality lead-free perovskite NCs, and provides valuable insights into the crystal structures, excitonic properties and their relationships of perovskite NCs.
Introduction
Moving away from lead-based perovskite materials driven by environmental and toxicity concerns, lead-free halide perovskite (LFHP) nanocrystals (NCs) have emerged as a promising family of materials that exhibits great potential in various applications owing to their unique optoelectronic properties and highly adaptable solution processibility.1–8 Within the family, double perovskites (DPs) and layered double perovskites (LDPs, also known as quadruple perovskites) are two widely-studied types of LFHPs with high structural similarity to lead-based perovskites in the cubic phase (Scheme 1).9–14 By replacing every two Pb2+ cations in APbX3 lead-halide perovskites with a pair of M(I) and M(III) cations, the DP crystal structure with balanced charge can be formed with a general chemical formula of A2M(I)M(III)X6 (A = Rb+, Cs+, etc.; X = Cl−, Br− or I−). The resulting DP lattice is composed of alternating [M(I)X6]5− (M(I) = Na+, K+, Ag+, etc.) and [M(III)X6]3− (M(III) = In3+, Sb3+, Bi3+, etc.) octahedra which construct a cubic framework through corner-shared halide X− anions (Scheme 1, left).10 One step further, when replacing every two M(I) cations in the DP with one M(II) cation and one vacancy (V), the LDP crystal structure with a chemical stoichiometry of A4M(II)M(III)2X12 can be obtained with an ordered sandwich structure of M(III)–M(II)–M(III) layers between two adjacent vacancy layers along the [111] direction of the original DP lattice (Scheme 1, right).13 Intriguing optical and optoelectronic properties have been reported for DP and LDP NCs.15–19 For example, Cs2M(I)InCl6 DP NCs, such as Cs2KInCl6 NCs and Cs2(Na1−xAgx)InCl6 NCs, show efficient white light photoluminescence (PL) through the recombination of the self-trapped excitons (STEs) due to the strong exciton–phonon coupling (EPC) strength.20–23 Vacancy-ordered double perovskites Cs2M(IV)X6, including Cs2SnX6 and Cs2ZrX6 NCs, exhibit a unique thermally activated delayed fluorescence with a tunable PL profile and high PL quantum yields (>60%).24–26 The Cs4CuSb2Cl12 LDP NCs possess a characteristic narrow direct band gap of ∼1.8 eV, making them a promising candidate material for applications such as photovoltaics, photoconductors and photocatalysts.27–29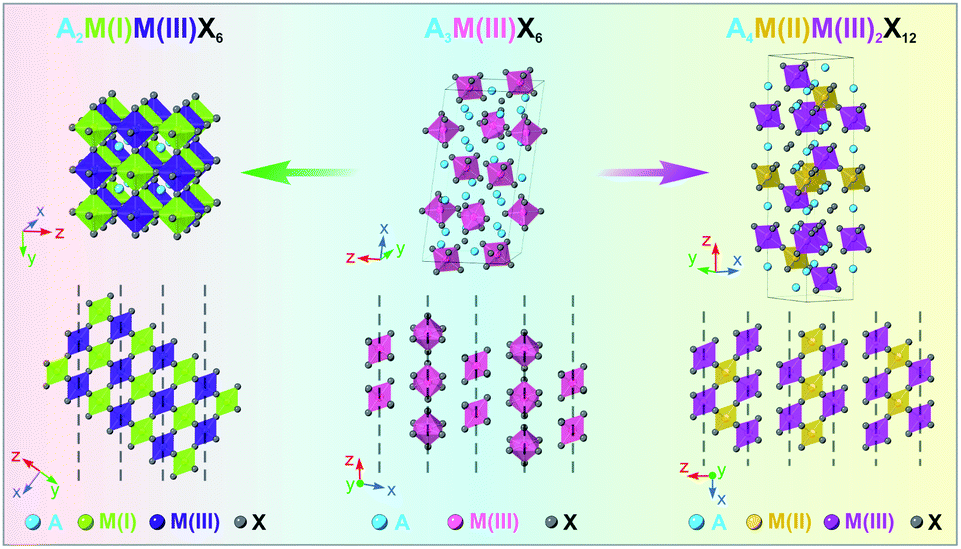 | ||
| Scheme 1 Schematic illustration of the crystal structures of the A2M(I)M(III)X6 DP phase (left), A3M(III)X6 0D perovskite phase (middle), and A4M(II)M(III)2X12 LDP phase (right). | ||
With regard to the material fabrication, current synthetic approaches that produce high-quality DP or LDP NCs mostly rely on a solution-based hot-injection method, where the needed precursors are rapidly mixed in a preheated organic solution to initiate the NC nucleation and growth.30–32 Yet, unlike lead-halide perovskite NCs, injection-based syntheses of DP and LDP NCs have encountered more problems due to the involvement of increased types of components as well as other possible reaction side-products, oftentimes leading to poor purities in the product composition and/or crystal phase.33–35 To alleviate this problem, indirect synthesis strategies for halide perovskite NCs involving converting the pre-existing NCs to targeted materials with desired compositions and crystal phases have been proposed and demonstrated.36–41 In this regard, zero-dimensional (0D) perovskite NCs with isolated metal-halide [MX6]3− octahedra have been prototyped as an ideal starting material for transformation reactions (Scheme 1, middle).42,43 The metal-deficient stoichiometry of 0D perovskites offers a strong tendency of transforming to other perovskite-type phases upon external stimulations such as varying solvent polarity or introducing additional metal halide precursors.44–56 For example, the transformation reaction of Cs4PbX6 0D perovskite NCs to obtain uniform CsPbX3 3D perovskite NCs has been successfully demonstrated.44,57–59 Unlike injection-based syntheses that rely on the rapid nucleation and growth rate to obtain NCs, the slow reaction kinetics and stop-on-demand feature of post-synthesis transformation are more beneficial for obtaining intermediate species during structural and compositional evolution. In addition, without using high temperature and reactive reagents, the post-synthesis transformation reaction could also be more suitable to obtain unstable species. Recently, we reported a colloidal synthesis of high-quality Cs3BiX6 (X = Cl, Br) 0D perovskite NCs which can be further transformed into Cs3Bi2X9 layered perovskite NCs post synthesis.50 To date, however, synthesizing lead-free DP or LDP NCs through a facile 0D-perovskite NC transformation reaction has not yet been reported.
Herein, we present a fundamentally new approach to access two important classes of LFHP NCs, i.e., Cs2AgBiX6 (X = Cl, Br) DP NCs and Cs4MBi2Cl12 (M = Cd, Mn) LDP NCs through post-synthetic transformation of Cs3BiX6 0D perovskite NCs by taking advantage of their structural flexibility and compositional tunability. The resulting LFHP NCs exhibit comparable qualities in terms of particle morphological uniformity and optical performances to the ones synthesized directly. Importantly, unlike the fast reactions during direct syntheses, the more controllable and slow reaction nature of the post-synthetic transformation process allows us to closely monitor the reaction evolution and capture intermediate products. Thus, the reaction mechanism can be studied and delineated as an alloy-intermediate-assisted intra-particle transformation process for both DP and LDP cases. In all, our study provides not only a new route towards fabrication of high-quality LFHP NCs, but also provides a powerful platform which enables a deeper understanding of composition–structure–property relationships of such fascinating materials.
Results and discussion
Zero-dimensional (0D) Cs3BiX6 (X = Cl, Br) NCs were first synthesized using our previously reported method.50 The crystal structure is composed of isolated [BiX6]3− octahedra (Scheme 1, middle), leading to a 0D electronic structure with molecule-like optical properties.60,61 The resulting 0D NCs showed characteristic sharp absorption peaks at 333 nm and 381 nm for Cs3BiCl6 and Cs3BiBr6 perovskite NCs, respectively (Fig. S1a†). Neither Cs3BiCl6 nor Cs3BiBr6 0D perovskite NCs exhibited any detectable PL signals at room temperature. The X-ray diffraction (XRD) patterns of both samples confirmed the monoclinic crystal structure of 0D perovskite (Fig. S1b†).50 Both Cs3BiCl6 and Cs3BiBr6 0D perovskite NCs exhibited a cubic shape with an average edge length of 7.7 ± 1.0 nm and 8.9 ± 1.3 nm, respectively (Fig. S1c–h†). The obtained high-quality Cs3BiX6 NCs were used as the starting material for the post-synthetic transformation reactions described below.Post-synthetic transformation reaction from Cs3BiX6 0D perovskite NCs to Cs2AgBiX6 3D DP NCs was carried out by adding an acetonitrile solution of silver nitrate (AgNO3) to the Cs3BiX6 NC hexane dispersion (see ESI† for details). The reaction was conducted at 50 °C for 24 hours and monitored by UV-vis absorption and PL spectroscopies. The low miscibility between hexane and acetonitrile facilitates the separation of NCs from the AgNO3 precursor, and the reaction temperature was chosen to avoid solvent evaporation. The occurrence of Cs3BiX6 NC transformation reaction was proved by the emergence of a new absorption peak at 362 nm (430 nm) and gradual disappearance of the initial 333 nm (381 nm) absorption peak for the Cl-based (Br-based) sample (Fig. 1a). In addition, a broad PL peak centered at 672 nm (670 nm) with a full-width-at-half-maximum (FWHM) of 0.69 eV (0.63 eV) was detected (Fig. 1a), which can be assigned to the STE emission of the Cs2AgBiCl6 (Cs2AgBiBr6) DP NCs.16 The PL excitation (PLE) spectra overlapped with the absorption profiles of the final Cs2AgBiX6 DP NC samples, which unambiguously confirmed that the PL peaks originate from the band gap absorption of Cs2AgBiX6 (Fig. 1a). The small shoulders of the PLE spectra on the longer wavelength side were likely caused by fine electronic substructures near the band edge of the Cs2AgBiX6 DP NCs.62 These optical properties of the Cs2AgBiX6 NCs synthesized via the post-synthesis transformation reaction showed nearly no difference from the ones synthesized via the direct hot-injection method (Fig. S2†).
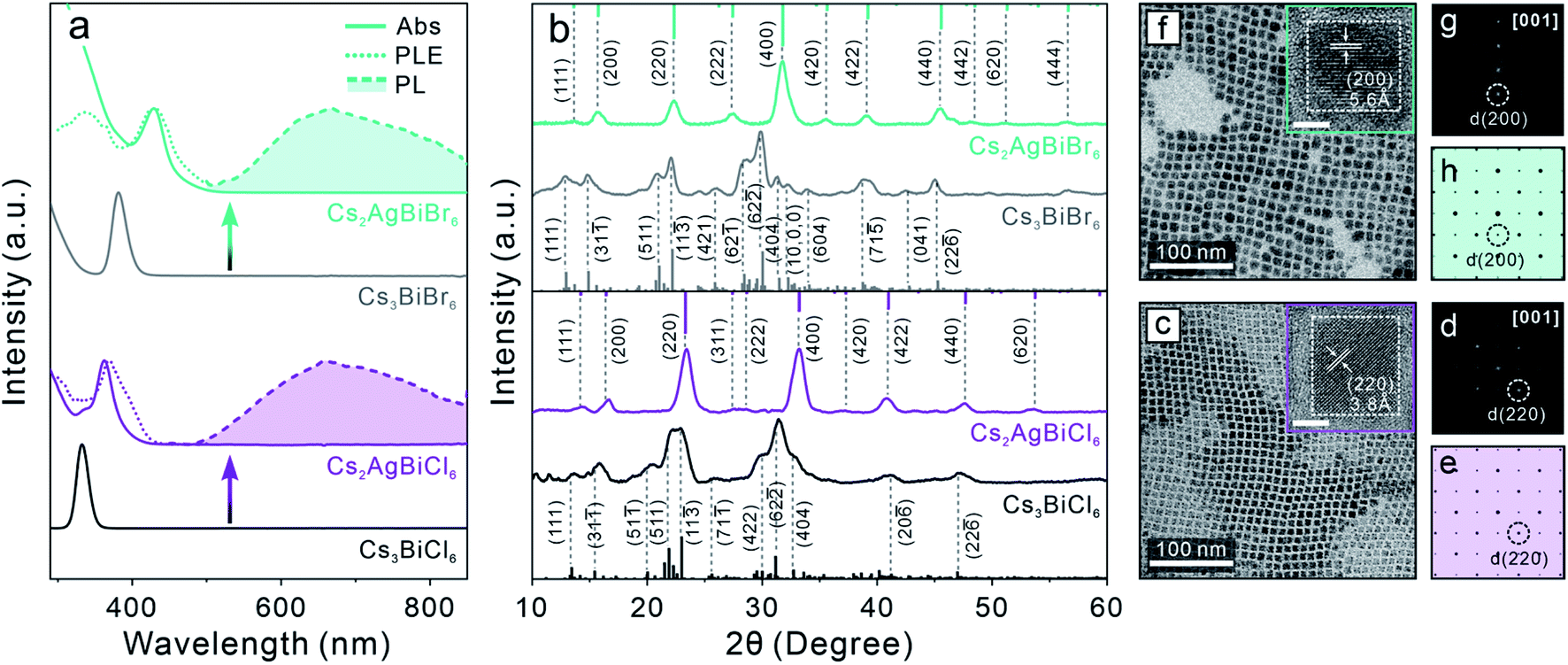 | ||
| Fig. 1 (a) Absorption spectra (solid lines), PL spectra (dashed lines with shade) and PLE spectra (dotted lines) of starting 0D perovskite NCs and the Cs2AgBiX6 DP NCs obtained by the transformation reactions. (b) XRD patterns of the starting 0D perovskite NCs and the Cs2AgBiX6 DP NCs. (c and f) TEM images of the resulting Cs2AgBiCl6 (c) and Cs2AgBiBr6 (f) DP NCs. Insets: HR-TEM images of the corresponding DP NCs. Scale bar = 5 nm. (d and g) Fast-Fourier transformation (FFT) patterns of the corresponding HR-TEM images. (e and h) Simulated electron-diffraction patterns of the DP crystal structure along the [001] zone axis. | ||
XRD patterns of the final samples confirmed the structural transformation from the 0D monoclinic phase to the 3D cubic DP phase (Fig. 1b). All the Bragg diffraction peaks of the final product matched the standard XRD pattern of the cubic DP structure. The disappearance of the characteristic 0D perovskite diffraction features – for example the absence of signals in the 2θ range of 30.0–32.8° corresponding to the (422) (62![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) and (404) characteristic Bragg diffractions of the monoclinic 0D phase (Fig. 1b) – proved the completion of transformation reaction, in accordance with optical measurements (Fig. 1a). Crystallite sizes of the produced DP NCs were estimated by Scherrer analyses to be 7.9 ± 1.0 nm and 7.8 ± 1.3 nm for Cs2AgBiCl6 and Cs2AgBiBr6 NCs, respectively (Fig. S3, Tables S1 and S2†), in agreement with TEM measurements (Fig. 1c, f and S4†). High-resolution TEM (HR-TEM) images and the corresponding fast-Fourier transformation (FFT) patterns of both samples showed the DP lattice viewed along its [001] zone direction (Fig. 1d, e, g and h).31,39 Nearly unchanged particle size and morphology between the starting and the final NCs suggested the preservation of individual NC integrity during the transformation reaction.44
) and (404) characteristic Bragg diffractions of the monoclinic 0D phase (Fig. 1b) – proved the completion of transformation reaction, in accordance with optical measurements (Fig. 1a). Crystallite sizes of the produced DP NCs were estimated by Scherrer analyses to be 7.9 ± 1.0 nm and 7.8 ± 1.3 nm for Cs2AgBiCl6 and Cs2AgBiBr6 NCs, respectively (Fig. S3, Tables S1 and S2†), in agreement with TEM measurements (Fig. 1c, f and S4†). High-resolution TEM (HR-TEM) images and the corresponding fast-Fourier transformation (FFT) patterns of both samples showed the DP lattice viewed along its [001] zone direction (Fig. 1d, e, g and h).31,39 Nearly unchanged particle size and morphology between the starting and the final NCs suggested the preservation of individual NC integrity during the transformation reaction.44
The drastic structural leap from 0D perovskite (with isolated octahedral units) to 3D perovskite (with connected octahedral units) motivated us to investigate the structural evolution mechanism. The relatively slow reaction kinetics and stop-on-demand feature of the transformation reactions allow us to fine-tune the Ag concentration as well as closely monitor the transformation process of the Cs3BiCl6 NC model system (Fig. 2), in contrast to the injection-based synthesis routes which exhibit rapid reactions kinetics that hinder real-time monitoring of the reaction progress. XRD measurements showed that with increasing the feeding amount of the Ag precursor (i.e., AgNO3), the crystal structure gradually changed from the initial 0D monoclinic phase to the 3D cubic DP structure (Fig. 2a). The cubic DP crystal structure started to emerge when the stoichiometry ratio between silver and bismuth (i.e., [Ag]/[Bi]) reached 15% (Fig. 2a, b and S5†). Given the low concentration of Ag+ cations, the Ag-doped 0D Cs3BiCl6 NCs coexisted with the Cs2(Cs1−xAgx)BiCl6 alloyed DP NCs (with the M(I) sites occupied by either Cs+ or Ag+ cations). Upon further increasing the Ag precursor amount, the DP crystal diffraction features became prominent, revealing an increased contribution from the DP crystal phase (Fig. 2a and b). Besides, all the DP diffraction peaks shifted to larger 2θ angles, indicating a continuous lattice contraction during the reaction (Fig. 2b), in line with the insertion of small Ag+ cations (ionic radius: 1.15 Å) to the M(I) site and replacing the larger Cs+ cation (ionic radius: 1.67 Å).63 For instance, the (400) peak shifted from 32.6° to 33.2° (Fig. 2b, and S5, Tables S1 and S3–S7†), corresponding to an ∼1% lattice constant shrinkage from 10.9 Å to 10.8 Å. The insertion of Ag+ cations and the corresponding local structural change were further supported by the X-ray photoelectron spectroscopy (XPS) measurements, where the Ag 3d peaks (3d5/2 and 3d3/2) of the sample showed enhanced intensity with increasing the [Ag]/[Bi] ratio (Fig. 2c and S6†). An ∼0.3 eV downshift in binding energy suggested an increase of the Ag–Cl bond strength,64 corresponding to the evolution from the Ag-doped 0D Cs3BiCl6 phase (where Ag+ cations occupy the original Cs-sites with the resulting Ag–Cl bond length of ∼3.7 Å) to the Cs2(Cs1−xAgx)BiCl6 alloyed DP phase (Ag–Cl bond length ∼ 2.7 Å). Such gradual insertion of Ag+ cations was also confirmed by the energy dispersive X-ray spectroscopy measurements (Fig. S7 and S8†). This structural change during the transformation reaction can also be reflected by the Raman spectral evolution (Fig. 2d). The Raman peak at 259 cm−1 corresponds to the symmetric stretching mode (A1g) of the [BiCl6]3− octahedra (Fig. S9†).65,66 After introducing the Ag component, a shoulder-like Raman feature emerged on the higher binding energy side, which was due to the Bi–Cl bond length shortening of the [BiCl6]3− octahedra upon lattice contraction. With increasing Ag concentration, the intensity of the newly emerged peak became more pronounced along with a gradual intensity decrease of the initial Raman peak (Fig. 2d). In addition, the new Raman peak continuously shifted to a larger wavenumber from 274 cm−1 to 281 cm−1, suggesting a Bi–Cl bond shortening process (Fig. 2d). Such results agreed well with the XRD measurements showing a continuous shrinkage of the [BiCl6]3− octahedra upon increasing the Ag+ concentration (Bi–Cl bond length decreased from 2.72 Å to 2.68 Å, Fig. S5, Tables S1 and S3–S7†). Moreover, the peak of [BiCl6]3− octahedral scissoring mode (T2g) became more defined (Fig. 2d), further supporting the increased crystallography symmetry with a simplified neighboring Cs+ environment (involved in the [BiCl6]3− scissoring vibration mode) in the final DP structure.66 TEM images showed that the perovskite NCs kept a similar size and size distribution for all the Cs2(AgxCs1−x)BiCl6 NCs with different amounts of Ag, proving the preservation of NC integrity during the reaction (Fig. 2e–h and S10†). This observation indicated an intra-particle transformation mechanism in contrast to the dissolution–recrystallization process reported for the case of Cs4PbBr6 0D perovskite NCs to CsPbBr3 perovskite NW transformation.51 Taking these results together, the transformation reaction can be described by the following chemical equations:
| Cs3BiCl6 + xAg+ → Cs(3−x)AgxBiCl6 + xCs+ | (1) |
| 2Cs(3−x)AgxBiCl6 + (y − x)Ag+ → Cs(3−x)AgxBiCl6·Cs2Cs(1−y)AgyBiCl6 + (y − x)Cs+ | (2) |
| Cs(3−x)AgxBiCl6·Cs2Cs(1−y)AgyBiCl6 + (y − x)Ag+ → 2Cs2Cs(1−y)AgyBiCl6 + (y − x)Cs+ | (3) |
| Cs2Cs(1−y)AgyBiCl6 + (1 − y)Ag+ → Cs2AgBiCl6 + (1 − y)Cs+ | (4) |
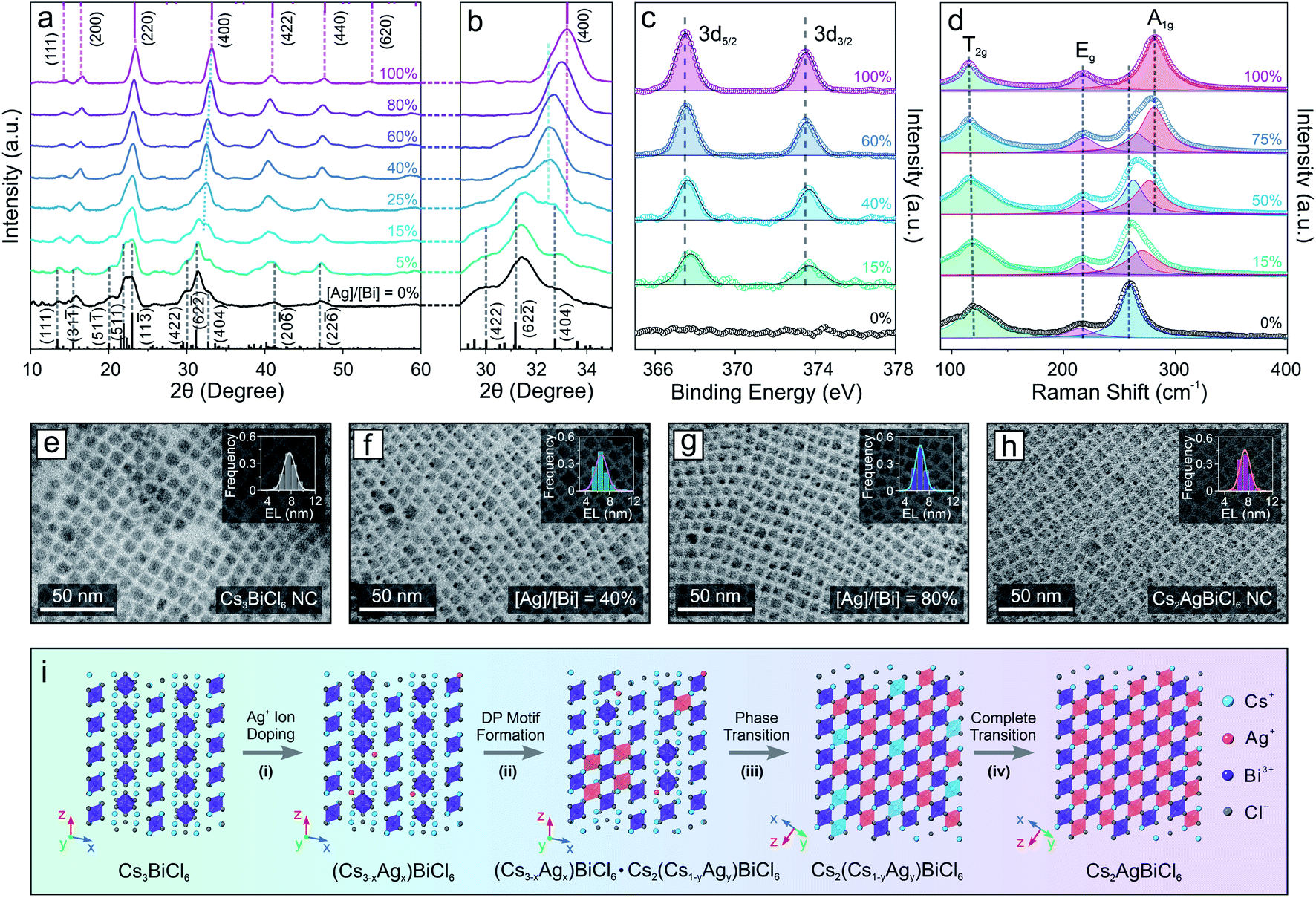 | ||
| Fig. 2 (a) XRD evolution of the DP transformation reaction from the Cs3BiCl6 0D perovskite NCs. Black bars represent the Cs3BiCl6 standard peak positions, and the purple bars represent the Cs2AgBiCl6 standard peak positions. (b) Zoomed-in area of the XRD patterns for clear visualization. (c) XPS spectra of Ag for the NCs during the transformation reaction. (d) Raman spectra of the NCs during the transformation reaction. (e–h) TEM images of the starting Cs3BiCl6 0D perovskite NCs, intermediate Cs2(Cs1−xAgx)BiCl6 NCs, and the final Cs2AgBiCl6 NCs. Insets: size-distribution histograms (EL: edge length). (i) Schematic illustration of the proposed transformation reaction mechanism. | ||
The entire structural transformation process of Cs3BiCl6 0D perovskite to Cs2AgBiCl6 3D DP can be delineated as shown in Fig. 2i. Upon introducing the Ag+ cations, the Ag-doped 0D perovskite phase is formed initially with Ag+ occupying the Cs-sites (Fig. 2i, step (i)). Increasing the Ag concentration leads to the partial phase transition from the 0D A3BiCl6 phase to the alloyed DP phase where both [AgCl6]5− and [CsCl6]5− octahedral units are formed by sharing the corner Cl− ions with the [BiCl6]3− octahedra inside each NC (Fig. 2i, step (ii)). In this process, the cubic DP crystal motif emerges and coexists with the pristine 0D perovskite structure. With further increasing the Ag concentration, the intermediate Cs2(AgxCs1−x)BiCl6 alloyed DP crystal motif progressively dominates the crystal phase of the NCs (Fig. 2i, step (iii)). When the Ag concentration reaches a sufficient level to fully occupy the M(I) sites of the DP crystal structure, pure Cs2AgBiCl6 DP NCs are obtained as the final product (Fig. 2i, step (iv)). Similar crystal structural evolution was also observed when transforming Cs3BiBr6 perovskite NCs to Cs2AgBiBr6 DP NCs (Fig. S11†), further proving the proposed transformation mechanism. It is worth mentioning that recent theoretical studies showed that the monoclinic 0D perovskite structure of the Cs3BiCl6 is thermodynamically more stable than the Cs2CsBiCl6 cubic DP structure (where one third of Cs+ ions occupy the M(I) site of the cubic DP crystal lattice, Scheme 1).67 However, the experimental results demonstrated here reveal that by introducing a smaller monovalent cation, the thermodynamically unfavored DP structure with both cations (e.g., Cs+ and Ag+) co-occupying the M(I) site of DP phase can be stabilized, which is at least partly because of the improved tolerance factor (t, increased from 0.78 for Cs2CsBiCl6 DP to 0.85 for Cs2AgBiCl6 DP).68
Doping of alkali metal cations (e.g., Na+ or K+) into the M(I) site of DPs was utilized to tune and optimize the optical performances of DP NCs by lifting the parity forbidden selection rule and partially reducing the electronic dimensionality.21,69 However, such effect in the DPs doped with large monovalent cations such as Cs+ has been rarely reported due to synthetic challenges.32,70,71 The post-synthetic transformation reaction discussed above allows for the preparation of Cs2(Cs1−xAgx)BiCl6 alloyed DP NCs, which leads us to further study the optical property evolution of the reaction. Upon increasing the Ag+ concentration, a new absorption peak at 362 nm (Fig. 3a) emerged along with a gradual decrease of the 333 nm peak from the initial Cs3BiCl6 NCs.31,33 Meanwhile, a broad emission feature centered at 765–785 nm (FWHM of 250–300 nm, or 0.55–0.65 eV) was observed for the Cs2(Cs1−xAgx)BiCl6 intermediates (Fig. 3b), which can be assigned to the radiative recombination of the STEs evidenced by the large Stokes shift and wide emission profile.72 The energy transfer from [BiCl6]3− is evident based on the PLE and absorption profile overlapping (Fig. 3a). The intensity of this emission peak increased drastically and reached its maximum at [Ag]/[Bi] = 50% (Fig. 3c). This initial PL intensity increase can be attributed to the increase of the Ag+ cation concentration, which can serve as centers for STE formation. Further increasing the Ag concentration ([Ag]/[Bi] > 50%) leads to a decrease of the PL intensity due to the enforced parity selection rule and the loss of dimension-reduction effect while forming DP lattices (Fig. 3c).21,73 In addition, the obvious ionic size difference between Cs+ (1.67 Å) and Ag+ (1.15 Å) can result in local structure deformation with an enhanced EPC effect, thus enlarging the STE trapping energy (Etrap),74 which was proved by the increased Stokes shift of the intermediates as compared to that of the final Cs2AgBiCl6 DP NCs (2.1 eV vs. 1.6 eV, Fig. 3c and d). The enlarged Etrap favors the radiative recombination of STEs by suppressing the non-radiative detrapping processes, in good accordance with the PL intensity measurements as well as time-resolved PL lifetime study results (Fig. 3c, d, S12 and Table S8†).75 The longer emission lifetime (∼1 μs) of the Cs2(AgxCs1−x)BiCl6 intermediates than that of the final DP NCs supports the suppression of STE non-radiative detrapping-induced processes (Fig. S12 and Table S8†).32 Also, the Cs2(Cs1−xAgx)BiCl6 alloyed DP NCs showed high stability as minimal variations in the absorption spectra were observed upon storage under ambient conditions for at least 42 days (Fig. S13†). No such red-shifted PL signal was observed for any intermediates (i.e., Cs2(Cs1−xAgx)BiBr6 alloyed DP NCs) in the Cs3BiBr6 transformation reactions (Fig. S14†), which can be attributed to the weaker EPC effect of the Br-based system as reported previously.76,77
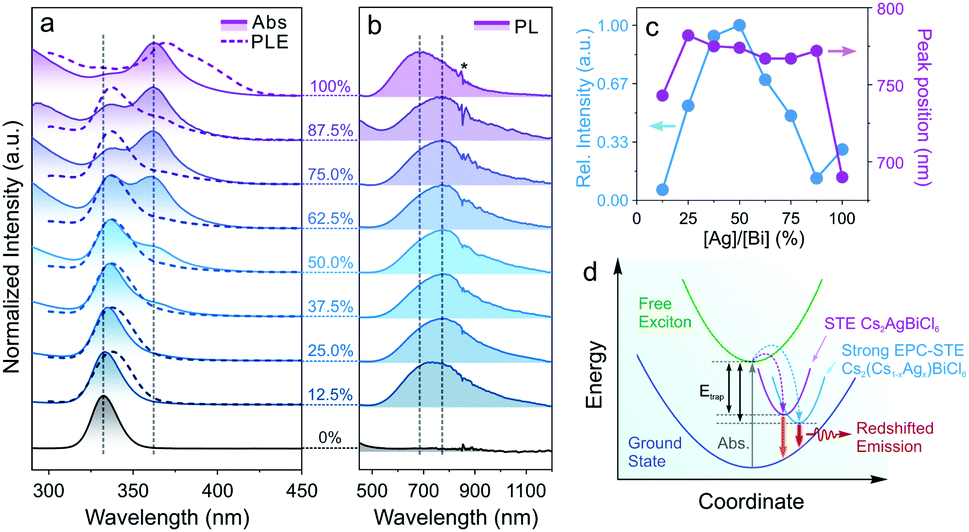 | ||
| Fig. 3 (a) Evolution of the absorption spectra (solid lines with shade) and the PLE (dashed lines) spectra of the Cs2(Cs1−xAgx)BiCl6 NCs. (b) PL spectral evolution of the Cs2(Cs1−xAgx)BiCl6 NCs (the asterisk marks the artifact caused by switching detectors). (c) The PL peak position and the relative PL intensity as a function of the [Ag]/[Bi] stoichiometry ratio. (d) Schematic illustration of the STE-based electronic transition and emission mechanism. | ||
To further generalize this post-synthetic transformation method to fabricate other types of LFHP NCs, we performed the transformation reaction targeting 2D LDP NCs. In this case, a biphasic reaction was performed by adding excess amounts of MnCl2 or CdCl2 metal halide powders to the Cs3BiCl6 NC colloidal solution (2 mg mL−1 in toluene) at 50 °C. The reaction was terminated after 24 hours, and the samples were purified and collected through centrifugation (see details in the ESI†). The UV-vis absorption peaks of the final Cs4MnBi2Cl12 and Cs4CdBi2Cl12 LDP NCs showed a subtle blueshift (∼3 nm) with direct band gaps of 3.38 eV (for Cs4MnBi2Cl12 LDP NCs) and 3.42 eV (for Cs4CdBi2Cl12 LDP NCs) determined by Tauc plot analyses (Fig. 4a and S15†), in good accordance with the samples directly synthesized using a hot-injection method.30 An appreciable peak broadening effect was observed for both cases (Fig. 4a), which may be attributed to the [BiCl6]3− octahedral distortion induced by connecting to the neighboring [M(II)Cl6]4− (M(II) = Mn or Cd) octahedral units through formation of the bridging Bi–Cl–M(II) bonds (Fig. S16†).30 In addition, a weak PL peak at 614 nm (FWHM: 98 nm or 0.32 eV, PL quantum yield < 1%) was detected for the Cs4MnBi2Cl12 LDP NC sample (Fig. 4a). The corresponding PLE spectrum showed a good match with the sample absorption profile (Fig. 4a), proving that the energy transfer process from the [BiCl6]3− (origin of the absorption) to the neighboring Mn2+ ion centers was responsible for the observed emission (4T1g → 6A1g electronic transition of the Mn2+ ion).30,78,79 Markedly, the absorption and PL spectra of the LDP NCs synthesized via the post-synthesis transformation method are comparable to those of the directly synthesized LDP NCs (Fig. S17†). XRD measurements of the final products confirmed the trigonal LDP crystal structure (space group: R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with the lattice parameters of a = 7.56 Å, c = 37.2 Å for Cs4MnBi2Cl12 and a = 7.60 Å, c = 37.2 Å for Cs4CdBi2Cl12 LDP NCs (Fig. 4b and S18, Tables S9 and S10†).30,80 TEM images showed that both Cs4MnBi2Cl12 and Cs4CdBi2Cl12 LDP NCs possessed a sphere-like shape with average diameters of 9.4 ± 1.6 nm and 10.8 ± 1.7 nm, respectively (Fig. 4c, f and S19†). The deviation from the cubic shape of starting Cs3BiCl6 NCs suggested an extensive reorganization of the crystal structure within individual NCs. HR-TEM images and the corresponding FFT patterns further confirmed the LDP structure with high crystallinity of the samples (Fig. 4d, e, g and h). Together, all these results unambiguously exhibited the successful syntheses of high-quality LDP NCs through the post-synthetic transformation from Cs3BiCl6 0D perovskite NCs. Absorption spectra of the LDP NCs synthesized by transformation reaction showed no observable changes upon storage under ambient conditions for over a month, suggesting high stability of the NCs synthesized by this method (Fig S20†).
m) with the lattice parameters of a = 7.56 Å, c = 37.2 Å for Cs4MnBi2Cl12 and a = 7.60 Å, c = 37.2 Å for Cs4CdBi2Cl12 LDP NCs (Fig. 4b and S18, Tables S9 and S10†).30,80 TEM images showed that both Cs4MnBi2Cl12 and Cs4CdBi2Cl12 LDP NCs possessed a sphere-like shape with average diameters of 9.4 ± 1.6 nm and 10.8 ± 1.7 nm, respectively (Fig. 4c, f and S19†). The deviation from the cubic shape of starting Cs3BiCl6 NCs suggested an extensive reorganization of the crystal structure within individual NCs. HR-TEM images and the corresponding FFT patterns further confirmed the LDP structure with high crystallinity of the samples (Fig. 4d, e, g and h). Together, all these results unambiguously exhibited the successful syntheses of high-quality LDP NCs through the post-synthetic transformation from Cs3BiCl6 0D perovskite NCs. Absorption spectra of the LDP NCs synthesized by transformation reaction showed no observable changes upon storage under ambient conditions for over a month, suggesting high stability of the NCs synthesized by this method (Fig S20†).
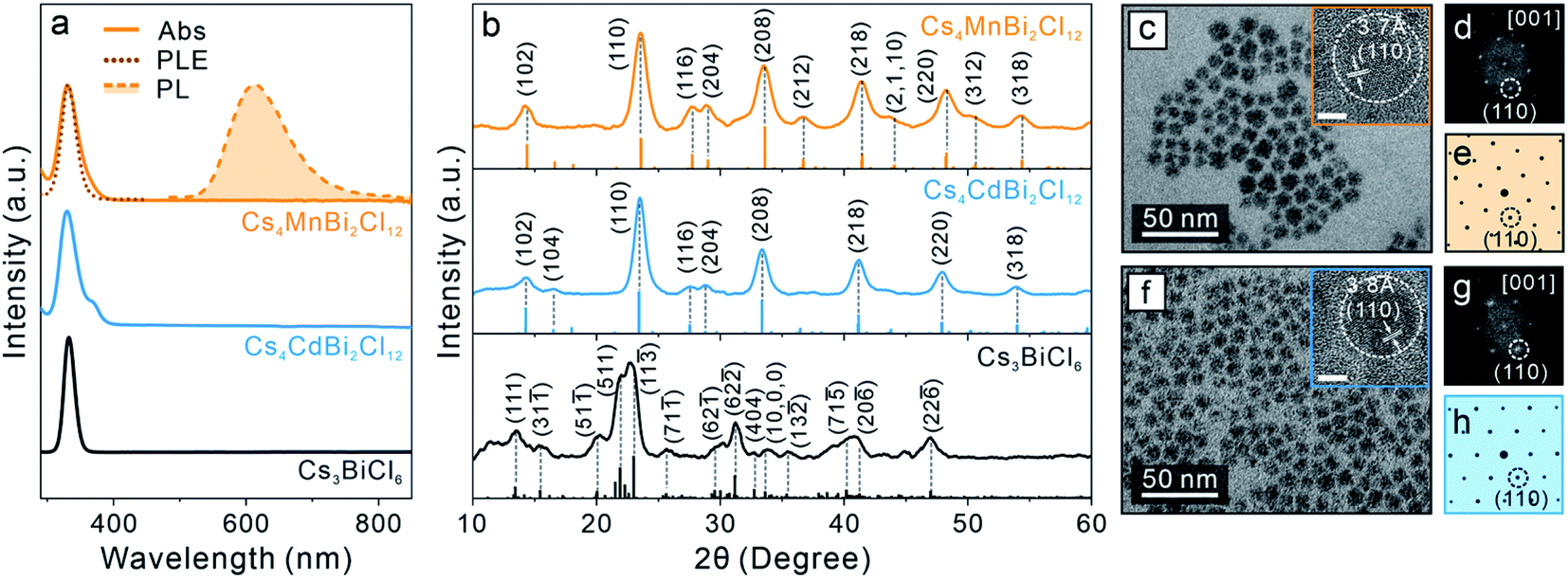 | ||
| Fig. 4 (a) Absorption (solid lines), PL (dashed line with shade) and PLE (dotted line) of the starting Cs3BiCl6 0D perovskite NCs and the final LDP NCs obtained by the transformation reaction. (b) XRD patterns of the starting 0D perovskite NCs and the final LDP NCs. Bars represent standard diffraction peak positions. (c and f) TEM images of the final Cs4MnBi2Cl12 (c) and Cs4CdBi2Cl12 (f) LDP NCs. Insets: HR-TEM images of the corresponding LDP NCs. Scale bar = 5 nm. (d and g) The FFT patterns of the corresponding HR-TEM images. (e and h) Simulated electron diffraction patterns of the LDP crystal structure along the [001] zone axis. | ||
To understand the detailed transformation mechanism, we monitored the optical property evolution for the Mn-containing reaction. The absorption peak showed both red-shifting and broadening effects as described for the final Cs4MnBi2Cl12 LDP NCs within 3 hours of the transformation reaction and remained nearly unchanged afterwards (Fig. 5a). Meanwhile, the PL peak started to emerge at around 2 hours followed by blue-shifting from ∼630 nm to ∼610 nm along with a dramatic intensity increase within 4 hours of the reaction (Fig. 5a and S21†). Both PL peak position and intensity showed negligible changes for the rest of the reaction (Fig. S21†). The PL peak blueshift indicated that the incorporated Mn ions migrated from the NC surface to the inside (diluting Mn2+ ions with reduced Mn–Mn coupling interactions) through a dopant inward diffusion process, similar to the post-synthetic Mn doping into CsPbCl3 NCs reported previously.81 Time-resolved PL lifetime measurements further supported the occurrence of inward diffusion by showing the average lifetime change from 0.25 ms for the 2 hour sample (relaxed spin-forbidden 4T1g to 6A1g transition by strong Mn–Mn coupling) to 0.44 ms (reenforced spin-forbidden transition after diluting the Mn2+ ion concentration), and finally to 0.25 ms for the final LDP NCs (intralayer antiferromagnetic ordering effect of the 2D LDP lattices) (Fig. S22 and Table S11†).80,82 The significantly longer PL lifetime (average PL lifetime of 0.25–0.44 ms for the post-synthesis transformed NCs vs. ∼ 10 μs for the directly synthesized ones) suggests that the defect-induced non-radiative recombination routes are greatly suppressed in the Cs4MnBi2Cl12 LDP NC synthesized via the post-synthesis transformation reaction as compared to the directly synthesized ones.30
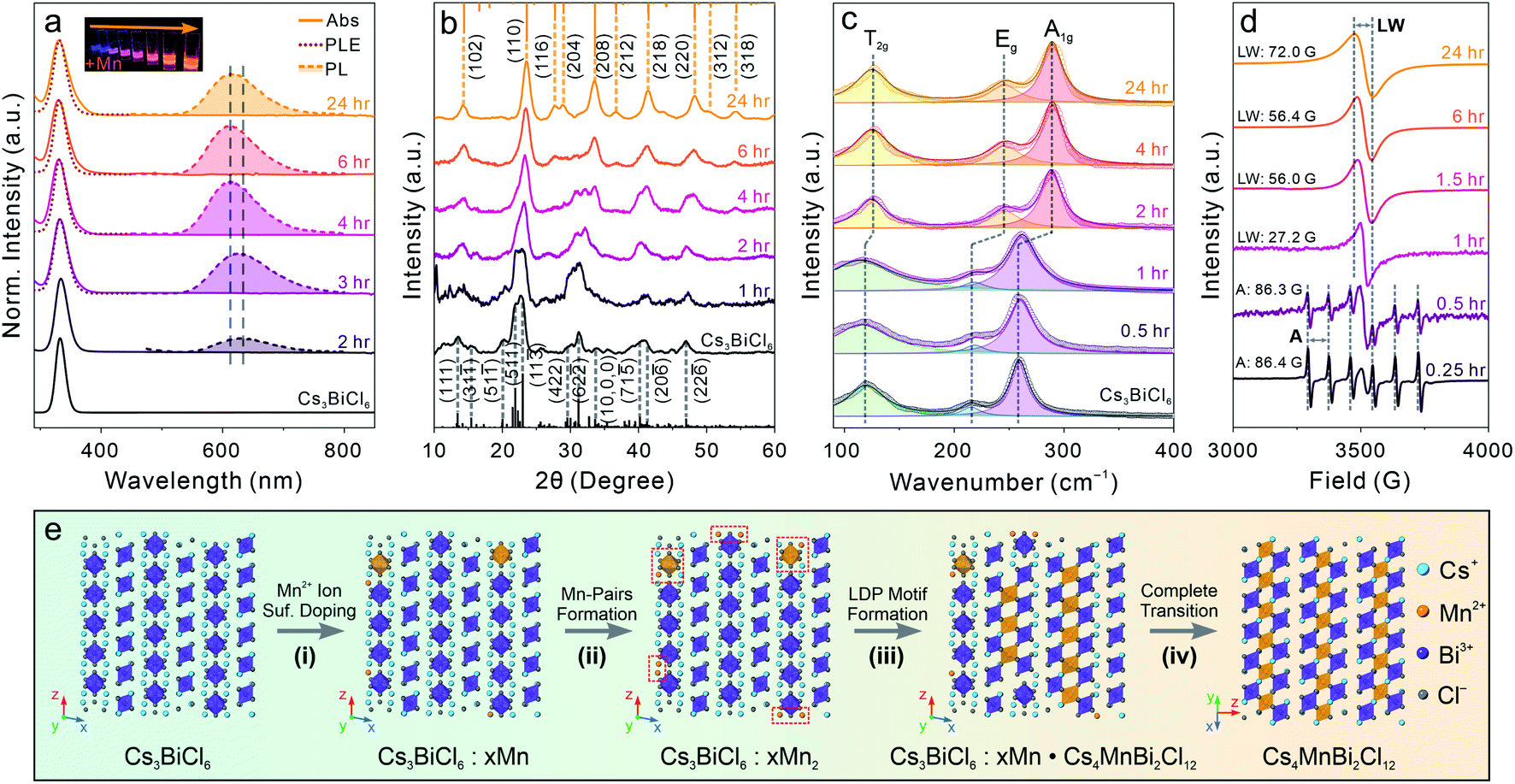 | ||
| Fig. 5 (a) Evolution of the absorption spectra (solid lines), PLE spectra (dotted lines), and PL spectra (dashed lines with shade). Inset: photograph of the NCs with different reaction times under UV light. (b) XRD pattern evolution during the transformation reaction. (c) Raman spectra of the NCs during the transformation reaction. (d) EPR spectral evolution during the transformation reaction. LW: linewidth; and A: hyperfine splitting constant. (e) Schematic illustration of the proposed transformation reaction mechanism. | ||
XRD measurements showed that at the early stage of the reaction, the initial 0D perovskite remained as the dominant crystal phase, followed by a gradual transition to the Cs4MnBi2Cl12 LDP phase starting from 2 hours after the reaction started (Fig. 5b). This structural evolution was consistent with the optical measurements and was also supported by the Raman spectroscopy characterization (Fig. 5c). Raman spectra showed the emergence of a new Raman peak at a higher bonding energy of ∼290 cm−1 at around 2 hours of the reaction, which can be assigned to the symmetric stretching (A1g) of the Bi–Cl bonds with a shortened bond length of 2.62 Å in an octahedral coordination environment (Fig. S16†). The narrowing of Raman peaks, especially for the low-energy peak associated with the T2g mode ([BiCl6]3− scissoring vibration mode), reflected the high symmetry of the LDP crystal structure as compared to the Cs3BiCl6 structure.66 The electron paramagnetic resonance (EPR) spectra showed a six-fold hyperfine splitting pattern with an average splitting constant of ∼86.3–86.4 G at the initial reaction stage (within one hour), confirming that the Mn2+ ions were present in the isolated [MnCl6]4− octahedral environment (replacing Bi3+ centers, Fig. 5d).83,84 The sextet pattern was later replaced by a single EPR peak with the linewidth gradually broadening from 27.2 G (at one hour) to 72.0 G (at 24 hours) for the final Cs4MnBi2Cl12 LDP NCs (Fig. 5d). We recently demonstrated that the symmetry-orientated spin-exchange interaction played the determining role in narrowing the EPR linewidth of Cs4(Cd1−xMnx)Bi2Cl12 alloyed LDP NCs with increasing the Mn concentration.30,85 However, the opposite trend observed here indicated that the EPR linewidth evolution, in this case, was predominated by the Mn–Mn dipole–dipole interactions over the spin-exchange interactions.30,85,86 For the initial intermediate NCs, a strong localized spin-exchange interaction overwhelmed the global dipole–dipole interaction among the dispersed Mn2+ ion pairs, resulting in a relatively narrow EPR peak (Fig. 5d). Upon increasing the Mn concentration and transforming the crystal phase to the LDP structure containing in-plane [MnCl6]4− octahedral layers (aligned in the (001) plane of LDP), the Mn–Mn dipole–dipole interactions enhanced and gradually outperformed the spin-exchange interactions, subsequently leading to a broadened EPR linewidth as observed in the experiments (Fig. 5d).
Taking all the results together, a complete transformation scheme to LDP NCs can be proposed as shown in Fig. 5e. Initially, the Mn2+ ions from the dissolved MnCl2 precursor (assisted by oleylamine ligands) reach surfaces of the starting Cs3BiCl6 NCs and subsequently form the inhomogeneously surface-doped Cs3BiCl6 NCs (replacing the surface Cs+ or Bi3+ ions, Fig. 5e, step (i)). When the Mn concentration is low, the Mn2+ ions preferentially replace the neighboring Cs+ ions (due to the relatively weak Cs–Cl bond) and occupy the A-site of the 0D perovskite NCs to form Mn–Mn pairs within proximity (shortest Mn–Mn distance of 4.6 Å vs. 7.5 Å in Cs4MnBi2Cl12 LDP) (Fig. 5e, step (ii)). The reaction is then allowed to proceed by inward diffusion of surface Mn dopants while incorporating more Mn2+ ions onto the particle surface (Fig. 5e, step (iii)). The inward-diffused Mn2+ ions start to produce internal [MnCl6]4− octahedral units and connect with the neighboring [BiCl6]3− octahedra, forming the Cs4MnBi2Cl12 LDP motif that coexists with the 0D structure in individual NCs. Upon further increasing the Mn concentration, the 0D perovskite motif was gradually replaced by the 2D LDP structure driven by the latter's enhanced structural stability with balanced charge.87 Finally, when the Mn concentration reaches the stoichiometry ratio of [Mn]/[Bi] = 50%, the final Cs4MnBi2Cl12 LDP NCs are obtained (Fig. 5e, step (iv)). The overall transformation reaction can be expressed by the following chemical equations:
| Cs3BiCl6 + xMn2+ → Cs3BiCl6:xMn2+ | (5) |
| 3Cs3BiCl6:xMn2+ + (1 − 2x)Mn2+ → Cs3BiCl6:xMn2+·Cs4MnBi2Cl12 + 2Cs+ | (6) |
| 2Cs3BiCl6:xMn2+·Cs4MnBi2Cl12 + (1 − 2x)Mn2+ → 3Cs4MnBi2Cl12 + 2Cs+ | (7) |
Conclusions
In conclusion, we report a facile synthetic strategy towards the fabrication of lead-free Bi-based perovskite NCs based on a post-synthetic transformation reaction of the Cs3BiX6 (X = Cl, Br) 0D perovskite NCs under mild conditions. Both high-quality Cs2AgBiX6 3D DP NCs and Cs4M(II)Bi2Cl12 (M(II) = Cd, Mn) 2D LDP NCs can be obtained using this method, resulting in LFHP NCs with uniform size and comparable optical properties to the ones obtained from conventional hot-injection syntheses. The evolution of optical properties and NC structure was investigated, based on which we propose an intra-particle transformation mechanism involving cation-alloyed intermediates. The slow reaction kinetics and stop-on-demand feature of the transformation reaction facilitate the synthesis of Cs2(Cs1−xAgx)BiCl6 intermediate DP NCs which possess unique STE emission. We anticipate that this post-synthetic NC transformation method can be further expanded to other perovskite and/or even non-perovskite systems. Our study presented here demonstrates a low-energy-input and controllable pathway towards the production of high-quality LFHP NCs post synthesis and paves the road for future exploration of novel perovskite and perovskite-analogue materials and understanding their composition- and structure-related properties.Data availability
All the relevant data discussed in the manuscript are provided within the article and in the ESI.†Author contributions
H. Y. and O. C. conceived and designed the experiments. H. Y. performed NC synthesis and the transformation reaction. T. C. performed DP NC synthesis and XPS measurements. H. Y. and L. D. performed optical spectra measurements. H. Y. performed XRD, TEM, Raman, and EPR measurements. O. C. supervised the entire projects. The manuscript was written through the contributions of all authors. All authors have given approval to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
O. C. acknowledges the supports from the National Science Foundation (CMMI-1934314), and NASA-RI EPSCoR Research Seed Grant. O. C. also thanks for the supports of TEM, XRD, Raman and XPS measurements performed at the Electron Microscopy Facility and NanoTools Facility at Brown UniversityNotes and references
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X.-g. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C.-J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed.
- Q. Fan, G. V. Biesold-McGee, J. Ma, Q. Xu, S. Pan, J. Peng and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 1030–1046 CrossRef CAS PubMed.
- Y. Wang, L. Song, Y. Chen and W. Huang, ACS Photonics, 2020, 7, 10–28 CrossRef CAS.
- W. Ke and M. G. Kanatzidis, Nat. Commun., 2019, 10, 965 CrossRef PubMed.
- I. Infante and L. Manna, Nano Lett., 2021, 21, 6–9 CrossRef CAS PubMed.
- S. Khalfin and Y. Bekenstein, Nanoscale, 2019, 11, 8665–8679 RSC.
- Z. Xiao, Z. Song and Y. Yan, Adv. Mater., 2019, 31, 1803792 CrossRef CAS PubMed.
- H. Yang, Y. Zhang, K. Hills-Kimball, Y. Zhou and O. Chen, Sustainable Energy Fuels, 2018, 2, 2381–2397 RSC.
- Y. Liu, A. Nag, L. Manna and Z. Xia, Angew. Chem., Int. Ed., 2021, 60, 11592–11603 CrossRef CAS PubMed.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- G. Volonakis, A. A. Haghighirad, R. L. Milot, W. H. Sio, M. R. Filip, B. Wenger, M. B. Johnston, L. M. Herz, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2017, 8, 772–778 CrossRef CAS PubMed.
- W. Pan, H. Wu, J. Luo, Z. Deng, C. Ge, C. Chen, X. Jiang, W.-J. Yin, G. Niu, L. Zhu, L. Yin, Y. Zhou, Q. Xie, X. Ke, M. Sui and J. Tang, Nat. Photonics, 2017, 11, 726–732 CrossRef CAS.
- B. Vargas, E. Ramos, E. Pérez-Gutiérrez, J. C. Alonso and D. Solis-Ibarra, J. Am. Chem. Soc., 2017, 139, 9116–9119 CrossRef CAS PubMed.
- J.-H. Wei, J.-F. Liao, X.-D. Wang, L. Zhou, Y. Jiang and D.-B. Kuang, Matter, 2020, 3, 892–903 CrossRef.
- B. A. Connor, L. Leppert, M. D. Smith, J. B. Neaton and H. I. Karunadasa, J. Am. Chem. Soc., 2018, 140, 5235–5240 CrossRef CAS PubMed.
- N. Chen, T. Cai, W. Li, K. Hills-Kimball, H. Yang, M. Que, Y. Nagaoka, Z. Liu, D. Yang, A. Dong, C.-Y. Xu, R. Zia and O. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 16855–16863 CrossRef CAS PubMed.
- M.-M. Yao, L. Wang, J.-S. Yao, K.-H. Wang, C. Chen, B.-S. Zhu, J.-N. Yang, J.-J. Wang, W.-P. Xu, Q. Zhang and H.-B. Yao, Adv. Opt. Mater., 2020, 8, 1901919 CrossRef CAS.
- H. Tang, Y. Xu, X. Hu, Q. Hu, T. Chen, W. Jiang, L. Wang and W. Jiang, Adv. Sci., 2021, 8, 2004118 CrossRef CAS PubMed.
- X. Zhu, L. Bian, H. Fu, L. Wang, B. Zou, Q. Dai, J. Zhang and H. Zhong, Light: Sci. Appl., 2020, 9, 73 CrossRef CAS PubMed.
- M. Cong, Q. Zhang, B. Yang, J. Chen, J. Xiao, D. Zheng, T. Zheng, R. Zhang, G. Qing, C. Zhang and K.-l. Han, Nano Lett., 2021, 21, 8671–8678 CrossRef CAS PubMed.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- P. Han, X. Zhang, C. Luo, W. Zhou, S. Yang, J. Zhao, W. Deng and K. Han, ACS Cent. Sci., 2020, 6, 566–572 CrossRef CAS PubMed.
- D. Manna, T. K. Das and A. Yella, Chem. Mater., 2019, 31, 10063–10070 CrossRef CAS.
- S. Liu, B. Yang, J. Chen, D. Wei, D. Zheng, Q. Kong, W. Deng and K. Han, Angew. Chem., Int. Ed., 2020, 59, 21925–21929 CrossRef CAS PubMed.
- A. Abfalterer, J. Shamsi, D. J. Kubicki, C. N. Savory, J. Xiao, G. Divitini, W. Li, S. Macpherson, K. Gałkowski, J. L. MacManus-Driscoll, D. O. Scanlon and S. D. Stranks, ACS Mater. Lett., 2020, 2, 1644–1652 CrossRef CAS PubMed.
- L. Tan, W. Wang, Q. Li, Z. Luo, C. Zou, M. Tang, L. Zhang, J. He and Z. Quan, Chem. Commun., 2020, 56, 387–390 RSC.
- T. Cai, W. Shi, S. Hwang, K. Kobbekaduwa, Y. Nagaoka, H. Yang, K. Hills-Kimball, H. Zhu, J. Wang, Z. Wang, Y. Liu, D. Su, J. Gao and O. Chen, J. Am. Chem. Soc., 2020, 142, 11927–11936 CrossRef CAS PubMed.
- T. Cai, W. Shi, D. J. Gosztola, K. Kobbekaduwa, H. Yang, N. Jin, Y. Nagaoka, L. Dube, J. Schneider, S. Hwang, J. Gao, X. Ma and O. Chen, Matter, 2021, 4, 2936–2952 CrossRef CAS.
- A. P. P, M. Joshi, D. Verma, S. Jadhav, A. R. Choudhury and D. Jana, ACS Appl. Nano Mater., 2021, 4, 1305–1313 CrossRef.
- H. Yang, W. Shi, T. Cai, K. Hills-Kimball, Z. Liu, L. Dube and O. Chen, Nanoscale, 2020, 12, 23191–23199 RSC.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed.
- D. Zhu, J. Zito, V. Pinchetti, Z. Dang, A. Olivati, L. Pasquale, A. Tang, M. L. Zaffalon, F. Meinardi, I. Infante, L. De Trizio, L. Manna and S. Brovelli, ACS Energy Lett., 2020, 5, 1840–1847 CrossRef CAS PubMed.
- Y. Bekenstein, J. C. Dahl, J. Huang, W. T. Osowiecki, J. K. Swabeck, E. M. Chan, P. Yang and A. P. Alivisatos, Nano Lett., 2018, 18, 3502–3508 CrossRef CAS PubMed.
- S. E. Creutz, H. Liu, M. E. Kaiser, X. Li and D. R. Gamelin, Chem. Mater., 2019, 31, 4685–4697 CrossRef CAS.
- S. Levy, S. Khalfin, N. G. Pavlopoulos, Y. Kauffmann, G. Atiya, S. Shaek, S. Dror, R. Shechter and Y. Bekenstein, Chem. Mater., 2021, 33, 2370–2377 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- G. P. Li, H. Wang, Z. F. Zhu, Y. J. Chang, T. Zhang, Z. H. Song and Y. Jiang, Chem. Commun., 2016, 52, 11296–11299 RSC.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Nano Lett., 2018, 18, 1118–1123 CrossRef CAS PubMed.
- Y. Lou, M. Fang, J. Chen and Y. Zhao, Chem. Commun., 2018, 54, 3779–3782 RSC.
- M. Que, Z. Dai, H. Yang, H. Zhu, Y. Zong, W. Que, N. P. Padture, Y. Zhou and O. Chen, ACS Energy Lett., 2019, 4, 1970–1975 CrossRef CAS.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- F. Palazon, G. Almeida, Q. A. Akkerman, L. De Trizio, Z. Dang, M. Prato and L. Manna, Chem. Mater., 2017, 29, 4167–4171 CrossRef CAS PubMed.
- F. Palazon, C. Urso, L. De Trizio, Q. Akkerman, S. Marras, F. Locardi, I. Nelli, M. Ferretti, M. Prato and L. Manna, ACS Energy Lett., 2017, 2, 2445–2448 CrossRef CAS PubMed.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- Y. Li, H. Huang, Y. Xiong, S. V. Kershaw and A. L. Rogach, CrystEngComm, 2018, 20, 4900–4904 RSC.
- X. Yu, L. Wu, H. Hu, M. Chen, Y. Tan, D. Yang, Q. Pan, Q. Zhong, T. Supasai and Q. Zhang, Langmuir, 2018, 34, 10363–10370 CrossRef CAS PubMed.
- H. Yang, T. Cai, E. Liu, K. Hills-Kimball, J. Gao and O. Chen, Nano Res., 2020, 13, 282–291 CrossRef CAS.
- H. Yang, T. Cai, L. Dube, K. Hills-Kimball and O. Chen, Cryst. Growth Des., 2021, 21, 1924–1930 CrossRef CAS.
- X. Sun, X. Shi, W. Zhang, B. Xu, Z. Gao, Z. Wang, X. Wang and X. Meng, Nanoscale, 2021, 13, 18647–18656 RSC.
- B. A. H. Huisman, F. Palazon and H. J. Bolink, Inorg. Chem., 2021, 60, 5212–5216 CrossRef CAS PubMed.
- F. Palazon, Q. A. Akkerman, M. Prato and L. Manna, ACS Nano, 2016, 10, 1224–1230 CrossRef CAS PubMed.
- B. Billstrand, K. Bian, C. Karler, D. Ye, A. Hwang and H. Fan, MRS Adv., 2018, 3, 2825–2831 CrossRef CAS.
- K. Hills-Kimball, H. Yang, T. Cai, J. Wang and O. Chen, Adv. Sci., 2021, 8, 2100214 CrossRef CAS PubMed.
- D. Baranov, G. Caputo, L. Goldoni, Z. Dang, R. Scarfiello, L. De Trizio, A. Portone, F. Fabbri, A. Camposeo, D. Pisignano and L. Manna, Chem. Sci., 2020, 11, 3986–3995 RSC.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- J. Ren, X. Zhou and Y. Wang, Nano Res., 2020, 13, 3387–3395 CrossRef CAS.
- J. Yin, P. Maity, M. De Bastiani, I. Dursun, O. M. Bakr, J.-L. Brédas and O. F. Mohammed, Sci. Adv., 2017, 3, e1701793 CrossRef PubMed.
- Y. Tang, M. Liang, B. Chang, H. Sun, K. Zheng, T. Pullerits and Q. Chi, J. Mater. Chem. C, 2019, 7, 3369–3374 RSC.
- A. Schmitz, L. L. Schaberg, S. Sirotinskaya, M. Pantaler, D. C. Lupascu, N. Benson and G. Bacher, ACS Energy Lett., 2020, 5, 559–565 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. Lee, J. H. Park, B. R. Lee, E. D. Jung, J. C. Yu, D. Di Nuzzo, R. H. Friend and M. H. Song, J. Phys. Chem. Lett., 2017, 8, 1784–1792 CrossRef CAS PubMed.
- W. M. A. Smit, G. J. Dirksen and D. J. Stufkens, J. Phys. Chem. Solids, 1990, 51, 189–196 CrossRef CAS.
- S. J. Zelewski, J. M. Urban, A. Surrente, D. K. Maude, A. Kuc, L. Schade, R. D. Johnson, M. Dollmann, P. K. Nayak, H. J. Snaith, P. Radaelli, R. Kudrawiec, R. J. Nicholas, P. Plochocka and M. Baranowski, J. Mater. Chem. C, 2019, 7, 8350–8356 RSC.
- W. Shi, T. Cai, Z. Wang and O. Chen, J. Chem. Phys., 2020, 153, 141101 CrossRef CAS PubMed.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- Z. Xiao, W. Meng, J. Wang, D. B. Mitzi and Y. Yan, Mater. Horiz., 2017, 4, 206–216 RSC.
- B. Ke, R. Zeng, Z. Zhao, Q. Wei, X. Xue, K. Bai, C. Cai, W. Zhou, Z. Xia and B. Zou, J. Phys. Chem. Lett., 2020, 11, 340–348 CrossRef CAS PubMed.
- R. S. Lamba, P. Basera, S. Bhattacharya and S. Sapra, J. Phys. Chem. Lett., 2019, 10, 5173–5181 CrossRef CAS PubMed.
- B. M. Benin, D. N. Dirin, V. Morad, M. Wörle, S. Yakunin, G. Rainò, O. Nazarenko, M. Fischer, I. Infante and M. V. Kovalenko, Angew. Chem., Int. Ed., 2018, 57, 11329–11333 CrossRef CAS PubMed.
- F. Locardi, E. Sartori, J. Buha, J. Zito, M. Prato, V. Pinchetti, M. L. Zaffalon, M. Ferretti, S. Brovelli, I. Infante, L. De Trizio and L. Manna, ACS Energy Lett., 2019, 4, 1976–1982 CrossRef CAS.
- D. Cortecchia, S. Neutzner, A. R. Srimath Kandada, E. Mosconi, D. Meggiolaro, F. De Angelis, C. Soci and A. Petrozza, J. Am. Chem. Soc., 2017, 139, 39–42 CrossRef CAS PubMed.
- S. D. Stranks, V. M. Burlakov, T. Leijtens, J. M. Ball, A. Goriely and H. J. Snaith, Phys. Rev. Appl., 2014, 2, 034007 CrossRef CAS.
- J. A. Steele, P. Puech, M. Keshavarz, R. Yang, S. Banerjee, E. Debroye, C. W. Kim, H. Yuan, N. H. Heo, J. Vanacken, A. Walsh, J. Hofkens and M. B. J. Roeffaers, ACS Nano, 2018, 12, 8081–8090 CrossRef CAS PubMed.
- L. Zhang, Y. Fang, L. Sui, J. Yan, K. Wang, K. Yuan, W. L. Mao and B. Zou, ACS Energy Lett., 2019, 4, 2975–2982 CrossRef CAS.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS PubMed.
- T. Cai, J. Wang, W. Li, K. Hills-Kimball, H. Yang, Y. Nagaoka, Y. Yuan, R. Zia and O. Chen, Adv. Sci., 2020, 7, 2001317 CrossRef CAS PubMed.
- B. Vargas, D. T. Reyes-Castillo, E. Coutino-Gonzalez, C. Sánchez-Aké, C. Ramos, C. Falcony and D. Solis-Ibarra, Chem. Mater., 2020, 32, 9307–9315 CrossRef CAS.
- K. Hills-Kimball, M. J. Pérez, Y. Nagaoka, T. Cai, H. Yang, A. H. Davis, W. Zheng and O. Chen, Chem. Mater., 2020, 32, 2489–2500 CrossRef CAS.
- J. Xu, C. Xu, J.-B. Liu, L. Bellaiche, H. Xiang, B.-X. Liu and B. Huang, npj Comput. Mater., 2019, 5, 114 CrossRef CAS.
- F. D. Tsay and L. Helmholz, J. Chem. Phys., 1969, 50, 2642–2650 CrossRef CAS.
- A. H. Davis, S. Li, H. Lin, C. Chu, J. M. Franck, G. Leem, M. M. Maye and W. Zheng, J. Mater. Chem. C, 2021, 9, 14226–14235 RSC.
- Y. Liu, J. Zhang, B. Han, X. Wang, Z. Wang, C. Xue, G. Bian, D. Hu, R. Zhou, D.-S. Li, Z. Wang, Z. Ouyang, M. Li and T. Wu, J. Am. Chem. Soc., 2020, 142, 6649–6660 CrossRef CAS PubMed.
- N. Abhyankar, S. Bertaina and N. S. Dalal, J. Phys. Chem. C, 2015, 119, 28143–28147 CrossRef CAS.
- Y.-P. Lin, S. Hu, B. Xia, K.-Q. Fan, L.-K. Gong, J.-T. Kong, X.-Y. Huang, Z. Xiao and K.-Z. Du, J. Phys. Chem. Lett., 2019, 10, 5219–5225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00574c |
| This journal is © The Royal Society of Chemistry 2022 |