
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiaryliodonium salts facilitate metal-free mechanoredox free radical polymerizations†
Sarah M.
Zeitler‡
 ,
Progyateg
Chakma‡
and
Matthew R.
Golder
*
,
Progyateg
Chakma‡
and
Matthew R.
Golder
*
Department of Chemistry, Molecular Engineering & Science Institute, University of Washington, 36 Bagley Hall, Seattle, WA 98195, USA. E-mail: goldermr@uw.edu
First published on 16th March 2022
Abstract
Mechanically-induced redox processes offer a promising alternative to more conventional thermal and photochemical synthetic methods. For macromolecule synthesis, current methods utilize sensitive transition metal additives and suffer from background reactivity. Alternative methodology will offer exquisite control over these stimuli-induced mechanoredox reactions to couple force with redox-driven chemical transformations. Herein, we present the iodonium-initiated free-radical polymerization of (meth)acrylate monomers under ultrasonic irradiation and ball-milling conditions. We explore the kinetic and structural consequences of these complementary mechanical inputs to access high molecular weight polymers. This methodology will undoubtedly find broad utility across stimuli-controlled polymerization reactions and adaptive material design.
Introduction
Mechanochemistry1–5 is an interdisciplinary field spanning small molecule methodology, crystal engineering, and polymer science. Chemical processes induced by mechanical force offer advantages over other stimuli. For example, undesirable thermal byproducts can be avoided and limited stimuli penetration (e.g., photochemistry) into solutions can be mitigated.1,5 For macromolecules, force-responsive polymers with engineered mechanophores6,7 can change color,8 alter bulk electronic9 or structural10–13 properties, and release cargo14,15 on demand via mechanochemically-driven processes; such systems find utility in sensing,10 additive manufacturing,16,17 and therapeutic delivery18,19 applications. One common form of mechanochemical input is ultrasound (US) irradiation.20 Ultrasonic waves generate cavitation bubbles21 that collapse and produce forces that fuel subsequent processes. US itself is readily accessible and has numerous biological applications22–24 in diagnostic imaging25 and targeted drug release.26,27 A complimentary method to generate force in mechanochemical systems is ball milling, a sustainable and economical technique due to the removal of nearly all solvent.3 This industrially scalable technology was originally developed for the breakdown of minerals and biopolymers28 but recently became a widespread method for solid-state synthesis.3,29 The ubiquity of force-induced macromolecular deconstruction largely dominates the field of mechanochemistry. Counterintuitively, analogous systems also exist that construct matter30via covalent bond formations under mechanical stimulation.One mechanism to facilitate the use of force in chemical synthesis is mechanoredox catalysis31 (Fig. 1); the piezoelectric effect32–35 converts mechanical energy into a useable electric potential. Many applications mirror bone growth36 and mechanogenetics24,37 mechanisms found in biological systems. Other uses of piezoelectric materials and molecules include water splitting and treatment;38–41 wearable devices have even been developed to use piezoelectricity as a means to couple human motion with energy storage.42 Only recently have chemists utilized this concept in modern synthetic polymer chemistry (i.e., mechanoredox catalysis). Several examples now exist that harness US and the piezoelectric effect to drive mechanoredox polymerization processes (Fig. 1A).31 Both the Esser-Kahn and Matyjaszewski groups have developed systems where piezoelectric nanoparticles (PNP) facilitate either free radical polymerization (FRP) or atom transfer radical polymerization (ATRP) in the presence of US.43–46 While both works demonstrate the feasibility of mechanoredox polymerizations, these methods require transition metal additives (e.g., copper or iron salts)44,45 and have significant non-mechanoredox background reactions.43,44 The current state of the art leaves vast opportunities to further refine mechanoredox polymerization methodology. Such fundamental developments are imperative to the development and implementation of next-generation stimuli-responsive soft materials.
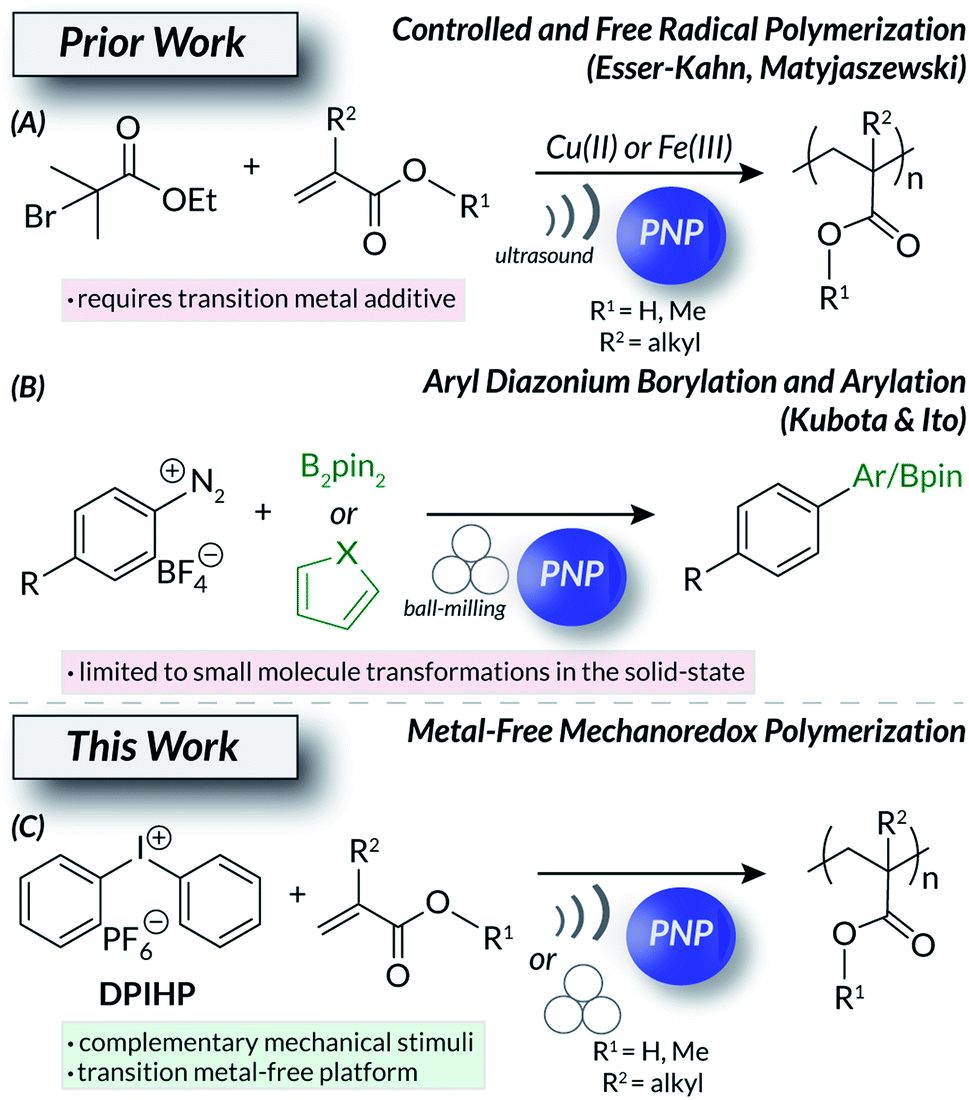 | ||
| Fig. 1 Evolution of (A) mechanoredox polymerizations and (B) small molecule transformations (e.g., borylation, arylation) as an inspiration for (C) our metal-free ultrasonic irradiation and ball-milling mechanoredox polymerization methodology. | ||
PNPs can also be used to drive small molecule mechanoredox transformations. In recent work by Kubota and Ito, ball-milling with PNPs (e.g., ZnO, BaTiO3) initiates C–H borylations and arylation reactions (Fig. 1B).47 Similar solid-state techniques also effect aryl trifluoromethylation48 and atom-transfer radical cyclization reactions.49 Interestingly, mechanoredox radical polymerizations have never been initiated under ball-milling conditions. In the initial small molecule mechanoredox study by Kubota and Ito, aryl diazonium salts were used as “initiators” to drive the subsequent radical C–H functionalization reactions.47 These salts, along with other aryl onium salts (e.g., diaryl iodoniums and triaryl sulfoniums) have been used in photoredox systems as tunable aryl radical50 surrogates with varying reduction potentials;51 aryl diazoniums are extremely labile while diaryl iodoiniums and triaryl sulfoniums (Ered = −0.3 V to −1.0 V vs. SCE) are significantly less susceptible to reduction. The reactivity of onium salts can be further manipulated through substituent effects, leading to a wide array of reactivity.52 Many onium salts are also either commercially available or readily synthesized, making them accessible, bench-stable building blocks for synthetic manipulations. Furthermore, onium salts can be used to integrate soft materials with surfaces, presenting opportunities for tuneable polymer grafting and composites.53,54
Using these recent works as inspiration, we now report the mechanoredox onium salt-initiated FRP of (meth)acrylates under both ultrasonic irradiation and ball-milling conditions (Fig. 1C). This work details the first examples of metal-free mechanoredox polymerizations and compares the consequences of these conditions under complementary reaction conditions (i.e., ultrasonic irradiation and ball-milling). Overall, we demonstrate the broad application of mechanical force to access industrially relevant high molecular weight polymeric materials without relying on traditional thermal55–57 or photochemical58–61 inputs.
Results & discussion
Ultrasonic irradiation (US) mechanoredox polymerizations
Inspired by the work47 of Kubota and Ito, we initially wondered whether aryl diazonium salts could be used as mechanoredox initiators for FRP of commercially available acrylates. To investigate, 4-bromobenzenediazonium tetrafluoroborate (BBDT) and 4-methoxybenzenediazoium tetrafluoroborate (MBDT) were used as initiators in the presence of a suitable PNP (e.g., ZnO or BaTiO3) and acrylate monomer. Degassed reaction mixtures were immersed into a thermostated ultrasonic bath (see Fig. S1† for a typical reaction setup); to our surprise, while high monomer conversion was achieved in a variety of different organic solvents in the presence of both US and PNPs, similar results were obtained in the absence of US (Table S1†). Despite numerous examples employing BBDT or MBDT as aryl radical precursors in photoredox processes,51 we found after extensive studies that aryl diazoniums were too capricious to use in this system. The background reactions in the absence of force affirm that even a low concentration of radicals, presumably from aryl diazonium decomposition in solution, can initiate FRP. These early studies prompted us to investigate alternative onium salts with more negative reduction potentials; we rationalized that such initiators would exhibit superior solution-state stability.We hypothesized that diaryliodonium salts,62 which are more difficult to reduce (Ered = ca. −0.5 V vs. SCE) and thus should be more stable in solution than aryl diazonium salts, would be more suitable for mechanoredox FRP. To explore this new system, we first studied the polymerization of tert-butyl acrylate (tBA) in DMF by using diphenyl iodonium hexafluorophosphate (DPIHP) as our initiator (Table 1) with either BaTiO3 or ZnO (Table 1, entries 1–5) as the PNP. With both PNPs, the reaction mixtures became visibly viscous within 8 h of sonication, suggesting high monomer conversion (see Fig. S2† representative photographs). In the presence of 7 wt% BaTiO3 and ZnO (relative to the combined mass of the monomer, solvent, and DPIHP), 1H NMR analysis of the resulting polymers showed 92% and 68% monomer conversion after 20 h, respectively. In the absence of US or PNP, however, little to no conversion was seen over the same time period. A direct correlation of PNP loading to monomer conversion was observed, indicating the pivotal role of nanoparticles. Maximum monomer conversion was achieved with 7 wt% PNP, so this loading was used for all future experiments. Results remained consistent with high monomer conversion on larger scale (10x scale = 5 g tBA monomer) using BaTiO3 (Fig. S5†), highlighting the potential scalability of ultrasonic irradiation mechanoredox polymerizations.
|
|
|||
|---|---|---|---|
| Entryb | Nanoparticle | Ultrasound? | Conversionc (%) |
a Reaction conditions: [monomer]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[DPIHP]0 = 100:1.
b [tBA] = 7.3 M, [DPIHP] = 0.073 M in DMF.
c Conversion was determined by 1H NMR spectroscopy. Ultrasonic bath (40 kHz, 70 W, 20 °C); reaction time: 20 h. :[DPIHP]0 = 100:1.
b [tBA] = 7.3 M, [DPIHP] = 0.073 M in DMF.
c Conversion was determined by 1H NMR spectroscopy. Ultrasonic bath (40 kHz, 70 W, 20 °C); reaction time: 20 h.
|
|||
| 1 | BaTiO3 (1.5 wt%) | Yes | 35 |
| 2 | BaTiO3 (3.5 wt%) | Yes | 80 |
| 3 | BaTiO3 (7 wt%) | Yes | 92 |
| 4 | BaTiO3 (7 wt%) | No | <5 |
| 5 | ZnO (7 wt%) | Yes | 70 |
| 6 | TiO2 (7 wt%) | Yes | 17 |
| 7 | None | Yes | 14 |
Intriguingly, in the absence of either US (Table 1, entry 4) or PNP (Table 1, entry 7), no visual change to the reaction mixture was observed and little to no conversion was measured by 1H NMR spectroscopy. Similar results (Fig. S2†) were obtained when a neutral non-PNP (TiO2) was used (Table 1, entry 6). Based on these results, we surmised that solvent played a key role in the background processes (Table 1, entries 6 & 7) observed. While the exact nature of all initiating species is not fully clear, these collective data demonstrate the importance of iodonium salt for efficient polymerization.
We postulated that the trivial conversion (ca. 15%) measured in the absence of PNPs could be attributed to solvent initiated polymerization (Fig. S3 and S4†). Organic solvent radicals are known to form in response to high frequency US (ca. 500 kHz); subsequent homolytic C–C, C–N, or C–H bond cleavage forms species that can initiate radical polymerization.63–67 Unfortunately, the role of solvent radicals in background reactions are often overlooked in recent mechanoredox polymerization reports. To examine whether low frequency (40 kHz) US-mediated solvent radical generation can also induce significant polymerization, control experiments were conducted where tBA was sonicated in different organic solvents without PNP and DPIHP. A maximum monomer conversion of 15% was observed over 20 h (Table S2†), suggesting that although certain solvents can generate radicals in response to low frequency US (40 kHz), the local concentration of active initiator is not sufficient to achieve high monomer conversion.
While the importance of PNPs is already established (Table 2, entry 1), additional control experiments were necessary to further probe the polymerization mechanism. When the DPIHP initiator was removed from the reaction mixture, less than 15% monomer conversion was observed in 20 h (Table 2, entry 2). The trivial monomer conversion can be attributed to US-mediated solvent radical polymerization (vide supra) and confirms the significant role the diaryliodonium salt plays in the observed US-mechanoredox FRPs. This result was corroborated with a simple kinetics experiment; tBA and BaTiO3 were sonicated in DMF without DPIHP for 8 h and ∼10% conversion was observed by 1H NMR spectroscopy. Upon addition of DPIHP to this reaction mixture, >90% monomer conversion was observed after an additional 12 h (20 h total reaction time) (Fig. S6†). Additionally, when polymerizations were run without exclusion of air (Table 2, entry 3) or with 1 equiv. of radical inhibitor 4-methoxyphenol (MEHQ) (Table 2, entry 4), <5% conversion was observed, supporting the envisaged free-radical mechanism.
| Entry | Conditions | Conversionc (%) |
|---|---|---|
|
a Reaction conditions: [monomer]0:[DPIHP]0 = 100:1.
b [tBA] = 7.3 M, [DPIHP] = 0.073 M in DMF, 7 wt% BaTiO3.
c Conversion was determined by 1H NMR spectroscopy. Ultrasonic bath (40 kHz, 70 W, 20 °C); reaction time: 20 h.
|
||
| 1b | Standard reaction | 92 |
| 2 | Without DPIHP | 14 |
| 3 | Under air | <5 |
| 4 | MEHQ (1:1 eq. to DPIHP) |
<5 |
To further optimize reaction conditions, we then evaluated solvent scope in the mechanoredox polymerization of tBA (Table S3†). In general, >30% monomer conversion was observed in polar aprotic organic solvents (i.e., DMF, DMAc, DMSO, 1,4-dioxane) but no conversion was observed in non-polar solvents (i.e., toluene, anisole). These differences are likely in part due to poor DPIHP solubility in less polar solvents. In all cases, <5% conversion was observed in the absence of US (Table S4†). Additionally, when US-mechanoredox FRP was carried out in anhydrous solvent, no significant difference in monomer conversion was observed, confirming minimal radical generation from water.67–69 Overall, higher monomer conversion was achieved using BaTiO3 than with ZnO. Based upon these cumulative results (Table S3†), BaTiO3/DMF and ZnO/DMAc were used for the remainder of the studies reported below.
Next, to study the viability of the optimized conditions with other monomers, the mechanoredox polymerizations of butyl acrylate (BA), ethyl acrylate (EA), methyl acrylate (MA), and methyl methacrylate (MMA) were studied. The resulting monomer conversions (Table 3 and Fig. 3A) reveal consistently higher conversions of BaTiO3 reactions over ZnO reactions (Table 3 and Fig. 3B) and acrylates over methacrylates. The number average molecular weights (Mn), and dispersity (Đ = Mw/Mn) data measured by gel permeation chromatography coupled with a multi-angle light scattering detector (GPC-MALS) reveals high molecular weight (>100 kDa) polymer with little control over dispersity as is expected from a conventional mechanoredox FRP process (Fig. 2). Again, no polymerization was observed when reactions were carried out in the absence of US (Table S5†).
| Entry | Monomer | Nanoparticle | Conversione (%) | M n (kDa) | Đ |
|---|---|---|---|---|---|
|
a Reaction conditions: [monomer]0:[DPIHP]0 = 100:1.
b [monomer] = 7.3 M, [DPIHP] = 0.073 M.
c [monomer] = 9.3 M, [DPIHP] = 0.093 M.
d [monomer] = 10.9 M, [DPIHP] = 0.109 M. DMF and DMAc were used as solvents for mechanoredox reactions with BaTiO3 and ZnO, respectively.
e Conversion was determined by 1H NMR spectroscopy.
f
M
n and Đ were determined by GPC-MALS. 7 wt% nanoparticle loading was used for all reactions. Ultrasonic bath (40 kHz, 70 W). Reaction time: 20 h.
|
|||||
| 1b | tBA | BaTiO3 | 92 | 284 | 1.7 |
| 2b | tBA | ZnO | 68 | 347 | 1.6 |
| 3b | BA | BaTiO3 | 82 | 431 | 1.8 |
| 4b | BA | ZnO | 56 | 358 | 1.7 |
| 5c | EA | BaTiO3 | 64 | 491 | 1.5 |
| 6c | EA | ZnO | 51 | 533 | 1.5 |
| 7d | MA | BaTiO3 | 78 | 1230 | 1.8 |
| 8d | MA | ZnO | 32 | 357 | 2.0 |
| 9c | MMA | BaTiO3 | 38 | 105 | 1.7 |
| 10c | MMA | ZnO | 35 | 107 | 1.5 |
 | ||
| Fig. 2 GPC-RI traces of US-mechanoredox (meth)acrylate FRP using (A) BaTiO3 or (B) ZnO as the PNP (see Table 3). | ||
Finally, tBA mechanoredox polymerization kinetics were studied, and the resulting data were analysed by 1H NMR spectroscopy and GPC-MALS. Within the 20 h reaction window, a time dependent progression of polymer formation was observed. Polymerizations were faster with BaTiO3 than with ZnO at all analysed time points. In both cases, high molecular weight polymer was observed from GPC-MALS traces, indicating fast propagation rates relative to initiation rates. To study whether any decrease in Mn over time (Fig. 3) was due to mechanochemical polymer cleavage,70 US-mediated chain scission experiments were carried out on DMF solutions of freshly synthesized poly(tBA) and poly(MMA). After sonication for 24 h, analysis of the resulting materials by GPC-MALS (Fig. S7†) indicated that mechanochemical chain scission is operative at extended reaction times. For example, 170 kDa poly(tBA) synthesized through US-mechanoredox FRP was reduced to 21 kDa after prolonged ultrasonication. Hence, we believe that the observed molecular weight evolutions (Fig. 3) likely are complicated through competing propagation and chain scission pathways.
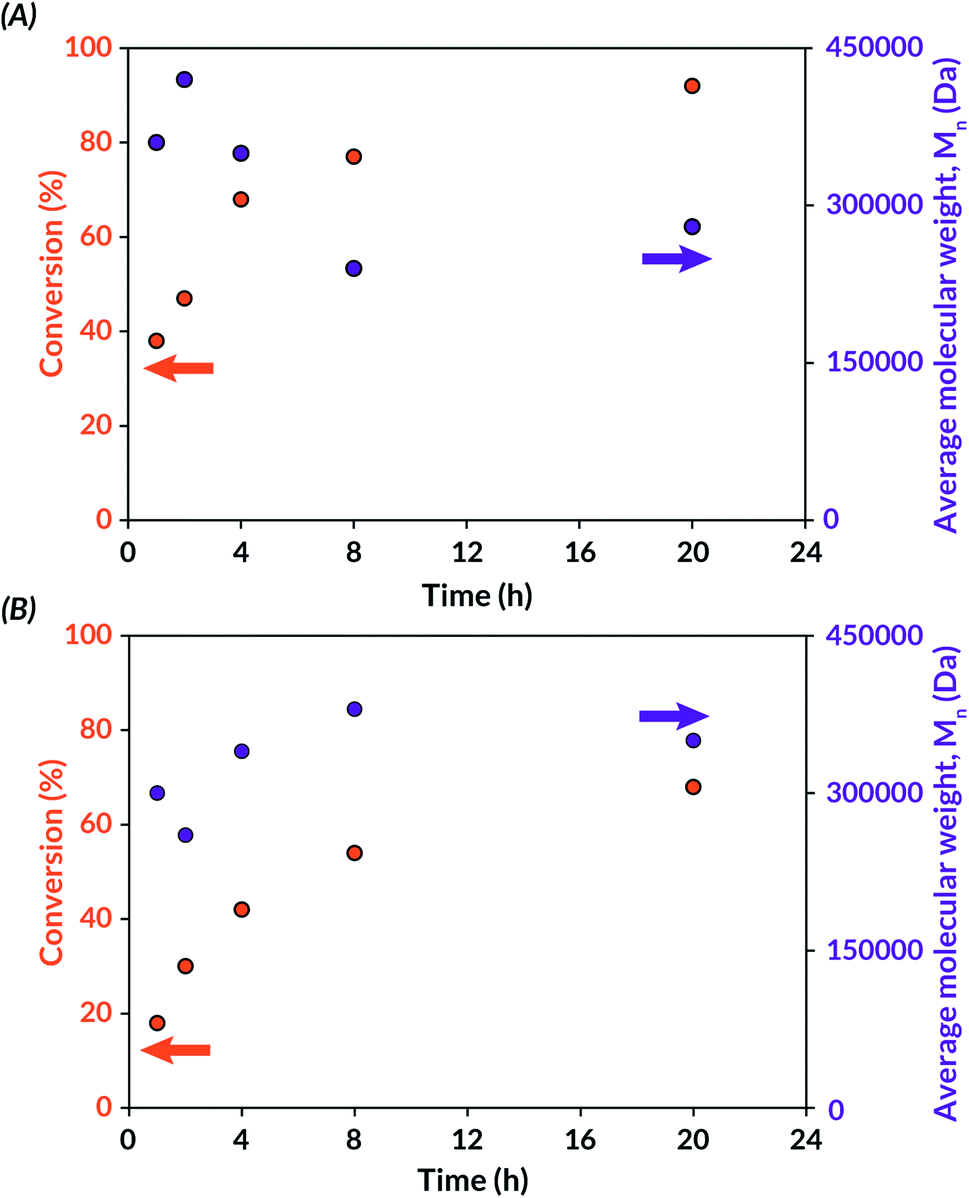 | ||
| Fig. 3 Conversion and molar mass progression during US-mechanoredox tBA polymerizations under optimized conditions (see Table 3): (A) with BaTiO3 (7 wt%) in DMF; (B) with ZnO (7 wt%) in DMAc. | ||
Additionally, an “on–off” experiment was conducted to assess the role of US on the FRP reaction profile (Fig. S8†). As assessed by 1H NMR spectroscopy, tBA conversion reached 35% after 1 h of US and remained stagnant (<5% further conversion) during the first “off” period of 2 h. During the second 2 h “on” period, monomer conversion increased to 57%. Interestingly, after a longer “off” period (15 h), monomer conversion increased to 73%. These data collectively suggest that while FRP rates are higher in the presence of US, likely due to continuous mechanoredox generation of initiating radicals and/or thermal cavitation effects under US irradiation, propagating chain ends still remain active even in the absence of US.
Ball-milling (BM) mechanoredox polymerizations
As the diaryl iodonium chemistry evolved in solution using ultrasound, we were intrigued to find out if mechanoredox FRP could transition into the ball mill. The advantages of ball-milling, including reducing solvent usage, mitigating reagent insolubility,71 and facilitating the use of incompatible and/or immiscible reagents,72 are apparent from prior BM step-growth,73 ring-opening,74 and iterative75 polymerizations. Although poly(meth)acrylates have been accessed under ball-milling conditions via mechanochemical radical generation on quartz surfaces76 and a recapitulated solid-state ATRP process,77 the use of a tuneable mechanoredox pathway has remained unrealized under ball milling conditions prior to our work.To begin, we translated our optimized US-mechanoredox FRP conditions for tBA (50:1 tBA:DPIHP, 7 wt% BaTiO3) into the ball mill using minimal DMF (0.12 mL, 0.030% v/w = volume of DMF relative to total mass of all other reaction components) as is required for liquid assisted grinding (LAG).78 LAG is a procedure that enhances mechanochemical reactivity through the addition of small quantities of a solvent.3 Upon initial investigation, >95% monomer conversion was observed by 1H NMR spectroscopy (Fig. S9†) after only 3 h in the ball mill (30 Hz) compared to 20 h in solution with US; the resulting material in the stainless steel milling jar was a visibly viscous material (Fig. S10†) with a high molecular weight (Mn = 165 kDa) as determined by GPC-MALS. Unlike the US-mediated reactions, ball-milling was tolerant of oxygen and rigorous exclusion of air was not required. Our observations are in stark contrast to Bielawski's recent work on solid-state ATRP77 where polymerization only occurred in an inert atmosphere. Other common acrylates also showed high conversions (Table 4) under our BM-mechanoredox FRP conditions, but importantly, monomer conversion was seen only when samples were subjected to ball-milling. As with the US processes, both onium salt and PNP were required; when DPIHP and/or BaTiO3 were removed from the reaction mixture, no monomer conversion was observed (Table S6†). Similarly, when the PNP was replaced with TiO2, no conversion was seen after ball-milling for 3 h (Table S7†).
|
|
||||
|---|---|---|---|---|
| Entry | Monomer | Conversionb (%) | M n (kDa) | Đ |
| a Reaction conditions: monomer = 2.0 mmol, DPIHP = 0.040 mmol, BaTiO3 = 0.60 mmol, DMF (for LAG) = 0.12 mL (0.030% v/w). b Conversion was determined by 1H NMR spectroscopy. c M n and Đ were determined by GPC-MALS. Ball mill (1.5 mL stainless steel jar, 5 mm stainless steel grinding ball, 30 Hz). Reaction time: 3 h. | ||||
| 1 | tBA | >95 | 416 | 1.3 |
| 2 | BA | 90 | 556 | 1.6 |
| 3 | EA | >95 | 751 | 1.4 |
| 4 | MA | >95 | 937 | 1.2 |
| 5 | MMA | 86 | 56.0 | 1.7 |
We then investigated the kinetics of BM-mechanoredox FRP due to their expedited rates compared to our US-mediated FRP. Interestingly, these studies revealed that there is an incubation period; no conversion is observed prior to 140 minutes (Fig. 4). Since these reactions are conducted under air in sealed vessels, we hypothesized that the limited oxygen would eventually be removed from the atmosphere, potentially through reduction under mechanoredox conditions. Once the atmosphere is “scrubbed” of oxygen, nearly quantitative monomer conversion is quickly observed after just an additional ca. 10 min. To test this hypothesis, we set up a tBA BM-mechanoredox FRP reaction under an inert N2 atmosphere in a glovebox and observed that FRP proceeded without a noticeable incubation period (Table S8†). 14% conversion was observed after just 10 minutes while nearly 70% conversion was observed after 90 minutes. Oxygen seems to perturb the onset of monomer conversion but does not significantly affect the overall time to full conversion (ca. 150 min). We surmise that mixing is the rate limiting factor under inert conditions; in the presence of oxygen, sufficient mixing becomes competitive with atmosphere scrubbing so monomer consumption appears to be nearly instantaneous once all oxygen is removed from the system.
 | ||
| Fig. 4 Comparison of BM-mechanoredox kinetics (tBA conversion) in an inert atmosphere and under air. | ||
The overall physical data collected from GPC-MALS analysis (Table 4 and Fig. S11†) of BM-mechanoredox FRP polymers show similar trends to what was measured for US-mechanoredox FRP. Importantly, the similarities in Mn and Đ between the two methods suggest that ball-milling leads to uniform rate enhancements (i.e. rates of initiation, propagation, and termination) relative to US; drastic disparities in Mn and/or Đ would suggest non-uniform rate enhancements in the ball mill. Under an inert atmosphere, >90% monomer consumption is achieved in under 3 h via ball-milling conditions, while almost a full day (20 h) reaction is needed under ultrasonic irradiation conditions. Hence, solvating conditions (i.e. US-mechanoredox FRP) slow the overall rate of monomer consumption compared to that of BM-mechanoredox FRP, but do not significantly alter the makeup of the final poly(meth)acrylates. Interestingly, resubjection of BM-mechanoredox polymers (e.g., poly(tBA), poly(MMA)) to the original reaction conditions (0.030% v/w DMF, ball-milling at 30 Hz, 3 h) led to only a small decrease in molar mass as assessed by GPC-MALS (Fig. S12 and S13†). Hence, while mechanochemical chain scission pathways were operative under ultrasonication conditions, they were less prevalent, at least on this shorter time scale, under ball-milling conditions.
Based on work from the ultrasonic irradiation reactions, we hypothesized that PNP identity may also influence polymerization efficiency. Upon testing this hypothesis, we determined that the difference in reactivity between the two PNPs in ball-milling is much more significant than with US. Under otherwise identical conditions (Table 4), no conversion was observed when BaTiO3 was replaced with ZnO (Table S7†). These results parallel the trends observed in C–H borylation and arylation reactions studied by Kubota and Ito;47 the specific mechanistic underpinnings behind such a stark reactivity difference is unclear at this time.
Conclusions
In summary, we developed mechanoredox methodology for the synthesis of poly(meth)acrylates under complementary reaction conditions. The fields of self-healing and strain-strengthening materials will undoubtedly benefit from fundamental processes that can forge chemical bonds in response to mechanical inputs. In fact, several examples of mechanoredox polymer crosslinking79–81 already exist, providing compelling arguments for accessing thermoset materials that may be inaccessible under thermal conditions. Furthermore, as photons are readily absorbed by chromophores or scattered by insoluble additives,82,83 the ability to spatially focus mechanical energy will allow for the development of advanced, responsive macromolecular networks. The diversity of mechanical inputs provides the opportunity for divergent applications as the field evolves. Additional mechanistic studies are ongoing in our lab to probe experimental differences observed in these mechanoredox FRP studies and the exact identity of initiating species.Author contributions
S. M. Z. and P. C. contributed equally. S. M. Z. and P. C. conducted ultrasonic irradiation mechanoredox polymerizations; S. M. Z. conducted ball-milling mechanoredox polymerizations. All authors analysed and discussed experimental data. S. M. Z., P. C., and M. R. G. wrote the manuscript; all authors discussed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Washington for generous startup funds. This work was supported in part by a seed grant from UW MEM-C, an NSF MRSEC funded under DMR-1719797.Notes and references
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- J. A. Leitch and D. L. Browne, Chem.–Eur. J., 2021, 27, 9721–9726 CrossRef CAS PubMed.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2019, 2–14 Search PubMed.
- R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef.
- J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- M. E. McFadden and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 11388–11392 CrossRef CAS PubMed.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS PubMed.
- C. L. Brown and S. L. Craig, Chem. Sci., 2015, 6, 2158–2165 RSC.
- C. E. Diesendruck, G. I. Peterson, H. J. Kulik, J. A. Kaitz, B. D. Mar, P. A. May, S. R. White, T. J. Martínez, A. J. Boydston and J. S. Moore, Nat. Chem., 2014, 6, 623–628 CrossRef CAS PubMed.
- T. G. Hsu, J. Zhou, H. W. Su, B. R. Schrage, C. J. Ziegler and J. Wang, J. Am. Chem. Soc., 2020, 142, 2100–2104 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 2105–2109 CrossRef CAS PubMed.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, ACS Cent. Sci., 2021, 7, 1216–1224 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189–8192 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, Macromol. Chem. Phys., 2016, 217, 354–364 CrossRef CAS.
- M. A. Ghanem, A. Basu, R. Behrou, N. Boechler, A. J. Boydston, S. L. Craig, Y. Lin, B. E. Lynde, A. Nelson, H. Shen and D. W. Storti, Nat. Rev. Mater., 2021, 6, 84–98 CrossRef CAS.
- Z. Shi, J. Wu, Q. Song, R. Göstl and A. Herrmann, J. Am. Chem. Soc., 2020, 142, 14725–14732 CrossRef CAS PubMed.
- Y. Zhang, J. Yu, H. N. Bomba, Y. Zhu and Z. Gu, Chem. Rev., 2016, 116, 12536–12563 CrossRef CAS PubMed.
- K. S. Suslick, Science, 1990, 247, 1439–1445 CrossRef CAS PubMed.
- G. Cravotto, E. C. Gaudinoa and P. Cintas, Chem. Soc. Rev., 2013, 42, 7521–7534 RSC.
- K. Kooiman, H. J. Vos, M. Versluis and N. De Jong, Adv. Drug Delivery Rev., 2014, 72, 28–48 CrossRef CAS PubMed.
- S. Ibsen, A. Tong, C. Schutt, S. Esener and S. H. Chalasani, Nat. Commun., 2015, 6(6), 8264 CrossRef CAS PubMed.
- Y. Pan, S. Yoon, J. Sun, Z. Huang, C. Lee, M. Allen, Y. Wu, Y. J. Chang, M. Sadelain, K. Kirk Shung, S. Chien and Y. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 992–997 CrossRef CAS PubMed.
- P. N. T. Wells, Phys. Med. Biol., 2006, 51, R83 CrossRef CAS PubMed.
- J. Gonzales, R. K. Nair, S. J. Madsen, T. Krasieva and H. Hirschberg, J. Biomed. Opt., 2016, 21, 078002 CrossRef PubMed.
- I. Lentacker, S. C. De Smedt and N. N. Sanders, Soft Matter, 2009, 5, 2161–2170 RSC.
- S. Dabral, H. Wotruba, J. G. Hernández and C. Bolm, ACS Sustainable Chem. Eng., 2018, 6, 3242–3254 CrossRef CAS.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- J. Ayarza, Z. Wang, J. Wang, C.-W. Huang and A. P. Esser-Kahn, ACS Macro Lett., 2020, 1237–1248 CrossRef CAS.
- S. Tu, Y. Guo, Y. Zhang, C. Hu, T. Zhang, T. Ma and H. Huang, Adv. Funct. Mater., 2020, 30, 1–31 Search PubMed.
- M. B. Starr and X. Wang, Nano Energy, 2015, 14, 296–311 CrossRef CAS.
- M. Wang, B. Wang, F. Huang and Z. Lin, Angew. Chem., Int. Ed., 2019, 58, 7526–7536 CrossRef CAS PubMed.
- A. Cafarelli, A. Marino, L. Vannozzi, J. Puigmartí-Luis, S. Pané, G. Ciofani and L. Ricotti, ACS Nano, 2021, 15, 11066–11086 CrossRef CAS PubMed.
- P. Christen, K. Ito, R. Ellouz, S. Boutroy, E. Sornay-Rendu, R. D. Chapurlat and B. Van Rietbergen, Nat. Commun., 2014, 5, 4855 CrossRef CAS PubMed.
- R. J. Nims, L. Pferdehirt, N. B. Ho, A. Savadipour, J. Lorentz, S. Sohi, J. Kassab, A. K. Ross, C. J. O'conor, W. B. Liedtke, B. Zhang, A. L. Mcnulty and F. Guilak, Sci. Adv., 2021, 7, 9858–9885 CrossRef PubMed.
- S. Li, Z. Zhao, J. Zhao, Z. Zhang, X. Li and J. Zhang, ACS Appl. Nano Mater., 2020, 3, 1063–1079 CrossRef CAS.
- M. B. Starr, J. Shi and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 5962–5966 CrossRef CAS PubMed.
- Z. Liang, C. F. Yan, S. Rtimi and J. Bandara, Appl. Catal., B, 2019, 241, 256–269 CrossRef CAS.
- J. M. Wu, Y.-G. Sun, W.-E. Chang and J.-T. Lee, Nano Energy, 2018, 46, 372–382 CrossRef CAS.
- R. Caliò, U. Rongala, D. Camboni, M. Milazzo, C. Stefanini, G. de Petris and C. Oddo, Sensors, 2014, 14, 4755–4790 CrossRef PubMed.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- Z. Wang, J. Ayarza and A. P. Esser-Kahn, Angew. Chem., Int. Ed., 2019, 58, 12023–12026 CrossRef CAS PubMed.
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS.
- Z. Wang, X. Pan, J. Yan, S. Dadashi-Silab, G. Xie, J. Zhang, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2017, 546–549 CrossRef CAS.
- K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS PubMed.
- Y. Pang, J. W. Lee, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 22570–22576 CrossRef CAS PubMed.
- C. Schumacher, J. G. Hernández and C. Bolm, Angew. Chem., Int. Ed., 2020, 59, 16357–16360 CrossRef CAS PubMed.
- N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- P. P. Romańczyk and S. S. Kurek, Electrochim. Acta, 2020, 136404 CrossRef.
- M. M. Chehimi, A. Lamouri, M. Picot and J. Pinson, J. Mater. Chem. C, 2014, 2, 356–363 RSC.
- R. Steeno, M. C. Rodríguez González, S. Eyley, W. Thielemans, K. S. Mali and S. De Feyter, Chem. Mater., 2020, 32, 5246–5255 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 1–9 Search PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed.
- B. M. Teo, M. Ashokkumar and F. Grieser, J. Phys. Chem. B, 2008, 112, 5265–5267 CrossRef CAS PubMed.
- B. M. Teo, F. Grieser and M. Ashokkumar, Macromolecules, 2009, 42, 4479–4483 CrossRef CAS.
- T. G. McKenzie, F. Karimi, M. Ashokkumar and G. G. Qiao, Chem.–Eur. J., 2019, 1–18 Search PubMed.
- J. Collins, T. G. McKenzie, M. D. Nothling, S. Allison-Logan, M. Ashokkumar and G. G. Qiao, Macromolecules, 2018, 52, 185–195 CrossRef.
- J. Collins, T. G. McKenzie, M. D. Nothling, M. Ashokkumar and G. G. Qiao, Polym. Chem., 2018, 9, 2562–2568 RSC.
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 CrossRef CAS PubMed.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski, J. Yan, A. E. Enciso, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2018, 275–280 CrossRef.
- J. Collins, T. G. McKenzie, M. D. Nothling, S. Allison-Logan, M. Ashokkumar and G. G. Qiao, Macromolecules, 2019, 52, 185–195 CrossRef CAS.
- J. B. Ravnsbæk and T. M. Swager, ACS Macro Lett., 2014, 3, 305–309 CrossRef.
- G. S. Lee, B. R. Moon, H. Jeong, J. Shin and J. G. Kim, Polym. Chem., 2019, 10, 539–545 RSC.
- S. Grätz and L. Borchardt, RSC Adv., 2016, 6, 64799–64802 RSC.
- N. Ohn, J. Shin, S. S. Kim and J. G. Kim, ChemSusChem, 2017, 10, 3529–3533 CrossRef CAS PubMed.
- O. Maurin, P. Verdié, G. Subra, F. Lamaty, J. Martinez and T. X. Métro, Beilstein J. Org. Chem., 2017, 13, 2087–2093 CrossRef CAS PubMed.
- M. Hasegawa, Y. Akiho and Y. Kanda, J. Appl. Polym. Sci., 1995, 55, 297–304 CrossRef CAS.
- H. Y. Cho and C. W. Bielawski, Angew. Chem., Int. Ed., 2020, 59, 13929–13935 CrossRef CAS PubMed.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC.
- H. Mohapatra, J. Ayarza, E. Sanders, A. M. Scheuermann, P. Griffin and A. P. Esser-Kahn, Angew. Chem., Int. Ed., 2018, 11208–11212 CrossRef CAS PubMed.
- Z. Wang, J. Wang, J. Ayarza, T. Steeves, Z. Hu, S. Manna and A. P. Esser-Kahn, Nat. Mater., 2021, 20, 869–874 CrossRef CAS PubMed.
- J. Ayarza, Z. Wang, J. Wang and A. P. Esser-Kahn, ACS Macro Lett., 2021, 10, 799–804 CrossRef CAS.
- A. Bagheri and J. Jin, ACS Appl. Polym. Mater., 2019, 1, 593–611 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional control experiments, photographs of experimental setups, NMR spectra. See DOI: 10.1039/d2sc00313a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |