
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater enables diastereodivergency in bispidine-based chiral amine-catalyzed asymmetric Mannich reaction of cyclic N-sulfonyl ketimines with ketones†
Gonglin
Li
,
Yan
Zhang
,
Hongkun
Zeng
,
Xiaoming
Feng
 ,
Zhishan
Su
* and
Lili
Lin
*
,
Zhishan
Su
* and
Lili
Lin
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: suzhishan@scu.edu.cn; lililin@scu.edu.cn; Web: http://www.scu.edu.cn/chem_asl/
First published on 22nd March 2022
Abstract
Tuning diastereoselectivity is a great challenge in asymmetric catalysis for the inherent stereochemical bias of the substrates. Here, we report a diastereodivergent asymmetric Mannich reaction of cyclic N-sulfonyl ketimines with ketones catalyzed by a bispidine-based chiral amine catalyst, in which additional water switches the diastereoselectivity efficiently. Both chiral anti- and syn-benzosultams with potential anti-HIV-1 activity are obtained in excellent yields and good to excellent ee values. Control experiments and density functional theory (DFT) calculations were applied to study the diastereodivergent mechanism, which reveal that the diastereodivergent catalysis should be state-determined, and the water reverses the energies of states to realize the diastereodivergency. The findings are quite new and might inspire more diastereodivergent asymmetric synthesis.
Introduction
Multiple stereocenters are widely spread in natural products and drug molecules. Both the diastereomers and enantiomers of a molecule with multiple stereocenters might have distinct or even opposing biological activities because enzymes and receptors provide chiral environments in biological systems.1,2 So, all stereoisomers of pharmaceutical candidates need to be obtained for evaluating their bioactivities during the drug discovery and development process.2 After rapid development of asymmetric catalysis, highly dia- and enantioselective reactions or cascades have been developed to deliver one diastereoisomer of chiral products with two or more stereocenters in one step or in one pot, and the enantiomers can normally be achieved with equal ease by applying the quasi-enantiomeric catalyst. However, other diastereomers are often unavailable efficiently because of the inherent stereochemical bias of the substrates.Diastereodivergent asymmetric catalysis3 is attractive and challenging because it aims to generate different chiral diastereomers starting from the same substrates just by small variation of reaction conditions, which is undoubtedly a starting material-economy process with minimum possible expenditure. Diastereodivergent dual catalysis4 and cycle-specific catalysis,5 controlling different stereocenters in one step or in sequential steps with two different chiral catalysts, have developed as novel concepts. Diastereodivergency with a single catalyst is also developed. Besides elegant studies in metal-based diastereodivergent catalysis,6 organocatalysis can achieve diastereodivergency by modulating catalysts7 or additives.8 Barbas, III,7a Shao,7b Singh,7c Kesavan7d and Chen7e realized the diastereodivergent asymmetric Mannich reactions of imines with aldehydes, hydroxyketones, benzofuran-3-ones and α,α-dicyanoolefins by varying organocatalysts based on amino acids, diamines and cinchona alkaloids. Though some progresses have been achieved, compared with the rapid development of asymmetric catalysis, diastereodivergent asymmetric catalysis is still in its infancy, and more diastereodivergent reactions need to be realized, more strategies need to be developed.
Chiral benzosultams are important compounds possessing interesting biological activities, such as γ-secretase inhibitors, HIV-1 inhibitors and aldose reductase inhibitors (Scheme 1).9 Among all the synthetic methods,10 the catalytic asymmetric reactions about cyclic N-sulfonyl ketimines are undoubtedly one of the most convenient and atom-economic one. Up to now, many asymmetric methodologies, including Mannich reaction,11 aza-Friedel–Crafts reaction,12 annulation,13 C(sp3)–H functionalization reaction,14 addition of organometallic reagents15 or unsaturated hydrocarbons16 to imines and so on,17 have been developed to synthesize various functionalized chiral benzosultams. However, there is still no diastereodivergent example in this area.
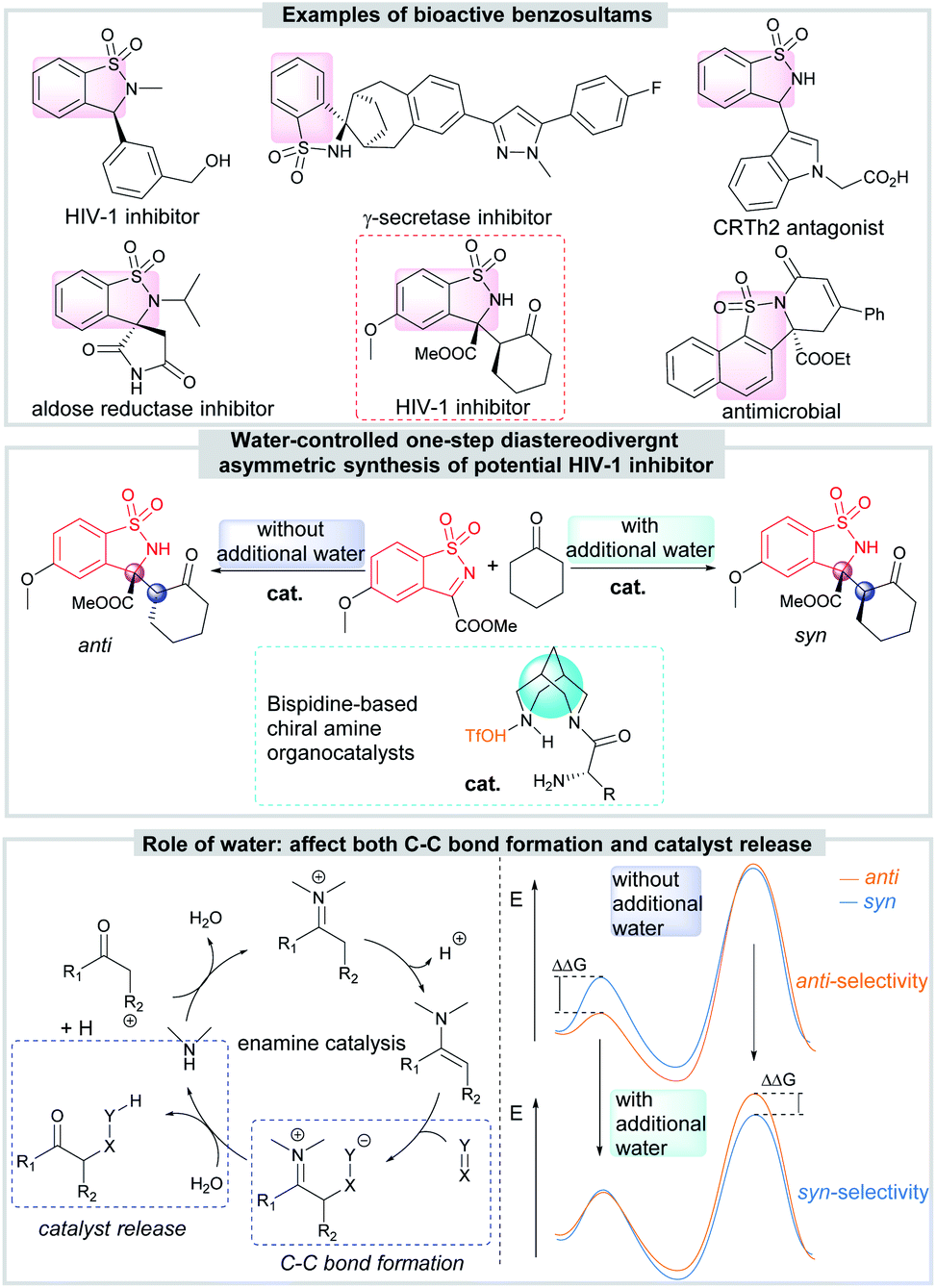 | ||
| Scheme 1 Diastereodivergent asymmetric Mannich reaction for synthesis of bioactive benzosultams. | ||
The asymmetric Mannich reaction of cyclic N-sulfonyl ketimines with cyclohexanone attracts our attention because it affords directly the chiral benzosultams containing vicinal tetrasubstituted and tertiary carbon stereocenters, more importantly, with anti-HIV-1 activity. Though syn-selective asymmetric reaction has been achieved by applying a bifunctional amino sulfonohydrazide as catalyst,11a the diastereodivergent asymmetric version is still not achieved. Developing efficient method to diversify the diastereochemical outcome of the reaction, obtaining both syn- and anti-products by small change of reaction conditions, is undoubtedly meaningful for further research on drug development. Bispidine-based chiral amines developed by our group show good ability in promoting asymmetric reactions through enamine catalysis.18 We envisaged such organocatalysts might have the potential to achieve the diastereodivergent goal since they possess unique core and multiple hydrogen-bonding donors and acceptors.
Herein, we report our finding that water can switch the enforced sense of diastereoselectivity when bispidine-based primary amine catalyzes the reaction of cyclic N-sulfonyl ketimines with ketones. DFT calculations reveal the diastereodivergent catalysis should be state-determined, and the water reverses the energies of states (Scheme 1).
Results and discussion
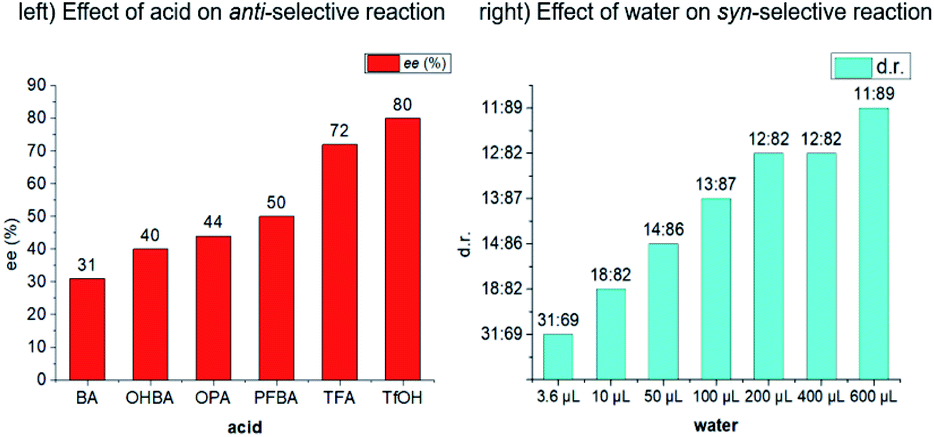
Initially, cyclic N-sulfonyl ketimine 1a and cyclohexanone 2a were selected as the model substrates to optimize the reaction conditions (Table 1). In our initial study, bispidine-based chiral amine catalysts derived from various amino acids could promote the reaction smoothly with ortho-phthalic acid (OPA) as cocatalyst at 35 °C. However, the dia- and enantioselectivities were moderate (for details, see the ESI†). Representatively, C1 derived from cyclohexyl substituted glycine could gave 3aa in near equivalent yield but only 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24 dr and 67% ee (entry 1). Excitedly, when 4 Å molecular sieves (MS) were added to the system, only anti-selective Mannich reaction occurred (dr > 19:1, entry 2). When various acids as co-catalysts were detected, regularly, the enantioselectivity increased with the increase of acidity (bottom, left histogram). When trifluoroacetic acid (TFA) was applied as co-catalyst, the ee value of major product was improved to 72% (entry 3). Exceptionally, trifluoromethanesulfonic acid (TfOH) gave 85:15 dr although it could further improve ee value (entry 4). With in situ prepared C1-TfOH as catalyst and trifluoroacetic acid as cocatalyst, the ee value was improved to 83% (entry 5). Undergoing in dichloromethane, the reaction afforded anti-3aa in 98% yield with >19:1 dr and 92% ee (entry 6). Analyzing the experimental data, we guess the influence of 4 Å MS and TfOH on dr might be caused by their water-absorbing quality. The 4 Å MS decreased the effect of water on diastereoselectivity, differently, TfOH increased the effect of water on diastereoselectivity.
:24 dr and 67% ee (entry 1). Excitedly, when 4 Å molecular sieves (MS) were added to the system, only anti-selective Mannich reaction occurred (dr > 19:1, entry 2). When various acids as co-catalysts were detected, regularly, the enantioselectivity increased with the increase of acidity (bottom, left histogram). When trifluoroacetic acid (TFA) was applied as co-catalyst, the ee value of major product was improved to 72% (entry 3). Exceptionally, trifluoromethanesulfonic acid (TfOH) gave 85:15 dr although it could further improve ee value (entry 4). With in situ prepared C1-TfOH as catalyst and trifluoroacetic acid as cocatalyst, the ee value was improved to 83% (entry 5). Undergoing in dichloromethane, the reaction afforded anti-3aa in 98% yield with >19:1 dr and 92% ee (entry 6). Analyzing the experimental data, we guess the influence of 4 Å MS and TfOH on dr might be caused by their water-absorbing quality. The 4 Å MS decreased the effect of water on diastereoselectivity, differently, TfOH increased the effect of water on diastereoselectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst/acid | Solvent | Additive | Yieldb (%) | drc (3/4) | eec (%) |
|
a Unless otherwise noted, all reactions were performed with catalyst/acid (1:1, 20 mol%), 1a (0.20 mmol), 2a (0.6 mL) at 35 °C for 20 h. If additive was added, the amount of 4 Å MS was 20 mg and H2O was 0.2 mL. If solvent was added, 2a (0.2 mL) in solvent (0.6 mL) for 12–16 h.
b Isolated yields of two diastereomers.
c Determined by SFC analysis on a chiral stationary phase.
d The reaction was performed at 30 °C for 24 h. BA = benzoic acid; OHBA = o-hydroxybenzoic acid; OPA = o-phthalic acid; PFBA = pentafluorobenzoic acid; TfOH = trifluoromethanesulfonic acid; TFA = trifluoroacetic acid.
|
||||||
| 1 | C1/OPA | — | — | 99 | 76:24 |
67/— |
| 2 | C1/OPA | — | 4 Å MS | 95 | >19:1 |
44/— |
| 3 | C1/TFA | — | 4 Å MS | 93 | >19:1 |
72/— |
| 4 | C1/TfOH | — | 4 Å MS | 95 | 85:15 |
80/— |
| 5 | C1-TfOH/TFA | — | 4 Å MS | 99 | >19:1 |
83/— |
| 6 | C1-TfOH/TFA | CH2Cl2 | 4 Å MS | 98 | >19:1 |
92/— |
| 7 | C1-TfOH/TFA | CH2Cl2 | H2O | 99 | 21:79 |
—/93 |
| 8 | C1-TfOH/TFA | Et2O | H2O | 98 | 12:88 |
—/93 |
| 9 | C1-TfOH/TFA | H2O | H2O | 98 | 11:89 |
—/88 |
| 10d | C1-TfOH/TFA | Et2O | H2O | 98 | 10:90 |
—/94 |
|
||||||

Realizing trace amount of water might greatly affect the diastereoselectivity, we wondered whether syn-selective Mannich reaction could be achieved if additional water was added to the system. Exhilaratingly, reversion of diastereoselectivity really happened. When 0.2 mL water was added, syn-4aa was obtained as the major product with 79:21 dr and 93% ee (entry 7). The diastereoselectivity could be improved to 88:12 when the solvent changed from dichloromethane to ether (entry 8). Regularly, the diastereoselectivity increased with the increase of the amount of water (bottom, right histogram). It is worth mentioning that this reaction could take place even in aqueous phase though the ee value of 4aa was slightly decreased (entry 9). Finally, the diastereoselectivity further increased to 90:10 when the temperature was adjusted to 30 °C (entry 10).
With the optimal conditions in hand, the substrate scope was then investigated. For the anti-selective Mannich reaction, all anti-3 were obtained with dr > 19:1 (Scheme 2). The steric hindrance of ester group in cyclic N-sulfonyl ketimine displayed a limited influence on either the reactivity or the stereoselectivity (3ba–3ea). Next, various substituents on aromatic ring of N-sulfonyl ketimines were tested. Regardless of the steric or electronic effect of the substituents at C5 position, all the corresponding anti-benzosultams could be obtained smoothly with good results (3fa–3ja, 98% yields, >19:1 dr, and 86–95% ee). Limited by the synthesis method, N-sulfonyl ketimines with electron-donating substituent at C6 position and with electron-withdrawing substituent at C7 position could be obtained. They transformed to the corresponding 3ka–3na in excellent yields and excellent ee values. Naphthyl 3o could also transform to the corresponding product in 99% yield, >19:1 dr and 96% ee. Varation of ketones showed that six-membered cyclic ketones 2b–2d containing heteroatom and 2e–2h with substituents at C4 position were all suitable substrates. By reacting with N-sulfonyl ketimine 1a, the corresponding 3ab–3ah were delivered in good to excellent yields with good dr and ee. When 2g and 2h were applied, desymmetrization occurred and three stereocenters formed in excellent stereoselectivity. Regretfully, other ketones, such as cyclobutanone, cyclopentanone, cycloheptanone and acyclic ketones, were not suitable. Low stereoselectivities or trace amounts of products were given (for details, see ESI†). The absolute configuration of 3aa was determined to be (3R,2′R) by X-ray crystallography analysis,19 and the configurations of other products were assigned to be (3R,2′R) by comparison with the CD spectrum of compound 3aa. The newly generated chiral centers of 3ag and 3ah were determined to be (S) by X-ray crystallography analysis or NMR analysis.19
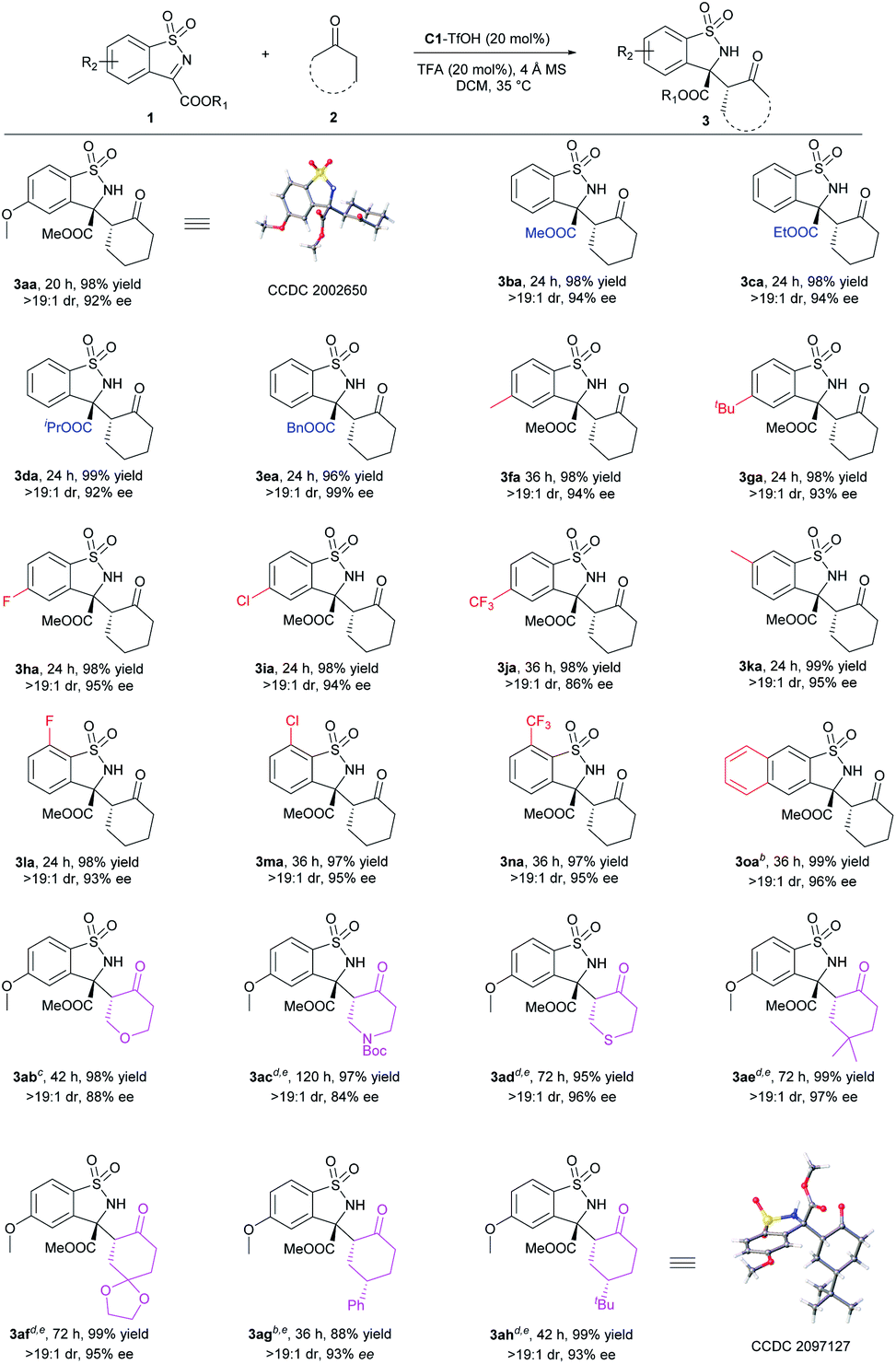 | ||
| Scheme 2 Substrate scope of anti-selective Mannich reaction. a Unless otherwise noted, all reactions were performed with catalyst/acid (1:1, 20 mol%), 1 (0.20 mmol), 2 (0.2 mL) and 4 Å MS (20 mg) in CH2Cl2 (0.6 mL) at 35 °C. All yields were the isolated products of the two diastereomers. The ee values were detected by SFC analysis on a chiral stationary phase and dr values were determined by 1H NMR analysis. b CH2Cl2 (1.0 mL) as the solvent. c CH2ClCH2Cl (0.6 mL) as the solvent at 40 °C. d CH2ClCH2Cl (1.0 mL) as the solvent at 40 °C. e Solid ketones were 1.0 mmol. | ||
Subsequently, the substrate scope of syn-selective Mannich reaction was investigated (Scheme 3). Similarly, ester groups and both electron-donating and electron-withdrawing substituents on phenyl ring of ketimines had no significant effect on the reactivity and stereoselectivity. Various syn-benzosultams 4aa–4oa were obtained in excellent yields with good to excellent dr and ee values (96–99% yields, 85:15–95:5 dr, and 86–95% ee). When cyclic ketones were examined, the heterocyclic tetrahydrothiopyran-4-one 2d, 4-dimethyl cyclohexanone 2e and 4-phenyl cyclohexanone 2g could smoothly form syn-4ad–4ag in good to excellent yields, good dr and excellent ee. Ketones 2b, 2c, 2f with O or N hetero atom and 2h with bulky tBu group proceeded mainly anti-selective Mannich reaction under this condition (for details, see the ESI†), which might be caused by the more H acceptor or larger steric hindrance. The absolute configuration of 4aa was determined to be (3R,2′S) by X-ray crystallography analysis.19 The newly generated chiral center of 4ag was determined to be (R) by NMR analysis.
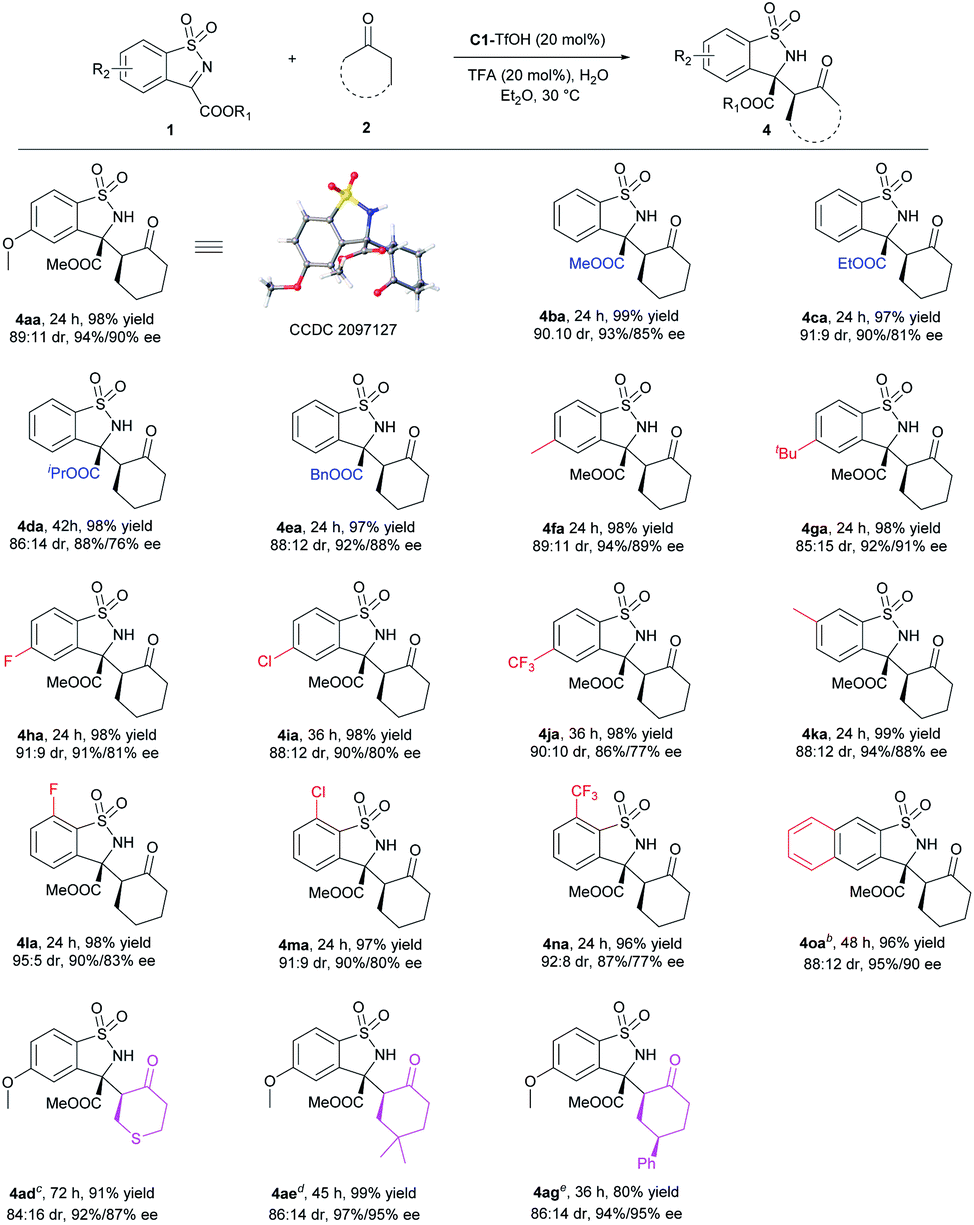 | ||
| Scheme 3 Substrate scope of syn-selective Mannich reaction. a Unless otherwise noted, all reactions were performed with catalyst/acid (1:1, 20 mol%), 1 (0.20 mmol), 2 (0.2 mL) and H2O (0.2 mL) in Et2O (0.6 mL) at 30 °C. All yields were the isolated products of the two diastereomers. The ee values were detected by SFC analysis on a chiral stationary phase and dr values were determined by 1H NMR analysis. b Et2O (1.0 mL) as the solvent. c2d was 1.0 mmol, Et2O (1.0 mL) as the solvent at 35 °C. d2e was 1.0 mmol, Et2O (1.0 mL) as the solvent at 30 °C. e2g was 1.0 mmol, methyl tertiary butyl ether (MTBE, 0.6 mL) and H2O (0.6 mL) as the solvent at 35 °C. | ||
To evaluate the synthetic potential of the diastereodivergent method, gram-scale synthesis of potential HIV-1 inhibitors anti-3aa and syn-4aa were carried out. As shown in Scheme 4, under respective optimized conditions, 4.0 mmol of cyclic N-sulfonyl ketimine 1a reacted smoothly with 4.0 mL cyclohexanone 2a, giving 1.38 g (98% yield) of anti-3aa with >19:1 dr and 90% ee (97% ee after single recrystallization), and 1.38 g (98% yield) of syn-4aa with 83:17 dr and 87% ee (99% ee after single recrystallization), separately. Reduction of anti-3aa in the presence of NaBH4, both ketone and ester group were reduced, gaving the compound 5 in 70% yield with maintained dr and ee value. Spirocyclic product 6 was also obtained in 27% yield, which might be formed through transesterification. The newly generated chiral center was determined to be (S) by NMR analysis. Reduction of syn-product 4aa under the same condition gave the compound 9 with ester group in 57% yield and spirocyclic product 10 in 35% yield with maintained dr and ee value. The ester group was more difficult to be reduced, which might be due to the larger steric hindrance of syn-4aa compared with that anti-3aa. The product 3aa could also be converted to oxime 7 in the presence of hydroxylammonium chloride and sodium acetate, and the configuration was determined to be E by X-ray crystallography analysis.19 The oxime 7 could be further converted to hydrazone 8 by tosyl chloride and 4-dimethylaminopyridine. When syn-4aa was converted in the same condition, anti-7 was obtained, suggesting configuration reversal occurred in the reaction condition.
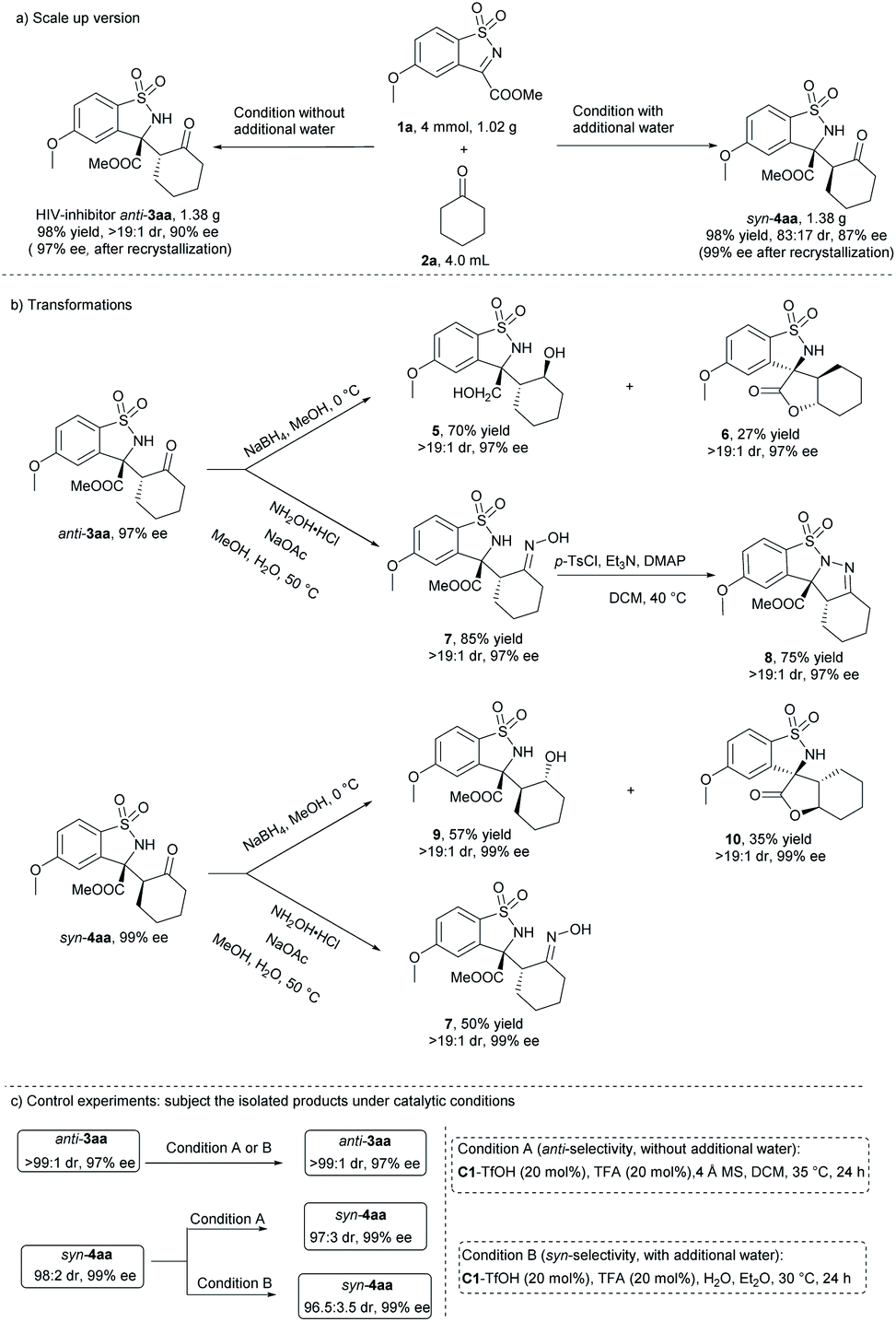 | ||
| Scheme 4 Scale-up synthesis, further transformations and control experiments. | ||
To understand the mechanism of the reaction, control experiments were conducted (Scheme 4c). Highly pure anti-3aa (>19:1 dr and 97% ee) and syn-4aa (47:1 dr and 99%), which were obtained through recrystalization of the corresponding products isolated from the catalytic system, were subjected under both catalytic conditions. After 24 hours, there was not any or very little change about the dr and ee values of both anti- and syn-products, which indicated that the diverse diastereochemical outcomes of the reaction came from the catalytic process rather than isomerization of the products in the catalytic conditions.
In addition, density functional theory (DFT) calculations were performed at the M062X-D3/6-31G(d,p) (SMD, CH2Cl2) level of theory (Scheme 5). In the catalytic system without additional water, 1b interacts with the enamine species generated by condensation between the primary amine of catalyst and cyclohexanone 2a. Then, the C–C bond is constructed via transition state 3-TS1 and 4-TS1. For 3-TS1, the H-bonds between protonated hydrogen on piperidine of catalyst and the nitrogen atom on the imine as well as the oxygen atom on a sulfonyl group, fix the N-sulfonyl ketimine so that the bispidine C1 can interact with two substrates at the same time. The regeneration of catalyst is predicted to be the rate-determining step (RDS). So, the pre-steps should be fully equilibrated, and the diastereoselectivity should be determined by more stable states. By calculation, the states to form anti-products are more stable, typically, the ΔG of 3-TS1 is lower than that of 4-TS1 by 6.4 kcal mol−1. In 3-TS1, the enamine and the N-sulfonyl ketimine react both with Si-faces, leading to (3R,2′R)-3ba.
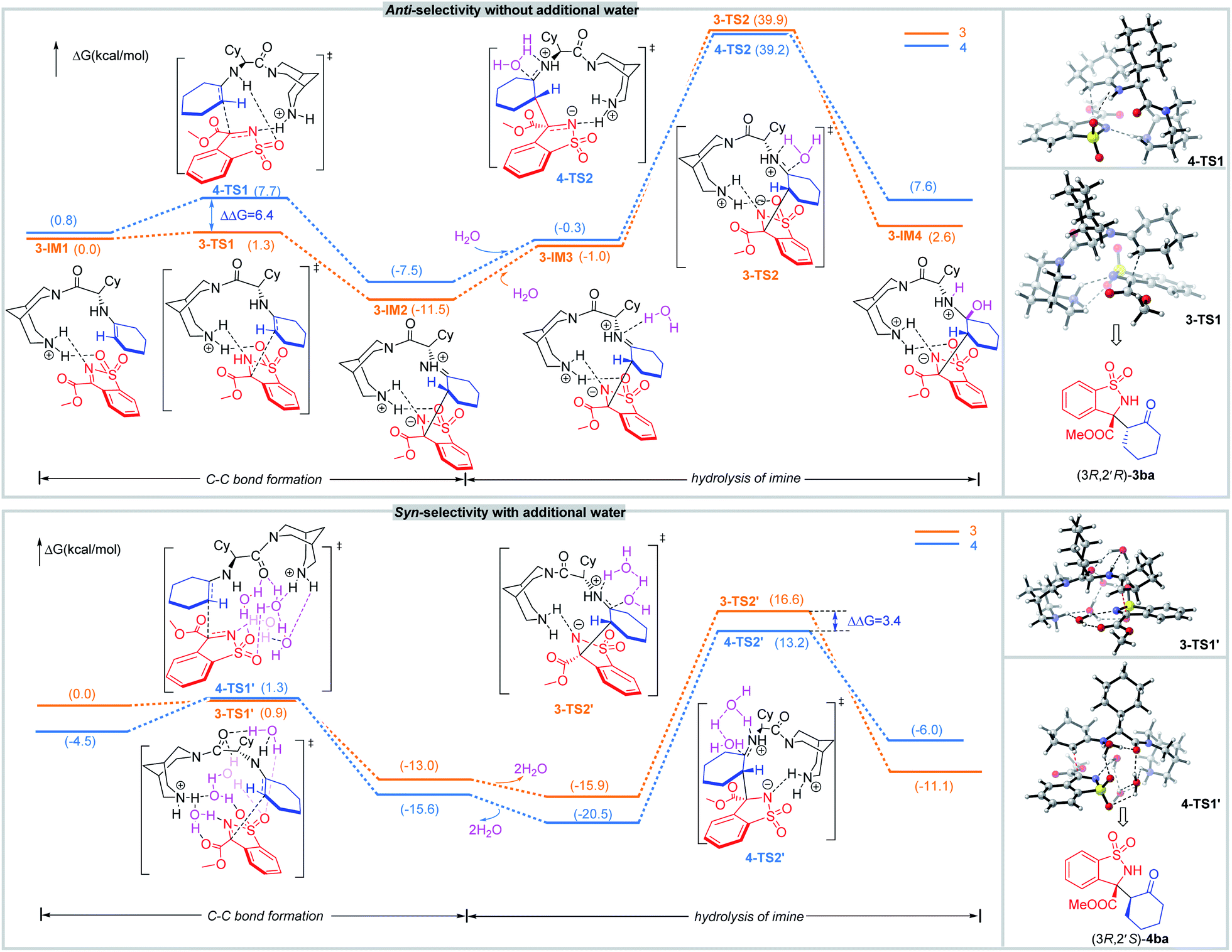 | ||
| Scheme 5 DFT calculations for diastereodivergent and enantioselective mechanism. | ||
To get insight into the effect of water on the diastereoselectivity, the reaction mechanism in the presence of additional water was studied. In the chiral controlling C–C bond formation step, the optimized geometries of key intermediates and transition states with one to four water molecules in the structures were located (for details, see the ESI†). The relative energy difference of the two competing transition states 3-TS1′ and 4-TS1′ decreased, especially for those with four waters. In addition, waters can also accelerate the re-generation of catalyst by decreasing the activation barrier in the hydrolysis of imine step. The states to form syn-product are more stable, for example, the ΔG of 4-TS2′ is lower than that of 3-TS2′ by 3.4 kcal mol−1, so syn-4 is predominantly formed in the presence of water. In 4-TS1′, the enamine reacts with the N-sulfonyl ketimine with its Re-face from the Si-face of the latter, leading to (3R,2′S)-4ba.
Conclusions
We realize a bispidine-based amine-catalyzed diastereodivergent asymmetric Mannich reaction of cyclic N-sulfonyl ketimines with ketones through additional water switching the enforced sense of diastereoselectivity. Both syn- and anti-benzosultams with potential anti-HIV-1 activity are obtained in good to excellent yields, good to excellent dr and ee. DFT calculations support that the additional water is more likely through reversing the energies of states in the C–C bond formation and hydrolysis of imine steps to switch the diastereoselectivity. The methodology offers a new idea for diastereoselective modulation, which is valuable for organic synthesis and drug research. Further efforts will be devoted to realizing more diastereodivergent catalytic asymmetric reactions.Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, SFC spectra, CD spectra, computational methods and X-ray crystallographic data are available in the ESI.†Author contributions
G. L. L. performed the experiments. H. K. Z. repeated data. Y. Z. and Z. S. S. conducted the DFT calculations. X. M. F. and L. L. L. supervised the project. G. L. L. and L. L. L. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. 22171189, 21871188 and 21973066) for financial support. We are grateful to Dr Yuqiao Zhou from College of Chemistry, Sichuan University for the X-ray single crystal diffraction analysis.Notes and references
- (a) E. M. Carreira and L. Kvaerno, Classics in Stereoselective Synthesis, Wiley, Weinheim, 2009 Search PubMed; (b) E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Supplements I and II, Springer, New York, 1999, vol. I–III Search PubMed; (c) E. M. Carreira and H. Yamamoto, Comprehensive Chirality, Elsevier Science, Amsterdam, 2012, vol. 1–9 Search PubMed.
- (a) K. Jozwiak, W. J. Lough and I. W. Wainer, Drug Stereochemistry: Analytical Methods and Pharmacology, Informa, New York, 2012 CrossRef; (b) K. M. Rentsch, J. Biochem. Biophys. Methods, 2002, 54, 1 CrossRef CAS; (c) B. M. Trost, Science, 1983, 219, 245 CrossRef CAS PubMed.
- For selected reviews of diastereodivergent synthesis, see: (a) L. L. Lin and X. M. Feng, Chem.–Eur. J., 2017, 23, 6464 CrossRef CAS PubMed; (b) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS PubMed; (c) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534 CrossRef CAS; (d) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675 RSC; (e) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed.
- For selected examples of stereodivergent dual catalysis, see: (a) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed; (b) S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3020 CrossRef CAS PubMed; (c) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093 CrossRef CAS PubMed; (d) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029 CrossRef CAS PubMed; (e) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87 CrossRef CAS PubMed; (f) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 CrossRef CAS PubMed; (g) Q. Zhang, H. Yu, L. Shen, T. Tang, D. Dong, W. Chai and W. Zi, J. Am. Chem. Soc., 2019, 141, 14554 CrossRef CAS PubMed; (h) M.-M. Zhang, Y.-N. Wang, B.-C. Wang, X.-W. Chen, L.-Q. Lu and W.-J. Xiao, Nat. Commun., 2019, 10, 2716 CrossRef PubMed; (i) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48 CrossRef CAS; (j) B. Kim, Y. Kim and S. Y. Lee, J. Am. Chem. Soc., 2021, 143, 73 CrossRef CAS PubMed; (k) S.-Q. Yang, Y.-F. Wang, W.-C. Zhao, G.-Q. Lin and Z.-T. He, J. Am. Chem. Soc., 2021, 143, 7285 CrossRef CAS PubMed.
- For selected examples of cycle-specific catalysis, see: (a) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS PubMed; (b) B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS PubMed; (c) Y. Chi, S. T. Scroggins and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 6322 CrossRef CAS PubMed; (d) Y. Wang, R.-G. Han, Y.-L. Zhao, S. Yang, P.-F. Xu and D. J. Dixon, Angew. Chem., Int. Ed., 2009, 48, 9834 CrossRef CAS PubMed; (e) J. Jiang, H.-D. Xu, J.-B. Xi, B.-Y. Ren, F.-P. Lv, X. Guo, L.-Q. Jiang, Z.-Y. Zhang and W.-H. Hu, J. Am. Chem. Soc., 2011, 133, 8428 CrossRef CAS PubMed; (f) N. K. Rana, H. Huang and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2014, 53, 7619 CrossRef CAS PubMed; (g) S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353 CrossRef CAS PubMed; (h) D. Lotter, A. Castrogiovanni, M. Neuburger and C. Sparr, ACS Cent. Sci., 2018, 4, 656 CrossRef CAS PubMed; (i) Y. Kwon, A. J. Chinn, B. Kim and S. J. Miller, Angew. Chem., Int. Ed., 2018, 57, 6251 CrossRef CAS PubMed; (j) B. M. Trost, C.-I. (Joey) Hung, T. Saget and E. Gnanamani, Nat. Catal., 2018, 1, 523 CrossRef CAS; (k) X.-J. Liu, S. Jin, W.-Y. Zhang, Q.-Q. Liu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2020, 59, 2039 CrossRef CAS PubMed; (l) D.-X. Zhu, J.-G. Liu and M.-H. Xu, J. Am. Chem. Soc., 2021, 143, 8583 CrossRef CAS PubMed.
- For selected examples of metal-based diastereodivergent catalysis, see: (a) X.-X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X.-L. Hou and Y. D. Wu, J. Am. Chem. Soc., 2008, 130, 14362 CrossRef CAS PubMed; (b) A. Nojiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 3779 CrossRef CAS PubMed; (c) G. Lu, T. Yoshino, H. Morimoto, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4382 CrossRef CAS PubMed.
- For selected examples of diastereodivergent synthesis by modulating catalysts, see: (a) S. Mitsumori, H. Zhang, P. H.-Y. Cheong, K. N. Houk, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 1040 CrossRef CAS PubMed; (b) J. Dai, D. Xiong, T. Yuan, J. Liu, T. Chen and Z. Shao, Angew. Chem., Int. Ed., 2017, 56, 12697 CrossRef CAS PubMed; (c) V. Bisai, R. A. Unhale, A. Suneja, S. Dhanasekaran and V. K. Singh, Org. Lett., 2015, 17, 2102 CrossRef CAS PubMed; (d) K. Sivamuthuraman and V. Kesavan, J. Org. Chem., 2018, 83, 8936 CrossRef CAS PubMed; (e) X.-F. Xiong, Z.-J. Jia, W. Du, K. Jiang, T.-Y. Liu and Y.-C. Chen, Chem. Commun., 2009, 6994 RSC; (f) Y. Wang, H. Li, Y.-Q. Wang, Y. Liu, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2007, 129, 6364 CrossRef CAS PubMed; (g) X. Feng, Z. Zhou, R. Zhou, Q.-Q. Zhou, L. Dong and Y.-C. Chen, J. Am. Chem. Soc., 2012, 134, 19942 CrossRef CAS PubMed; (h) C. Verrier and P. Melchiorre, Chem. Sci., 2015, 6, 4242 RSC; (i) Y. Wei, S. Wen, Z. Liu, X. Wu, B. Zeng and J. Ye, Org. Lett., 2015, 17, 2732 CrossRef CAS PubMed; (j) D. Uraguchi, K. Yoshioka and T. Ooi, Nat. Commun., 2017, 8, 14793 CrossRef PubMed; (k) S. B. J. Kan, H. Maruyama, M. Akakura, T. Kano and K. Maruoka, Angew. Chem., Int. Ed., 2017, 56, 9487 CrossRef CAS PubMed; (l) P.-G. Ding, F. Zhou, X. Wang, Q.-H. Zhao, J.-S. Yu and J. Zhou, Chem. Sci., 2020, 11, 3852 RSC.
- For selected examples of diastereodivergent synthesis by modulating additives, see: (a) J. Gao, S. Bai, Q. Gao, Y. Liu and Q. Yang, Chem. Commun., 2011, 47, 6716 RSC; (b) X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 17934 CrossRef CAS PubMed; (c) A. Martinez-Castaneda, H. Rodriguez-Solla, C. Concellon and V. del Amo, J. Org. Chem., 2012, 77, 10375 CrossRef CAS PubMed.
- For selected examples of various bioactive molecules and natural products, see: (a) J. Wrobel, A. Dietrich, S. A. Woolson, J. Millen, M. McCaleb, M. C. Harrison, T. C. Hohman, J. Sredy and D. Sullivan, J. Med. Chem., 1992, 35, 4613 CrossRef CAS PubMed; (b) R. J. Cherney, R. Mo, D. T. Meyer, K. D. Hardman, R.-Q. Liu, M. B. Covington, M. Qian, Z. R. Wasserman, D. D. Christ, J. M. Trzaskos, R. C. Newton and C. P. Decicco, J. Med. Chem., 2004, 47, 2981 CrossRef CAS PubMed; (c) J. De Vicente, R. T. Hendricks, D. B. Smith, J. B. Fell, J. Fischer, S. R. Spencer, P. J. Stengel, P. Mohr, J. E. Robinson, J. F. Blake, R. K. Hilgenkamp, C. Yee, G. Adjabeng, T. R. Elworthy, J. Li, B. H. Wang, J. T. Bamberg, S. F. Harris, A. Wong, V. J. Leveque, I. Najera, S. Le Pogam, S. Rajyaguru, G. Ao-Ieong, L. Alexandrova, S. Larrabee, M. Brandl, A. Briggs, S. Sukhtankar and R. Farrell, Bioorg. Med. Chem. Lett., 2009, 19, 5652 CrossRef CAS PubMed; (d) G. J. Wells, M. Tao, K. A. Josef and R. Bihovsky, J. Med. Chem., 2001, 44, 3488 CrossRef CAS PubMed; (e) Y. Takeuchi and Z. Liu, Heterocycles, 2002, 56, 693 CrossRef; (f) A. Jirgensons, G. Leitis, I. Kalvinsh, D. Robinson, P. Finn, N. Khan, Patent Appl. WO2008142376 A1, 2008; (g) J. Mao and D. C. Baker, US pat., 6458962B1, 2003 Search PubMed.
- For selected review of synthesis of benzosultams, see: Q.-Q. Zhao and X.-Q. Hu, Molecules, 2020, 25, 4367 CrossRef CAS PubMed.
- For selected examples of Mannich reaction, see: (a) S. Zhang, L. J. Li, Y. B. Hu, Z. G. Zha, Z. Y. Wang and T.-P. Loh, Org. Lett., 2015, 17, 1050 CrossRef CAS PubMed; (b) B. K. Qiao, Y.-J. Huang, J. Nie and J.-A. Ma, Org. Lett., 2015, 17, 4608 CrossRef CAS PubMed; (c) H.-X. Duan, Y. Zhang, Z.-Z. Zhang and Y.-Q. Wang, J. Org. Chem., 2020, 85, 11331 CrossRef CAS PubMed; (d) J. Lv, J. Xu, X. Pan, Z. Jin and Y. R. Chi, Sci. China: Chem., 2021, 64, 985 CrossRef CAS; (e) L. Xie, H. Ma, J. Li, Y. Yu, Z. Qin and B. Fu, Org. Chem. Front., 2017, 4, 1858 RSC; (f) Q. Shao, L. Wu, J. Chen, I. D. Gridnev, G. Yang, F. Xie and W. Zhang, Adv. Synth. Catal., 2018, 360, 4625 CrossRef CAS; (g) M. Zhou, Q. Su, Y. Addepalli, Y. He and Z. Wang, Org. Biomol. Chem., 2018, 16, 2923 RSC; (h) S. Nakamura, M. Sano, A. Toda, D. Nakane and H. Masuda, Chem.–Eur. J., 2015, 21, 1 CrossRef; (i) H. Zhang, C. Jiang, J.-P. Tan, H.-L. Hu, Y. Chen, X. Ren, H.-S. Shang and T. Wang, ACS Catal., 2020, 10, 5698 CrossRef CAS; (j) S. Zhang, L. Li, L. Xin, W. Liu and K. Xu, J. Org. Chem., 2017, 82, 2399 CrossRef CAS PubMed.
- For selected examples of the aza- Friedel−Crafts reaction, see: (a) L. Wu, R. R. Liu, G. Zhang, D. J. Wang, H. Wu, J. Gao and Y. X. Jia, Adv. Synth. Catal., 2015, 357, 709 CrossRef CAS; (b) L. Wang, A. Rahman and X. Lin, Org. Biomol. Chem., 2017, 15, 6033 RSC; (c) B. B. Huang, L. Wu, R. R. Liu, L. L. Xing, R. X. Liang and Y. X. Jia, Org. Chem. Front., 2018, 5, 929 RSC.
- For selected examples of addition of annulation, see: (a) R. R. Liu, J.-P. Hu, J.-J. Hong, C.-J. Lu, J.-R. Gao and Y.-X. Jia, Chem. Sci., 2017, 8, 2811 RSC; (b) P. Wonneberger, N. König, F. B. Kraft, M. B. Sárosi and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2019, 58, 3208 CrossRef CAS PubMed; (c) B. S. Li, Y. Wang, Z. Jin, P. Zheng, R. Ganguly and Y. R. Chi, Nat. Commun., 2015, 6, 6207 CrossRef PubMed; (d) X. Wang, M. Lu, Q. Su, M. Zhou, Y. Addepalli, W. Yao, Z. Wang and Y. Lu, Chem.–Asian J., 2019, 14, 3409 CrossRef CAS PubMed; (e) M. Rommel, T. Fukuzumi and J. W. Bode, J. Am. Chem. Soc., 2008, 130, 17266 CrossRef CAS PubMed; (f) A. G. Kravina, J. Mahatthananchai and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9433 CrossRef CAS PubMed; (g) X. Y. Chen, Z. H. Gao, C. Y. Song, C. L. Zhang, Z. X. Wang and S. Ye, Angew. Chem., Int. Ed., 2014, 53, 11611 CrossRef CAS PubMed; (h) P. Zheng, S. Wu, C. Mou, W. Xue, Z. Jin and Y. R. Chi, Org. Lett., 2019, 21, 5026 CrossRef CAS PubMed; (i) Y.-H. Chen, X.-J. Lv, Z.-H. You and Y.-K. Liu, Org. Lett., 2019, 21, 5556 CrossRef CAS PubMed.
- For selected examples of C(sp3)–H functionalization reaction, see: (a) Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016 CrossRef CAS; (b) W. K. Weigel, H. T. Dang, H.-B. Yang and D. B. C. Martin, Chem. Commun., 2020, 56, 9699 RSC.
- For selected examples of addition of organometallic reagents to imines, see: (a) T. Nishimura, A. Noishiki, G. C. Tsui and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 5056 CrossRef CAS PubMed; (b) H. Wang, T. Jiang and M. H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed; (c) C. Jiang, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2014, 53, 9936 CrossRef CAS PubMed; (d) Y. Álvarez-Casao, D. Monge, E. Álvarez and R. Fernández, Org. Lett., 2015, 17, 5104 CrossRef PubMed; (e) M. Quan, X. Wang, L. Wu, I. D. Gridnev, G. Yang and W. Zhang, Nat. Commun., 2018, 9, 2258 CrossRef PubMed; (f) Z. Qiu, Y. Li, Z. Zhang and D. Teng, Transition Met. Chem., 2019, 44, 649 CrossRef CAS; (g) M. F. Li, A.-Q. Miao, H.-Y. Zhu, R. Wang, W.-J. Hao, S.-J. Tu and B. Jiang, J. Org. Chem., 2020, 85, 13602 CrossRef CAS PubMed.
- For selected examples of addition of unsaturated hydrocarbons to imines, see: (a) R.-R. Liu, D.-J. Wang, L. Wu, B. Xiang, G.-Q. Zhang, J.-R. Gao and Y.-X. Jia, ACS Catal., 2015, 5, 6524 CrossRef CAS; (b) K. Morisak, M. Sawa, R. Yonesaki, H. Morimoto, K. Mashima and T. Ohshima, J. Am. Chem. Soc., 2016, 138, 6194 CrossRef PubMed; (c) Z. Liang, S. Singh, F. Xie, L. Wu and W. Zhang, Chem. Commun., 2017, 53, 5364 RSC.
- For selected examples, see: (a) J. Pan, J.-H. Wu, H. Zhang, X. Ren, J. P. Tan, L. Zhu, H. S. Zhang, C. Jiang and T. Wang, Angew. Chem., Int. Ed., 2019, 58, 7425 CrossRef CAS PubMed; (b) Y. Yao, J.-L. Li, Q.-Q. Zhou, L. Dong and Y.-C. Chen, Chem.–Eur. J., 2013, 19, 9447 CrossRef CAS PubMed; (c) Y. Li, K. Zhou, Z. Wen, S. Cao, X. Shen, M. Lei and L. Gong, J. Am. Chem. Soc., 2018, 140, 15850 CrossRef CAS PubMed; (d) L. Z. Hou, T. F. Kang, L. K. Yang, W. D. Cao and X. M. Feng, Org. Lett., 2020, 22, 1390 CrossRef CAS PubMed; (e) H. B. Hepburn and H. W. Lam, Angew. Chem., Int. Ed., 2014, 53, 11605 CrossRef CAS PubMed; (f) C. A. Osborne, T. B. D. Endean and E. R. Jarvo, Org. Lett., 2015, 17, 5340 CrossRef CAS PubMed; (g) J. Kim, M. Shin and S. H. Cho, ACS Catal., 2019, 9, 8503 CrossRef CAS; (h) J. Wang, Q. Zhang, B. Zhou, C. Yang, X. Li and J.-P. Cheng, iScience, 2019, 16, 511 CrossRef CAS PubMed.
- For selected examples of bispidine-based chiral amines, see: (a) J. Liu, Z. G. Yang, Z. Wang, F. Wang, X. H. Chen, X. H. Liu, X. M. Feng, Z. S. Su and C. W. Hu, J. Am. Chem. Soc., 2008, 130, 5654 CrossRef CAS PubMed; (b) Z. G. Yang, J. Liu, X. H. Liu, Z. Wang, X. M. Feng, Z. S. Su and C. W. Hu, Adv. Synth. Catal., 2008, 350, 2001 CrossRef CAS; (c) J. Liu, Z. G. Yang, X. H. Liu, Z. Wang, Y. L. Liu, S. Bai, L. L. Lin and X. M. Feng, Org. Biomol. Chem., 2009, 7, 4120 RSC; (d) G. L. Li, M. H. Z. Liu, S. J. Zou, X. M. Feng and L. L. Lin, Org. Lett., 2020, 22, 8708 CrossRef CAS PubMed.
- CCDC number 2058446 (C1-2TfOH); CCDC 2002650 (3aa); CCDC 2084361 (4aa); CCDC 2097127 (3ah); CCDC 2090581 (7) contain the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, X-ray crystallographic data for 6. CCDC 2058446, 2002650, 2084361, 2097127 and 2090581. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00446a |
| This journal is © The Royal Society of Chemistry 2022 |