
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress and prospects of electrolyte chemistry of calcium batteries
Qianshun
Wei
a,
Liping
Zhang
a,
Xiaohua
Sun
b and
T. Leo
Liu
 *a
*a
aDepartment of Chemistry and Biochemistry, Utah State University, Logan, UT, USA. E-mail: Leo.Liu@usu.edu
bCollege of Materials and Chemical Engineering, China Three Gorges University, Yichang, 443002, China
First published on 20th April 2022
Abstract
The increasing energy storage demand of portable devices, electric vehicles, and scalable energy storage has been driving extensive research for more affordable, more energy dense battery technologies than Li ion batteries. The alkaline earth metal, calcium (Ca), has been considered an attractive anode material to develop the next generation of rechargeable batteries. Herein, the chemical designs, electrochemical performance, and solution and interfacial chemistry of Ca2+ electrolytes are comprehensively reviewed and discussed. In addition, a few recommendations are presented to guide the development and evaluation of Ca2+ electrolytes in future.
1. Introduction
The last few decades have witnessed the tremendous economic and societal impacts of rechargeable batteries, especially Li ion batteries, with their increasing applications in portable devices and electric vehicles.1,2 However, the Li ion battery technology has already reached its maximal storage capability in terms of energy density (around 400 W h kg−1 theoretical energy density) and will not meet escalating energy storage demands in future.1,2 In addition, battery technologies will play essential roles in scalable energy storage to achieve “carbon neutrality”.3 Because of the limited abundance of lithium in the Earth's crust (<0.1 pg kg−1), there is a strong concern that there might not be sufficient Li resources for grid energy storage and electric vehicles.4,5 To move beyond Li ion batteries, abundant elemental alkali and alkaline earth metals including Na, K, Mg, and Ca have been considered as attractive anode materials to develop the next generation of rechargeable batteries.4–8 Multivalent Mg batteries have received increasing research attention in the last two decades. The electrolyte research of Mg batteries has been most fertile, including versatile designs of Mg–Cl complex electrolytes and advanced Cl-free Mg electrolytes and the in-depth understanding of electrolyte solution and interfacial chemistry.7,9 However, due to the strong Lewis acidity of Mg2+ ions expressed by ion electronegativity (47.6 eV, Fig. 1),10 developing Mg2+ ion intercalation cathode materials remains very challenging and significantly hampers the development of Mg batteries for practical applications.11 So far, Chevrel phase Mo6S8 has been the only cathode material that facilitates facile and stable Mg ion intercalation at room temperature.11,12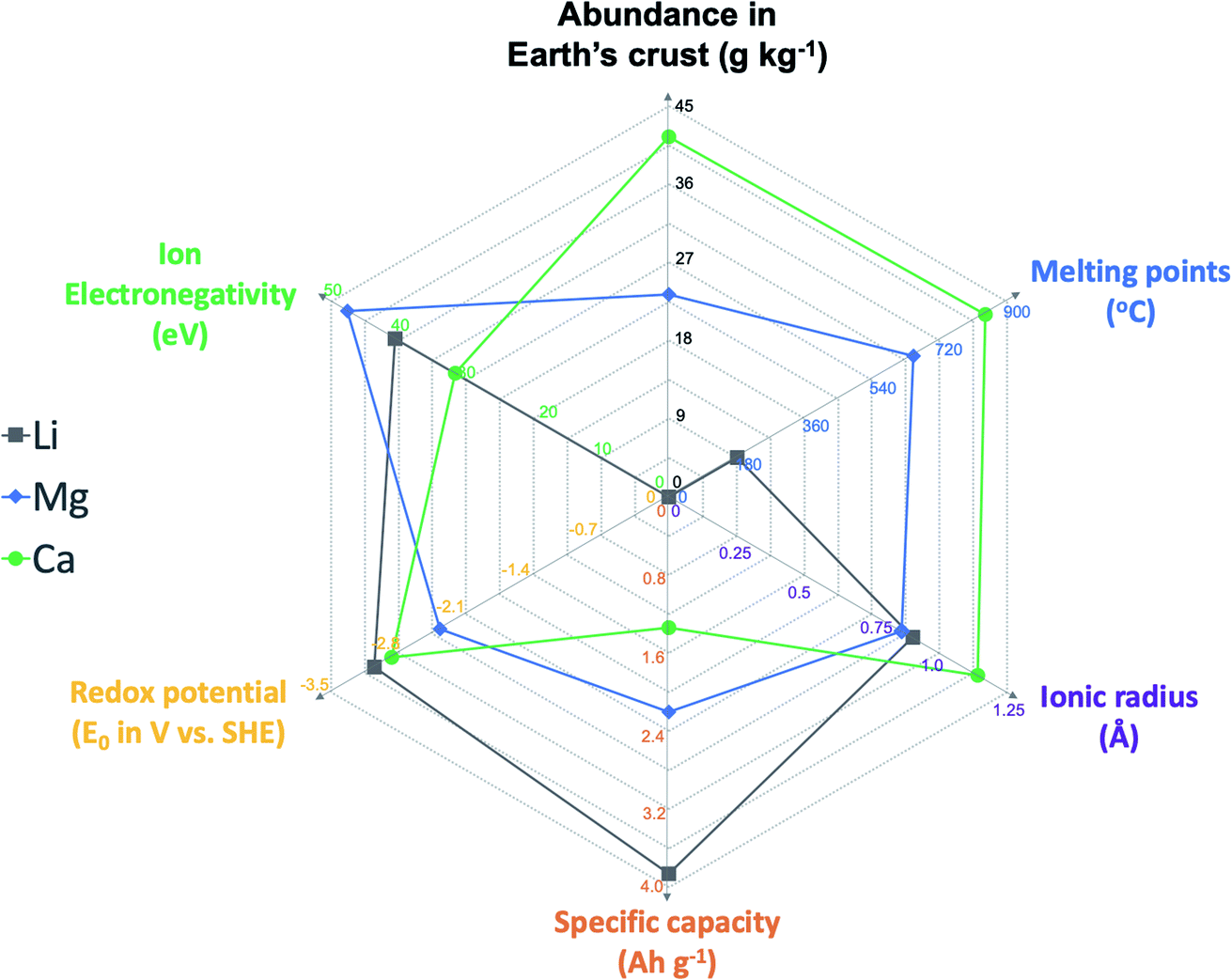 | ||
| Fig. 1 Physical properties of Li, Mg, and Ca elements, including abundance in the Earth's crust (g kg−1), melting point (°C), ionic radius (Å), specific capacity (A h g−1), redox potential (V vs. SHE), and ion electronegativity (eV). | ||
More recently, the research wave of multivalent batteries has also aroused interest in calcium rechargeable batteries because of the attractive material properties of the Ca anode, including the high abundance (41.5 g kg−1), low redox potential (−2.87 V vs. SHE), high capacity benefitting from two electron storage (1.34 A h kg−1), and high melting point (842 °C) for safe operations (Fig. 1).13–15 Compared to the Mg2+ ion, the Ca2+ ion with its lower Lewis acidity (31.6 eV) is proposed to feasibly operate with existing insertion cathode materials.9,13,16 Ca electrolytes are pivotal to the operation of Ca batteries as they enable reversible plating and stripping of Ca2+ ions and shuttle the ions between the cathode and the Ca anode. Early studies on high-temperature, solid state Ca batteries (e.g., Ca‖KCl–LiCl–AgCl–K2CrO4‖Ag cells) date back to the 1970s.15,17 In the past several years, there has been renewed interest in developing stable Ca2+ electrolytes for reversible Ca deposition to enable room temperature rechargeable Ca batteries.13 In this review, the chemical composition, physicochemical properties, and interfacial chemistry of Ca electrolytes, as well as their correlation with electrochemical performance, are reviewed and discussed. A few suggestions are also provided to guide the development and evaluation of Ca2+ electrolytes in future. We hope that this review will promote the development of advanced calcium electrolytes and their applications in developing energy dense, long cycling rechargeable Ca batteries.
2. Early attempts using traditional calcium salt electrolytes for Ca deposition
Early studies on liquid calcium electrolytes were motivated by the development of safer Ca‖SOCl2 primary batteries than Li‖SOCl2 primary batteries, which dates back to the 1980s.18–20 Ca‖SOCl2 batteries have a cell voltage of 3.64 V and an energy density of 1230 mW h g−1 that are comparable to those of Li‖SOCl2 batteries, 3.65 V and 1474 mW h g−1. Stankiewicz proposed that the higher melting point (839 °C) of calcium metal than that of lithium (180 °C) would obviate the safety concern of anode melting, which could be caused by short-circuit currents.18 In this early study, a Ca(AlCl4)2-SOCl2 electrolyte with a concentration of up to 2.0 M was prepared by combining CaCl2 and AlCl3 in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in an inorganic solvent, SOCl2. The Ca(AlCl4)2-SOCl2 electrolyte was evaluated for the corrosion of the Ca anode. It was confirmed that Ca metal corrosion with SOCl2 followed the battery reaction and led to the formation of CaCl2 on the metal surface (Fig. 2), which was characterized by XRD and IR spectroscopy. Gaseous SO2 and sulfur (S) were proposed to be other byproducts. It was noted that the corrosion rate in the SOCl2 solvent without Ca(AlCl4)2 was much slower. We believe that the solvation of SOCl2 with Ca2+ can promote the reduction of the solvent by the Ca anode. Because of the solvent corrosion, it was observed that the capacity of Ca‖SOCl2 batteries decreased to 50% after ca. one month of storage. The author reminded that excess AlCl3 and the presence of water in the electrolyte could cause additional corrosion reactions of Ca metal. The author also prepared two other electrolytes, Ca(FeCl4)2-SOCl2 and Ca(SbCl6)2-SOCl2, and found that these two electrolytes underwent more rapid corrosion with Ca.
:2 in an inorganic solvent, SOCl2. The Ca(AlCl4)2-SOCl2 electrolyte was evaluated for the corrosion of the Ca anode. It was confirmed that Ca metal corrosion with SOCl2 followed the battery reaction and led to the formation of CaCl2 on the metal surface (Fig. 2), which was characterized by XRD and IR spectroscopy. Gaseous SO2 and sulfur (S) were proposed to be other byproducts. It was noted that the corrosion rate in the SOCl2 solvent without Ca(AlCl4)2 was much slower. We believe that the solvation of SOCl2 with Ca2+ can promote the reduction of the solvent by the Ca anode. Because of the solvent corrosion, it was observed that the capacity of Ca‖SOCl2 batteries decreased to 50% after ca. one month of storage. The author reminded that excess AlCl3 and the presence of water in the electrolyte could cause additional corrosion reactions of Ca metal. The author also prepared two other electrolytes, Ca(FeCl4)2-SOCl2 and Ca(SbCl6)2-SOCl2, and found that these two electrolytes underwent more rapid corrosion with Ca.
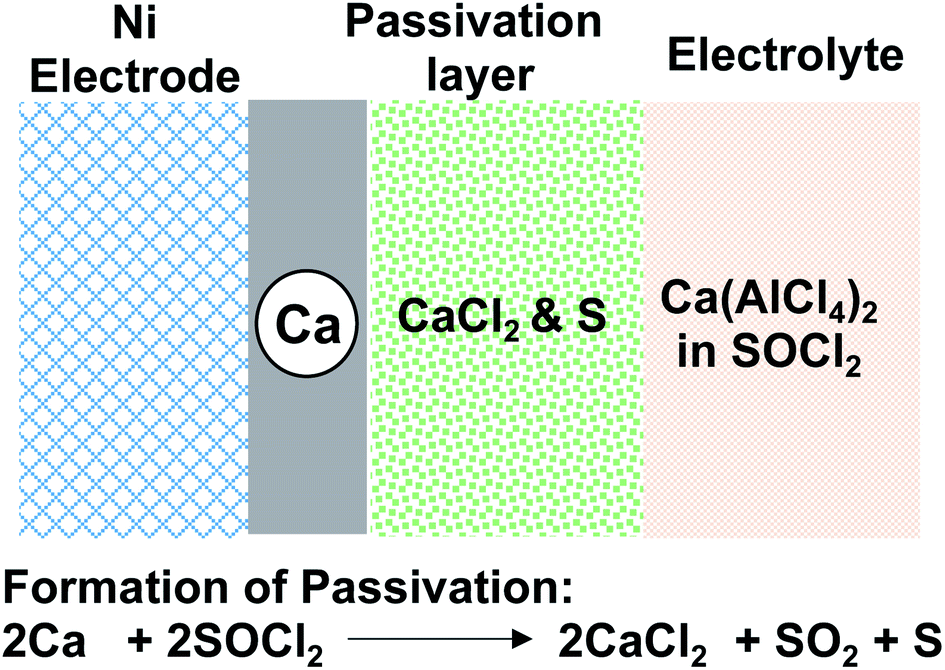 | ||
| Fig. 2 Model for the formation of CaCl2 passivation of the Ca metal in the Ca(AlCl4)2-SOCl2 electrolyte. | ||
In Staniericz's work, there was little evidence of Ca deposition through the CaCl2 layer (Fig. 2). Stankiewicz proposed that CaCl2 functions as a solid electrolyte interphase (SEI) layer where the Cl− ion is a charge carrier through defect sites and leaves Ca2+ for deposition at a very negative potential (−10 V vs. Ca at 1.24 mA cm−2). Peled and co-workers also investigated Ca deposition using Ca(AlCl4)2-SOCl2 electrolytes.19–21 However, their results also revealed CaCl2 as a passivation layer (<100 Å) with poor Ca2+ conductivity (Fig. 2). In their studies, they also observed that the electrolysis of the CaCl2 passivation layer only occurred at a large overpotential. They concluded that Ca‖SOCl2 batteries could not be charged, and such characteristics could be considered an additional safety advantage over Li‖SOCl2 batteries. They also found that the electrolyte conductivities and the battery capacities were affected by both the concentration of CaCl2 and temperature. At 60 °C, a Ca‖SOCl2 battery using an electrolyte consisting of 2.8 M AlCl3 and 1.2 M CaCl2 could be discharged at 50 mA cm−2 and deliver a capacity of 12 mA h cm−2 with a cutoff voltage of 1.5 V.
In 1991, Aurbuch and co-workers examined the electrochemistry of the Ca electrode in a variety of combinations of supporting electrolytes (Ca(ClO4)2, Ca(BF4)2, TBA(ClO4), and TBA(BF4); TBA stands for tetrabutylammonium) and solvents (acetonitrile, γ-butyrolactone (BL), THF, and propylene carbonate (PC)).22 It is noted that not too much information was disclosed for Ca(BF4)2. The dissolution current and morphology of the Ca anode is strongly dependent on the supporting electrolytes, and solvents contribute to the formation of different passivation films. Based on X-ray spectroscopic studies, it is found that ClO4− is much more prone to cathodic reduction than BF4− and presumably, leads to the formation of CaCl2. As a result, the Ca anode displayed a much higher stripping current in a Ca(ClO4)2-BL electrolyte than a TBA(ClO4)2-BL electrolyte. The reactivity of the Ca anode with the studied solvents is quite complicated and was assessed using FTIR through chemical reactions. Ca metal can react with BL to form ester and carboxylate species, derivatives of butyric acid, and the cyclic β-ketoester. The reduced product of PC by Ca is mainly inorganic CaCO3 which is different from the product with Li metal, ROCO2Li. The reduction products of CH3CN were assigned to the derivatives of CH3C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NLi)CH2-CN or CH3C(Li)N-CH2CN. Ca can reduce THF to form alkoxide. This study also reported that the Ca anode was highly reactive even with trace water (20 ppm) in electrolytes resulting in the formation of inorganic Ca(OH)2 and was unstable in air as it reacts with CO2 to form CaCO3. The spectroscopic data on the interfacial chemistry of the Ca anode can be highly valuable for future studies. An important conclusion of this work is that there was no evidence of calcium deposition in all tested electrolyte solutions at room temperature because of the formation of non-conductive passivation films (Table 1), highlighting the challenge of developing reversible Ca deposition/stripping chemistry.
NLi)CH2-CN or CH3C(Li)N-CH2CN. Ca can reduce THF to form alkoxide. This study also reported that the Ca anode was highly reactive even with trace water (20 ppm) in electrolytes resulting in the formation of inorganic Ca(OH)2 and was unstable in air as it reacts with CO2 to form CaCO3. The spectroscopic data on the interfacial chemistry of the Ca anode can be highly valuable for future studies. An important conclusion of this work is that there was no evidence of calcium deposition in all tested electrolyte solutions at room temperature because of the formation of non-conductive passivation films (Table 1), highlighting the challenge of developing reversible Ca deposition/stripping chemistry.
| Electrolytes | Stripping current (mA cm−2) | Species in passivation films |
|---|---|---|
| 0.5 M Ca(ClO4) in BL | 3.2 | CaCl2, ester, and carboxylate |
| TBA(BF4)2 in BL | ca. 50 | Ester and carboxylate |
| TBA(BF4)2 in PC | 5 | CaCO3 |
| TBA(BF4)2 in CH3CN | 5 | Derivatives of CH3C(NLi)CH2-CN or CH3C(Li)N-CH2CN |
| TBA(BF4)2 in THF | 10 | Alkoxide |
3. Renewed interest in traditional calcium salt electrolytes
As discussed above, early studies on traditional simple Ca electrolytes for reversible Ca deposition were not promising.18–22 According to these early studies, the passivation layer of the Ca anode is attributed to the severe electrolyte decomposition by the highly reducing Ca anode and has a very poor conductivity for Ca2+ ions at room temperature. Despite these known challenges, in 2016, Palacín and co-workers revisited the electrochemistry of Ca(BF4)2 and Ca(ClO4)2 along with Ca(TFSI)2 in carbonate solvents.23 The mixture of conventional ethylene carbonate (EC) and propylene carbonate (PC) solvents was selected because EC could form stable SEI layers, and the mixture has a wide liquidus range from −90 to 240 °C and a stable electrochemical operation window up to 4.0 V vs. Ca. They observed quasi-reversible Ca metal plating/stripping of Ca(BF4)2 in EC/PC (50/50 wt%) at 100 °C (Fig. 3a, red curve) using stainless steel (SS) as the working electrode (WE). Calcium deposition using Ca(ClO4)2 (Fig. 3a, black curve) and Ca(TFSI)2 (Fig. 3a, blue curve) was negligible, emphasizing the impact of counter anions on the deposition/stripping efficiency. A 0.45 M Ca(BF4)2 electrolyte exhibited the best deposition/stripping performance at 100 °C with the smallest overpotential (0.10 V) and highest ionic conductivity (Fig. 3b). However, the coulombic efficiency was measured to be only ca. 40% and it suggests undesired side reactions of the electrolyte during the Ca deposition.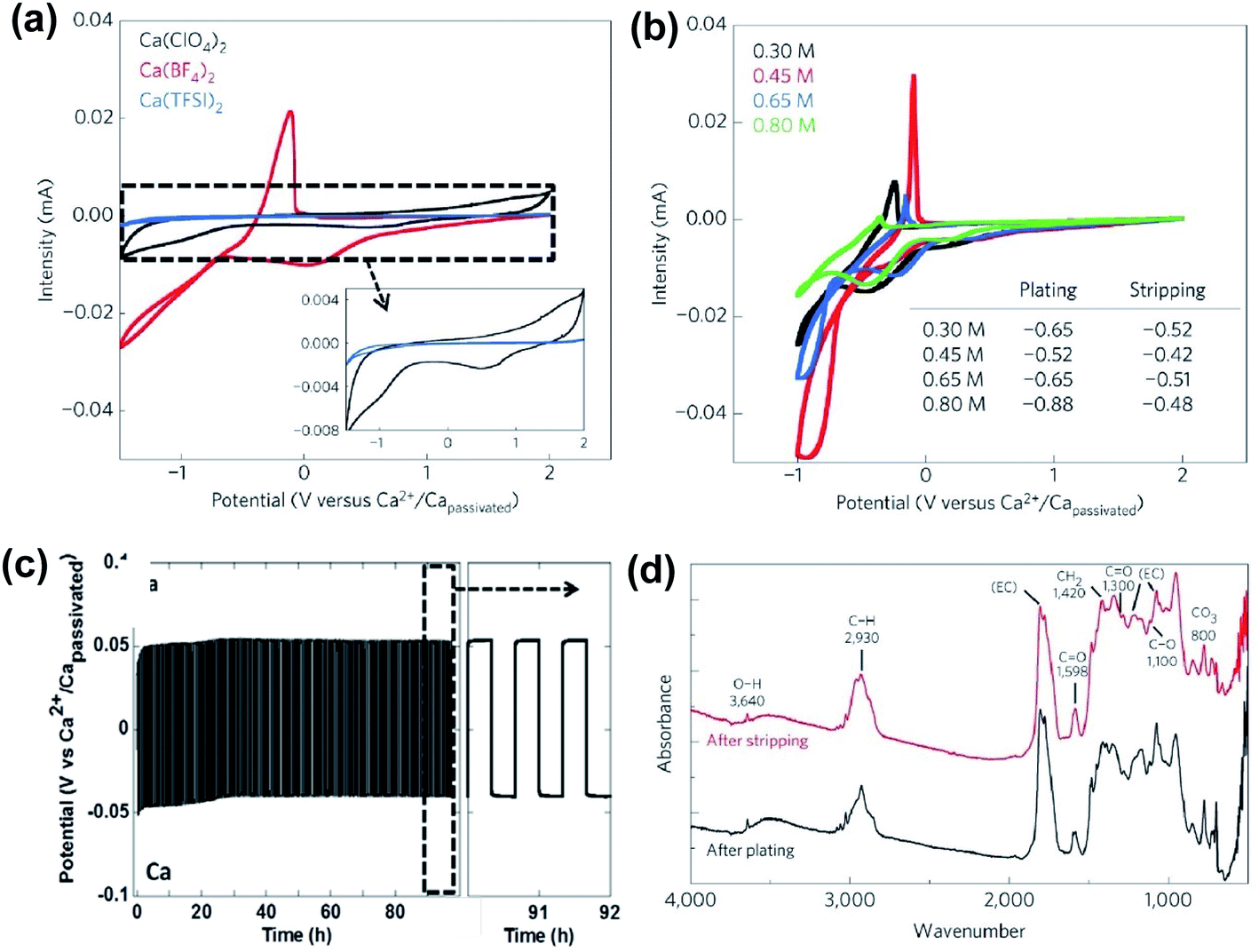 | ||
| Fig. 3 Electrochemical studies of Ca(BF4)2 electrolytes at elevated temperatures. (a) Cyclic voltammograms of 0.3 M Ca(BF4)2, Ca(ClO4)2, and Ca(TFSI)2 in EC:PC at 100 °C; 0.5 mV s−1 scan rate, stainless steel (SS) as the working electrode (WE), and Ca as the reference electrode (RE) and counter electrode (CE); inset: expanded current scale for Ca(BF4)2 and Ca(TFSI)2 electrolytes. (b) Cyclic voltammograms of 0.3 to 0.8 M Ca(BF4)2 electrolytes at 75 °C or 100 °C. (c) Charge/discharge curves (50 μA cm−2, 150 cycles) of a symmetric Ca‖Ca cell with 0.45 M Ca(BF4)2 in EC:PC at 100 °C. (d) Infrared spectra on calcium deposits after plating and stripping. Adapted with permission from ref. 23. Copyright 2016 Springer Nature. | ||
A symmetric Ca‖Ca cell with the 0.45 M Ca(BF4)2 electrolyte was demonstrated for 100 cycles at 100 °C (Fig. 3c). According to scanning electron microscopy (SEM) images, the gray deposits scratched from the copper substrate disks using Ca(BF4)2 were dense and thick. Further synchrotron X-ray diffraction (XRD) and energy-dispersive X-ray analysis (EDX) analysis of the deposit confirmed the major phases of Ca metal and CaF2, as well as their relative molar ratio. Infrared spectra of electrodeposited calcium electrodes after plating (Fig. 3d) indicated a very small amount of hydroxide (3640 cm−1) and some ROCO2− species (CO asymmetric stretching (ca. 1600 cm−1), CH2 bending (1420 cm−1), CO symmetric stretching (ca. 1300 cm−1), C–O stretching (ca. 1100 cm−1), and CO3 bending (ca. 800 cm−1)). The complexity of the SEI components and the low coulombic efficiencies highlight the drastic electrolyte decomposition under the highly reducing conditions of Ca deposition. Additionally, the high operating temperature of the Ca(BF4)2 electrolytes indicates a high energy barrier for the Ca2+ ion transport in the SEI, which is consistent with the poor conductivity of the SEI at room temperature reported in early studies. This work aroused research interest in Ca rechargeable batteries by showing the possibility of Ca deposition using traditional Ca electrolytes.
Subsequently, Hosein and co-workers re-investigated the same Ca(BF4)2-EC/PC electrolyte aiming for Ca deposition at room temperature as Ca deposition at 100 °C is unrealistic for practical applications.24 They observed Ca deposition/stripping using a 1.0 M Ca(BF4)2-EC/PC electrolyte within a wide overpotential window from −1.7 to 2.0 V (Fig. 4a). The large electrochemical window suggests poor electron transfer kinetics at room temperature in comparison with the data reported at 100 °C.23 The coulombic efficiency was found to be up to 95%, likely attributed to reduced side reactions of the electrolyte at room temperature. The deposited Ca was confirmed by SEM and X-ray diffraction (XRD) studies (Fig. 4b and c). According to energy-dispersive X-ray spectroscopy (EDX), XRD, and Fourier-transform infrared spectroscopy (FTIR) studies, the authors proposed that the SEI is mainly composed of organic components derived from the carbonate solvents and lacks a significant amount of CaF2, as observed in the high temperature studies. This work suggests the viability of ionic liquids as electrolyte solvents for Ca batteries.
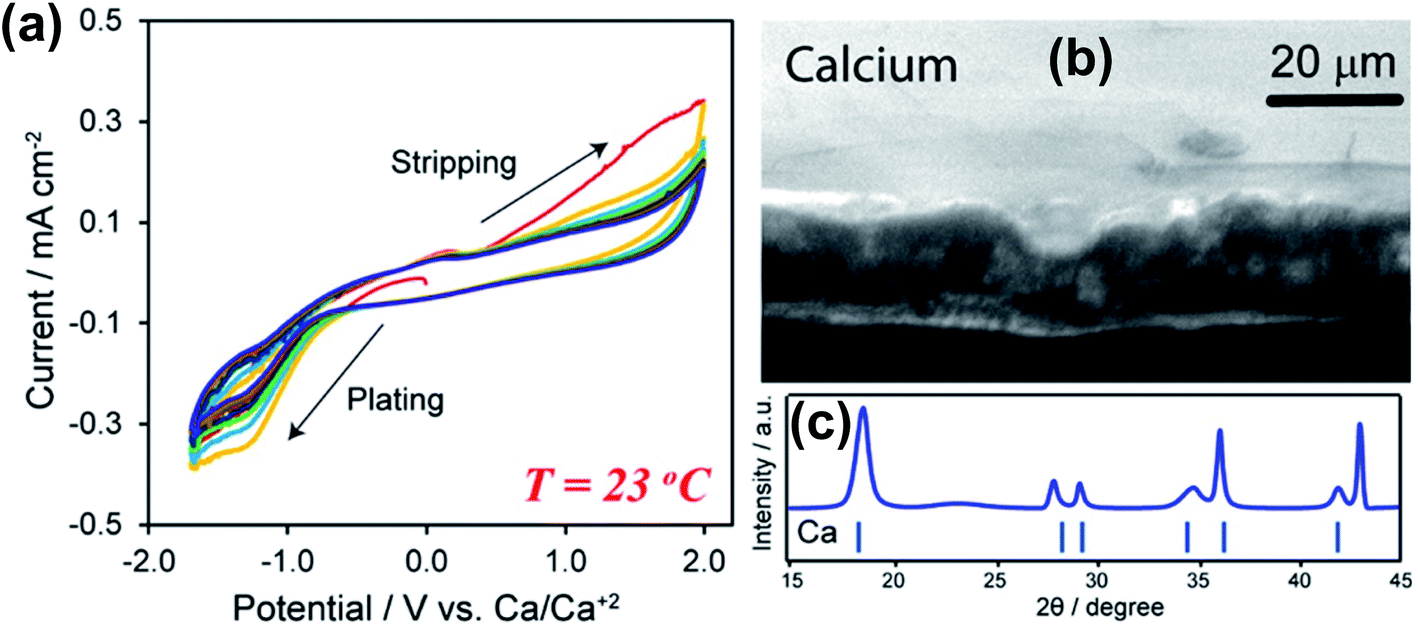 | ||
| Fig. 4 Electrochemical studies of Ca(BF4)2-EC/PC electrolytes at room temperature. (a) Cyclic voltammograms of a 1.0 M Ca(BF4)2 electrolyte. (b) A SEM image of deposited Ca metal. (c) XRD pattern of deposited Ca metal. Adapted with permission from ref. 24. Copyright 2019 American Chemical Society. | ||
In order to unveil the SEI components enabling facile Ca plating, Ponrouch et al. reported detailed experimental studies of the passivation layer of the Ca metal electrode formed in Ca(BF4)2-EC/PC (50:50 wt%) and Ca(TFSI)2-EC/PC electrolytes.25 TEM images visualized that the passivation layers were ca. 12 nm for the Ca(BF4)2 electrolyte (Fig. 5a and c) and ca. 80 nm for the Ca(TFSI)2 electrolyte (Fig. 5b and d). The passivated Ca disks prepared in the Ca(BF4)2 electrolyte seemed to be completely covered by a dark deposit, while the disk passivated in the Ca(TFSI)2 electrolyte remained shiny and visually unaltered (Fig. 5e, insets). XPS spectra of the calcium disk in the Ca(TFSI)2 electrolyte revealed that there were ca. 34% CaCO3, 2% CaF2, and 7% Ca(TFSI)2 at the surface of the Ca dick based on quantitative XPS analysis. However, a much thicker layer was observed for the BF4− anion and a much greater proportion of organic species (C–C, C–H, and CO) at the surface of the Ca(BF4)2 sample (35%) instead of 16% observed for the Ca(TFSI)2 electrolyte. The quantitative analysis revealed ca. 14% boron in an oxygenated environment and ca. 15% CaF2 at the surface of the Ca electrode in the Ca(BF4)2 electrolyte. According to FTIR and electron energy loss spectroscopy (EELS) studies, the authors proposed that the BO3 species is a key component to facilitate Ca2+ transport for the SEI using the Ca(BF4)2 electrolyte.
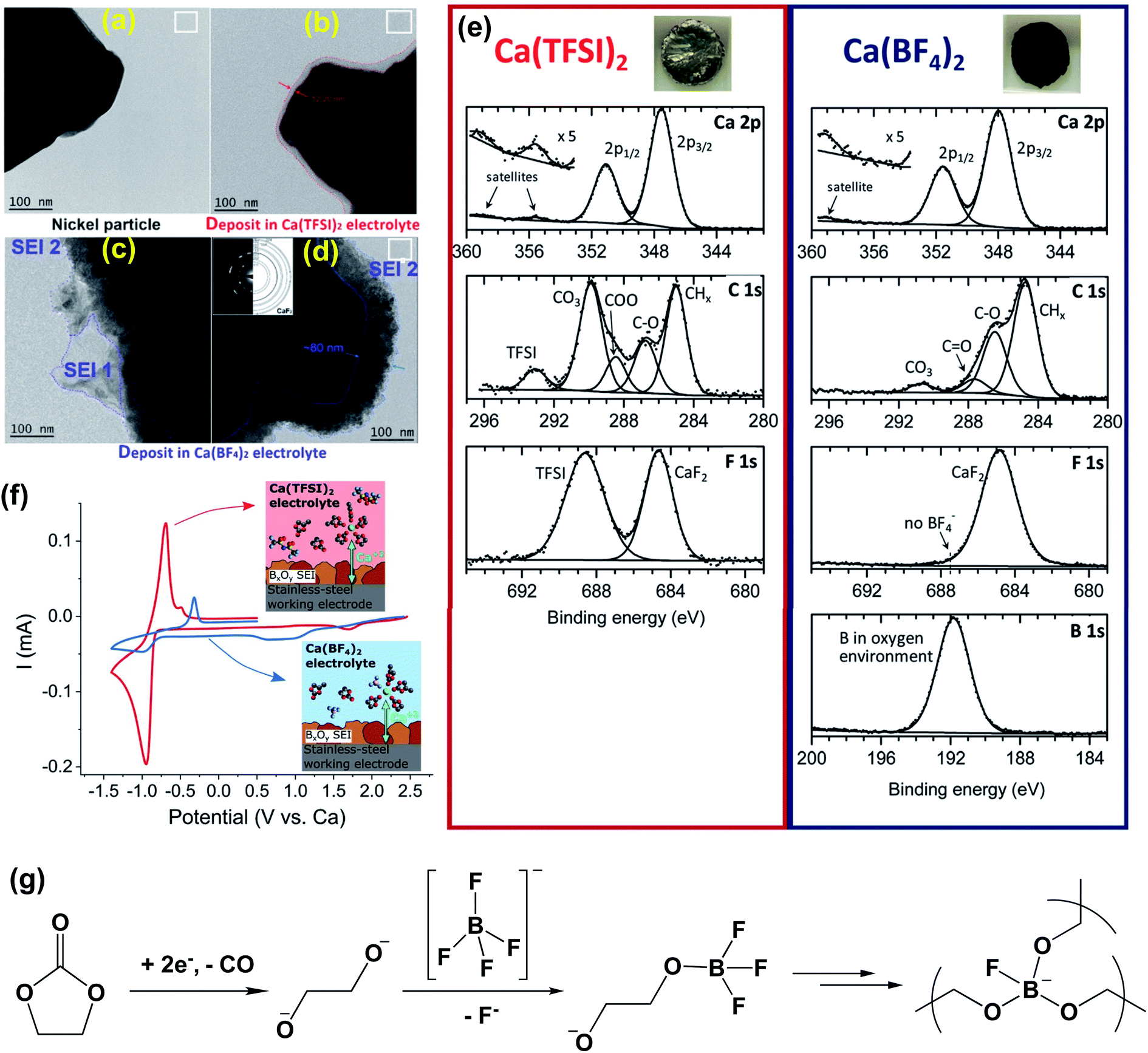 | ||
| Fig. 5 Surface characterization of the Ca anode in Ca(BF4)2-EC/PC (50:50 wt%) and Ca(TFSI)2-EC/PC electrolytes. (a–d) Bright field TEM images of Ni particles (working electrode) before and after the formation of a surface layer. (e) XPS spectra of the passivation layers formed on a calcium disk in the Ca(TFSI)2 electrolyte (left panel) and the Ca(BF4)2 electrolyte (right panel), respectively; insets show the appearance of each calcium disk after negative polarization for 48 hours. (f) Cyclic voltammograms (0.1 mV s−1) of pre-passivated stainless-steel electrodes (covered with borate films) using either Ca(TFSI)2 (red curve) or Ca(BF4)2 (blue curve) electrolytes. Insets show the proposed cation solvation structures in each electrolyte. (g) Proposed decomposition mechanism for the Ca(BF4)2-EC/PC electrolyte to form a proposed BO3 polymer. Adapted with permission from ref. 25. Copyright 2020 Royal Society of Chemistry. | ||
To support the hypothesis, they also demonstrated that the borate-rich passivation layer effectively works as a solid electrolyte interphase (SEI) to enable calcium migration and subsequent calcium plating in the Ca(TFSI)2 electrolyte. After pre-passivation of the stainless steel working electrode by a borate containing passivation layer using the Ca(BF4)2 electrolyte, apparently improved Ca plating/stripping reversibility of the Ca(TFSI)2 electrolyte was observed (Fig. 5f). The proposed mechanism for the formation of the BO3 based polymer SEI is shown in Fig. 5g. Most intermediate products (e.g., alkoxides) were generated by the reduction of the carbonate solvents. The charged-oxygen species is sufficiently nucleophilic to substitute the fluorine ligands of the BF4− anion and can lead to a crosslinked boron polymer upon further reduction. The DFT studies were also performed to compare the migration energy of Ca2+ ions in a number of calcium compounds, including CaH2, CaB6, CaO, CaF2, Ca3(BO3)2, Ca2B2O5, and CaCO3. It was found that CaH2 (541 meV) and CaO (997 meV) have lower barriers for Ca2+ transport.
In 2018, Bruce and co-workers reported the highly reversible Ca plating/stripping of a 1.5 M Ca(BH4)2 electrolyte in THF, representing the first Ca electrolyte operating at room temperature (Fig. 6). Cyclic voltammetry (CV) on an Au electrode displayed a high Ca plating/stripping efficiency (94.8%) with anodic stability up to 3.0 V using three-electrode cells (Fig. 6a and c). An overpotential of ca. 250 mV was observed on the first plating and then it was reduced to ca. 100 mV on repeated plating and stripping cycles (Fig. 6b). The coulombic efficiency for the first cycle was 93% and was gradually increased to 94–96% over 25 cycles (Fig. 6b). Powder X-ray diffraction (PXRD, Fig. 6d) and FTIR spectra were utilized to study anode samples extracted from the cell after Ca deposition and indicated the Ca metal as a dominant product with a small amount of CaH2. Scanning electron microscopy (SEM), time-of-flight secondary ion mass spectroscopy (TOF-SIMS), and gas chromatography mass spectrometry (GC-MS) studies suggested that the CaH2 was formed by the reaction of the freshly deposited Ca and THF solvent during the electrochemical deposition process. This byproduct was believed to be a passivation layer to prevent further solvent decomposition, but it does not act as an SEI component to facilitate Ca plating/stripping.
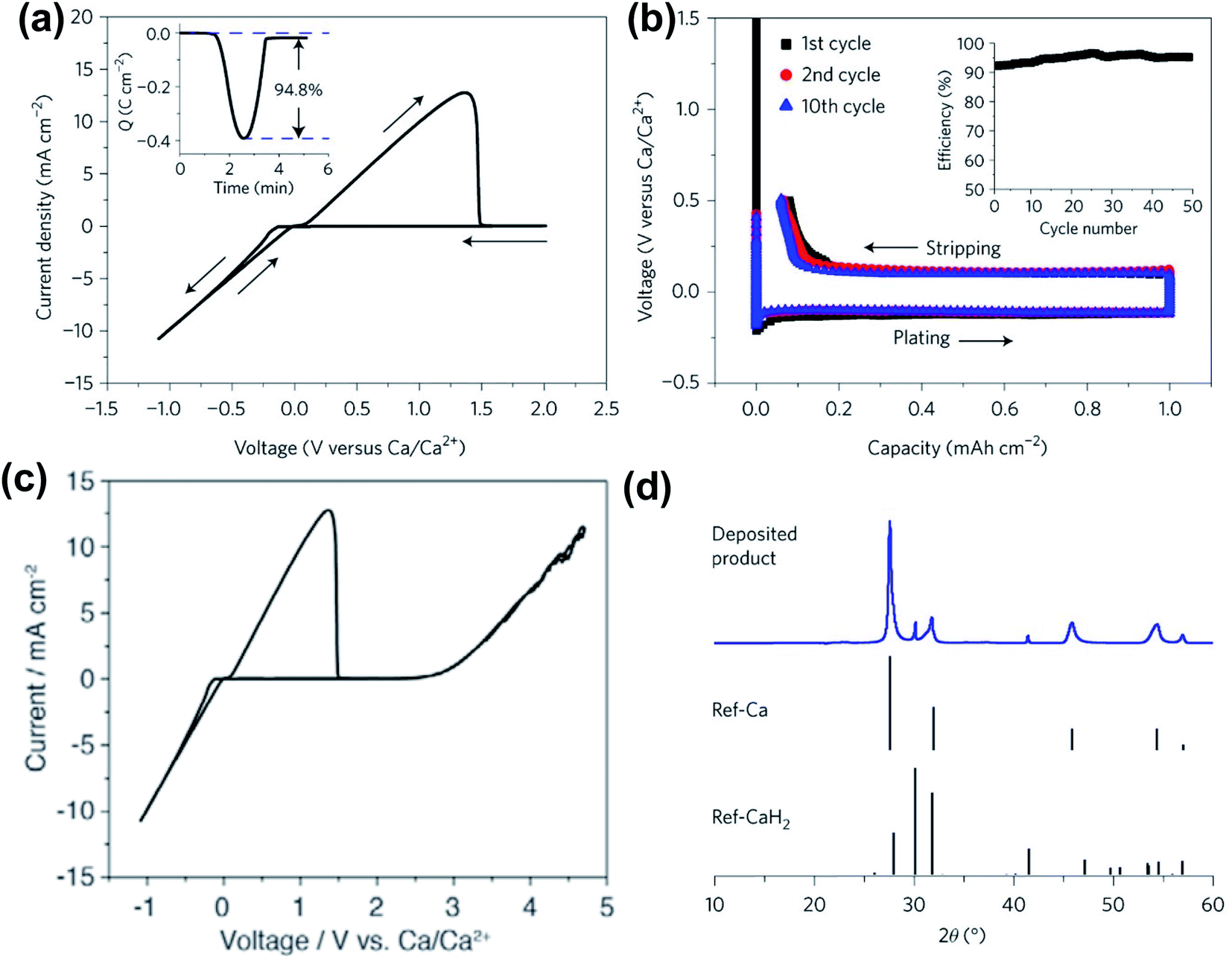 | ||
| Fig. 6 Electrochemical results of calcium plating/stripping of 1.5 M Ca(BH4)2 in THF reported by Bruce and co-workers. (a) Cyclic voltammogram of calcium plating/stripping in 1.5 M Ca(BH4)2-THF. The working, reference, and counter electrodes are Au, Ca, and Pt, respectively. Scan rate 25 mV s−1. Inset shows charge passed on plating/stripping from the CV. (b) Galvanostatic calcium plating/stripping in 1.5 M Ca(BH4)2-THF at a rate of 1.0 mA cm−2. Inset shows the variation of coulombic efficiency with cycle number. (c) Cyclic voltammogram showing the electrochemical window of the electrolyte. (d) Powder X-ray diffraction patterns showing calcium as the dominant product on plating with a small amount of CaH2. Adapted with permission from ref. 26. Copyright 2018 Springer Nature. | ||
Later, Gewirth and co-workers also examined the coordination chemistry and electrochemistry of the Ca(BH4)2-THF electrolyte.27 According to Raman spectroscopic studies, the Ca2+ cation is primarily coordinated by THF and does not strongly interact with the BH4− anion. They used Au and Pt microelectrodes to study the Ca deposition at scan rates from 2 to 20 mV s−1. They found that the deposition current intensities were inversely dependent on the scan rates (Fig. 7) with both electrodes. They suggested that a chemical step involving the dehydrogenation of the BH4− anion takes place before the Ca deposition. Then they detected the formation of BH3(THF) using liquid injection field desorption ionization mass spectrometry (LIFDI-MS). They proposed that the leaving H− species is adsorbed at the electrode surface to assist the Ca deposition. Then CaH2 is formed on the top of the deposited Ca. This mechanism is different from what Bruce et al. proposed. It was noted the deposition current at the Au electrode was more sensitive to that at the Pt electrode. Numerical simulation of electrochemical data suggested that Pt (38.7 s−1) is more active than Au (3.0 s−1) to catalyze the dehydrogenation of the BH4− anion. As a result, they believe that the kinetics of the chemical step can affect the deposition morphology of Ca. With the slow rate, hydride has enough time to diffuse at the surface of the Au electrode surface and enables uniform deposition of Ca. In contrast, the lack of hydride diffusion leads to patchy Ca deposition on the Pt electrode.
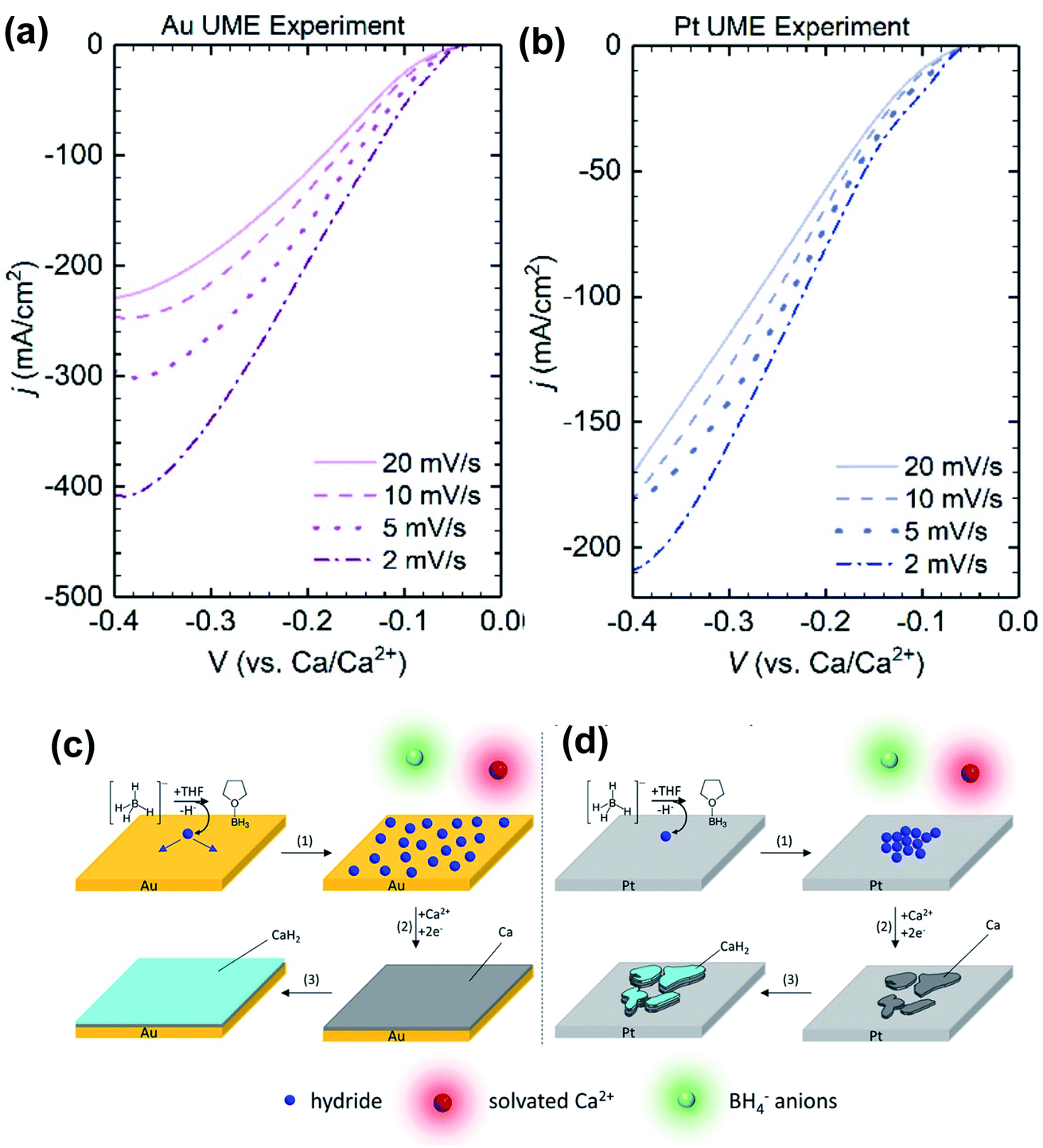 | ||
| Fig. 7 Electrochemical studies of the Ca(BH4)2-THF electrolyte by Gewirth and co-workers. (a) and (b) Dependence of deposition current with scan rate using Au and Pt working electrodes, respectively. (c) and (d) Proposed deposition mechanisms using Au and Pt working electrodes, respectively. Adapted with permission from ref. 27. Copyright 2020 American Chemical Society. | ||
Jiao et al. reported that a mixed Ca(BH4)2 and LiBH4 electrolyte in THF could achieve a high coulombic efficiency of up to 99.1% for galvanostatic plating/stripping of the calcium–metal anode for over 200 cycles in Ca‖Au and Ca‖Cu coin cells at room temperature (Fig. 8a).28 In full Ca‖lithium titanate (LTO) batteries, a capacity retention of roughly 80% was obtained after 200 cycles with the first discharge specific capacity of ca. 170 mA h g−1. The corresponding voltage–capacity profile for the galvanostatic Ca-metal plating/stripping in the Ca(BH4)2/LiBH4-THF electrolyte shows an overpotential of ca. 200 mV (vs. Ca) for the first plating process and about 130 mV for the first stripping process, which was stabilized at ca. 97 mV after 5 cycles. The scanning electron microscopy (SEM) analysis of deposits on Au electrodes in the Ca(BH4)2/LiBH4-THF electrolyte showed that the Ca particle has a micro-sized spherical shape with a golf-ball-like dimpled surface (Fig. 8c and d), which reduces the possibility of penetrating through the separator. The deposited Ca particles with a small amount of carbon and oxygen on the surface were confirmed by EDX studies. Further investigation of the composition of the SEI layer by XPS indicates that the deposits formed in Ca‖Au coin cells contain inorganic CaCO3, CaO, and organic carbonyl CO and C–O species derived from THF (Fig. 8e). Further investigation with 43Ca NMR spectroscopy (Fig. 8f) and MD simulations (Fig. 8g and h) of the Ca(BH4)2 and Ca(BH4)2/LiBH4 electrolytes revealed that the introduction of LiBH4 salt decreases the coordination number of Ca2+ ions in the first solvation shell, which weakens the solvation energy of Ca2+ in the Ca(BH4)2/LiBH4-THF electrolyte. The results indicate that LiBH4 functions as a solvation structure modulator. This work suggests that a salt additive can be useful in improving electrolyte performance.
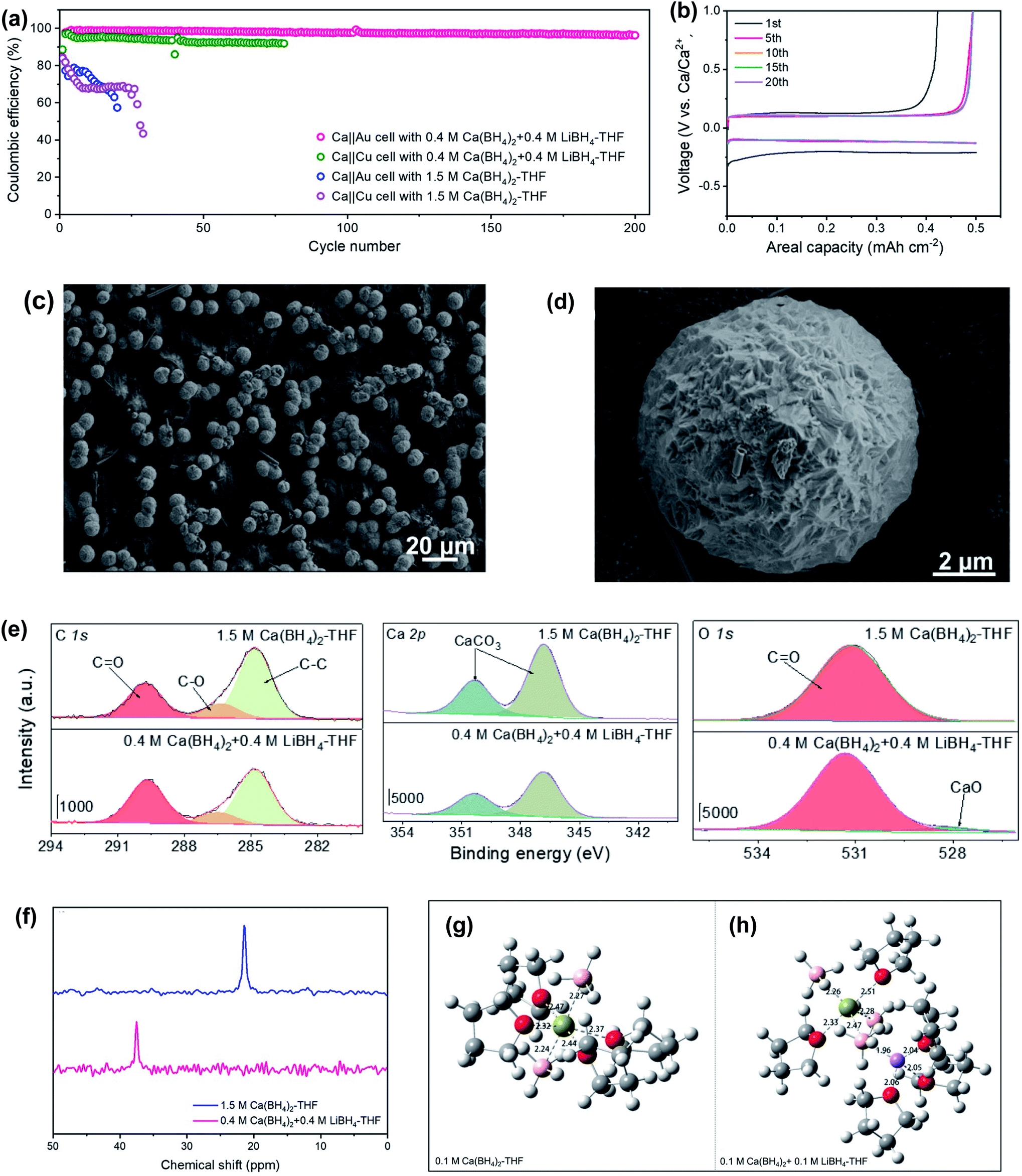 | ||
| Fig. 8 Electrochemical and spectroscopic studies of Ca(BH4)2/LiBH4-THF electrolytes and control Ca(BH4)2-THF electrolytes. (a) CE values of Ca‖Au and Ca‖Cu coin cells using Ca(BH4)2/LiBH4-THF and Ca(BH4)2-THF electrolytes at 1.0 mA cm−2 and 0.5 mA h cm−2. (b) Voltage–capacity profiles of selected cycles on Au electrodes in the Ca(BH4)2/LiBH4-THF electrolyte (c and d). Ca plating morphology in the Ca(BH4)2/LiBH4-THF electrolyte. (e) XPS studies of the Au working electrode. (f) 43Ca NMR spectra of two electrolytes. The calculated structures of (g) pure Ca(BH4)2-THF and (h) Ca(BH4)2/LiBH4-THF electrolytes (Ca, green; O, red; Li, purple, C, gray, and B, pink). Adapted with permission from ref. 28. Copyright 2020 Wiley-VCH. | ||
4. Advanced calcium electrolytes with low-coordinating anions
One of the fundamental problems for the practical application of traditional Ca salts used in non-aqueous Ca electrolytes mentioned above is the incompatibility of the Ca metal in aprotic solvents due to the formation of passivation surface layers which prevents the transportation of Ca2+ ions. Engineering counter-anions is an effective strategy to enhance the electrochemical performance of battery electrolytes. The concept of bulky weakly coordinating anions has been successfully applied to the development of advanced magnesium electrolytes for highly reversible Mg deposition, including Mg(CB11H12)2,29–31 Mg(CB11H11F)2,32 Mg[Al(HFIP)4]2,33 and Mg[B(HFIP)4]2 (HFIP = −OCH(CF3)2),34,35 Mg(FPB)2 (FPB = B(O2C2(CF3)4)2]2),36 and Mg[Al(OC(CF3)3)4]2.37 It is a logical option to develop calcium electrolytes based on these low-coordinating, chemically stable anions.13 In addition, weakly coordinating anions that lack donor atoms can enable more favored ion dissociation and higher cation mobility.Zhao-Karger et al. reported the synthesis of Ca[B(hfip)4]2 by reacting Ca(BH4)2 with hexafluoroisopropanol (CF3CHOHCF3) in dimethoxy ethane (DME) and characterized it as [Ca(DME)4][B(hfip)4]2 with X-ray single crystal crystallography (Fig. 9a).38 The thermal stability analysis of [Ca(DME)4][B(hfip)4]2 by simultaneous thermogravimetric analysis-differential scanning calorimetry-mass spectroscopy (TGA-DSC-MS) confirmed that this Ca salt is thermally stable up to 120 °C. A high ionic conductivity of 8.3 mS cm−1 at 23 °C was reported for a 0.25 M Ca[B(hfip4)2]-DME electrolyte. It is believed that the delocalized charge in the bulky alkoxyl borate anion due to the high electronegativity of the fluorine atom and the high stability of C–F bonds contributes to the weak cation–anion interactions plus a high ionic conductivity and the high electrochemical stability, respectively. The cyclic voltammogram (CV) scans of the 0.25 M Ca[B(hfip)4]2 in DME on a Pt working electrode demonstrated high reversibility for Ca plating/stripping with a voltage range of −0.35 to 4 V at room temperature (Fig. 9b). The overpotential for plating remained at −0.22 V and 0.10 V for stripping in the extended CV cycles (Fig. 9b). The coulombic efficiencies increased within the first few cycles and were stabilized at ∼80% in the continuous cycles (Fig. 9c). The oxidative stability of 0.25 M Ca[B(hfip)4]2-DME electrolyte on current collectors such as stainless steel (SS 316), Pt, and Al was examined by linear sweep voltammetry (LSV), e.g., 3.9 V for Pt and 4.5 V for Al (Fig. 9d). However, the deposition/stripping reversibility with non-Pt electrodes was not reported.
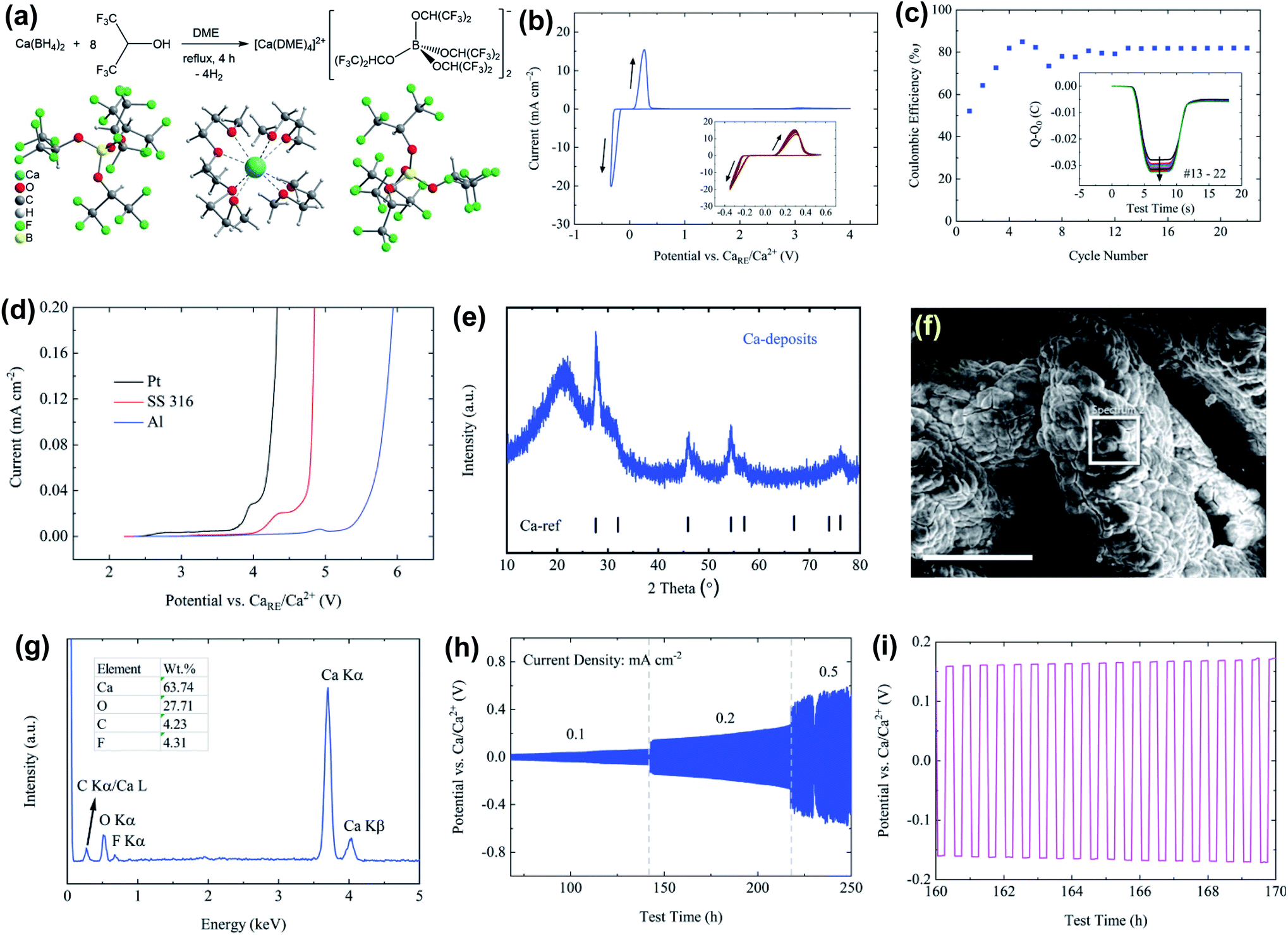 | ||
| Fig. 9 Synthesis and electrochemical studies of the Ca[B(hfip)4]2-DME electrolyte reported by Zhao-Karger et al. (a) Synthesis route and X-ray single-crystal structure of [Ca(DME)4][B(hfip)4]2. Electrochemical characterization of the Ca[B(hfip)4]2-DME electrolyte: (b) CV of Ca plating/stripping after conditioning cycles at a scan rate of 80 mV s−1 using Pt as a WE and Ca as a RE and CE, respectively. (c) Coulombic efficiencies determined from CVs. The inset presents the charge balance within each cycle. (d) LSVs on various WEs with a three-electrode configuration using Ca as a RE and CE, respectively, at a scan rate of 1 mV s−1. (e) PXRD pattern of Ca deposits. (f) SEM image of the deposits; the scale bar is 250 μm. (g) EDX spectrum within the white squared area in the SEM image. (h) Cycling performance of the electrolyte at various current densities. (i) A typical voltage profile at a current density of 0.2 mA cm−2. Adapted with permission from ref. 38. Copyright 2019 Royal Society of Chemistry. | ||
As depicted in Fig. 9e, the PXRD pattern of Ca deposits indicates a dominant cubic Ca phase. The broad peaks with a low intensity stand for a lower crystallinity and a smaller size of Ca deposits. The SEM image of the deposits also indicates micro-sized agglomerates without obvious crystalline features (Fig. 9f). The energy dispersive X-ray spectroscopy (EDX) spectrum (Fig. 9g) shows that the deposits are mainly composed of Ca and O with trace amounts of carbon (C) and fluorine (F). The atomic ratio of F/Ca is ca. 14% and corresponds to ca. 7% CaF2 in the Ca deposits. The presence of O, C, and F elements suggests electrolyte and solvent decomposition for the formation of an SEI layer. In addition, the electrolyte was further tested in symmetric Ca‖Ca cells at constant current densities from 0.1 to 0.5 mA cm−2, displaying a moderate polarization of ca. 60 mV at 0.1 mA cm−2 and ca. 200 mV at 0.2 mA cm−2 (Fig. 9h and i).
The exact same Ca[B(hfip)4]2 electrolyte was simultaneously reported by Nazar's group using a different synthetic route between calcium hexafluoroisopropoxide (Ca(hfip)2·xTHF) and tris-hexafluoroisopropoxy borate (B(hfip)3).39 The CV studies show calcium plating and stripping with ca. 92% coulombic efficiency in the fourth cycle using an Au electrode (Fig. 10a). Further galvanostatic cycling in the window from −1.5 to 1.2 V vs. Ca showed stable stripping and plating of calcium to a capacity of 1.0 mA h cm−2 at a current density of 0.2 mA cm−2 with an overpotential of 300 mV for plating and ca. 180 mV for stripping (Fig. 10b). A Bu4NCl additive was added to increase the ionic conductivity from 3.2 to 6.7 mS cm−1 and also enhanced the cycle life of the Ca[B(hfip)4]2 electrolyte (Fig. 10c). Anodic stability measurement of the Ca(B(hfip)4)2-DME electrolyte with linear sweep voltammetry (LSV) exhibited an oxidative stability of 3.8 V on Au and achieved the highest anodic stability of ca. 4.1 V on Al. Further synchrotron XRD experiments and SEM-EDX analysis confirmed the Ca metal deposition and stripping in the CV studies, and the nature of the passivating layer has been identified. The full profile fitting of the XRD pattern indicates two distinct phases (cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and orthorhombic Cmcm) of Ca deposits and calcium fluoride. EDX elemental analysis revealed a Ca/F ratio of 3:1 in the Ca deposits, which corresponds to a Ca/CaF2 ratio of 5:1. Overall, these results are generally consistent with those reported by Zhao-Karger et al.38
m and orthorhombic Cmcm) of Ca deposits and calcium fluoride. EDX elemental analysis revealed a Ca/F ratio of 3:1 in the Ca deposits, which corresponds to a Ca/CaF2 ratio of 5:1. Overall, these results are generally consistent with those reported by Zhao-Karger et al.38
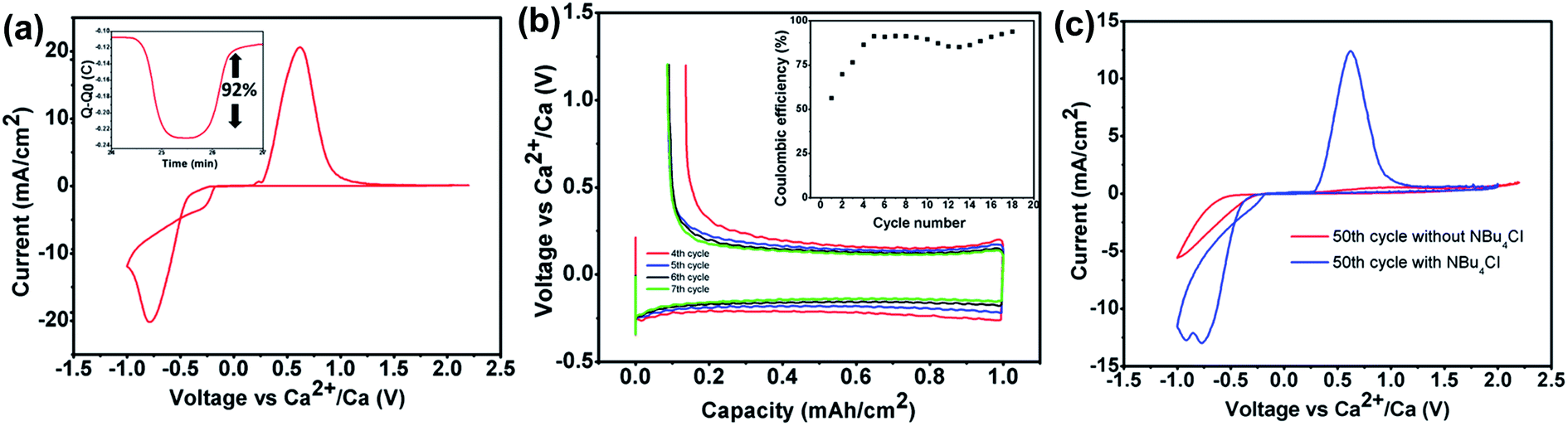 | ||
| Fig. 10 Electrochemical studies of the Ca[B(hfip)4]2-DME electrolyte reported by Nazar et al. (a) CV of a 0.5 M Ca(B(hfip)4)2 electrolyte on an Au electrode with calcium as the reference and counter electrodes at a scan rate of 25 mV s−1. The inset shows the charge passed on plating and stripping from the voltammogram. (b) Galvanostatic stripping and plating of calcium on an Au electrode with Ca counter and reference electrodes from a 500 mM Ca[B(hfip)4]2 electrolyte at 0.2 mA cm−2 to a capacity of 1.0 mA h cm−2, with the inset showing the coulombic efficiency. (c) Comparison of the 50th CV cycles with a 500 mM Ca[B(hfip)4]2 electrolyte (red curve) and a 0.5 M Ca[B(hfip)4]2/0.1 mM Bu4NCl electrolyte (blue curve). Adapted with permission from ref. 39. Copyright 2020 American Chemical Society. | ||
Subsequently, our group also studied the Ca[B(hfip)4]2 electrolyte by examining the effects of working electrodes and solvents on Ca deposition.40 We first studied a 0.25 M Ca[B(hfip)4]2-DME electrolyte using glassy carbon (GC), Al, Pt, and Cu working electrodes. The electrolyte using Pt achieved a coulombic efficiency of ca. 80%, consistent with the value reported in Zhao-Karger's work.38 However, reversibility for Ca deposition using GC (a coulombic efficiency of 30%), Al (a coulombic efficiency of 20%), and Cu (a coulombic efficiency of less than 15%) was much worse while overpotential was also increased. It is not clear what are causes of different coulombic efficiencies observed with different working electrodes. To examine the effect of solvents on the reversibility of Ca deposition, tridentate diglyme (DGM) and mono-dentate THF solvents were studied using the same working electrodes. In THF, the reversibility was even worse than in DME. Much lower reversibility of a Ca[B(hfip)4]2-THF electrolyte was observed only up to 68% coulombic efficiency on a Pt electrode. In stark contrast, the Ca[B(hfip)4]2 in DGM displayed apparently improved reversibility, i.e., Pt (82.1%), GC (85.5%), Al (61%), and Cu (76%) (Fig. 11). For comparison, the electrochemical properties of Ca[B(hfip)4]2 and other reported calcium electrolytes in organic solvents are summarized in Table 2.
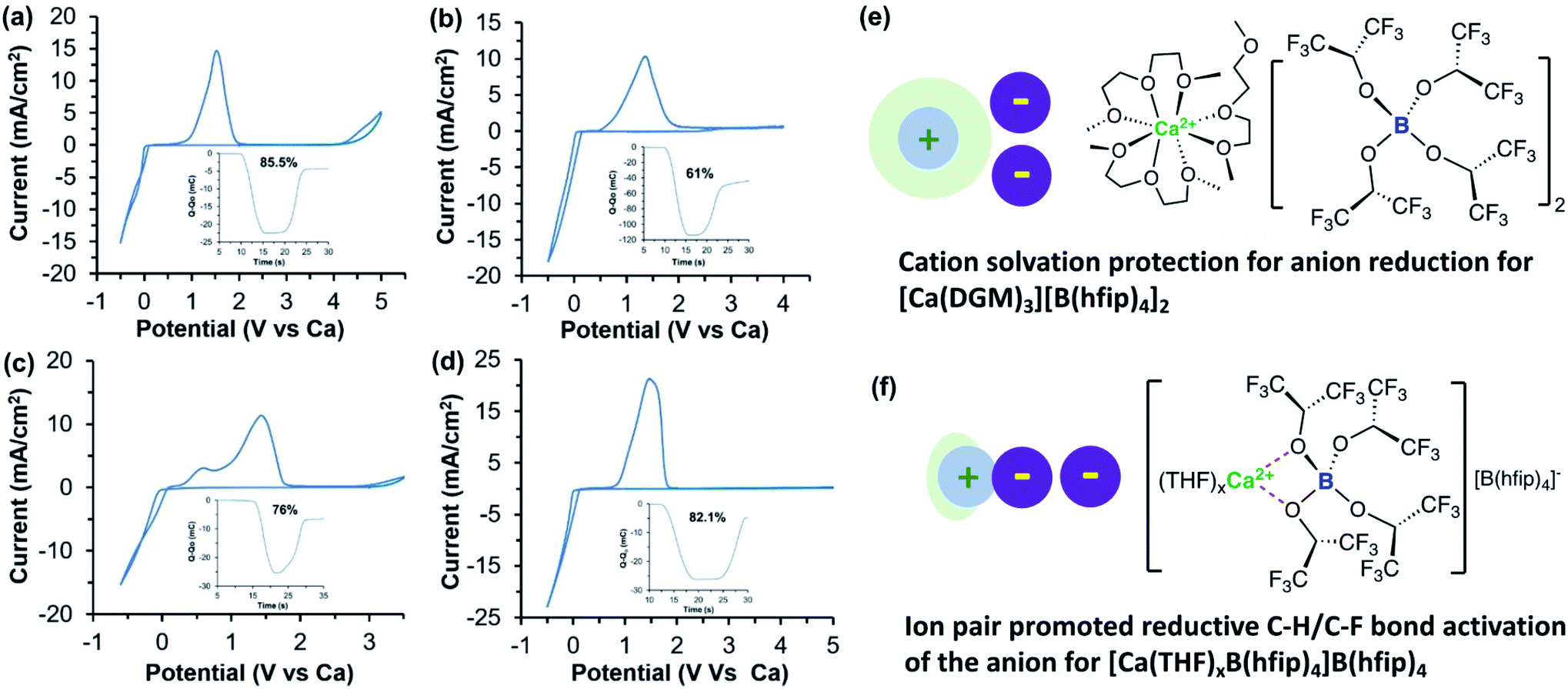 | ||
| Fig. 11 Cyclic voltammograms of a 0.25 M Ca[B(hfip)4]2-DGM electrolyte solution with different working electrodes. (a) Glassy carbon working electrode. (b) Al working electrode with the inset showing accumulation of charge and coulombic efficiency. (c) Cu working electrode with the inset showing coulombic efficiency. (d) Pt working electrode with the inset showing coulombic efficiency. All samples were cycled at 100 mV s−1 scan rate. (e) Proposed structure of Ca[B(hfip)4]2-DGM where Ca2+ and [B(hfip)4] are fully dissociated. (f) Proposed structure of partly solvated Ca[B(hfip)4]2-THF electrolyte where Ca2+ and [B(hfip)4] form an ion pair. Adapted with permission from ref. 40. Copyright 2020 Wiley-VCH. | ||
| Electrolytes | CE (%) | Deposition/stripping potential (V) (scan rate) | Anodic stability (V) | Working electrode | Temperature (°C) | Ref. |
|---|---|---|---|---|---|---|
| 0.45 M Ca(BF4)2-EC/PC (50/50 wt%) | 40% | −0.50/−0.42, (0.5 mV s−1) | 3.5 (Al) | SS | 100 | 23 |
| 1 M Ca(BF4)2-EC/PC (50/50 v/v) |
95% | −0.7/0.5 (25 mV s−1) | n.a. | Cu | 23 | 24 |
| 1.5 M Ca(BH4)2-THF | 95% | −0.25/−0.10 | 3.0 | Au | r.t. | |
| 0.4 M Ca(BH4)2/0.4 M LiBH4-THF | 97.6% | −0.10/0.10 (coin cells) | 2.8 | Au | r.t. | 28 |
| 0.25 M Ca[B(hfip)4]-DME | 80% | −0.22/0.10 (80 mV s−1) | 3.9 | Pt | r.t. | 38 |
| 0.5 M Ca[B(hfip)4]/0.1 M Bu4NCl-DME | 92% | −0.50/0.18 (25 mV s−1) | 3.8 | Au | r.t. | 39 |
| 0.25 M Ca[B(hfip)4]-THF | 68% | −0.15/0.1 (100 mV s−1) | 3.3 | Pt | r.t. | 40 |
| 0.25 M Ca[B(hfip)4]-DME | 30% | −0.01/0.65 (100 mV s−1) | 4.0 | GC | r.t. | 40 |
| 40% | −0.01/1.1 (100 mV s−1) | 4.9 | Al | r.t. | ||
| 25% | −0.18/0.30 (100 mV s−1) | 2.6 | Cu | r.t. | ||
| 80.5% | −0.01/0.60 (100 mV s−1) | 4.1 | Pt | r.t. | ||
| 0.25 M Ca[B(hfip)4]-diglyme | 85.5% | −0.20/0.70 (100 mV s−1) | 4.2 | GC | r.t. | 40 |
| 61% | −0.20/0.40 (100 mV s−1) | >4.0 | Al | r.t. | ||
| 76% | −0.20/0.30 (100 mV s−1) | 2.9 | Cu | r.t. | ||
| 82.1 | −0.20/0.60 (100 mV s−1) | >5 | Pt | r.t. |
There were two stripping waves observed at ca. 0.5 and 1.5 V vs. Ca, respectively, which are tentatively assigned to the oxidation of pure Ca and the Ca/Cu alloy (Fig. 11c). It was interpreted that the tridentate DGM solvent can stabilize Ca2+ by avoiding the Ca2+-B(hfip)4− ion pair (Fig. 11e). According to the solvation structure of Ca2+ with DME, [Ca(DME)4]2+,38 the solvation of Ca2+ with DGM is proposed as two tridentate DMG and one bidentate DMG coordination. In the Ca[B(hfip)4]2-THF electrolyte, the formation of the Ca2+-B(hfip)4− ion pair is proposed to activate the [B(hfip)4]− anion to undergo reductive C–F and C–H bond decomposition (Fig. 11f). In addition, DGM is more reductively stable than DME and THF to further improve the electrolyte reversibility. The first-sphere coordination effect was also later observed in Ca(TFSI)2 electrolytes using N-donor additives.41
Then the Ca[B(hfip)4]2-DGM electrolyte in the three solvents was tested in symmetric Ca‖Ca cells with current densities of 1.0, 2.0, 4.0, and 8.0 mA cm−2. If noted for all solvents, voltage spikes were observed above 2.0 mA cm−2 with a magnitude order of THF (up to 5 V) > DME (>3 V) > DGM (<0.5 V, Fig. 12a). In the THF electrolyte, the cell failed to cycle at 8.0 mA cm−2 after 170 h. In the DME electrolyte, the cell stopped cycling after 220 h. In the DGM electrolyte, the cell was cycled up to 300 h with stable voltage polarization at 1.0 mA cm−2 after the rate performance (Fig. 12a). Consistently, SEM and EDX studies revealed that the DGM electrolyte exhibits much smoother and cleaner Ca deposition than the other two electrolytes. Finally, the DGM electrolyte was evaluated in a Ca‖FePO4 full battery for 10 cycles (Fig. 12b). A discharge capacity of 120 mA h g−1 and a coulombic efficiency of 70% were recorded. The representative charge/discharge profiles indicate a 3.5 V cell voltage (Fig. 12c). EDX and ICP-MS studies confirmed the Ca intercalation in a discharged state formula of Ca0.43FePO4. The full-cell study promises further electrolyte studies to develop high voltage, high energy density Ca batteries.
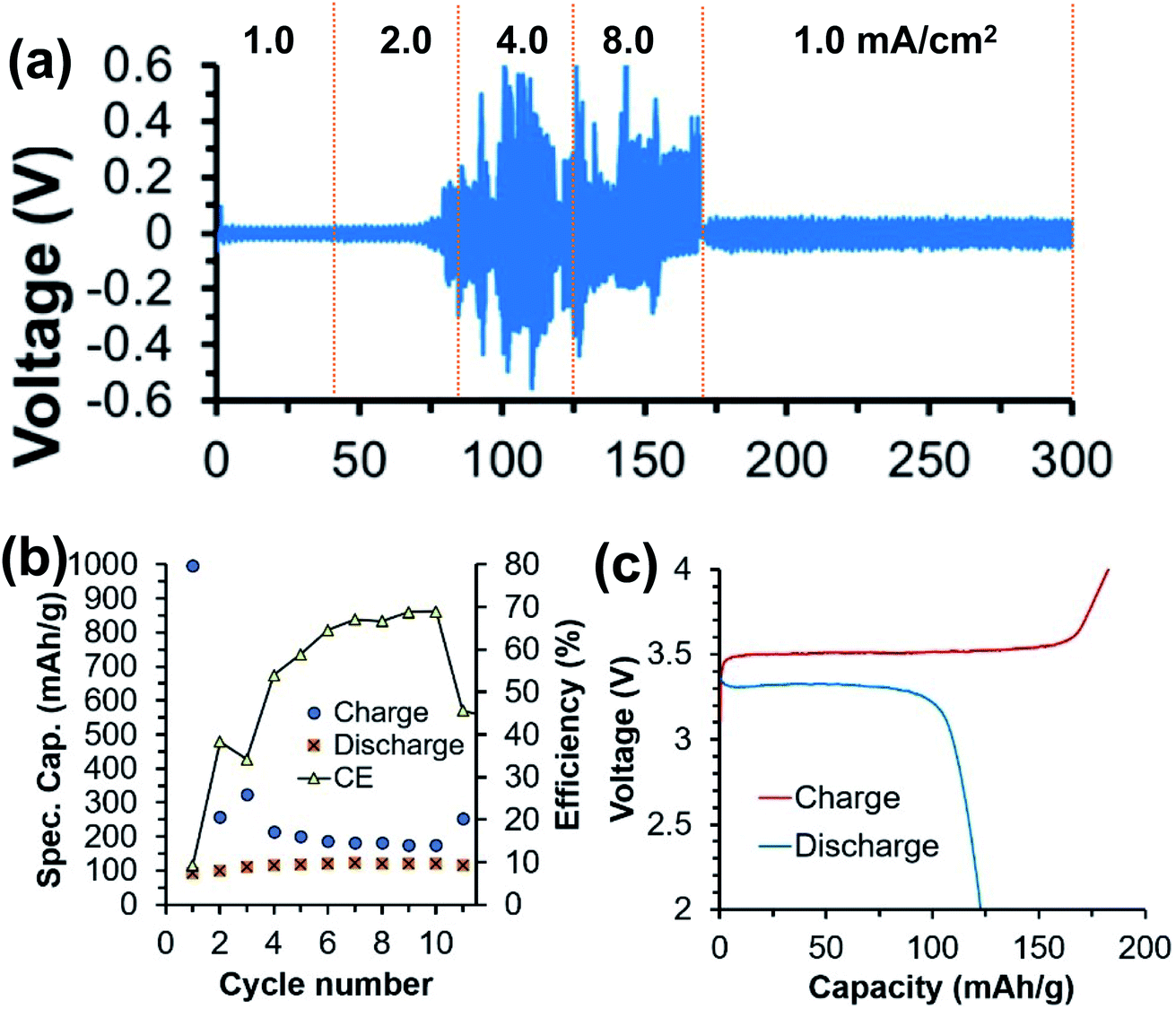 | ||
| Fig. 12 Half-cell and full-cell studies of a 0.25 M Ca[B(hfip)4]2-DGM electrolyte. (a) A Ca‖Ca symmetric cell using a Ca[B(hfip)4]2-DGM electrolyte. (b) Cycling performance and (c) representative charge/discharge profiles of a Ca‖FePO4 full battery using the Ca[B(hfip)4]2-DGM electrolyte. Adapted with permission from ref. 40. Copyright 2020 Wiley-VCH. | ||
5. Polymer electrolytes
In comparison to liquid electrolytes, solid state electrolytes are advantageous in improving battery safety by avoiding flammable organic solvents and boosting battery cycling performance by improving stability with electrode materials. There are ongoing efforts in developing calcium solid polymer electrolytes (SPEs). These early studies of Ca polymer electrolytes have a primary focus on formulation and conductivity studies. These studies provide a good foundation for the development of more conductive and more stable polymer electrolytes to develop energy dense and safe Ca batteries.Hosein et al. reported the synthesis and properties of crosslinked poly(ethylene glycol) diacrylate (PEGDA) network based calcium nitrate (Ca(NO3)2) solid electrolytes.42 Ionic conductivities of 3.4 × 10−4 S cm−1 at 110 °C and 3.0 × 10−6 S cm−1 at room temperature were achieved, with good thermal stability at 136 °C or higher decomposition temperatures. The same group subsequently studied another polymer electrolyte with an improved conductivity of 0.1 mS cm−1 at room temperature using a copolymer of polytetrahydrofuran (PTHF) with 3,4-epoxycyclohexylmethyl-3′,4′-epoxycyclohexane carboxylate and Ca(NO3)2 as a Ca2+ ion source.43 This SPE also displayed suitable thermal (stable over a temperature range of 30–120 °C) and mechanical stability. Sanchez et al. reported a series of homogenous calcium salt-PEO electrolytes, including Ca(CF3SO3)2, Ca(TFSI)2, and CaI2 (Fig. 13).44 The Ca(CF3SO3)2-PEO and CaTFSI2-PEO SPEs with a moderate molar concentration ratio of PEO/Ca of 40 could even reach a conductivity of above 0.47 mS cm−1 at 90 °C. The group reported that the higher solvation or donor number of PEO and the higher degree of salt dissociation of Ca(TFSI)2 resulted in the higher conductivities of Ca(CF3SO3)2-PEO and CaTFSI2-PEO SPEs than those reported by Hosein's group.
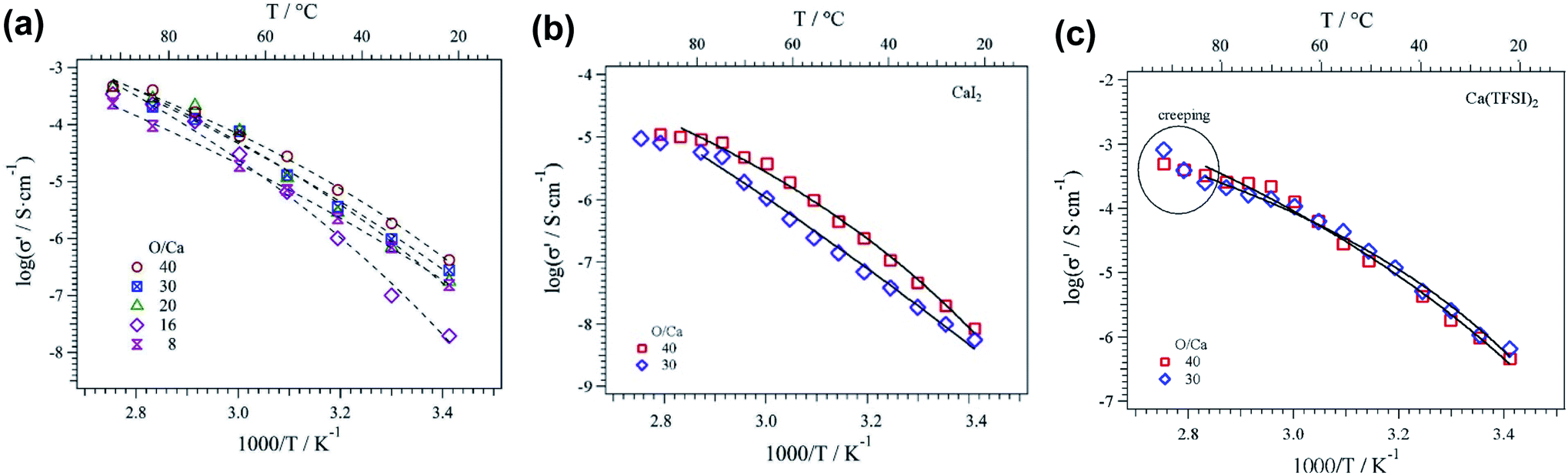 | ||
| Fig. 13 Ionic conductivity as a function of temperature for POE-Ca(CF3SO3)2 (a), POE-CaI2 (b), and POE-Ca(TFSI)2 (c) electrolytes. Continuous lines correspond to Vogel–Tamman–Fulcher fitting. Adapted with permission from ref. 44. Copyright 2020 Elsevier. | ||
More recently, Hosein's group developed a series of ionic-liquid polymer gel electrolytes (ILGEs) by swelling a poly(ethylene glycol) diacrylate (PEGDA) host matrix in an ionic liquid (1-ethyl-3-methylimidazolium trifluoromethanesulfonate) solution of different calcium salts, Ca(ClO4)2, Ca(BF4)2, and Ca(TFSI)2 (Fig. 14a).45 The IL-GEs showed conductivities on the order of 10−4 S cm−1 at room temperature, reaching maximal conductivities on the order of 10−3 S cm−1 (Fig. 14b), which also demonstrated excellent thermal stability up to 275 °C. The cationic transference number (t+) was determined as 0.17 based on chronoamperometry and impedance studies. The high anodic stability of ca. 4 V vs. Ca/Ca2+ from linear sweep voltammetry may be attributed to the high oxidative stability imparted by the crosslinked polymer host and the high degree of salt dissociation. Then the electrolytes were tested in full battery studies in CR2032 coin cells using calcium cobalt oxide (Ca3Co4O9) as a positive electrode and vanadium oxide (V2O5) as a negative electrode. Particularly, Ca(TFSI)2 based IL-GEs were capable of operating at room temperature and delivering a discharge capacity of 140 mA h g−1 in the first cycle with an open-circuit voltage of 1.2 V (Fig. 14c). However, the rapid fading of charge and discharge capacities over cycling was observed (Fig. 14d), which may result from the difficult reinsertion of Ca2+ into Ca3Co4O9, changes in the crystal structure of V2O5 upon cycling, and electrolyte decomposition.
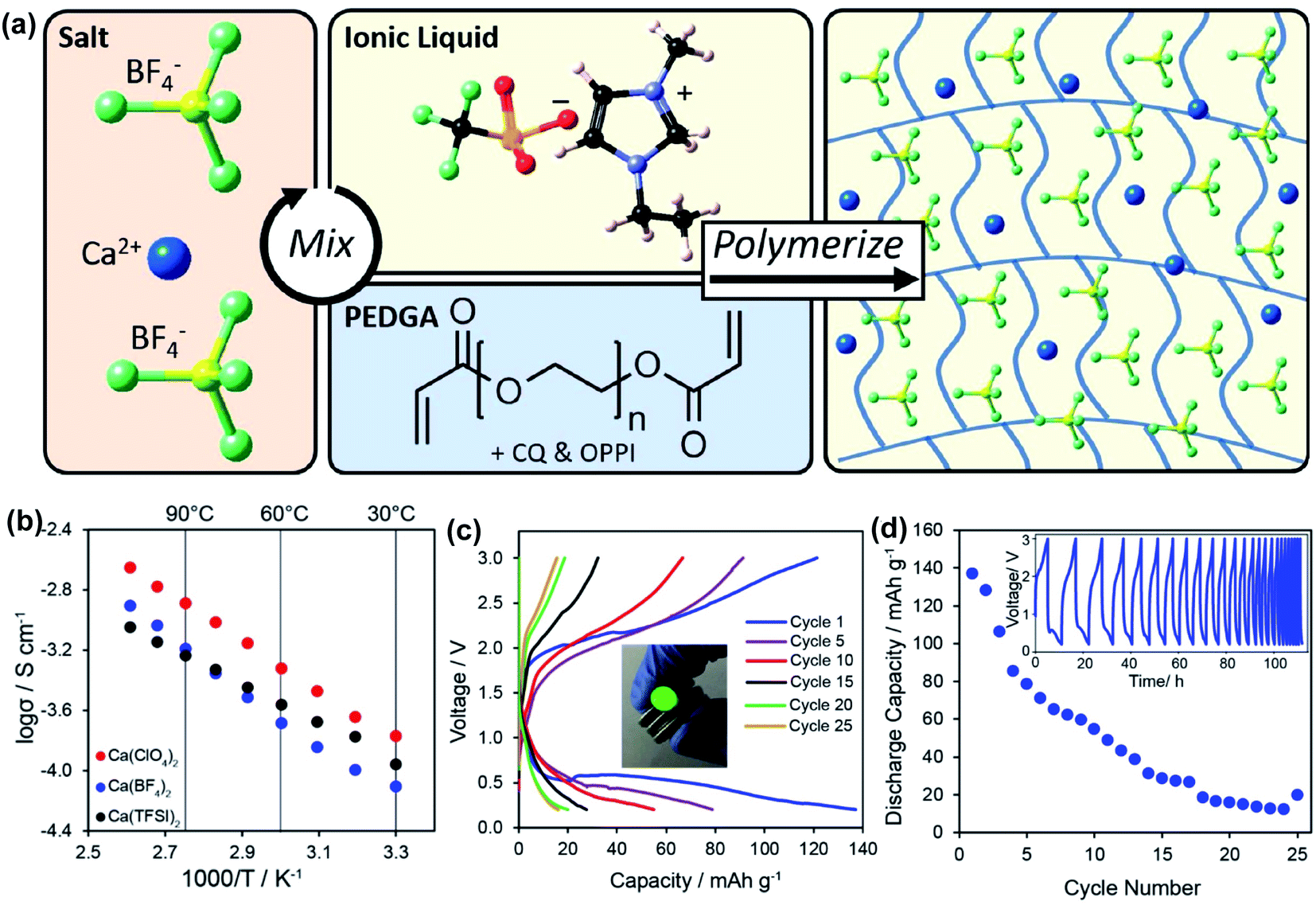 | ||
| Fig. 14 Electrochemical studies of polymer and ionic liquid based Ca2+ electrolytes. (a) Illustration of the synthesis of an exemplary Ca(BF4)2-PEGDA/IL gel electrolyte. (b) Conductivity studies of a series of Ca salt gel electrolytes. (c) A V2O5‖Ca3Co4O9 battery study using the Ca(TFSI)2-PEGDA/IL electrolyte. (d) Cycling performance of the battery. Adapted with permission from ref. 45. Copyright 2020 American Chemical Society. | ||
6. Perspectives and conclusions
We summarized and discussed the historical development and current status of liquid and polymer calcium electrolytes for developing rechargeable Ca batteries. Undoubtedly, the field of Ca batteries is still in its infancy stage. We would like to share a number of recommendations in evaluating and developing new Ca2+ ion electrolytes in future.First, performance metrics of Ca2+ electrolytes, including coulombic efficiencies, deposition and stripping overpotential, anodic stability, and charge transport kinetics should be comprehensively evaluated by cyclic voltammetry, bulk electrolysis, electrochemical impedance spectroscopy, and other electrochemical methods, which are preferably performed using common current collector electrodes such as stainless steel (SS), Cu, and Al. The durability with the Ca anode, and more reliable coulombic efficiencies should be further tested using Ca‖Cu or SS coin cells (Fig. 15a). In the literature, most studies reported half-cell studies using the Ca‖Ca configuration. The coulombic efficiency on a Ca electrode is not trustworthy as the Ca anode will provide excess Ca to increase the stripping current and thus result in higher coulombic efficiencies. In addition, it is also worth noting that graphite46 and alloys47 can also serve as anode materials and provide new opportunities for developing Ca batteries.
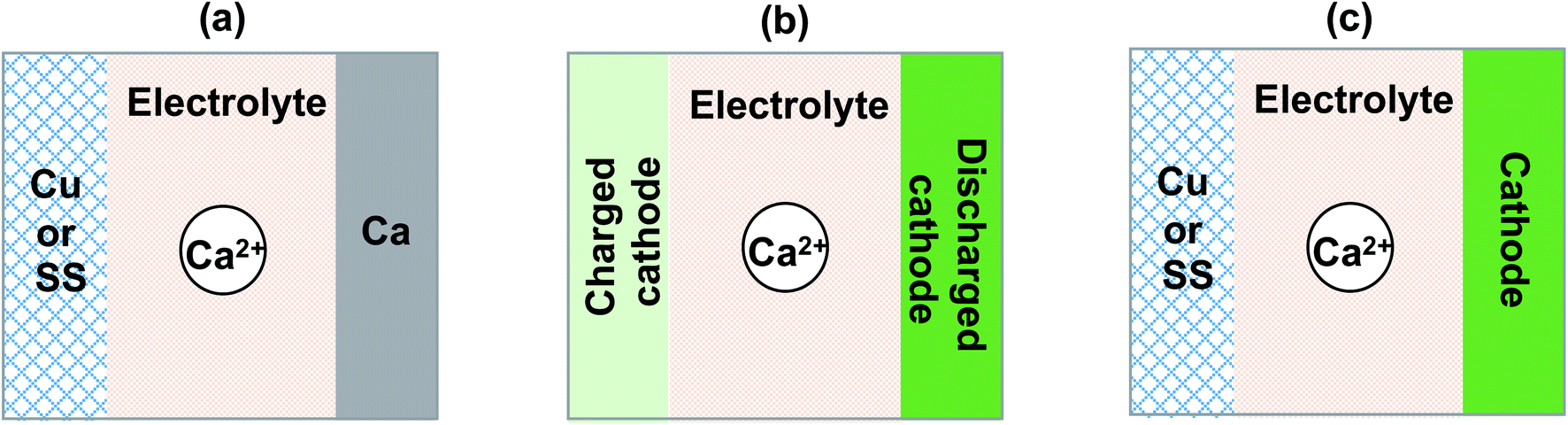 | ||
| Fig. 15 Battery configurations for evaluating Ca electrolytes. (a) Ca‖Cu or SS configuration to evaluate the coulombic efficiency and anode stability of Ca electrolytes. (b) Charged cathode‖discharged cathode configuration to evaluate the coulombic efficiency and cathode stability. (c) Anode-free configuration to evaluate electrolytes in full batteries. | ||
Second, the interfacial chemistry of Ca2+ electrolytes and the Ca anode is still not fully understood. With the deployment of new Ca2+ electrolytes, it is expected that rich interfacial chemistry will be gained. A very fundamental question to address is how to elucidate the exact chemical composition and roles of organic and inorganic components of the SEI, which is highly valuable to design high-performance Ca2+ electrolytes. In addition, the SEI and electrolyte solvation are also critical to the Ca deposition morphology and are worth systematic investigation. Definitely, we can get inspiration and intellectual support from the known knowledge of Li batteries and Mg batteries.
Third, there are very few studies reported on the interfacial chemistry of Ca electrolytes and cathodes. Existing Li ion cathodes can serve as a good material library to survey Ca2+ intercalation chemistry. Following Dahn's work on elevating Li ion cathodes,48–50 we recommend using the cathode (discharged)‖cathode (charged) half cell configuration (Fig. 15b) to study the interfacial chemistry of Ca electrolytes and cathodes without the complication of the Ca anode. Even in full battery studies, the anode free configuration (cathode‖Cu or SS, Fig. 15c) is also recommended to report more reliable cycling performance of a Ca battery, which has been advocated for Li battery studies.
Fourth, regarding new electrolyte development, a fundamental, effective strategy to improve electrolyte performance is to engineer the chemical structure of electrolyte active species as evident from the recent advances of the Ca[B(hfip)4]2 electrolytes. One is to modulate the first sphere coordination structure of Ca2+ ions using different solvents and additives. Another strategy is to design chemically and electrochemically stable counter anions. It is worth noting that the interplay of Ca2+ cation and counter anions is very important to achieve a stable, highly conductive SEI (Fig. 11e and f). For example, strong solvation can avoid the formation of the cation–anion ion pair to stabilize anions from excessive decomposition. In contrast, a slight degree of the formation of the cation–anion ion pair could be beneficial to enable controlled decomposition of anions to form a stable SEI that can inhibit further electrolyte decomposition. In addition, additives that are commonly applied to improve the energy storage performance of Li ion batteries can also be highly effective in modulating and improving the chemical components and performance of the SEI for the Ca anode. A good Ca2+ electrolyte should be simultaneously compatible with both the Ca anode and a high redox potential cathode material.
In conclusion, successful demonstration of reversible Ca deposition at room temperature has been achieved with the rapid development of Ca2+ electrolytes in the last few years. There are many opportunities ahead for experimental and theoretical scientists to develop new Ca2+ electrolytes, understand electrolyte/electrode interfacial chemistry, and discover cathode materials. It is firmly believed that understanding Ca2+ electrolyte chemistry is key to developing practical Ca batteries and can also be beneficial to the development of other metal batteries.
Author contributions
T. Leo Liu conceived the writing plan of this review manuscript. Qianshun Wei drafted the mansucript. All authors revised the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are thankful for the Utah State University faculty startup fund and Utah Science Technology and Research initiative (USTAR) research awards for supporting our multivalent battery research. X. S. acknowledges the support by the 111 Project of the Ministry of Education of China (D20015).References
- J. B. Goodenough and K.-S. Park, The Li-Ion Rechargeable Battery: A Perspective, J. Am. Chem. Soc., 2013, 135(4), 1167–1176 CrossRef CAS PubMed.
- M. S. Whittingham, Lithium Batteries and Cathode Materials, Chem. Rev., 2004, 104(10), 4271–4302 CrossRef CAS PubMed.
- J. Luo, B. Hu, M. Hu, Y. Zhao and T. L. Liu, Status and Prospects of Organic Redox Flow Batteries towards Sustainable Energy Storage, ACS Energy Lett., 2019, 4, 2220–2240 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Electrical Energy Storage for the Grid: A Battery of Choices, Science, 2011, 334(6058), 928–935 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Towards greener and more sustainable batteries for electrical energy storage, Nat. Chem., 2015, 7(1), 19–29 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Quest for Nonaqueous Multivalent Secondary Batteries: Magnesium and Beyond, Chem. Rev., 2014, 114, 11683–11720 CrossRef CAS PubMed.
- S. He, K. V. Nielson, J. Luo and T. L. Liu, Recent advances on MgCl2 based electrolytes for rechargeable Mg batteries, Energy Storage Materials, 2017, 8, 184–188 CrossRef.
- K. Xu, Electrolytes and Interphases in Li-Ion Batteries and Beyond, Chem. Rev., 2014, 114(23), 11503–11618 CrossRef CAS.
- A. Ponrouch, J. Bitenc, R. Dominko, N. Lindahl, P. Johansson and M. R. Palacin, Multivalent rechargeable batteries, Energy Storage Materials, 2019, 20, 253–262 CrossRef.
- R. G. Parr and R. G. Pearson, Absolute hardness: companion parameter to absolute electronegativity, J. Am. Chem. Soc., 1983, 105(26), 7512–7516 CrossRef CAS.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Odyssey of Multivalent Cathode Materials: Open Questions and Future Challenges, Chem. Rev., 2017, 117(5), 4287–4341 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Prototype Systems for Rechargeable magnesium Batteries, Nature, 2000, 407(6805), 724–727 CrossRef CAS PubMed.
- K. V. Nielson and T. L. Liu, Dawn of Calcium Batteries, Angew. Chem., Int. Ed., 2020, 59(9), 3368–3370 CrossRef CAS PubMed.
- I. D. Hosein, The Promise of Calcium Batteries: Open Perspectives and Fair Comparisons, ACS Energy Lett., 2021, 6(4), 1560–1565 CrossRef CAS.
- G. E. McManis, M. H. Miles and A. N. Fletcher, Performance and Discharge Characteristics of Ca/LiCl, LiNO3/LiNO3, AgNO3/Ni Thermal Battery Cells, J. Electrochem. Soc., 1984, 131(2), 283–286 CrossRef CAS.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder and K. A. Persson, Spinel compounds as multivalent battery cathodes: a systematic evaluation based on ab initio calculations, Energy Environ. Sci., 2015, 8(3), 964–974 RSC.
- S. M. Selis, J. P. Wondowski and R. F. Justus, A High-Rate, High-Energy Thermal Battery System, J. Electrochem. Soc., 1964, 111(1), 6 CrossRef CAS.
- R. J. Staniewicz, A Study of the Calcium-Thionyl Chloride Electrochemical System, J. Electrochem. Soc., 1980, 127(4), 782–789 CrossRef CAS.
- A. Meitav and E. Peled, Solid Electrolyte Interphase (SEI) Electrode: II. The Formation and Properties of the SEI on Magnesium in Solutions, J. Electrochem. Soc., 1981, 128(4), 825–831 CrossRef CAS.
- E. Peled, A. Meitav and M. Brand, Calcium Thionyl Chloride High-Rate Reserve Cell, J. Electrochem. Soc., 1981, 128(9), 1936–1938 CrossRef CAS.
- A. Meitav and E. Peled, Calcium – Ca (AlCl4) 2 – Thionyl Chloride Cell: Performance and Safety, J. Electrochem. Soc., 1982, 129(3), 451–457 CrossRef CAS.
- D. Aurbach, R. Skaletsky and Y. Gofer, The Electrochemical Behavior of Calcium Electrodes in a Few Organic Electrolytes, J. Electrochem. Soc., 1991, 138(12), 3536–3545 CrossRef CAS.
- A. Ponrouch, C. Frontera, F. Bardé and M. R. Palacín, Towards a calcium-based rechargeable battery, Nat. Mater., 2016, 15(2), 169–172 CrossRef CAS PubMed.
- S. Biria, S. Pathreeker, H. Li and I. D. Hosein, Plating and Stripping of Calcium in an Alkyl Carbonate Electrolyte at Room Temperature, ACS Appl. Energy Mater., 2019, 2(11), 7738–7743 CrossRef CAS.
- J. Forero-Saboya, C. Davoisne, R. Dedryvère, I. Yousef, P. Canepa and A. Ponrouch, Understanding the nature of the passivation layer enabling reversible calcium plating, Energy Environ. Sci., 2020, 13(10), 3423–3431 RSC.
- D. Wang, X. Gao, Y. Chen, L. Jin, C. Kuss and P. G. Bruce, Plating and stripping calcium in an organic electrolyte, Nat. Mater., 2018, 17(1), 16–20 CrossRef CAS PubMed.
- K. Ta, R. Zhang, M. Shin, R. T. Rooney, E. K. Neumann and A. A. Gewirth, Understanding Ca Electrodeposition and Speciation Processes in Nonaqueous Electrolytes for Next-Generation Ca-Ion Batteries, ACS Appl. Mater. Interfaces, 2019, 11(24), 21536–21542 CrossRef CAS PubMed.
- Y. Jie, Y. Tan, L. Li, Y. Han, S. Xu, Z. Zhao, R. Cao, X. Ren, F. Huang, Z. Lei, G. Tao, G. Zhang and S. Jiao, Electrolyte Solvation Manipulation Enables Unprecedented Room-Temperature Calcium-Metal Batteries, Angew. Chem., Int. Ed., 2020, 59(31), 12689–12693 CrossRef CAS PubMed.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, An Efficient Halogen-Free Electrolyte for Use in Rechargeable Magnesium Batteries, Angew. Chem., Int. Ed., 2015, 54(27), 7900–7904 CrossRef CAS PubMed.
- S. G. McArthur, L. Geng, J. Guo and V. Lavallo, Cation reduction and comproportionation as novel strategies to produce high voltage, halide free, carborane based electrolytes for rechargeable Mg batteries, Inorg. Chem. Front., 2015, 2(12), 1101–1104 RSC.
- S. G. McArthur, R. Jay, L. Geng, J. Guo and V. Lavallo, Below the 12-vertex: 10-vertex carborane anions as non-corrosive, halide free, electrolytes for rechargeable Mg batteries, Chem. Commun., 2017, 53(32), 4453–4456 RSC.
- N. T. Hahn, T. J. Seguin, K.-C. Lau, C. Liao, B. J. Ingram, K. A. Persson and K. R. Zavadil, Enhanced Stability of the Carba-closo-dodecaborate Anion for High-Voltage Battery Electrolytes through Rational Design, J. Am. Chem. Soc., 2018, 140(35), 11076–11084 CrossRef CAS PubMed.
- J. T. Herb, C. A. Nist-Lund and C. B. Arnold, A Fluorinated Alkoxyaluminate Electrolyte for Magnesium-Ion Batteries, ACS Energy Lett., 2016, 1(6), 1227–1232 CrossRef CAS.
- Z. Zhao-Karger, M. E. Gil Bardaji, O. Fuhr and M. Fichtner, A new class of non-corrosive, highly efficient electrolytes for rechargeable magnesium batteries, J. Mater. Chem. A, 2017, 5(22), 10815–10820 RSC.
- J. Wen, W. Shi, F. Zhang, D. Liu, S. Tang, H. Wang, X.-M. Lin and A. Lei, Electrooxidative Tandem Cyclization of Activated Alkynes with Sulfinic Acids To Access Sulfonated Indenones, Org. Lett., 2017, 19(12), 3131–3134 CrossRef CAS PubMed.
- J. Luo, Y. Bi, L. Zhang, X. Zhang and T. L. Liu, A Stable, Non-Corrosive Perfluorinated Pinacolatoborate Mg Electrolyte for Rechargeable Mg Batteries, Angew. Chem., Int. Ed., 2019, 58(21), 6967–6971 CrossRef CAS PubMed.
- K.-C. Lau, T. J. Seguin, E. V. Carino, N. T. Hahn, J. G. Connell, B. J. Ingram, K. A. Persson, K. R. Zavadil and C. Liao, Widening Electrochemical Window of Mg Salt by Weakly Coordinating Perfluoroalkoxyaluminate Anion for Mg Battery Electrolyte, J. Electrochem. Soc., 2019, 166(8), A1510–A1519 CrossRef CAS.
- Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Towards stable and efficient electrolytes for room-temperature rechargeable calcium batteries, Energy Environ. Sci., 2019, 12, 3496–3501 RSC.
- A. Shyamsunder, L. E. Blanc, A. Assoud and L. F. Nazar, Reversible Calcium Plating and Stripping at Room Temperature Using a Borate Salt, ACS Energy Lett., 2019, 4(9), 2271–2276 CrossRef CAS.
- K. V. Nielson, J. Luo and T. L. Liu, Optimizing Calcium Electrolytes by Solvent Manipulation for Calcium Batteries, Batteries Supercaps, 2020, 3(8), 766–772 CrossRef CAS.
- S. Hou, X. Ji, K. Gaskell, P.-f. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Solvation sheath reorganization enables divalent metal batteries with fast interfacial charge transfer kinetics, Science, 2021, 374(6564), 172–178 CrossRef CAS PubMed.
- F. S. Genier, C. V. Burdin, S. Biria and I. D. Hosein, A novel calcium-ion solid polymer electrolyte based on crosslinked poly(ethylene glycol) diacrylate, J. Power Sources, 2019, 414, 302–307 CrossRef CAS.
- J. Wang, F. S. Genier, H. Li, S. Biria and I. D. Hosein, A Solid Polymer Electrolyte from Cross-Linked Polytetrahydrofuran for Calcium Ion Conduction, ACS Appl. Polym. Mater., 2019, 1(7), 1837–1844 CrossRef CAS.
- C. S. Martinez-Cisneros, A. Fernandez, C. Antonelli, B. Levenfeld, A. Varez, K. Vezzù, V. Di Noto and J. Y. Sanchez, Opening the door to liquid-free polymer electrolytes for calcium batteries, Electrochim. Acta, 2020, 353, 136525 CrossRef CAS.
- S. Biria, S. Pathreeker, F. S. Genier and I. D. Hosein, A Highly Conductive and Thermally Stable Ionic Liquid Gel Electrolyte for Calcium-Ion Batteries, ACS Appl. Polym. Mater., 2020, 2(6), 2111–2118 CrossRef CAS.
- J. Park, Z.-L. Xu, G. Yoon, S. K. Park, J. Wang, H. Hyun, H. Park, J. Lim, Y.-J. Ko, Y. S. Yun and K. Kang, Stable and High-Power Calcium-Ion Batteries Enabled by Calcium Intercalation into Graphite, Adv. Mater., 2020, 32(4), 1904411 CrossRef CAS PubMed.
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H.-M. Cheng, Reversible calcium alloying enables a practical room-temperature rechargeable calcium-ion battery with a high discharge voltage, Nat. Chem., 2018, 10(6), 667–672 CrossRef CAS PubMed.
- X. Zeng, G.-L. Xu, Y. Li, X. Luo, F. Maglia, C. Bauer, S. F. Lux, O. Paschos, S.-J. Kim, P. Lamp, J. Lu, K. Amine and Z. Chen, Kinetic Study of Parasitic Reactions in Lithium-Ion Batteries: A Case Study on LiNi0.6Mn0.2Co0.2O2, ACS Appl. Mater. Interfaces, 2016, 8(5), 3446–3451 CrossRef CAS PubMed.
- R. Petibon, N. N. Sinha, J. C. Burns, C. P. Aiken, H. Ye, C. M. VanElzen, G. Jain, S. Trussler and J. R. Dahn, Comparative study of electrolyte additives using electrochemical impedance spectroscopy on symmetric cells, J. Power Sources, 2014, 251, 187–194 CrossRef CAS.
- Y. Liu, I. Hamam and J. R. Dahn, A Study of Vinylene Carbonate and Prop-1-ene-1,3 Sultone Electrolyte Additives Using Polycrystalline Li[Ni0.6Mn0.2Co0.2]O2 in Positive/Positive Symmetric Cells, J. Electrochem. Soc., 2020, 167(11), 110527 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |