
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeometrical influence on the non-biomimetic heterolytic splitting of H2 by bio-inspired [FeFe]-hydrogenase complexes: a rare example of inverted frustrated Lewis pair based reactivity†
Lucile
Chatelain
 *a,
Jean-Baptiste
Breton
a,
Federica
Arrigoni
*b,
Philippe
Schollhammer
*a and
Giuseppe
Zampella
*b
*a,
Jean-Baptiste
Breton
a,
Federica
Arrigoni
*b,
Philippe
Schollhammer
*a and
Giuseppe
Zampella
*b
aUMR CNRS 6521 Chimie, Electrochimie Moléculaires et Chimie Analytique, Université de Bretagne Occidentale, UFR Sciences et Techniques, 6 Avenue Victor le Gorgeu, CS 93837, Brest-Cedex 3, 29238, France. E-mail: philippe.Schollhammer@univ-brest.fr
bDepartment of Biotechnology and Bioscience, University of Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy. E-mail: giuseppe.zampella@unimib.it
First published on 22nd March 2022
Abstract
Despite the high levels of interest in the synthesis of bio-inspired [FeFe]-hydrogenase complexes, H2 oxidation, which is one specific aspect of hydrogenase enzymatic activity, is not observed for most reported complexes. To attempt H–H bond cleavage, two disubstituted diiron dithiolate complexes in the form of [Fe2(μ-pdt)L2(CO)4] (L: PMe3, dmpe) have been used to play the non-biomimetic role of a Lewis base, with frustrated Lewis pairs (FLPs) formed in the presence of B(C6F5)3 Lewis acid. These unprecedented FLPs, based on the bimetallic Lewis base partner, allow the heterolytic splitting of the H2 molecule, forming a protonated diiron cation and hydrido-borate anion. The substitution, symmetrical or asymmetrical, of two phosphine ligands at the diiron dithiolate core induces a strong difference in the H2 bond cleavage abilities, with the FLP based on the first complex being more efficient than the second. DFT investigations examined the different mechanistic pathways involving each accessible isomer and rationalized the experimental findings. One of the main DFT results highlights that the iron site acting as a Lewis base for the asymmetrical complex is the {Fe(CO)3} subunit, which is less electron-rich than the {FeL(CO)2} site of the symmetrical complex, diminishing the reactivity towards H2. Calculations relating to the different mechanistic pathways revealed the presence of a terminal hydride intermediate at the apical site of a rotated {Fe(CO)3} site, which is experimentally observed, and a semi-bridging hydride intermediate from H2 activation at the Fe–Fe site; these are responsible for a favourable back-reaction, reducing the conversion yield observed in the case of the asymmetrical complex. The use of two equivalents of Lewis acid allows for more complete and faster H2 bond cleavage due to the encapsulation of the hydrido-borate species by a second borane, favouring the reactivity of each FLP, in agreement with DFT calculations.
Introduction
Metalloenzymes have, over the years, shown high catalytic efficacy for the conversion of small and inert molecules (N2, H2, O2, CO, etc.) into valuable products without requiring noble metals for their active sites.1 Among them, [FeFe]-hydrogenase enzymes have received considerable attention.2 This enzyme family demonstrates efficient catalytic activity for the reversible conversion of protons into H2 gas, which is recognised as a key driver of future power and energy sources.2 However this reactivity is only supported under limited working conditions owing to the enzymatic sensitivity to exogenous substrates/oxygen.3–10 That is why synthetic chemists are tackling the challenging task of designing more robust catalysts using bio-inspired approaches.11 A multitude of mimics of the H-cluster, the active site of [FeFe]-hydrogenases, have been developed containing two iron metals bridged by a dithiolate ligand, allowing proton reduction,12–14 but only a handful of these complexes are able to activate H2.15 Of the rare examples supporting the arduous process of H2 activation, some complexes involve classical reactions, such as oxidative addition16 and reduction,17 or permit H2/D2 exchange.18–21 Inspired by the structure of the H-cluster, mixed-valent FeIFeII diiron cores were envisioned to support an active redox state for H2 binding and splitting, but the stability of such FeIFeII species limits this approach.22–30 Associated with amine functionality localized at the bridgehead of the dithiolate bridging ligand, some diiron complexes, e.g., {FeIFeII(μ-azadithiolate)}, permit the stoichiometric heterolytic splitting of H2;31,32 to date only three reported examples have achieved catalytic H2 oxidation.29,30,33 The limited interaction between the amine function of the bridgehead and the distal oxidized FeII metallic centre of the active site results in what has been described as a frustrated Lewis pair (FLP),34–36 where the amine in the second coordination sphere acts as a Lewis base and the iron metal plays the role of a Lewis acid.Concomitantly to this bioinspired strategy, the heterolytic activation of H2 using FLPs has emerged quickly in the past 15 years, starting from main group elements37–41 and then being extended to most elements of the periodic table. Metallic elements, such as transition metals, lanthanides, and even actinides, are alternatively used as supporting complexes,42–48 Lewis acids,49–54 or even Lewis base partners.55–65 To the best of our knowledge, no heterolytic splitting of H2 by a FLP combining a dinuclear moiety with metal–metal interactions as a Lewis base with a borane Lewis acid has been reported. One rare example, reminiscent of FLP chemistry, suggests that the compound [Fe2(μ-edt)(PMe3)2(CO)4] (edt2−: 1,2-ethanedithiolate) acts as a Lewis base when associated with an external borane Lewis acid, and this induces H2 cleavage.16 Considering the very limited number of models of [FeFe]-hydrogenase active sites able to activate the H–H bond, we have explored a non-biomimetic strategy to develop H2 activation using diiron dithiolate complexes.
In this work, we propose a unique study of the influence of symmetrical or unsymmetrical substitution at a diiron core, inspired by the H-cluster, on stoichiometric H2 activation based on two rare FLP examples involving a bimetallic moiety and B(C6F5)3 as the Lewis base and acid, respectively. The activities towards H2 of two low-valent FeIFeI diiron complexes [Fe2(μ-pdt)(PMe3)2(CO)4] (1) (pdt2−: 1,3-propanedithiolate)18 and [Fe2(μ-pdt)(κ2-dmpe)(CO)4] (2) (dmpe: 1,2-bis(dimethylphosphino)ethane)66 (Scheme 1) have been explored. Mechanistic investigations were performed via DFT to rationalize the different behaviours.
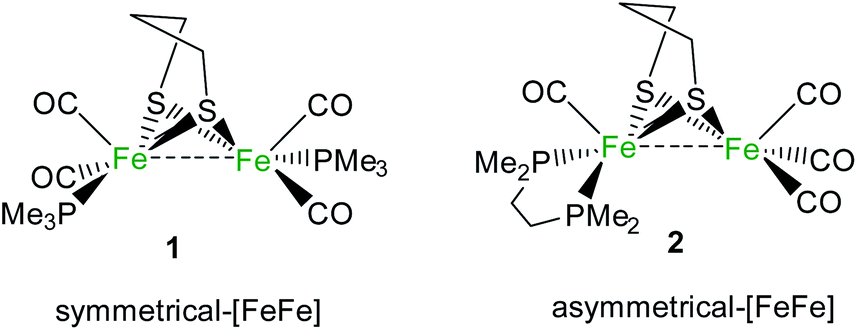 | ||
| Scheme 1 The [FeFe]-complexes studied as Lewis bases in the studied FLPs. | ||
Results and discussion
Formation of the FLPs
The addition of one equivalent of B(C6F5)3 to the [Fe2(μ-pdt)(PMe3)2(CO)4] (1) or [Fe2(μ-pdt)(κ2-dmpe)(CO)4] (2) complex (1 equiv.) yielded a red solution. Multinuclear (1H, 31P, 11B) NMR and IR analysis revealed specific spectroscopic fingerprints of each separated molecule, indicating the absence of any interaction between the diiron species and borane in solution (see the ESI†). These findings differ from reported examples of [FeFe]/[NiFe] complexes in which the addition of a borane molecule resulted in its binding to an available nitrogen-based ligand, such as cyanide67,68 or azadithiolate.69 Interactions between the carbonyl group and Lewis acid were also reported for the complex [Fe2(μ-pdt)(κ2-dppv)2(CO)2] (dppv = cis-1,2-C2H2(PPh2)2), leading to the diiron site adopting a rotated conformation.70 The different behaviours shown by 1 and 2 under these reaction conditions may arise from the less-electron-rich nature of disubstituted diiron complexes compared to related tetrasubstituted bis-dppv diiron compounds. On the basis of these observations, both the Lewis acid and base components in these complexes (B(C6F5)3 and 1/2, respectively) coexist in solution. According to FLP chemistry, these pairs may be predisposed to activating H2 through the heterolytic splitting of the H–H bond. Consequently, the exposure of 1 and 2 to H2 in the presence of B(C6F5)3 was performed.Activities of 1 and 2 towards H2 in the presence of B(C6F5)3
The bishydride salt [Fe2(μ-pdt)(μ-H)(PMe3)2(CO)4][HB(C6F5)3] (3) was isolated from an equimolar CH2Cl2 solution of [Fe2(μ-pdt)(PMe3)2(CO)4] (1) and B(C6F5)3 exposed to H2 (1 atm.) (Scheme 2). The slow formation of this complex was observed through spectroscopic analysis after 2 h. In solution, a maximum yield of 77% for the conversion of 1 into 3 was reached after 5 days. This bishydride salt was isolated, upon the addition of hexane to the CH2Cl2 solution, as a red powder in only 53% yield due to its partial solubility in a non-polar organic solvent. Spectroscopic characterization of 3 matched the previously reported data for the cation [Fe2(μ-pdt)(μ-H)(PMe3)2(CO)4]+ (ref. 18 and 19) and the anionic [HB(C6F5)3]− species (see the ESI†).71 Notably, a triplet at −15.3 ppm in the 1H-NMR spectrum with a 2JH–P coupling constant of 22 Hz is characteristic of a bridging hydride between the two iron atoms of the [Fe2(μ-pdt)(μ-H)(PMe3)2(CO)4]+ complex, while the borohydride [HB(C6F5)3]− was characterized based on a broad quadruplet at 3.6 ppm in the 1H-NMR spectrum, which is associated with a doublet in the 11B-NMR spectrum at −25.7 ppm (1JH–B = 82 Hz). IR spectroscopy revealed two strong bands at 2032 and 1992 cm−1 in CH2Cl2 solution in the carbonyl region of the IR spectrum, corresponding to the protonated iron complex [Fe2(μ-pdt)(μ-H)(PMe3)2(CO)4]+. In order to ascertain the origin of both hydride ligands, isotopic labelling experiments were performed using D2 gas. The salt [Fe2(μ-pdt)(μ-D)(PMe3)2(CO)4][DB(C6F5)3] (3D) was characterized unambiguously based on 2H-NMR analysis (see the ESI†).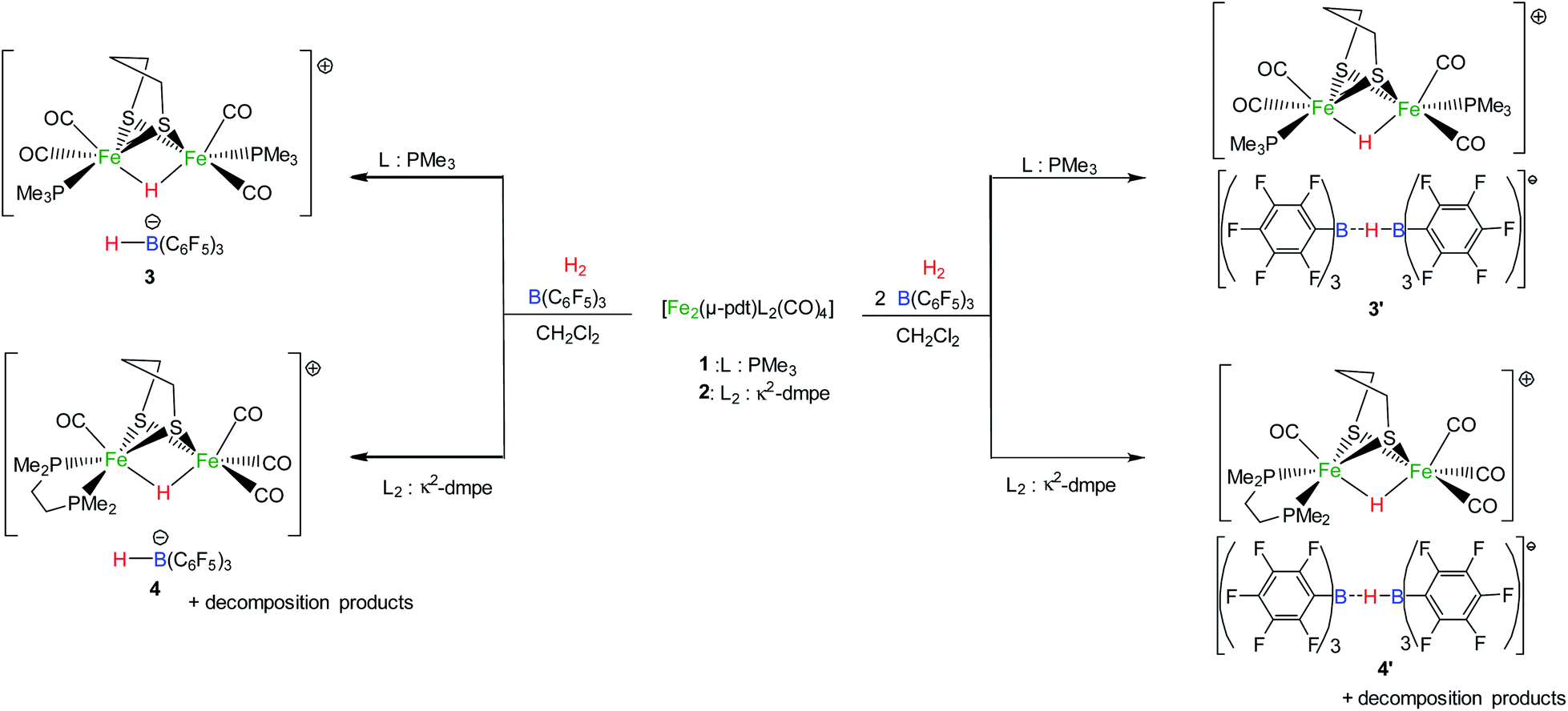 | ||
| Scheme 2 The reactivities of complexes 1 and 2 with H2 in the presence of B(C6F5)3. | ||
Similarly, the exposure to H2 of a solution containing 1 equivalent of [Fe2(μ-pdt)(κ2-dmpe)(CO)4] (2) and 1 equivalent of B(C6F5)3 in CH2Cl2 resulted in the slow formation of [Fe2(μ-pdt)(μ-H)(κ2-dmpe)(CO)4][HB(C6F5)3] (4) (Scheme 2). This salt was isolated after 5 days in 33% yield upon the addition of diethylether to the reaction mixture. The bridging hydride in the cationic complex [Fe2(μ-pdt)(μ-H)(κ2-dmpe)(CO)4]+ was characterized based on a triplet at −14.6 ppm (2JP–H = 21 Hz) in the 1H-NMR spectrum, and the IR spectrum showed three strong bands at 2095 (s), 2039 (s) and 1963 (s) cm−1 in the carbonyl region, which is in close agreement with the reported data for a sample of [Fe2(μ-pdt)(μ-H)(κ2-dmpe)(CO)4]+.66 A characteristic doublet in the 11B-NMR spectrum at −25.7 ppm corroborates the formation of the borohydride anion (see the ESI†).71
Additional unexpected resonances were observed in the NMR spectra of the crude reaction mixture. The triplet at −10.5 ppm (J = 50 Hz) and singlet at 67.8 ppm in the 1H- and 31P-NMR spectra, respectively, were assigned to the iron hydride complex [FeH(κ2-dmpe)(CO)3]+,72,73 while two resonances (at 0 ppm and −14 ppm) and the broad singlet (at 10.6 ppm) in the 11B- and 1H-NMR spectra, respectively, were attributed to the formyl-borate species [B(C6F5)3–CHO–B(C6F5)3]−.64,74
NMR monitoring showed that the species [FeH(κ2-dmpe)(CO)3]+ appears after at least 3 days of H2 exposure, and it is present in solution with 4 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio. The formyl borate anion is observed as soon as H2 is added, suggesting ready CO release, and this then decomposed over time, as reported in the literature.64,74
:3 ratio. The formyl borate anion is observed as soon as H2 is added, suggesting ready CO release, and this then decomposed over time, as reported in the literature.64,74
In order to investigate the possibility of the intermediate formation of a terminal hydride species, the pair formed from the complex 2 and B(C6F5)3 was exposed to H2 (1 atm.) at −70 °C (see the ESI†). A characteristic singlet at −4.5 ppm in the 1H-NMR spectrum revealed the presence of the cationic species [Fe2(μ-pdt)(Ht)(κ2-dmpe)(CO)4]+, featuring a terminal hydride ligand on the {Fe(CO)3} unit, in agreement with the literature.66 The sample was then allowed to warm to room temperature and above 253 K the terminal hydride complex converted into both apical-basal and dibasal bridging hydride isomers [Fe2(μ-pdt)(μ-H)(κ2-dmpe)(CO)4]+, with the dibasal isomer being the thermodynamically preferred species after several hours at room temperature.
Experiments carried out with D2 gas definitively established the origin of the hydride ligands in [Fe2(μ-pdt)(μ-H)(κ2-dmpe)(CO)4][HB(C6F5)3] (4). Moreover, the formation of the bis-deuteride salt [Fe2(μ-pdt)(μ-D)(κ2-dmpe)(CO)4][DB(C6F5)3] was accompanied by that of the mononuclear complex [FeD(κ2-dmpe)(CO)3]+. The detection of this latter species and the detection of the formyl-borate species in the reaction mixtures revealed the cleavage of 2 upon reaction with H2 in the presence of borane.
It should be noted that similar experiments with less acidic B(C6H5)3 borane do not allow H2 heterolytic splitting.
Influence of borane stoichiometry on heterolytic H2 splitting
The exposure to H2 of a 1:2 solution of [Fe2(μ-pdt)(PMe3)2(CO)4] (1) and B(C6F5)3 in CH2Cl2 leads to the complete conversion of the diiron complex into the protonated [Fe2(μ-pdt)(μ-H)(PMe3)2(CO)4]+ cation after 40 h (Scheme 2). Faster and more complete conversion is thus observed when using 2 equivalents of borane, as a maximum conversion yield of 77% was observed in the presence of one equivalent of B(C6F5)3. In the 11B{1H}-NMR spectrum, however, no signal was detected at room temperature. Reducing the temperature to 213 K allowed for the observation of a strong broad signal at −25 ppm (see the ESI†). The 1H-NMR spectrum displayed one broad resonance at 3.5 ppm without a well-resolved coupling constant with the boron atom. These observations strongly suggest the formation of the diborohydride anion [(C6F5)3B-(μ-H)-B(C6F5)3]−, whose NMR characterization requires low-temperature recording.75 The related diborohydride species [(C6H3(CF3)2)3B-(μ-H)-B(C6H3(CF3)2)3]− has already been reported, for which no 1H–11B coupling was detected until a donor solvent, such as pyridine or acetonitrile, was added, inducing the cleavage of the dimeric species and yielding the known [HB(C6H3(CF3)2)3]− anion.76,77 To highlight the formation of the diborohydride anion [(C6F5)3B-(μ-H)-B(C6F5)3]−, acetonitrile was added to the reaction mixture, resulting in the appearance of the characteristic resonances of [HB(C6F5)3]− together with a broad singlet at −10.8 ppm in the 11B-NMR spectrum, which is attributed to the Lewis adduct MeCN–B(C6F5)3.78
These observations were also made in relation to the dissymmetrical complex 2; the exposure to H2 of a solution of B(C6F5)3 (2 equiv.) and [Fe2(μ-pdt)(κ2-dmpe)(CO)4] (1 equiv.) in CH2Cl2 quantitatively yields a mixture of [Fe2(μ-pdt)(μ-H)(κ2-dmpe)(CO)4]+ and [FeH(κ2-dmpe)(CO)3]+ cations in a 2:1 ratio. 11B{1H}-NMR analysis revealed the presence of the formyl-borate [B(C6F5)3-CHO-B(C6F5)3]−, which degrades over time,64,74 and the diborohydride anion [(C6F5)3B-(μ-H)-B(C6F5)3]− in the anionic counterpart.
The difference in the conversion efficiencies of the reactions of 1 and 2 with one or two equivalents of borane may arise from the involvement of the hydride ligand as a bridge to the dimeric species [(C6F5)3B-(μ-H)-B(C6F5)3]−, thus preventing the reverse reaction between [HB(C6F5)3]− and the protonated diiron complexes.
Discussion of the different activities of 1 and 2 towards H2 in the presence of borane
1 and 2 display similar electronic properties.79,80 However, they behave differently towards the activation of H2 in the presence of B(C6F5)3. The differences, highlighted in this study, may arise from the symmetrical or dissymmetrical arrangement of the phosphine/diphosphine ligands at the diiron centre of the complex. In the symmetrically disubstituted complex 1, both iron metallic {Fe(PMe)3(CO)2} moieties have the same ligand environments, while in 2, the two iron moieties, {Fe(CO)3} and {Fe(κ2-dmpe)(CO)}, are electronically and sterically very different. The {Fe(CO)3} fragment is indeed less basic but also less sterically encumbered than the {Fe(κ2-dmpe)(CO)} part; thus, there are two different sites where the reaction can take place. DFT calculations have been performed in order to rationalize the different activities of 1 and 2 towards H2/B(C6F5)3. It is worth noting that in the presence of HBF4, it has previously been reported that the dissymmetry of 2 orientates the reaction path toward the formation of a transient terminal hydride species, while this was not observed in the case of 1 under similar reaction conditions.66,81Mechanistic investigations via theoretical calculations
In order to shed light on the mechanistic aspects of the heterolytic splitting of the H–H bond by 1 and 2 in the presence of B(C6F5)3 and on the experimental discrepancies observed between those two systems, DFT investigations using the BP86-D3/TZVP functional were performed. The theoretical rationalization of the experimental findings involves different challenges. The Lewis base component of the FLP formed between 1 or 2 and the borane Lewis acid has a bimetallic nature, which itself induces two possible iron sites for H2 interactions. Furthermore, the fluxionality of the coordination sphere around each iron center of such species is known, and various isomers should be taken into account when considering the reactivity pathway. The most stable isomers of 1 and 2 feature the phosphine ligands in a dibasal position, but the apical-basal isomers are only 0.7 kcal mol−1 and 3.0 kcal mol−1 higher in energy, respectively (see the ESI†). This is consistent with the isomers reported for 1 and 2 in the literature.18,66,81 In addition, isomers featuring a structure close to that of the active site of [FeFe]-hydrogenases were also considered, especially for 2. In such isomers, the square pyramidal coordination geometry of one Fe center is “inverted” compared to the other Fe center (see the ESI†). Considering these various geometries, the dibasal, basal-apical, and rotated diiron isomers for each FLP were examined. | ||
| Fig. 1 The H2 splitting mechanisms of 1 and B(C6F5)3. The optimized TS structures for H2 splitting of 1 basal–basal (1-TSbb) and 1 apical-basal (1-TSab) structures are represented in the insets. Selected distances are given in Å. NBO charges, calculated for the H1 and H2 atoms of 1-TSbb (blue values) and 1-TSab (red values), calculated at the BP86-D3 and B3LYP-D3 (in parentheses) levels, are reported in the respective insets. Colors: grey = carbon; red = oxygen; light blue = iron; purple = phosphorus; yellow = sulfur; orange = boron; and green = fluorine. | ||
The mechanisms shown in Fig. 2 involve the preliminary rotation of the {Fe(CO)3} moiety of 2. Indeed, 2 has a higher tendency, compared to 1, to rotate at the {Fe(CO)3} unit, which is consistent with the asymmetric coordination environments of the two iron ions.82,83 The isomer of 2 featuring a vacant coordination site in the apical position of a single iron is only 3.3 kcal mol−1 higher in energy with respect to the unrotated example. The dibasal form has been considered as the isomer that interacts first with H2, because the apical-basal isomer, at this stage, is predicted to be less stable than the dibasal one (see Fig. S20 in the ESI†). Only after H2 heterolysis has occurred do the two isomers become energetically equivalent (vide infra).
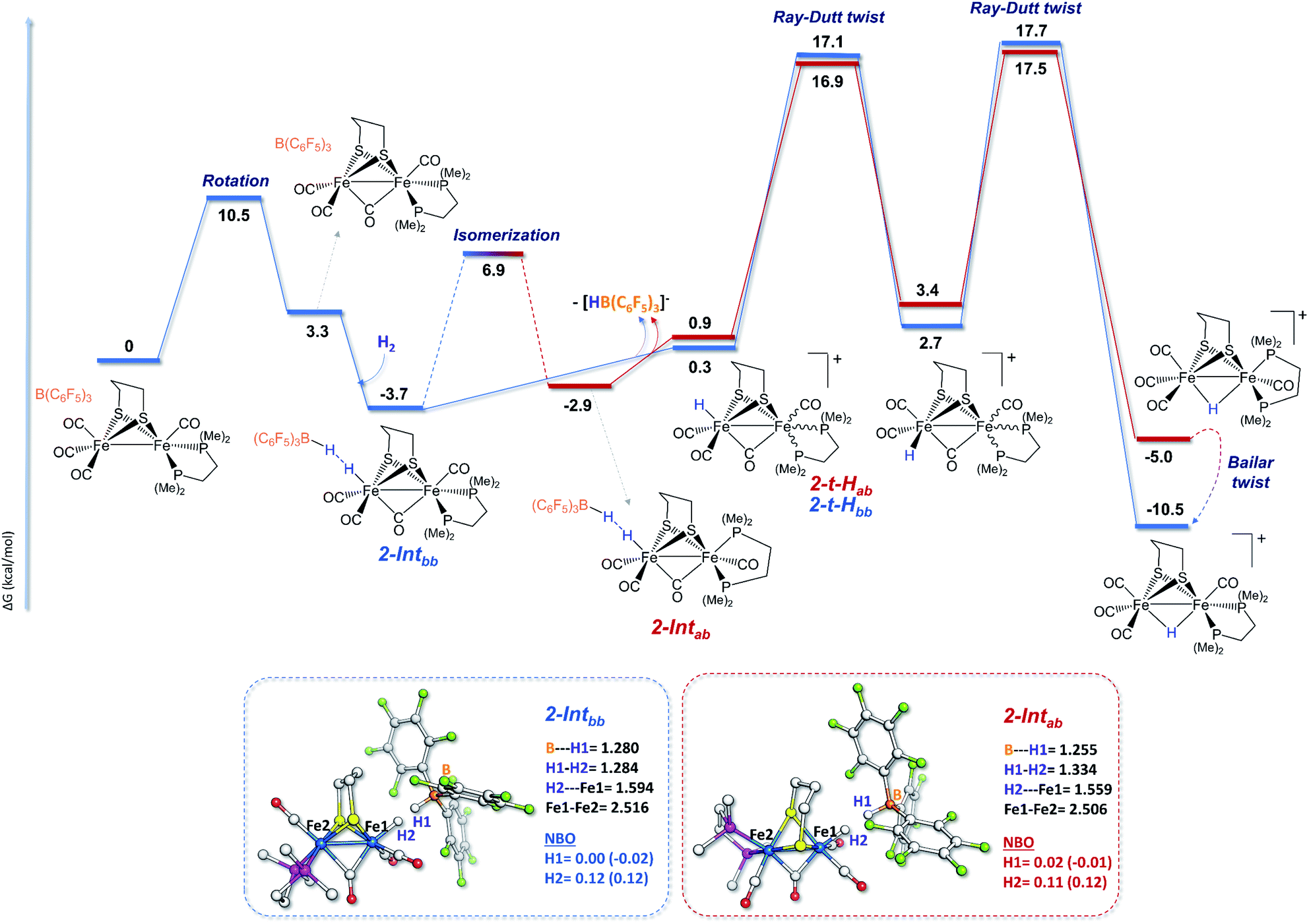 | ||
| Fig. 2 The H2 splitting mechanisms of 2 and B(C6F5)3 at the apical vacant site of rotated iron. The blue pathway shows the reactivity of the dibasal rotamer of 2, whereas the red one is related to the apical-basal rotamer. Selected distances given in Å and NBO charges, calculated for the H1 and H2 atoms of 2-Intbb (blue values) and 2-Intab (red values), calculated at the BP86-D3 and B3LYP-D3 (in parentheses) levels, are reported in the respective insets. Colors: grey = carbon; red = oxygen; light blue = iron; purple = phosphorus; yellow = sulfur; orange = boron; and green = fluorine. | ||
The H2 uptake between the diiron site and the borane molecule is a slightly exergonic process (−3.7 kcal mol−1), with the lengthening of the H–H bond between the diiron center and the borane moiety to around 1.28 Å. This step is predicted to be extremely fast, considering that our simulations show the immediate formation (i.e., with no detectable barrier of activation) of 2-Intbb once H2 is added to the 2/Lewis acid system. A terminal hydride cationic species is then formed after the loss of the anion [HB(C6F5)3]−. DFT calculations show that, at this stage, the two chelate orientations (dibasal and apical-basal) are mutually close to equilibrium, with quite a low barrier of interconversion. Such quasi-equilibrium results are basically unaffected by the presence of the R3BH anion, which implies that it equally involves both 2-Intbb/2-Intab and 2-t-Hbb/2-t-Hab.
The rate determining step of the entire mechanism of H2 cleavage mediated by the rotated structure of the diiron dithiolate is predicted to be the final step of the double Ray–Dutt twist (a pseudo-rhombic octahedral rearrangement)84 that converts the reactive terminally coordinated hydride into its thermodynamically more stable bridging counterpart with an overall energy barrier of around 17 kcal mol−1 (roughly independent of the chelate rotational isomer). A Bailar twist (trigonal-prismatic rearrangement) leads from the apical-basal μ-hydride to the thermodynamically more stable dibasal rotational isomer, as previously reported for strict analogues of 2.84 This DFT picture of the facile and kinetically accessible conversion of apical-basal μ-H is in full agreement with the experimental observation that dibasal μ-H is the thermodynamic product at room temperature.
 The newly formed almost-terminal Fe–H is stabilized by a long-range interaction with the boron hydride. This intermediate is less stable than the final bridging hydride, but it can be considered as the first species that is formed upon H2 splitting by 2 at the Fe–Fe site.
The newly formed almost-terminal Fe–H is stabilized by a long-range interaction with the boron hydride. This intermediate is less stable than the final bridging hydride, but it can be considered as the first species that is formed upon H2 splitting by 2 at the Fe–Fe site.
 | ||
| Fig. 3 The H2 splitting mechanisms by 2 and B(C6F5)3; the optimized structures of the H2-splitting TSs for 2 basal–basal (2-TSbb) and 2 apical-basal (2-TSab) isomers are represented in the insets. Selected distances are given in Å. NBO charges, calculated for the H1 and H2 atoms of 2-TSbb (blue values) and 2-TSab (red values), calculated at the BP86-D3 and B3LYP-D3 (in parentheses) levels, are also reported. Colors: grey = carbon; red = oxygen; light blue = iron; purple = phosphorus; yellow = sulfur; orange = boron; and green = fluorine. | ||
The occurrence of H2 cleavage at the non-substituted iron site makes this reactive event essentially unsensitive to the nature (dibasal or basal-apical) of the isomer that performs the reaction, as shown by the close energies of both transition states during the rate-determining step. The three mechanisms revealed significantly close energy barriers, but the one involving a terminal hydride isomer, observed experimentally, may be the most probable. Indeed, from the DFT viewpoint, the heights of the rate-determining step of each H2 cleavage pathway are very similar (differences within a 1 kcal mol−1 range), which makes them all theoretically possible. However, the experimental evidence points to a slight preference for the “enzyme-like mechanism”. This statement is further supported by the extremely facile H2 cleavage step at the apical site of a rotated iron atom (Fig. 2), in close analogy to what occurs in the H-cluster within the hydrogenase active site. The energy bottleneck of H2 splitting via the “rotated iron center mechanism” comes, in fact, after H2 undergoes heterolysis in the FLP system and consists of a two-step rearrangement that is impeded in the biological catalyst.85
The basal–apical forms of 1 and 2 and of the hydride species involved are relatively less stable than their dibasal isomers. This is due to steric bumps between the phosphine ligand and the propanedithiolate bridge, which are more emphasized with the more-rigid chelating dmpe ligand. Comparing the lowest energy profiles calculated for 1 and 2, it can be noticed that the former is preferred from a kinetic (by ca. 5.8 kcal mol−1) standpoint while the thermodynamics of both processes are similar. The steric hindrance at the Fe–Fe site was roughly similar for the apical-basal forms of both 1 and 2, and the experimentally observed difference can be ascribed to difference in the electron-rich natures of the iron sites directly involved in H2 splitting ({Fe(PMe3)(CO)2} for 1 and {Fe(CO)3} for 2). In the case of 1, the PMe3 ligand adopts a trans orientation with respect to the incoming hydrogen atom, which has protic character (that forming the B–H bond is hydridic). Analysis of the electron density distribution over the atomic centers based on classical partition schemes, such as NBO (see Fig. 1–3 and the ESI†) and Löwdin (see the ESI†), calculated at both the BP86-D3 and B3LYP-D3 levels on the H2 molecule with respect to 1-TSbb, 1-TSab, 2-TSbb, and 2-TSab corroborates the aforementioned considerations. Indeed, the extent of H2 heterolysis (corresponding to the polarization level of the H–H bond) is slightly higher for the apical-basal isomers than the dibasal examples. More relevant is that Hδ+–Hδ− polarization is more pronounced in 1 than in 2 as a result of the increased basicity of the metal center involved in splitting. Similar partial-charge-related arguments, based on the NBO data, have to be considered with some caution in the case of the splitting mechanism reported in Fig. 2 due to the actual absence of a transition state associated with such a step. However, considering the two intermediates arising just after H2 heterolysis (2-Intbb and 2-Intab, see the insets of Fig. 2), it can be concluded that the highest levels of Hδ+–Hδ− polarization is predicted to occur exactly when H2 splits in an enzyme-like fashion, which further supports such an activation pathway compared with the others.
The formation of the asymmetric semi-bridging hydride intermediate (not observed for 1) or the terminal hydride intermediate from 2 (Fig. 2 and 3) enhanced the reversibility of H2 splitting with respect to 1, and the backward reaction (namely H2 formation) could be easily triggered in the case of the former complex. This can also explain the similar behaviors of 2 and 1 in the presence of two Lewis acid equivalents, since [HB(C6F5)3]−, which is necessary to stabilize the higher-energy product in the case of 2, can be easily trapped by a second B(C6F5)3 molecule. Indeed, coupling between B(C6F5)3 and [HB(C6F5)3]− to form a [(C6F5)3B-(μ-H)-B(C6F5)3]− bridging hydride was calculated to be exergonic by −10.2 kcal mol−1 (see the ESI† for details).
Conclusions
In this work, we have established that FLPs consisting of a reduced FeIFeI complex and a borane molecule are able to activate H2, forming two hydride species, one connected to the diiron core and the other to the borane molecule. The higher reactivity of symmetrical [Fe2(μ-pdt)(PMe3)2(CO)4] (1) compared to that of asymmetrical chelated [Fe2(μ-pdt)(κ2-dmpe)(CO)4] (2) towards H2 in the presence of B(C6F5)3 has been elucidated based on theoretical calculations both in steric and electronic terms. The different symmetries of the two complexes lead to distinctive steric factors determining the selection of the iron center for performing the heterolytic cleavage of H2. For both complexes, the reaction takes place at the less sterically hindered iron center in order to favor the approach of both H2 and the Lewis acid. The reaction involving the symmetrical diiron complex 1 takes place at one of the two {Fe(PMe3)(CO)2} units, which are more basic than the {Fe(CO)3} part of the asymmetrical complex 2, thus favoring faster reactivity. In the case of 2, the direct observation of a terminal hydride intermediate in low-temperature experiments highlights a preferential mechanism that leads to [FeFe]-hydrogenase-like reactivity, occurring through the binding-activation of H2 at a free apical/terminal iron site via accessible rotated geometry at the {Fe(CO)3} site. The mechanistic differences emerging between 1 and 2 corroborate the importance of the coordination symmetry/asymmetry pattern when it comes to driving the regiochemistry of H2 heterolysis during hydrogenase biomimicry.Finally, this study may open up novel routes to H2 oxidation, and it offers a previously unexplored non-biomimetic strategy for H2 activation using diiron dithiolate complexes. In general, the use of transition-metal systems as Lewis acid/base components is less common than the use of main-group elements within the vast realm of FLP chemistry.45 Further experiments relating to the transfer of hydrogen atoms from H2 to unsaturated substrates and the oxidation of hydrido-borate are ongoing.
Experimental
Materials and methods
All the experiments were carried out under an inert atmosphere using Schlenk or glovebox techniques for synthesis. Solvents were deoxygenated and dried according to standard procedures. [Fe2(μ-pdt)(PMe3)2(CO)4] (1) and [Fe2(μ-pdt)(κ2-dmpe)(CO)4] (2) were prepared according to reported procedures.19,66 B(C6F5)3 was prepared according to the literature and sublimated.861H-, 31P-, 11B-, 19F-, and 13C-NMR spectra were respectively recorded at room temperature in deuterated solvents using a Bruker Avance 400 and Avance III HD 500 spectrometer at “Service RMN-SM de l’Université de Bretagne Occidentale”. They were referenced to SiMe4 (1H, 13C), H3PO4 (31P), BF3·OEt2 (11B), and CFCl3 (19F). Infrared spectra were recorded with a PerkinElmer spectrometer. Chemical analyses were carried out by the “Service d'Analyses – Chromato – Masse BioCIS – UMR 8076”, Châtenay-Malabry (France).Synthetic procedures
[FeH(κ2-dmpe)(CO)3]+: 1H NMR (500 MHz, 298 K, CD2Cl2): δ (ppm) 2.25 (m), 1.79 (m), 1.58 (m), −10.5 (t, 2JH–P = 50 Hz, 1H), integrals were not perfectly defined as the product was not isolated and resonances superimposed with the resonances of the complex [Fe2(μ-pdt)(μ-H)(κ2-dmpe)(CO)4]+. 31P{1H} NMR (202.46 MHz, 298 K, CD2Cl2): δ (ppm) 67.80 (s).
When the reaction was carried out in C6D6, no spectroscopic changes were observed in the 1H-, 31P-, and 11B-NMR spectra, even after 8 days at room temperature. However, a lot of solid precipitated in the NMR tube. After removing the supernatant and dissolving the residue in CD2Cl2, NMR spectra revealed the presence of the same two hydride complexes in a 1:3 ratio, together with residual traces of the starting complex.
2H NMR (76.73 MHz, 298 K, CD2Cl2): δ (ppm) 3.60 (br, B–D), −15.45 (t, 2JD–P = 3.1 Hz, μ-D) for [Fe2(μ-pdt)(μ-D)(PMe3)2(CO)4][DB(C6F5)3] and −14.7 (t, 2JD–P = 2.6 Hz, μ-D) for [Fe2(μ-pdt)(μ-D)(κ2-dmpe)(CO)4]+, −10.5 ppm (2JD–P = 7.0 Hz) for [Fe(κ2-dmpe)(CO)3D]+. 11B{1H} NMR (160.46 MHz, 298 K, CD2Cl2): δ (ppm) −25.74 (br s, B–D). 11B NMR (160.46 MHz, 298 K, CD2Cl2): δ (ppm) −25.74 (br s, B–D).
NMR characterization of [Fe2(μ-pdt)(μ-H)(PMe3)2(CO)4][(C6F5)3B-(μ-H)-B(C6F5)3] (3′). 1H NMR (400 MHz, 298 K, CD2Cl2): δ (ppm) 3.58 (br s., 1H, B–H–B), 2.58 (m, 4H, –S–CH2–), 2.19 (q, 2H, –CH2–), 1.56 (d, 2JH–P = 10 Hz, P–CH3), −15.37 (t, 2JH–P = 22 Hz, 1H, μ-H). 31P{1H} NMR (161.916 MHz, 298 K, CD2Cl2): δ (ppm) 21.26 (s, PMe3). 11B NMR (160.46 MHz, 213 K, CD2Cl2): δ (ppm) −26.4 (br s, B–H).
NMR characterization of [Fe2(μ-pdt)(μ-H)(κ2-dmpe)2(CO)4][(C6F5)3B-(μ-H)-B(C6F5)3] (4′). 1H NMR (400 MHz, 298 K, CD2Cl2): δ (ppm) 3.47 (br s, 1H, B–H–B), 2.83 (d, 13 Hz, 2H), 2.57 (d, 15 Hz, 1H), 2.47 (t, 13 Hz, 2H), 2.24 (m, 4H), 1.99 (m, 1H), 1.65 (d, 5 Hz, 6H, CH3), 1.30 (d, 5 Hz, 6H, CH3), −14.67 (t, 2JH–P = 21 Hz, 1H, μ-H). 31P{1H} NMR (161.916 MHz, 298 K, CD2Cl2): δ (ppm) 67.48 (s). 11B NMR (160.46 MHz, 213 K, CD2Cl2): δ (ppm) −26.4 (br s, B–H).
The addition of one drop of MeCN to a solution of 3′ or 4′ can provide spectroscopic data for the respective salt 3 or 4 and the adduct MeCN–B(C6F5)3 (11B{1H} NMR (160.46 MHz, 298 K, CD2Cl2): δ (ppm) −10.8 (br s)).
DFT
The TURBOMOLE 7.2 suite of programs87 was used to perform all calculations within the density functional theory (DFT) framework at the BP86/TZVP level.88–90 BP86 is a typical choice for the investigation of bioinorganic systems, particularly hydrogenase-related models, given its satisfactory performance in the reproduction of structural and energetic features.85,91–93 Dispersive interactions were accounted for using the Grimme D3 scheme.94 The solvent (CH2Cl2) was modelled implicitly according to the conductor-like screening model (COSMO) approach,95,96 by setting a dielectric constant of 8.9. We took advantage of the Resolution-of-Identity (RI) technique to speed up calculations.97 All species have been modeled as low-spin states, in light of the characteristics of the Fe coordination sphere. Full vibrational analysis has been performed with the aim of characterizing the nature of each stationary point (zero imaginary frequencies for pure minima and a single imaginary frequency for saddle points). A transition-state search was performed following a pseudo-Newton–Raphson procedure, as illustrated in detail in ref. 98.98 Free energy (G) values have been obtained from the electronic self-consistent field (SCF) energies via considering three contributions to the total partition function (Q): qtranslational, qrotational, and qvibrational, assuming that it can be written as a product of these. To evaluate enthalpy and entropy contributions, the values for temperature and pressure, and the scaling factor for the SCF wavenumbers were set to 298.15 K, 1 atm, and 0.9144 (as implemented in TURBOMOLE), respectively. Natural bond orbitals (NBO)99 and Löwdin100 population analysis were performed on selected intermediates and transition states, both at the BP86-D3 and B3LYP-D3 (single point calculations on BP86-D3 optimized structures) levels.Data availability
Experimental data have been provided in ESI.†Author contributions
The research project was defined by L. C. within the JCJC ANR program OxySplit-H2. J. B. B. and L. C. carried out the synthetic experiments and analysed the experimental data. F. A. and G. Z. carried out and analysed the DFT calculations. The manuscript was prepared through contributions from F. A., L. C., P. S., and G. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NMR experiments were performed at the ‘Service Général des Plateformes, Brest’ (SGPLAT). JBB is funded by the Region Bretagne. CNRS (Centre National de la Recherche Scientifique) and the Université de Bretagne Occidentale (Brest) are acknowledged for financial support. LC is thankful for the support of the Région Bretagne relating to the project ConvcatH (Stratégie d’Attractivité Durable) and the ANR (Agence Nationale de Recherche) for funding the project JCJC OxySplit-H2 (ANR-20-CE07-0027-01).References
- F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, Chem. –Eur. J., 2018, 24, 1471–1493 CrossRef PubMed.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- C. Baffert, M. Demuez, L. Cournac, B. Burlat, B. Guigliarelli, P. Bertrand, L. Girbal and C. Léger, Angew. Chem., Int. Ed., 2008, 47, 2052–2054 CrossRef CAS PubMed.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS PubMed.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17331–17336 CrossRef CAS PubMed.
- A. F. Wait, C. Brandmayr, S. T. Stripp, C. Cavazza, J. C. Fontecilla-Camps, T. Happe and F. A. Armstrong, J. Am. Chem. Soc., 2011, 133, 1282–1285 CrossRef CAS PubMed.
- A. Bachmeier, J. Esselborn, S. V. Hexter, T. Krämer, K. Klein, T. Happe, J. E. McGrady, W. K. Myers and F. A. Armstrong, J. Am. Chem. Soc., 2015, 137, 5381–5389 CrossRef CAS PubMed.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS PubMed.
- M. del Barrio, M. Sensi, L. Fradale, M. Bruschi, C. Greco, L. de Gioia, L. Bertini, V. Fourmond and C. Léger, J. Am. Chem. Soc., 2018, 140, 5485–5492 CrossRef CAS PubMed.
- M. Winkler, J. Duan, A. Rutz, C. Felbek, L. Scholtysek, O. Lampret, J. Jaenecke, U.-P. Apfel, G. Gilardi, F. Valetti, V. Fourmond, E. Hofmann, C. Léger and T. Happe, Nat. Commun., 2021, 12, 756 CrossRef CAS PubMed.
- Z. Thammavongsy, I. P. Mercer and J. Y. Yang, Chem. Commun., 2019, 55, 10342–10358 RSC.
- C. Elleouet, F. Y. Pétillon and P. Schollhammer, in Advances in Bioorganometallic Chemistry, ed. T. Hirao and T. Moriuchi, Elsevier, 2019, pp. 347–364 Search PubMed.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed.
- F. Wittkamp, M. Senger, S. T. Stripp and U.-P. Apfel, Chem. Commun., 2018, 54, 5934–5942 RSC.
- T. Xu, D. Chen and X. Hu, Coord. Chem. Rev., 2015, 303, 32–41 CrossRef CAS.
- Z. M. Heiden, G. Zampella, L. De Gioia and T. B. Rauchfuss, Angew. Chem., Int. Ed., 2008, 47, 9756–9759 CrossRef CAS PubMed.
- S. L. Matthews and D. M. Heinekey, Inorg. Chem., 2011, 50, 7925–7927 CrossRef CAS PubMed.
- X. Zhao, I. P. Georgakaki, M. L. Miller, J. C. Yarbrough and M. Y. Darensbourg, J. Am. Chem. Soc., 2001, 123, 9710–9711 CrossRef CAS PubMed.
- X. Zhao, I. P. Georgakaki, M. L. Miller, R. Mejia-Rodriguez, C.-Y. Chiang and M. Y. Darensbourg, Inorg. Chem., 2002, 41, 3917–3928 CrossRef CAS PubMed.
- I. P. Georgakaki, M. L. Miller and M. Y. Darensbourg, Inorg. Chem., 2003, 42, 2489–2494 CrossRef CAS PubMed.
- X. Yu, M. Pang, S. Zhang, X. Hu, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2018, 140, 11454–11463 CrossRef CAS PubMed.
- M. Razavet, S. J. Borg, S. J. George, S. P. Best, S. A. Fairhurst and C. J. Pickett, Chem. Commun., 2002, 700–701 RSC.
- Ö. F. Erdem, L. Schwartz, M. Stein, A. Silakov, S. Kaur-Ghumaan, P. Huang, S. Ott, E. J. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2011, 50, 1439–1443 CrossRef PubMed.
- T. Liu and M. Y. Darensbourg, J. Am. Chem. Soc., 2007, 129, 7008–7009 CrossRef CAS PubMed.
- A. K. Justice, T. B. Rauchfuss and S. R. Wilson, Angew. Chem., Int. Ed., 2007, 46, 6152–6154 CrossRef CAS PubMed.
- C. M. Thomas, T. Liu, M. B. Hall and M. Y. Darensbourg, Inorg. Chem., 2008, 47, 7009–7024 CrossRef CAS PubMed.
- A. K. Justice, L. De Gioia, M. J. Nilges, T. B. Rauchfuss, S. R. Wilson and G. Zampella, Inorg. Chem., 2008, 47, 7405–7414 CrossRef CAS PubMed.
- M. Karnahl, S. Tschierlei, Ö. F. Erdem, S. Pullen, M.-P. Santoni, E. J. Reijerse, W. Lubitz and S. Ott, Dalton Trans., 2012, 41, 12468–12477 RSC.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2012, 4, 26–30 CrossRef CAS PubMed.
- N. Wang, M. Wang, Y. Wang, D. Zheng, H. Han, M. S. G. Ahlquist and L. Sun, J. Am. Chem. Soc., 2013, 135, 13688–13691 CrossRef CAS PubMed.
- M. T. Olsen, B. E. Barton and T. B. Rauchfuss, Inorg. Chem., 2009, 48, 7507–7509 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, J. Am. Chem. Soc., 2011, 133, 8098–8101 CrossRef CAS PubMed.
- S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt, S. E. Kabir and B. E. Sanchez, Chem. Commun., 2013, 50, 945–947 RSC.
- R. M. Bullock and G. M. Chambers, Philos. Trans. R. Soc., A, 2017, 375, 20170002 CrossRef PubMed.
- F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. P. Huber, G. Kehr, K. Bergander, R. Fröhlich, G. Erker, S. Tanino, Y. Ohki and K. Tatsumi, Organometallics, 2008, 27, 5279–5284 CrossRef CAS.
- A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2010, 132, 3301–3303 CrossRef CAS PubMed.
- M. P. Boone and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 8508–8511 CrossRef CAS PubMed.
- S. R. Flynn and D. F. Wass, ACS Catal., 2013, 3, 2574–2581 CrossRef CAS.
- N. S. Lambic, R. D. Sommer and E. A. Ison, J. Am. Chem. Soc., 2016, 138, 4832–4842 CrossRef CAS PubMed.
- L. Chatelain, E. Louyriac, I. Douair, E. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11, 1–12 CrossRef PubMed.
- A. Paparo, A. L. P. Nguyen, J. S. Silvia, T. P. Spaniol, L. Maron, C. C. Cummins and J. Okuda, Dalton Trans., 2021, 50, 10692–10695 RSC.
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 8826–8829 CrossRef CAS PubMed.
- A. T. Normand, C. G. Daniliuc, B. Wibbeling, G. Kehr, P. Le Gendre and G. Erker, J. Am. Chem. Soc., 2015, 137, 10796–10808 CrossRef CAS PubMed.
- H. B. Hamilton, A. M. King, H. A. Sparkes, N. E. Pridmore and D. F. Wass, Inorg. Chem., 2019, 58, 6399–6409 CrossRef CAS PubMed.
- M. Carmona, J. Ferrer, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña, L. Cañadillas-Delgado and D. Carmona, Chem. –Eur. J., 2019, 25, 13665–13670 CrossRef CAS PubMed.
- P. H. M. Budzelaar, D. L. Hughes, M. Bochmann, A. Macchioni and L. Rocchigiani, Chem. Commun., 2020, 56, 2542–2545 RSC.
- M. Isegawa, T. Matsumoto and S. Ogo, RSC Adv., 2021, 11, 28420–28432 RSC.
- J. B. Bonanno, T. P. Henry, P. T. Wolczanski, A. W. Pierpont and T. R. Cundari, Inorg. Chem., 2007, 46, 1222–1232 CrossRef CAS PubMed.
- N. Tsoureas, Y.-Y. Kuo, M. F. Haddow and G. R. Owen, Chem. Commun., 2010, 47, 484–486 RSC.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS PubMed.
- H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053–3062 CrossRef CAS PubMed.
- B. E. Cowie and D. J. H. Emslie, Chem. –Eur. J., 2014, 20, 16899–16912 CrossRef CAS PubMed.
- B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262–10265 CrossRef CAS PubMed.
- S. J. K. Forrest, J. Clifton, N. Fey, P. G. Pringle, H. A. Sparkes and D. F. Wass, Angew. Chem., Int. Ed., 2015, 54, 2223–2227 CrossRef CAS PubMed.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS PubMed.
- N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem. –Eur. J., 2020, 26, 5982–5993 CrossRef CAS PubMed.
- K. Mistry, P. G. Pringle, H. A. Sparkes and D. F. Wass, Organometallics, 2020, 39, 468–477 CrossRef CAS.
- H. Tinnermann, C. Fraser and R. D. Young, Dalton Trans., 2020, 49, 15184–15189 RSC.
- S. Ezzaher, J.-F. Capon, F. Gloaguen, N. Kervarec, F. Y. Pétillon, R. Pichon, P. Schollhammer and J. Talarmin, C. R. Chimie, 2008, 11, 906–914 CrossRef CAS.
- B. C. Manor and T. B. Rauchfuss, J. Am. Chem. Soc., 2013, 135, 11895–11900 CrossRef CAS PubMed.
- B. C. Manor, M. R. Ringenberg and T. B. Rauchfuss, Inorg. Chem., 2014, 53, 7241–7247 CrossRef CAS PubMed.
- N. Lalaoui, T. Woods, T. B. Rauchfuss and G. Zampella, Organometallics, 2017, 36, 2054–2057 CrossRef CAS PubMed.
- A. K. Justice, G. Zampella, L. D. Gioia and T. B. Rauchfuss, Chem. Commun., 2007, 2019–2021 RSC.
- E. J. Lawrence, V. S. Oganesyan, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, J. Am. Chem. Soc., 2014, 136, 6031–6036 CrossRef CAS PubMed.
- J. R. S. Jr, V. Zanotti, G. Facchin and R. J. Angelici, J. Am. Chem. Soc., 1992, 114, 160–165 CrossRef.
- O. R. Allen, S. J. Dalgarno and L. D. Field, Organometallics, 2008, 27, 3328–3330 CrossRef CAS.
- R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 4974–4977 CrossRef CAS PubMed.
- R. González-Hernández, J. Chai, R. Charles, O. Pérez-Camacho, S. Kniajanski and S. Collins, Organometallics, 2006, 25, 5366–5373 CrossRef.
- T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019–9022 RSC.
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. –Eur. J., 2012, 18, 16938–16946 CrossRef CAS PubMed.
- H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS.
- A. Jablonskytė, L. R. Webster, T. R. Simmons, J. A. Wright and C. J. Pickett, J. Am. Chem. Soc., 2014, 136, 13038–13044 CrossRef PubMed.
- F. Arrigoni, S. Mohamed Bouh, L. De Gioia, C. Elleouet, F. Y. Pétillon, P. Schollhammer and G. Zampella, Chem. –Eur. J., 2017, 23, 4364–4372 CrossRef CAS PubMed.
- J. A. Wright and C. J. Pickett, Chem. Commun., 2009, 5719 RSC.
- J. W. Tye, M. Y. Darensbourg and M. B. Hall, Inorg. Chem., 2006, 45, 1552–1559 CrossRef CAS PubMed.
- F. Arrigoni, L. Bertini, R. Breglia, C. Greco, L. De Gioia and G. Zampella, New J. Chem., 2020, 44, 17596–17615 RSC.
- G. Zampella, P. Fantucci and L. De Gioia, Chem. Commun., 2010, 46, 8824 RSC.
- A. R. Finkelmann, M. T. Stiebritz and M. Reiher, Chem. Sci., 2014, 5, 215–221 RSC.
- D. S. Lancaster, Alkylation of boron trifluoride with pentafluorophenyl Grignard reagent; Tris(pentafluorophenyl)boron, http://cssp.chemspider.com/215, accessed 14 October 2021 Search PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. Bruschi, R. Breglia, F. Arrigoni, P. Fantucci and L. De Gioia, Int. J. Quantum Chem., 2016, 116, 1695–1705 CrossRef CAS.
- G. M. Chambers, T. B. Rauchfuss, F. Arrigoni and G. Zampella, Organometallics, 2016, 35, 836–846 CrossRef CAS.
- S. Sicolo, M. Bruschi, L. Bertini, G. Zampella, G. Filippi, F. Arrigoni, L. De Gioia and C. Greco, Int. J. Hydrog. Energy, 2014, 39, 18565–18573 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154122 CrossRef PubMed.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1996, 100, 3349–3353 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- F. Arrigoni, S. Mohamed Bouh, C. Elleouet, F. Y. Pétillon, P. Schollhammer, L. De Gioia and G. Zampella, Chem. –Eur. J., 2018, 24, 15036–15051 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- P. Löwdin, J. Chem. Phys., 1950, 18, 365–375 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic, DFT and additional experimental data. See DOI: 10.1039/d1sc06975f |
| This journal is © The Royal Society of Chemistry 2022 |