
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular photochemical [2 + 1]-cycloadditions of nucleophilic siloxy carbenes†
Amanda
Bunyamin
a,
Carol
Hua
 ab,
Anastasios
Polyzos
ac and
Daniel L.
Priebbenow
*ad
ab,
Anastasios
Polyzos
ac and
Daniel L.
Priebbenow
*ad
aSchool of Chemistry, University of Melbourne, Parkville, Victoria, 3010 Australia. E-mail: daniel.priebbenow@unimelb.edu.au
bSchool of Life and Environmental Sciences, Deakin University, Waurn Ponds, Victoria, 3216 Australia
cCSIRO Manufacturing, Clayton, Victoria, 3168 Australia
dDepartment of Medicinal Chemistry, Monash Institute of Pharmaceutical Science, Monash University, Parkville, Victoria, 3052 Australia
First published on 25th February 2022
Abstract
Visible light induced singlet nucleophilic carbenes undergo rapid [2 + 1]-cycloaddition with tethered olefins to afford unique bicyclo[3.1.0]hexane and bicyclo[4.1.0]heptane scaffolds. This cyclopropanation process requires only visible light irradiation to proceed, circumventing the use of exogenous (photo)catalysts, sensitisers or additives and showcases a vastly underexplored mode of reactivity for nucleophilic carbenes in chemical synthesis. The discovery of additional transformations including a cyclopropanation/retro-Michael/Michael cascade process to afford chromanones and a photochemical C–H insertion reaction are also described.
Introduction
Cyclopropane derivatives including donor–acceptor cyclopropanes and cyclopropanols are valuable building blocks in chemical synthesis.1 These three-membered carbocyclic rings are widely prevalent in natural products for example peyssonnoside A (Fig. 1).2 Cyclopropanes also play a valuable role in medicinal chemistry,3 as exemplified by the potent indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor recently reported by Hamilton and co-workers who highlighted the significant advantages that conformationally constrained bicyclo[3.1.0]hexane scaffolds offered in terms of improving metabolic stability and limiting off-target effects when used as a cyclohexane isostere (Fig. 1).4 | ||
| Fig. 1 Cyclopropanes are a valued motif in natural products and medicinal chemistry however relative to electrophilic singlet carbenes, the [2 + 1]-cycloaddition reaction of olefins with nucleophilic carbenes remains vastly underexplored. | ||
Common strategies to construct cyclopropanes include the Johnson–Corey–Chaykovsky or Simmons–Smith reaction.5 However, the [2 + 1]-cycloaddition of olefins with carbenes generated from diazo derivatives remains the most widely employed cyclopropanation strategy.6 Such cycloadditions typically involve reaction of alkenes with electrophilic carbene (or carbenoid) intermediates formed from diazoacetates or hydrazones in the presence of catalytic rhodium, iron, copper and cobalt complexes or boron derivatives.7
In recent years, more economical and sustainable catalyst-free methods for chemical synthesis have emerged employing visible light irradiation to generate reactive intermediates in the absence of photocatalysts or photosensitisers.8 Within this context, catalyst-free [2 + 1]-photocycloadditions of olefins with carbenes generated from diazo compounds have been developed.9 For example, in 2018 Davies first demonstrated that electrophilic carbenes generated photochemically from diazoacetates underwent [2 + 1]-cycloaddition with unsaturated carbocycles and heterocycles (Fig. 1).10 Subsequently, the visible-light induced cyclopropanation of indoles using aryl(diazo)acetates was reported by Zhang and co-workers,11 and the Koenigs group discovered a [2 + 1]-photocycloaddition of olefins using donor–acceptor diazoalkanes or tosyl hydrazone reagents in both batch and continuous flow.12 Building on these pioneering works, in 2021, Barham and co-workers reported the visible-light included cyclopropanation of heteroarenes in continuous flow using dimethyl carbonate as a more environmentally friendly solvent.13
A limitation, however, for both the metal-catalyzed and photochemical [2 + 1]-cycloaddition reactions developed to date is the requirement that carbene intermediates be generated from unstable and toxic diazo reagents that typically require electron-withdrawing groups for stability. To circumvent this requirement, new catalyst- and diazo-free photochemical strategies have been recently discovered involving the cyclopropanation of styrenes via the photochemical generation of iodomethyl carbonyl radicals from diiodoacetates,14 diradicals from iodonium ylides,15 or pyridyl carbenes from triazoles (Fig. 1).16
It is known that nucleophilic siloxy carbene intermediates—generated photochemically from bench-stable acyl silanes via visible light irradiation—are valuable intermediates in chemical synthesis, participating in reactions including boron–hydrogen insertion and 1,2-carbonyl addition.17 We thus questioned whether a new catalyst- and diazo-free cyclopropanation strategy might be accessible via the [2 + 1]-cycloaddition reactions of visible light induced siloxy carbenes with olefins. It was envisaged that such a strategy would afford access to unique cyclopropanol derivatives highly amenable to subsequent functionalization.
Whilst the cyclopropenation of alkynes with siloxy carbenes is known (affording strained cyclopropenes that undergo spontaneous ring-opening and silicon transfer to afford vinyl silanes),17g,h only two isolated examples of the cyclopropanation of nucleophilic siloxy carbenes exist, both involving [2 + 1]-photocycloaddition with diethyl fumarate as reported by Brook in 1971 and Dalton in 1981.18 In fact, relative to electrophilic carbenes the cyclopropanation of nucleophilic carbenes is vastly underexplored and has been seldom reported.18,19 To address this, we herein report that visible light induced nucleophilic carbenes undergo rapid and stereospecific catalyst- and diazo-free [2 + 1]-cycloadditions with olefins including styrene, acrylamide, and vinyl phosphonate derivatives,20 showcasing a significantly underexplored reaction mode for nucleophilic carbenes.21
Results and discussion
To begin, we investigated the intermolecular cyclopropanation of phenethyl acyl silane 1a with olefins including styrene, ethyl acrylate, beta-nitrostyrene, N-benzylmaleimide and dimethyl fumarate using visible light irradiation at 427 nm (a wavelength commonly used to excite both aryl and alkyl acyl silanes).17d,j However, the only olefin sufficiently activated enough to undergo intermolecular cyclopropanation was the fumarate possessing two electron withdrawing groups on the alkene, yielding trans-cyclopropane 2 in 84% yield (Scheme 1). This lack of reactivity for many of the alkenes correlates with that observed previously for the intermolecular [2 + 1]-cycloaddition of siloxy carbenes with alkynes,17g,h where two electron withdrawing groups were required to sufficiently lower the LUMOalkene energy to enable interaction with the filled carbene sp2-orbital (HOMOcarbene).22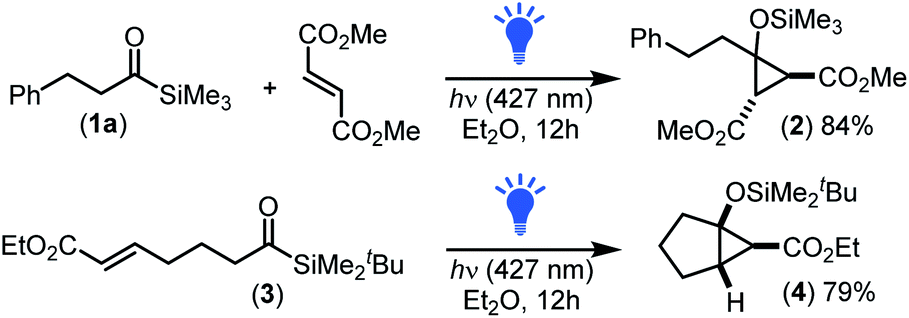 | ||
| Scheme 1 Carbenes generated from alkyl acyl silanes participated in both inter- and intramolecular photochemical [2 + 1]-cycloaddition processes. | ||
To further investigate the viability of a [2 + 1]-cycloaddition of siloxy carbenes with olefins other than fumarate, we next explored an intramolecular approach. This involved preparation and subsequent photochemical irradiation of acrylate derivative 3 (427 nm LED, 40 W) which afforded bicyclo[3.1.0]hexane 4 in good yield with exclusive formation of the exo-isomer (Scheme 1), demonstrating for the first time that the [2 + 1]-photocycloaddition of nucleophilic carbenes proceeded efficiently for olefins other than fumarates.
Building on this outcome, we considered that phenol or aniline derived aroyl silanes would best facilitate an in-depth investigation into this cyclopropanation process enabling rapid variation of the olefin tether using commercially available reagents and a common starting material. We thus prepared a sulfonamide derived benzoyl silane containing an electron-deficient olefin (7a).23 Exposure of a solution of aroyl silane 7a in diethyl ether to visible light irradiation (427 nm LED, 40 W) led to a colour change from bright yellow to colourless after only 10 minutes, affording tert-butyldimethylsiloxy azabicyclo[3.1.0]hexane (9a), readily identifiable by 1H NMR analysis from the distinctive cyclopropane C–H resonances at δ = 4.71 ppm and δ = 0.82 ppm.
Optimisation of the reaction conditions confirmed that several solvents were suitable including dimethyl carbonate, however diethyl ether remained the preferred choice (Table 1). X-ray crystallography of 9a (CCDC 2121946†) confirmed the structure (Scheme 2a), with the exo-isomer formed as the sole isomer as indicated by both NMR and X-ray analysis. Subsequently, ortho-sulfonamido aroyl silanes were reacted with various propiolates to afford a series of acyl silanes containing olefin tethers (7).
|
|
||
|---|---|---|
| Entry | Solvent | Conversionb (%) |
| a Reaction conducted on 0.2 mmol scale in 1 mL of solvent. b Conversion of acyl silane to product as assessed by 1H NMR. c 4 Å molecular sieves included in reaction vial. d Reaction conducted in the dark. e Isolated yield. | ||
| 1 | Et2O | 96% |
| 2 | CH2Cl2 | 92% |
| 3 | THFc | 91% |
| 4 | CHCl3c | 87% |
| 5 | MeCNc | 94% |
| 6 | Toluenec | 83% |
| 7 | Et2Oc | 99% (89%)e |
| 8 | Et2Oc,d | 0% |
| 9 | CH2Cl2c | 97% |
| 10 | Dimethyl carbonatec | 97% |

 | ||
| Scheme 2 A novel photochemical [2 + 1]-cycloaddition of nucleophilic carbenes was developed to access to a series of cyclopropyl-fused heterocyclic scaffolds including (a) azabicyclo[3.1.0]hexanes (b) oxabicyclo[4.1.0]heptanes and (c) azabicyclo[4.1.0]heptanes. Crystal structures of 9a (CCDC 2121946†), 10k (CCDC 2121948†) and 10ab (CCDC 2121949†) depicted at 50% probability (hydrogen atoms omitted for clarity): carbon (grey), oxygen (red), nitrogen (blue), sulfur (orange) and silicon (yellow). aReaction time of 2 hours required. | ||
Visible light irradiation of these substrates (427 nm LED, 40 W) produced a series of azabicyclo[3.1.0]hexanes (9a–9i, Scheme 2a). In each case, the exo-diastereoisomer was produced as the sole diastereoisomer, with variations in the silyl, aryl, and ester functional groups well tolerated. Advantageously, this cyclopropanation process is complete in less than 10 minutes and is operationally simple, requiring only visible light irradiation, circumventing the use of exogenous catalysts, or additives including photosensitisers.
To further probe the [2 + 1]-photocycloaddition, we set out to vary the heteroatom and extend the tether length to investigate if bicyclo[4.1.0]heptane frameworks were also accessible. To achieve this, we employed salicylaldehyde derived acyl silanes that were O-alkylated via reaction with various bromocrotonate derivatives. Subsequent irradiation of the functionalised acyl silanes (8) with visible light (427 nm LED, 40 W) afforded oxabicyclo[4.1.0]heptane scaffolds (10a–10k, Scheme 2b) with variation in the structure of both the crotonate ester group and benzoyl silane well tolerated. The oxabicyclo[4.1.0]heptane products were readily identified by 1H NMR analysis from the distinctive cyclopropane C–H resonances at δ = 2.60 ppm and δ = 2.20 ppm (J = 5.8 Hz). X-ray crystallographic analysis of 10k (CCDC 212194†) confirmed the structure and exo-configuration.
With the reaction performing well for ester derived electron deficient alkenes, we next explored the influence of alternative alkene substituents. Neutral olefins situated within allyl, methyl allyl and dimethyl allyl tethers all underwent photochemical cyclopropanation reaction with visible light induced nucleophilic carbenes, however these less activated alkenes required reaction times of up to 2 hours to achieve complete conversion (Scheme 2b, 10l–10n, 70–82%). The orbital interaction required to drive [2 + 1]-cycloaddition for less activated alkenes is that between the filled alkene π-orbital (HOMOalkene) and the vacant carbene 2p-orbital (LUMOcarbene). Thus, the fact that these relatively electron-rich olefins undergo cycloaddition with siloxy carbenes at all is impressive, given that the LUMOcarbene energy is considerably higher due to interaction of the oxygen atom's non-bonding valence electrons with the unoccupied 2p carbene orbital.17b
To further explore reactivity with less activated alkenes, styrene derivative 8r was prepared via alkylation of the acyl silane with cinnamyl bromide and again visible light irradiation (427 nm LED, 40 W) afforded cyclopropanol 10r in excellent yield (89%) in under 10 minutes. A cyclohexyl derived olefin tether was also installed which underwent cyclopropanation to afford spirocyclic derivative 10p (75%) generating two new ring systems and contiguous quaternary carbon centres (Scheme 2). Of note, the ability of nucleophilic carbenes to undergo cyclopropanation with relatively electron rich olefins produces highly substituted cyclopropanols that are not accessible using traditional diazo-based cyclopropanation strategies (where electron-withdrawing groups are typically required to stabilise both the diazo reagent and electrophilic carbene).
Further variation of the alkene substitution pattern revealed that phosphonate (10q, 83%) and amide (10r, 77%) derived cyclopropane frameworks were readily accessible via the photocycloaddition of nucleophilic carbenes and that tri-substituted alkenes could also be employed to yield penta-substituted cyclopropanes 10s (79%) and 10t (92%).
The alkylation of phenol derived acyl silanes with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (E)/(Z) mixture of 4-bromobut-2-enenitrile enabled us to gain additional insight into the reaction process as the (E)- and (Z)-alkenyl acyl silanes were readily separable. Irradiation of each of these substrates (427 nm LED, 40 W) resulted in rapid [2 + 1]-cycloaddition with the (E)-alkene leading to exclusive formation of the exo-isomer 10u while the (Z)-alkene afforded the endo-isomer as the major product 10v (Scheme 2b). This outcome infers that mechanistically, this [2 + 1]-cycloaddition occurs in a concerted and highly stereospecific fashion.
:1 (E)/(Z) mixture of 4-bromobut-2-enenitrile enabled us to gain additional insight into the reaction process as the (E)- and (Z)-alkenyl acyl silanes were readily separable. Irradiation of each of these substrates (427 nm LED, 40 W) resulted in rapid [2 + 1]-cycloaddition with the (E)-alkene leading to exclusive formation of the exo-isomer 10u while the (Z)-alkene afforded the endo-isomer as the major product 10v (Scheme 2b). This outcome infers that mechanistically, this [2 + 1]-cycloaddition occurs in a concerted and highly stereospecific fashion.
The opportunity to access azabicyclo[4.1.0]heptanes (Scheme 2c) was next explored. To achieve this, ortho-sulfonamido acyl silanes were alkylated using bromocrotonate derivatives with subsequent visible light irradiation (427 nm LED, 40 W) producing a series of azabicyclo[4.1.0]heptanes (10w–10ag, Scheme 2c). The cyclopropane adducts were identified by 1H NMR analysis from the distinctive cyclopropane C–H resonances at δ = 2.60 ppm and δ = 1.30 ppm (J = 5.6 Hz). Variations within the silyl group, aryl ring and alkene substituents were well tolerated to afford tetra- or penta-substituted cyclopropanes using only visible-light irradiation.
The structure and configuration of azabicyclo[4.1.0]heptane was again confirmed via X-ray crystallographic analysis of 10ab (CCDC 2121949†) and 10w (CCDC 2121947†).24 Furthermore, the use of the 1:1 (E)/(Z) mixture of 4-bromobut-2-enenitrile afforded access to both the endo and exo isomers of the cyano-derived cyclopropane derivatives 10af and 10ag (Scheme 2c). Intriguingly, it was observed that in deuterated chloroform the kinetically favoured endo-product 10ag produced from the (Z)-cyano-alkene isomerised over time to the thermodynamically favoured exo-isomer 10af (proposedly via a ring opening/closing process).24
During investigations into the cyclopropanation of less activated alkenes (to produce 10l–10n), up to 20% conversion of the acyl silane to the dihydrobenzofuran product arising from C–H insertion of the carbene into the allylic CH2 moiety was observed via NMR analysis. A related transformation involving C–H insertion of thermally generated siloxy carbenes (250 °C, microwave irradiation) to afford 2-phenyldihydrobenzofurans 13 was reported by the Dong group in 2009 with the cis-diastereoisomer formed as the major product (Scheme 3a).25
 | ||
| Scheme 3 (a) A highly diastereoselective C–H insertion process of visible-light induced carbenes was also discovered; (b) ring expansion via reaction with hydrazine afforded new heterocyclic scaffolds. | ||
Supported by computational analysis, the Wang group subsequently proposed that Dong's thermal carbene insertion process proceeded via an excited singlet carbene intermediate via abstraction of a hydrogen atom from the proximal benzylic position that afforded a diradical that could undergo diradical coupling to afford cis-dihydrofuran 13.26
Building on our observation that photochemically generated siloxy carbenes could undergo C–H insertion, we were intrigued by the possibility of replicating the process reported by Dong using visible light irradiation at ambient temperature. To explore this, we prepared benzyloxy aroyl silanes 11a and 11b and following irradiation with visible light (427 nm LED, 40 W, 6 h), we successfully isolated the corresponding 2-phenyldihydrofurans 12a (89% yield) and 12b (64% yield) (Scheme 3a). Intriguingly, the major product formed via our photochemical reaction in both cases was the trans-diastereomer in a ratio of 10:1, inferring that the photochemical process occurs via an alternative mechanism to the thermal process reported by Dong, an observation currently under further investigation within our group.
As is the case for donor–acceptor cyclopropanes, ring opening and expansion protocols exploiting the inherent ring strain of bicyclo[3.1.0]hexane and bicyclo[4.1.0]heptane derivatives have been reported.27 To this end, we also demonstrated that both aza- and oxa-bicyclo[4.1.0]heptanes prepared using the photochemical cyclopropanation process described herein readily underwent ring expansion reaction with hydrazine to afford valuable heterocyclic frameworks (Scheme 3b).28 Due to the facile and catalyst-free nature of the [2 + 1]-photocycloaddition process, the development of additional telescoping strategies to rapidly generate molecular complexity are currently under investigation.
During initial studies into the cyclopropanation of salicylaldehyde derived acyl silanes, acyl silanes 7j and 7k containing an electron deficient olefin were prepared via the DABCO catalysed reaction of the phenolic –OH with a propiolate.23 Visible-light irradiation (427 nm LED, 40 W) of a solution of diethyl ether containing acyl silane 7j led to a colour change from bright yellow to colourless after only 10 minutes. Analysis of the reaction mixture by NMR spectroscopy indicated that no cyclopropane was present and instead complete conversion of the acyl silane to a new product had occurred.
Further analysis revealed the structure of the new product to be that of the silyl enol ether derived pyran 16a (Scheme 4a). Formation of this product proposedly occurs via a photochemical cascade reaction initiated by rapid cyclopropanation of the carbene with the proximal alkene, followed by ring opening that generates an enolate intermediate that drives a retro-oxa-Michael reaction to afford a phenolate that undergoes Michael addition onto the activated acceptor producing 16a A related retro-Michael/Michael addition mechanism was previously reported by Tang and co-workers who in 2006 reported the tetrahydrothiophene catalysed synthesis of 2H-chromenes from benzyl bromides containing tethered acrylates.29 Subsequent hydrolysis of silyl enol ether 16a during silica-gel chromatography afforded chromanone 17a (Scheme 4a). Acyl silane 7k prepared from ethyl propiolate also underwent the photochemical cascade process to afford 16b and following hydrolysis, 17b.
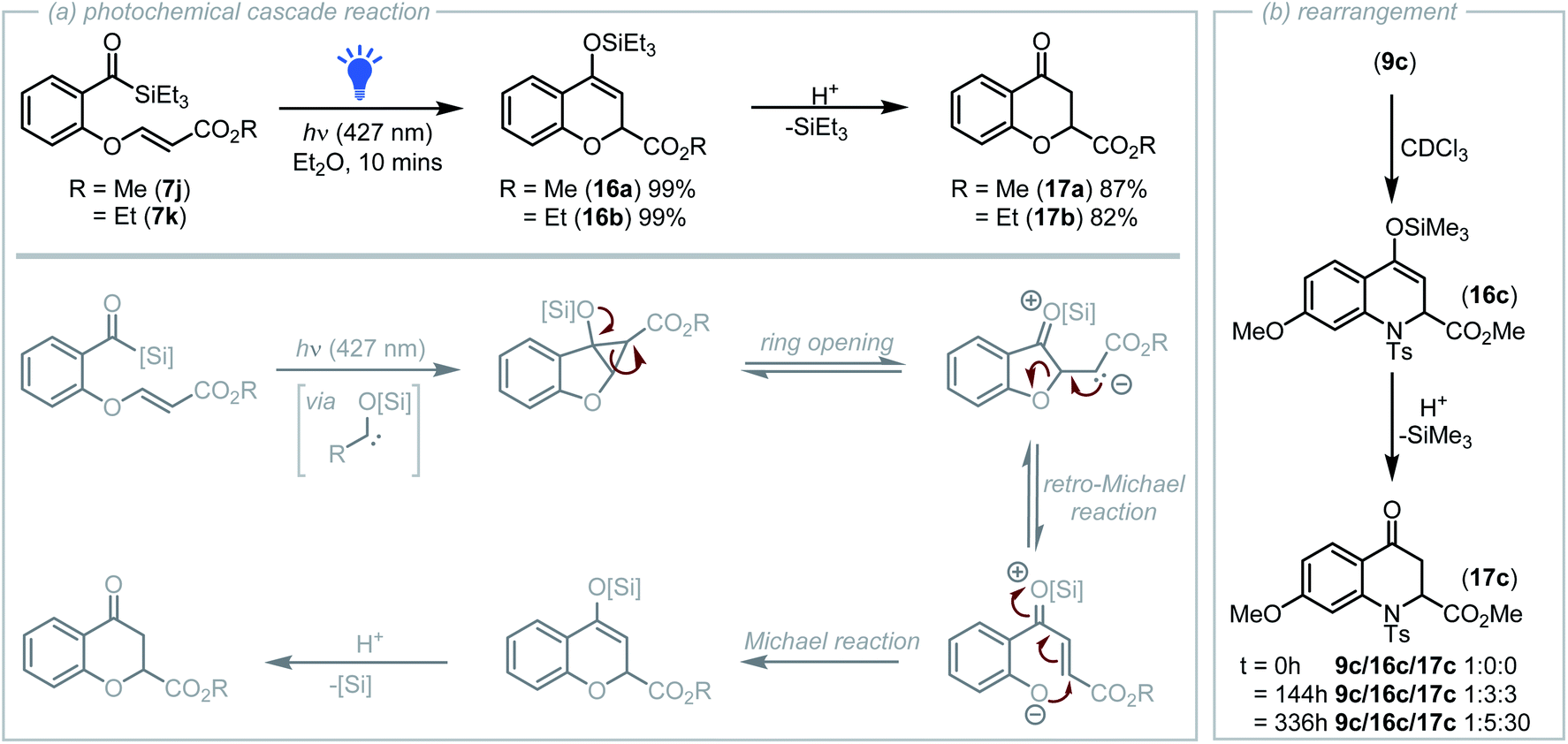 | ||
| Scheme 4 (a) A unique photochemical cascade reaction involving cyclopropanation, ring opening, retro-Michael and Michael addition to afford new heterocyclic derivatives was discovered. (b) In deuterated chloroform, cyclopropane 9c also gradually underwent a related ring opening, retro-Michael, Michael addition rearrangement process. | ||
For the photochemical cascade reaction outlined in Scheme 4a, it could be considered that the first step of this process is Michael-type addition of the nucleophilic carbene to the alkene rather than cyclopropanation. However, gradual isomerisation of the corresponding tosylamido cyclopropane 9c to quinoline 16c in deuterated chloroform and subsequent hydrolysis to afford 17c was observed by NMR analysis (Scheme 4b).24 This isomerisation proposedly occurs via the analogous ring-opening, retro-aza-Michael/Michael addition process, inferring that the first step of the rapid photochemical cascade reaction that produces 16a and 16b is indeed cyclopropanation.
Conclusions
It was discovered that visible light induced nucleophilic carbene intermediates underwent rapid [2 + 1]-cycloaddition with varying olefins generating valuable bicyclo[3.1.0]hexane and bicyclo[4.1.0]heptane scaffolds with simultaneous formation of two new ring systems (and in some cases contiguous quaternary carbon centres). Advantageously, this highly stereospecific reaction requires only visible light irradiation avoiding the requirement for exogenous additives, sensitisers or (photo)catalysts to afford a unique class of silicon-derived donor–acceptor cyclopropanes.Owing to the inherent nucleophilicity of the siloxy carbene intermediates, the reaction was highly compatible with electron-deficient olefins including acrylates, acrylamides, and vinyl phosphonates, yet also proceeded with less activated olefins including a cyclohexylidene derivative to afford a spirocyclic framework. Investigations into the reactivity of visible light induced nucleophilic carbenes also unveiled a novel photochemical C–H insertion process to afford 2-phenyldihydrofurans with high diastereoselectivity, and a unique photochemical cascade reaction involving sequential cyclopropanation/ring-opening/retro-Michael/Michael addition steps.
Overall, it is anticipated that the discoveries described herein will provide valuable insight into the reactivity of singlet nucleophilic carbenes and pave the way for the development of new synthetic transformations that capitalize on the ability of nucleophilic carbene intermediates to participate in synthetically useful catalyst- and diazo-free [2 + 1]-photocycloaddition processes.
Data availability
All the relevant data is contained within the ESI.†Author contributions
A. B. and D. L. P. performed the experiments, C. H. conducted the X-ray crystallographic analysis, A. P. and D. L. P. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. L. P. acknowledges the support of the Australian Research Council (DE200100949). A. P. acknowledges the University of Melbourne and CSIRO for the joint Establishment Grant and the Australian Research Council (IC1701000020). This research was undertaken in part using the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.Notes and references
- (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS PubMed; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed; (c) K. Ghosh and S. Das, Org. Biomol. Chem., 2021, 19, 965–982 RSC; (d) D. B. Werz and A. T. Biju, Angew. Chem., Int. Ed., 2020, 59, 3385–3398 CrossRef CAS PubMed; (e) T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3–79 CrossRef CAS PubMed; (f) L. R. Mills and S. A. L. Rousseaux, Eur. J. Org. Chem., 2019, 2019, 8–26 CrossRef CAS; (g) O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS PubMed; (h) A. Nikolaev and A. Orellana, Synthesis, 2016, 48, 1741–1768 CrossRef CAS; (i) O. Reiser, Isr. J. Chem., 2016, 56, 531–539 CrossRef CAS.
- (a) G. A. Chesnokov and K. Gademann, J. Am. Chem. Soc., 2021, 143, 14083–14088 CrossRef CAS PubMed; (b) M.-Y. Lyu, Z. Zhong, V. K.-Y. Lo, H. N. C. Wong and X.-S. Peng, Angew. Chem., Int. Ed., 2020, 59, 19929–19933 CrossRef CAS PubMed; (c) Y. H. Jin Wenbing and T. Gongli, Chin. J. Org. Chem., 2018, 38, 2324–2334 CrossRef; (d) L. A. Wessjohann, W. Brandt and T. Thiemann, Chem. Rev., 2003, 103, 1625–1648 CrossRef CAS PubMed; (e) D. Y. K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631–4642 RSC.
- (a) T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed; (b) S. J. Chawner, M. J. Cases-Thomas and J. A. Bull, Eur. J. Org. Chem., 2017, 2017, 5015–5024 CrossRef CAS PubMed; (c) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (d) J. Salaün, in Small Ring Compounds in Organic Synthesis VI, ed. A. de Meijere, Springer Berlin Heidelberg, Berlin, Heidelberg, 2000, pp. 1–67, DOI:10.1007/3-540-48255-5_1.
- M. M. Hamilton, F. Mseeh, T. J. McAfoos, P. G. Leonard, N. J. Reyna, A. L. Harris, A. Xu, M. Han, M. J. Soth, B. Czako, J. P. Theroff, P. K. Mandal, J. P. Burke, B. Virgin-Downey, A. Petrocchi, D. Pfaffinger, N. E. Rogers, C. A. Parker, S. S. Yu, Y. Jiang, S. Krapp, A. Lammens, G. Trevitt, M. R. Tremblay, K. Mikule, K. Wilcoxen, J. B. Cross, P. Jones, J. R. Marszalek and R. T. Lewis, J. Med. Chem., 2021, 64, 11302–11329 CrossRef CAS PubMed.
- (a) P. Helquist, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, pp. 951–997 Search PubMed; (b) H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 5323–5324 CrossRef CAS; (c) A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341–2372 CrossRef CAS PubMed.
- (a) W. Wu, Z. Lin and H. Jiang, Org. Biomol. Chem., 2018, 16, 7315–7329 RSC; (b) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS PubMed; (c) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed; (d) H. M. L. Davies and E. G. Antoulinakis, Intermolecular Metal-Catalyzed Carbenoid Cyclopropanations, in Organic Reactions, 2004, DOI:10.1002/0471264180.or057.01; (e) G. Bertrand, in Reactive Intermediate Chemistry, ed. R. A. Moss, M. S. Platz and M. Jones Jr, John Wiley & Sons, Inc., Hoboken, NJ, 2004, ch. 8 Search PubMed.
- (a) E. M. D. Allouche and A. B. Charette, Synthesis, 2019, 51, 3947–3963 CrossRef CAS; (b) A. Dasgupta, R. Babaahmadi, B. Slater, B. F. Yates, A. Ariafard and R. L. Melen, Chem, 2020, 6, 2364–2381 CrossRef CAS; (c) S. J. Hedley, D. L. Ventura, P. M. Dominiak, C. L. Nygren and H. M. L. Davies, J. Org. Chem., 2006, 71, 5349–5356 CrossRef CAS PubMed; (d) D. Marcoux, S. Azzi and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 6970–6972 CrossRef CAS PubMed; (e) M. Delgado-Rebollo, A. Prieto and P. J. Pérez, ChemCatChem, 2014, 6, 2047–2052 CrossRef CAS; (f) C. Deng, H.-K. Liu, Z.-B. Zheng, L. Wang, X. Yu, W. Zhang and Y. Tang, Org. Lett., 2017, 19, 5717–5719 CrossRef CAS PubMed; (g) H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 7697–7700 CrossRef CAS PubMed; (h) H. Wang, D. M. Guptill, A. Varela-Alvarez, D. G. Musaev and H. M. L. Davies, Chem. Sci., 2013, 4, 2844–2850 RSC; (i) C. G. Hamaker, G. A. Mirafzal and L. K. Woo, Organometallics, 2001, 20, 5171–5176 CrossRef CAS; (j) Y. Chen, J. V. Ruppel and X. P. Zhang, J. Am. Chem. Soc., 2007, 129, 12074–12075 CrossRef CAS PubMed.
- (a) M. Tavakolian and M. Hosseini-Sarvari, ACS Sustainable Chem. Eng., 2021, 9, 4296–4323 CrossRef CAS; (b) Z. Yang, Y. Liu, K. Cao, X. Zhang, H. Jiang and J. Li, Beilstein J. Org. Chem., 2021, 17, 771–799 CrossRef CAS PubMed.
- J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS.
- I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC.
- X. Zhang, C. Du, H. Zhang, X.-C. Li, Y.-L. Wang, J.-L. Niu and M.-P. Song, Synthesis, 2019, 51, 889–898 CrossRef CAS.
- (a) Y. Guo, C. Empel, C. Pei, I. Atodiresei, T. Fallon and R. M. Koenigs, Org. Lett., 2020, 22, 5126–5130 CrossRef CAS PubMed; (b) S. Jana, F. Li, C. Empel, D. Verspeek, P. Aseeva and R. M. Koenigs, Chem.–Eur. J., 2020, 26, 2586–2591 CrossRef CAS PubMed; (c) C. Empel and R. M. Koenigs, J. Flow Chem., 2020, 10, 157–160 CrossRef CAS; (d) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC; (e) Y. Guo, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2019, 21, 8814–8818 CrossRef CAS PubMed.
- V. Klöpfer, R. Eckl, J. Floß, P. M. C. Roth, O. Reiser and J. P. Barham, Green Chem., 2021, 23, 6366–6372 RSC.
- A. G. Herraiz and M. G. Suero, Chem. Sci., 2019, 10, 9374–9379 RSC.
- T. Chidley, I. Jameel, S. Rizwan, P. A. Peixoto, L. Pouységu, S. Quideau, W. S. Hopkins and G. K. Murphy, Angew. Chem., Int. Ed., 2019, 58, 16959–16965 CrossRef CAS PubMed.
- Z. Zhang, D. Yadagiri and V. Gevorgyan, Chem. Sci., 2019, 10, 8399–8404 RSC.
- (a) H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540–8571 RSC; (b) D. L. Priebbenow, Adv. Synth. Catal., 2020, 362, 1927–1946 CrossRef CAS; (c) D. L. Priebbenow, J. Org. Chem., 2019, 84, 11813–11822 CrossRef CAS PubMed; (d) D. L. Priebbenow, R. L. Pilkington, K. N. Hearn and A. Polyzos, Org. Lett., 2021, 23, 2783–2789 CrossRef PubMed; (e) P. Becker, R. Pirwerdjan and C. Bolm, Angew. Chem., Int. Ed., 2015, 54, 15493–15496 CrossRef CAS PubMed; (f) P. Becker, D. L. Priebbenow, R. Pirwerdjan and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 269–271 CrossRef CAS PubMed; (g) P. Becker, D. L. Priebbenow, H.-J. Zhang, R. Pirwerdjan and C. Bolm, J. Org. Chem., 2014, 79, 814–817 CrossRef CAS PubMed; (h) H.-J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F.-F. Pan and C. Bolm, Adv. Synth. Catal., 2012, 354, 2157–2161 CrossRef CAS; (i) J.-H. Ye, P. Bellotti, T. O. Paulisch, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 13671–13676 CrossRef CAS PubMed; (j) J.-H. Ye, L. Quach, T. Paulisch and F. Glorius, J. Am. Chem. Soc., 2019, 141, 16227–16231 CrossRef CAS PubMed; (k) K. Ishida, F. Tobita and H. Kusama, Chem.–Eur. J., 2018, 24, 543–546 CrossRef CAS PubMed; (l) K. Ishida, H. Yamazaki, C. Hagiwara, M. Abe and H. Kusama, Chem.–Eur. J., 2020, 26, 1249–1253 CrossRef CAS PubMed; (m) K. Ito, H. Tamashima, N. Iwasawa and H. Kusama, J. Am. Chem. Soc., 2011, 133, 3716–3719 CrossRef CAS PubMed; (n) J. Reimler and A. Studer, Chem.–Eur. J., 2021, 27, 15392–15395 CrossRef CAS PubMed; (o) C. Stuckhardt, M. Wissing and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 18605–18611 CrossRef CAS PubMed; (p) Z. Fan, Y. Yi, S. Chen and C. Xi, Org. Lett., 2021, 23, 2303–2307 CrossRef CAS PubMed; (q) L. Ma, Y. Yu, L. Xin, L. Zhu, J. Xia, P. Ou and X. Huang, Adv. Synth. Catal., 2021, 363, 2573–2577 CrossRef CAS.
- (a) J. C. Dalton and R. A. Bourque, J. Am. Chem. Soc., 1981, 103, 699–700 CrossRef CAS; (b) A. G. Brook, H. W. Kucera and R. Pearce, Can. J. Chem., 1971, 49, 1618–1621 CrossRef CAS.
- (a) R. A. Moss, M. Wlostowski, S. Shen, K. Krogh-Jespersen and A. Matro, J. Am. Chem. Soc., 1988, 110, 4443–4444 CrossRef CAS; (b) R. A. Moss, S. Shen, L. M. Hadel, G. Kmiecik-Lawrynowicz, J. Wlostowska and K. Krogh-Jespersen, J. Am. Chem. Soc., 1987, 109, 4341–4349 CrossRef CAS; (c) A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed., 1989, 28, 621–622 CrossRef; (d) S. Goumri-Magnet, T. Kato, H. Gornitzka, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 2000, 122, 4464–4470 CrossRef CAS.
- An earlier version of this work was published on the pre-print server ChemRxiv, A. Bunyamin, D. L. Priebbenow and A. Polyzos, ChemRxiv, 2021, DOI:10.26434/chemrxiv-22021-26430rfst.
- To note, during the processing of our manuscript, two related reports emerged regarding the cyclopropenation and cyclopropanation of carbene intermediates derived from acyl silanes; see: (a) G. Zhou and X. Shen, Angew. Chem., Int. Ed., 2022, 61, e202115334, DOI:10.1002/anie.202115334; (b) S. Sakurai, T. Inagaki, T. Kodama, M. Yamanaka and M. Tobisu, J. Am. Chem. Soc., 2022, 144, 1099–1105 CrossRef CAS PubMed.
- (a) A. E. Keating, S. R. Merrigan, D. A. Singleton and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 3933–3938 CrossRef CAS; (b) N. G. Rondan, K. N. Houk and R. A. Moss, J. Am. Chem. Soc., 1980, 102, 1770–1776 CrossRef CAS; (c) J.-L. Mieusset, M. Abraham and U. H. Brinker, J. Am. Chem. Soc., 2008, 130, 14634–14639 CrossRef CAS PubMed; (d) B. Lecea, M. Ayerbe, A. Arrieta, F. P. Cossío, V. Branchadell, R. M. Ortuño and A. Baceiredo, J. Org. Chem., 2007, 72, 357–366 CrossRef CAS PubMed.
- M.-J. Fan, G.-Q. Li and Y.-M. Liang, Tetrahedron, 2006, 62, 6782–6791 CrossRef CAS.
- Refer to the ESI† for additional details.
- Z. Shen and V. M. Dong, Angew. Chem., Int. Ed., 2009, 48, 784–786 CrossRef CAS PubMed.
- D. Cai, M. Wang, J. Wang and W. Duan, J. Phys. Org. Chem., 2012, 25, 400–408 CrossRef CAS.
- (a) J. Yedoyan, N. Wurzer, U. Klimczak, T. Ertl and O. Reiser, Angew. Chem., Int. Ed., 2019, 58, 3594–3598 CrossRef CAS PubMed; (b) N. Wurzer, U. Klimczak, T. Babl, S. Fischer, R. A. Angnes, D. Kreutzer, A. Pattanaik, J. Rehbein and O. Reiser, ACS Catal., 2021, 12019–12028, DOI:10.1021/acscatal.1c02564; (c) M. Yu and B. L. Pagenkopf, Org. Lett., 2003, 5, 5099–5101 CrossRef CAS PubMed; (d) M. Yu and B. L. Pagenkopf, J. Am. Chem. Soc., 2003, 125, 8122–8123 CrossRef CAS PubMed; (e) D. Gladow and H.-U. Reissig, Helv. Chim. Acta, 2012, 95, 1818–1830 CrossRef CAS; (f) J. Saadi, C. Bentz, K. Redies, D. Lentz, R. Zimmer and H.-U. Reissig, Beilstein J. Org. Chem., 2016, 12, 1236–1242 CrossRef CAS PubMed.
- (a) G. Cignarella, D. Barlocco, M. M. Curzu, G. A. Pinna, P. Cazzulani, M. Cassin and B. Lumachi, Eur. J. Med. Chem., 1990, 25, 749–756 CrossRef CAS; (b) G. Cignarella, D. Barlocco, S. Villa, M. M. Curzu, G. A. Pinna, A. Lavezzo and A. Bestetti, Eur. J. Med. Chem., 1992, 27, 819–823 CrossRef CAS.
- L.-W. Ye, X.-L. Sun, C.-Y. Zhu and Y. Tang, Org. Lett., 2006, 8, 3853–3856 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2121946–2121949. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00203e |
| This journal is © The Royal Society of Chemistry 2022 |