
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic C(sp3) radical generation via C–H, C–C, and C–X bond cleavage
Chia-Yu
Huang†
 ,
Jianbin
Li†
and
Chao-Jun
Li
*
,
Jianbin
Li†
and
Chao-Jun
Li
*
Department of Chemistry, FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke Street W., Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 18th April 2022
Abstract
C(sp3) radicals (R˙) are of broad research interest and synthetic utility. This review collects some of the most recent advancements in photocatalytic R˙ generation and highlights representative examples in this field. Based on the key bond cleavages that generate R˙, these contributions are divided into C–H, C–C, and C–X bond cleavages. A general mechanistic scenario and key R˙-forming steps are presented and discussed in each section.
1. Introduction
The C(sp3) radical (R˙) represents one of the fundamental organic species in synthetic chemistry, which is highly enabling in various settings. R˙ can be derived from feedstock chemicals such as alkanes, alkenes, alcohols, amines, aldehydes, ketones, carboxylic acids and their derivatives, making it a versatile option for different synthetic purposes. Besides, it features complementary reactivities to other alkyl intermediates (e.g., carbocation, carbanion, and carbene), providing flexible synthetic routes to build up the C(sp3)-rich scaffold and complexity.Historically, R˙ was rarely involved in reaction designs since it was often produced via energy-intensive or user-unfriendly pathways. In these cases, R˙ was less controllable, which could lead to non-productive quenching or other undesired side reactions (Scheme 1A). For instance, thermolysis of peroxides or persulfates at high temperatures exemplified one common practice of delivering R˙ via activating aliphatic C–H bonds or carboxyl groups. Utilizing toxic metals such as organotin reagents to fragment alkyl (pseudo)halides was another routine method for R˙ generation. Aside from the thermochemical protocols, direct irradiation of radical precursors in some earlier photochemical strategies could significantly lower the reaction temperature, although ultraviolet (UV) light was often required (Scheme 1B). Consequently, global excitation of reaction components was often inevitable, and radical dimerisation, disproportionation and other off-target processes were frequently observed.
 | ||
| Scheme 1 General approaches toward R˙ formation. | ||
In this context, developing efficient and controllable radical-generating methods for sp3 carbon functionalisations was long-sought-after. Pioneered by MacMillan, Stephenson, Yoon and others, photoredox catalysis experienced an impactful renaissance and stood out as a promising solution to previous limitations (Scheme 1C). Unlike conventional approaches, photocatalysis liberates radicals catalytically, usually under visible light, therefore, effectively managing a low concentration of radical species and minimizing the counterproductive radical accumulation. Besides, photocatalysts are commercially available in numerous forms, which could be organic or inorganic, homogeneous or heterogeneous. They are tunable in terms of redox potentials, excited-state energies and other photophysical properties, hence, suitable for multiple synthetic cases. More importantly, the resulting radicals were well orchestrated by the catalysts in these systems, which could elicit reactivities that were unattainable by conventional means, e.g., enantioselective alkylation, radical–radical cross-coupling and photoredox/transition metal dual catalysis.
Armed with these benign features, it is unsurprising that photocatalytic R˙ generation has become the mainstream for radical-based organic synthesis in recent years. Indeed, some of the aforementioned content has been reviewed in the literature, which was categorised either by types of the photocatalysts1–4 or bond formation.5–8
In this regard, we would like to contribute a review from a different perspective by focusing on the photocatalytic R˙ generation. These elegant reports will be organised based on the key bond cleavage during the R˙ generation, thus, highlighting the diversity of C(sp3) radical precursors and the corresponding photocatalytic bond-cleavage strategies and retrosynthetic possibilities (Scheme 2). We envision that this review could provide a quick overview of this field to the audiences, keep them updated with modern pathways to strategise R˙ generation, and facilitate the new design of photocatalysis reactions. Toward this goal, three types of bond cleavages that generate R˙ via the cleavage of (a) C–H bonds, (b) C–C bonds, and (c) C–X (X ≠ H and C) bonds will be covered. In the C–H cleavage section, the reactions will be arranged according to hydrogen atom abstractors, while the reactions in C–C and C–X parts will be grouped on a substrate basis.
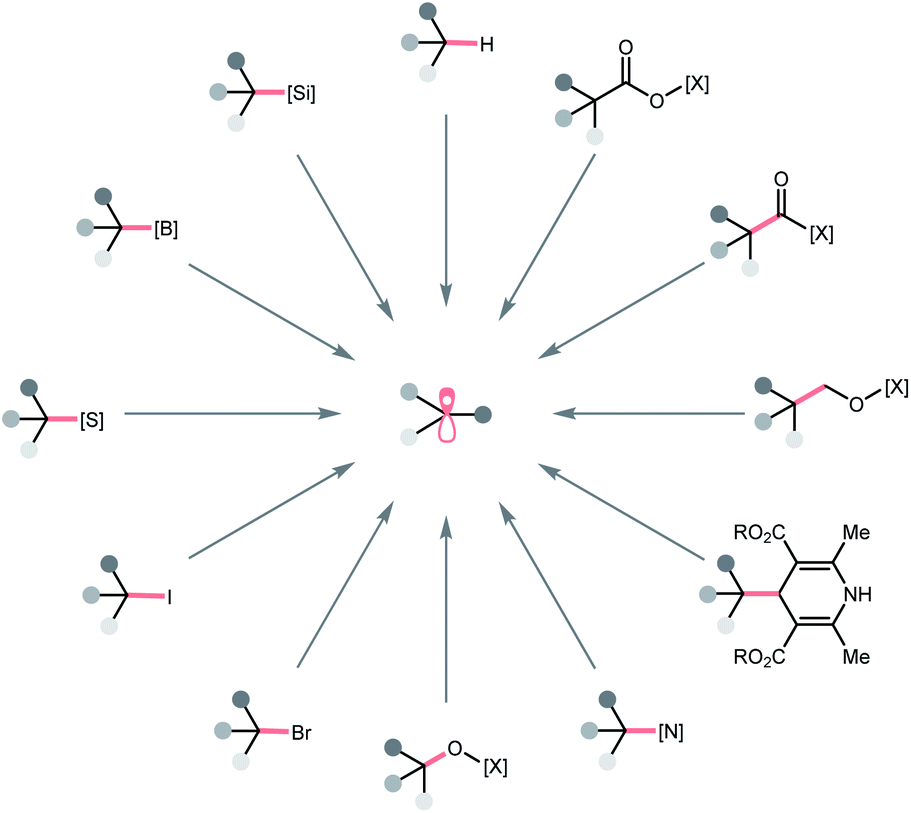 | ||
| Scheme 2 Representative precursors for photocatalytic R˙ formation. [X], activating group. | ||
To be noticed, R˙ in this review referred to all types of C(sp3) radicals, including hydrocarbon-based alkyl radicals or C-centred ones that contained at least one heteroatomic substituent (i.e., α-ethereal, α-amino). Giving a comprehensive list of all related literature in this evolving field is beyond our reach; therefore, only representative publications within a decade that featured new mechanistic insights or synthetic applications will be included. To keep this review concise, some photoenzymatic examples from Zhao's and Hyster's groups will not be included.9–13
2. C–H cleavage
C–H bonds are ubiquitous in organic molecules and readily available in nearly any synthetic stages; therefore, C(sp3)–H bonds have been conceived as ideal sources of C(sp3) radicals. However, due to their high bond dissociation energies (BDEC–H ∼85 to 105 kcal mol−1), the thermodynamic barrier of either direct or indirect hydrogen atom transfer (HAT) with C(sp3)–H bonds needs to be overcome.7,14,15 Electrophilic hydrogen atom abstractors are often required to cleave C(sp3)–H bond homolytically, which are typically electronegative-element-based radicals (X˙) featuring stronger H–X bonds (BDEX–H > BDEC–H, Scheme 3). Nonetheless, considering the instabilities of electrophilic radicals and difficulties in distinguishing similar C(sp3)–H bonds, enabling the generation of X˙ and selective HAT pose significant challenges in the HAT regime and encourage numerous efforts on designing new HAT reagents and reactions.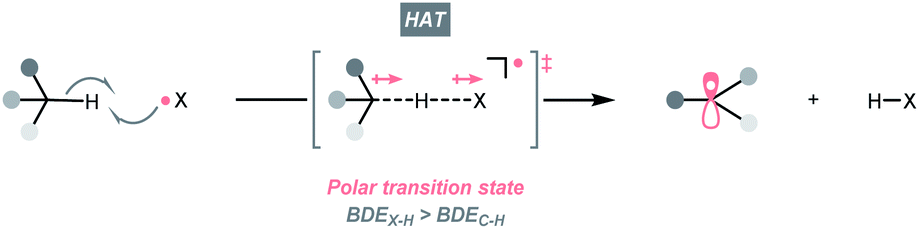 | ||
| Scheme 3 HAT-enabled R˙ generation. | ||
Among them, photocatalysis represents one of the state-of-the-art solutions, which could efficiently deliver the electrophilic radicals under mild conditions, set the stage for subsequent HAT and foster the C(sp3) radicals. Accordingly, the following sections will be organised based on the types of HAT agents.
2.1. HAT with oxy radicals
In light of the high electronegativity of oxygen and the strong O–H bond, oxy radicals should be a potent hydrogen atom abstractor for electron-rich C(sp3)–H bonds. Moreover, as oxygenous compounds are abundant, many of them could be used or engineered as oxy radical precursors. Among them, molecular oxygen (O2), peroxide and persulfate exemplified some classic options of oxy radical precursors, demonstrating their versatile HAT reactions with C(sp3)–H bonds under photocatalytic conditions.16,17 Other than these choices, using alkoxides, carboxylates and some inorganic oxides to generate oxy radicals photocatalytically is gaining popularity in recent years.Mindful of the tunable redox potentials of photoredox catalysts and the weak O–O bonds of peroxides/persulfates, their combination could be a facile method to release oxy radicals catalytically for HAT with C(sp3)–H bonds. In 2015, MacMillan's group employed Ir(III)-photocatalyst and persulfate for Minisci reaction between heteroarenes and ethers (Scheme 4).18 Mechanistically, in this photoredox cross-dehydrogenative coupling (CDC),19–21 K2S2O8 received an electron from the photoexcited Ir(III), generating the sulfate radical anion for ethereal α-C–H abstraction. The resulting R˙ was added to the electron-deficient heteroarene, followed by oxidative aromatisation with Ir(IV) to give the desired ethereal heteroarene.
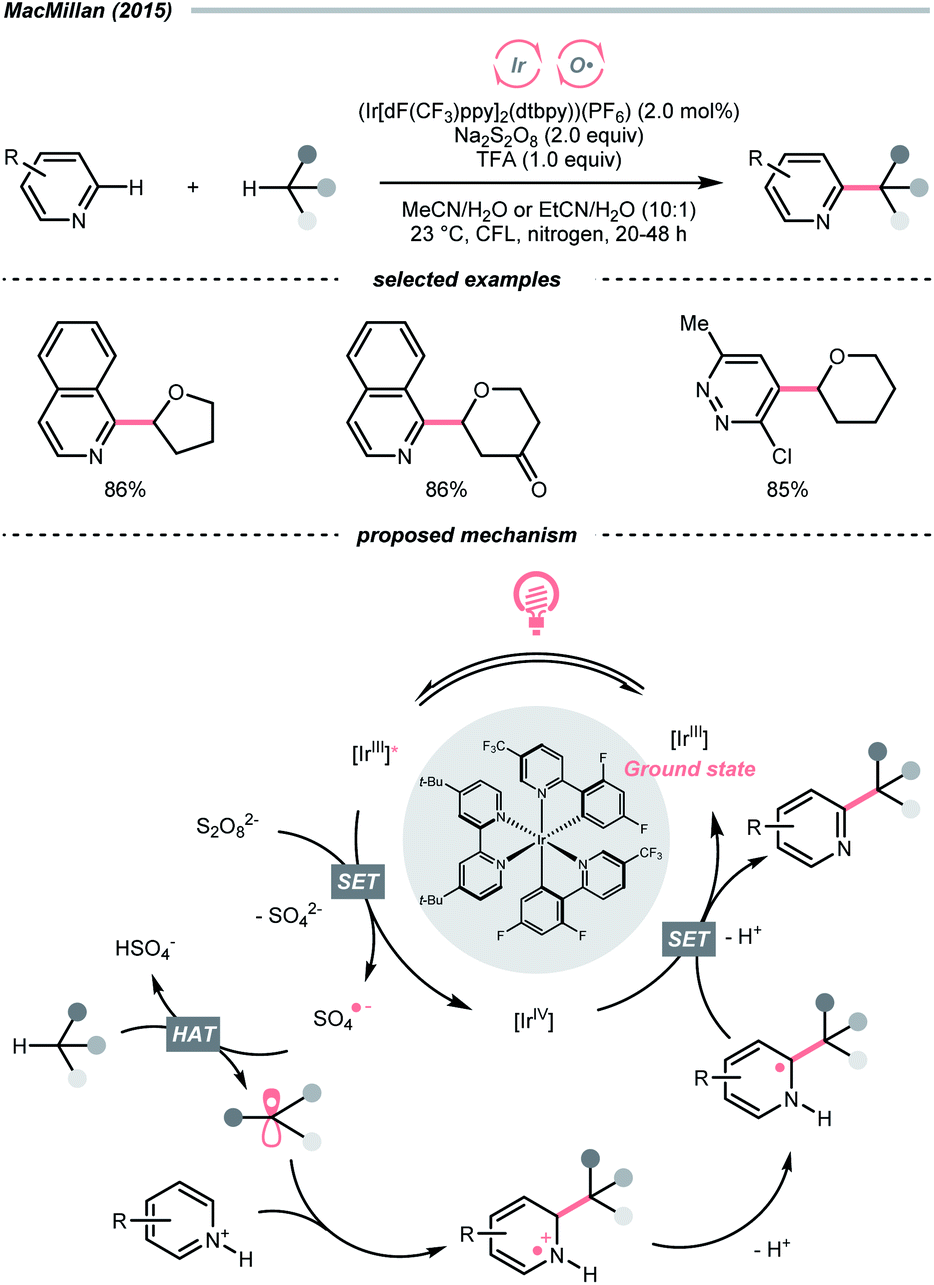 | ||
| Scheme 4 HAT with persulfate. | ||
With the same Ir-photocatalyst, the organic-based benzoate could also serve as the oxy radical precursor. In 2016, the group of Glorius reported site-selective C(sp3)–H trifluoromethylthiolations using a catalytic combination of Ir(III) polypyridyl complex and sodium benzoate under light irradiation (Scheme 5A).22 Based on the Stern–Volmer analysis, the benzoate was oxidised by the Ir(III) to form a benzoyloxy radical (kq = 5.6 × 107 M−1 s−1), which implemented the HAT with R–H to generate an R˙. The higher rate of HAT (k = 1.2 × 107 s−1) than decarboxylation (k = 1.4 × 106 s−1) is one crucial concern when choosing the benzoate as co-catalyst. The R˙ then coupled with N-trifluoromethylthiolated phthalimide to afford the product. Such an HAT protocol with photocatalytically generated oxy radical exhibited superior site selectivity. Tertiary C(sp3)–H was abstracted in preference over secondary and primary ones (>19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio), and the α-oxygenated secondary C(sp3)–H was more reactive than the unactivated tertiary ones.
:1 ratio), and the α-oxygenated secondary C(sp3)–H was more reactive than the unactivated tertiary ones.
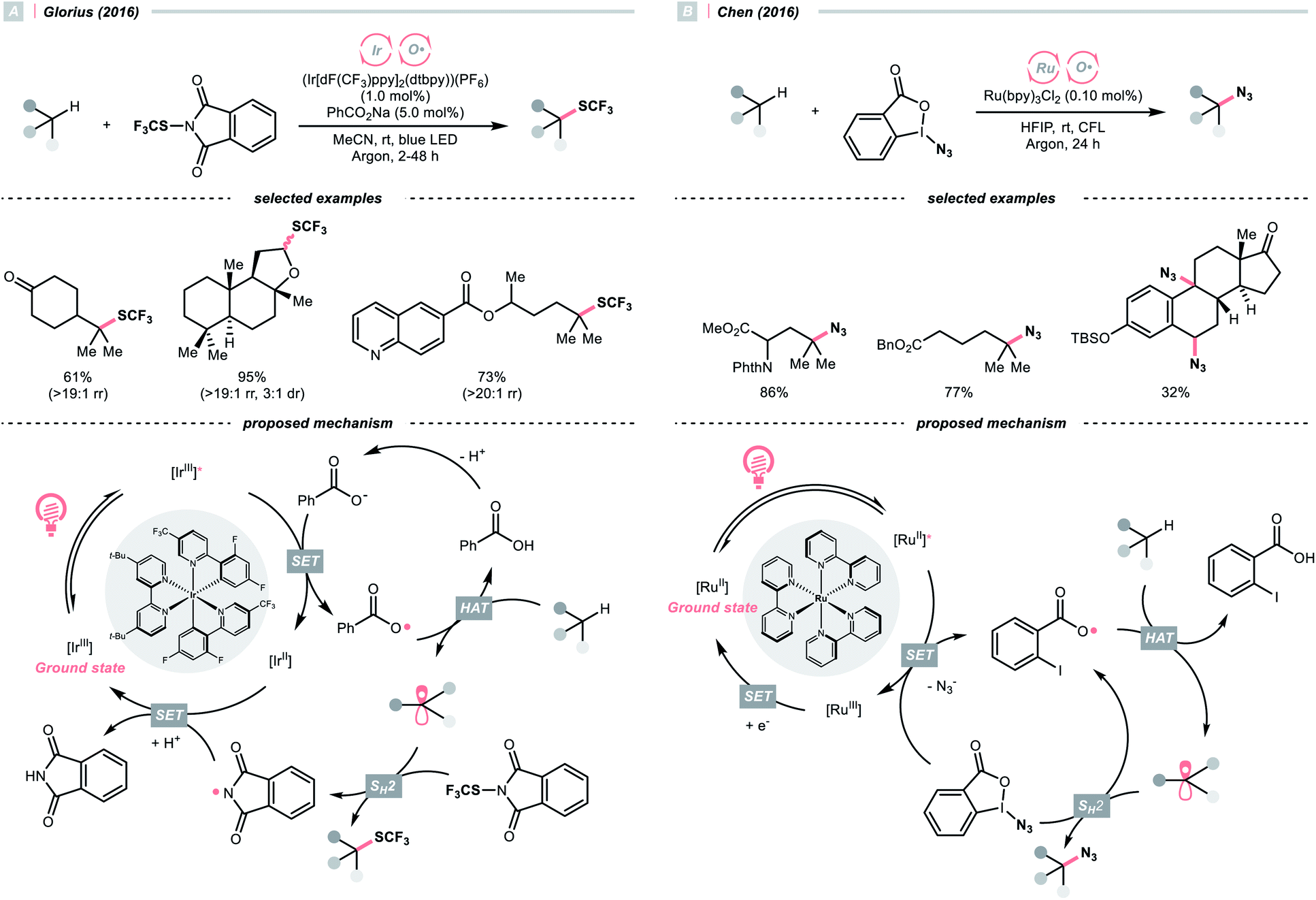 | ||
| Scheme 5 Benzoic acid derivatives as HAT agents. | ||
With opposite electronic demand, the aryloxy radical could also be acquired from the reductive SET of the 1,2-benziodoxol-3-(1H)-one, a benzoate-type hypervalent iodine. In 2016, Chen et al. showcased a C(sp3)–H azidation using 1-azido-1,2-benziodoxol-3-(1H)-one (BI–N3) as both HAT agent and azide source in the presence of a Ru(II)-photoredox catalyst (Scheme 5B).23 Unlike the example in Scheme 5A, single-electron reduction between Ru*(II) and BI–N3 occurred, and the latter underwent O–I cleavage to form the 2-iodobenzoyloxy radical, which was highly selective toward tertiary C(sp3)–H bonds. The generated R˙ initiated homolytic substitution with another BI–N3 and propagated the radical chain (quantum yield, ϕ ∼18). Notably, the authors showed that other than azidation, chlorination and bromination were also achievable by adding corresponding halide salts, which served as halogen sources through the azide-halide exchange with BI–N3.
Recently, alcohols were shown as effective organic precursors of oxy radicals, and various strategies were developed to tackle the challenging single-electron oxidation of alcohols. In 2018, Zhu's group documented a photocatalysed remote C(sp3)–H heteroarylation reaction (Scheme 6A).24 With K2S2O8 as the terminal oxidant, α-heteroaryl tertiary alcohols were converted to γ-heteroaryl ketones through sequential HAT/migratory arylation. An intramolecular proton-coupled electron transfer (PCET) might be operative to facilitate the sluggish oxidation of the –OH group, which was enabled by the cooperative interaction of Ir(IV) species and internal heteroaromatic base. 1,5-Hydrogen atom transfer (1,5-HAT) ensued to give the long-chain R˙ and triggered the heteroarene transfer. The ketyl radical resulted from the migration was then oxidised to the ketone product. In addition to this work, migratory C(sp3)–H cyanation, alkenylation and alkynylation were achieved by replacing heteroaromatic moiety under similar photochemical conditions.25–28
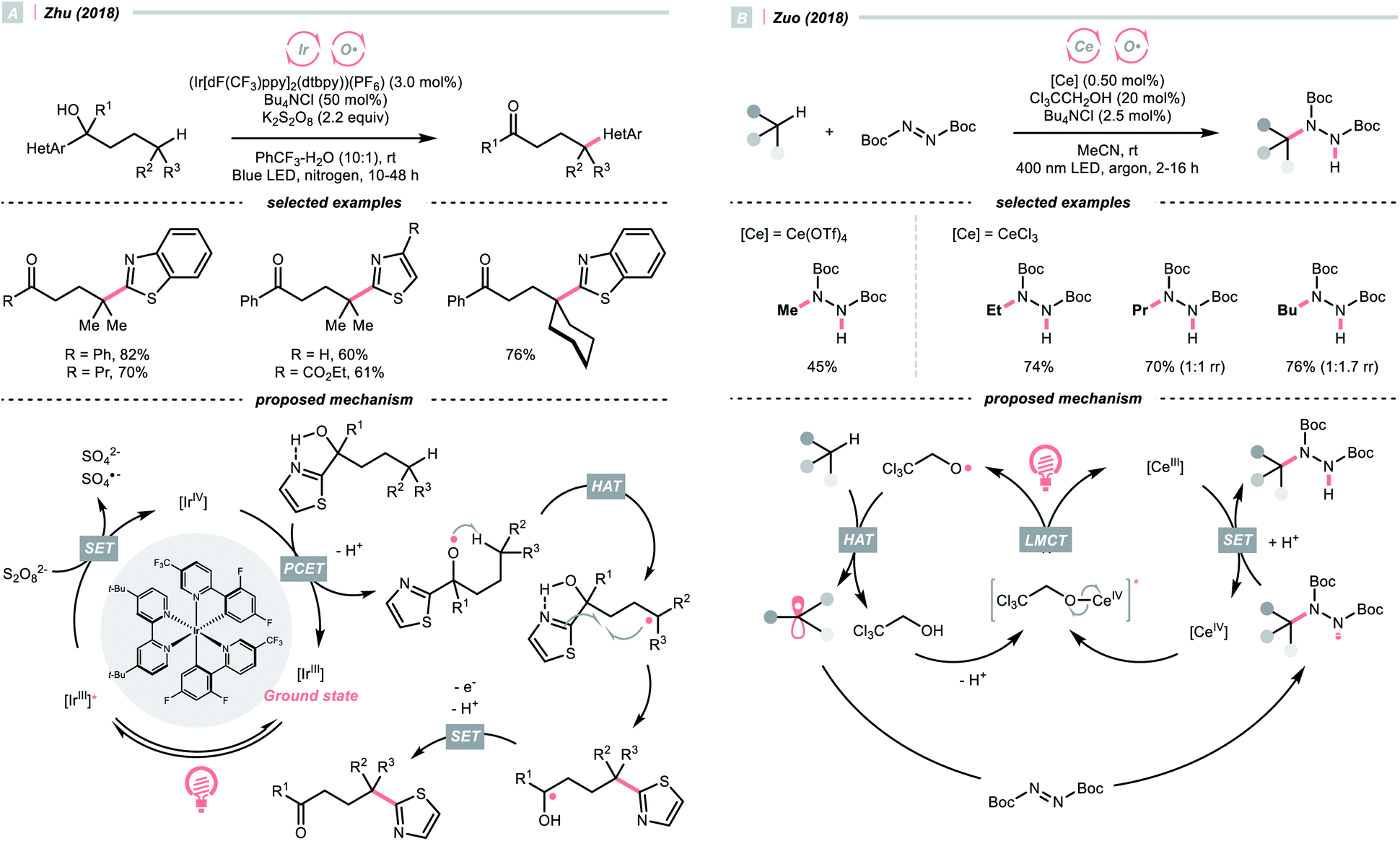 | ||
| Scheme 6 Employing alcohols as HAT agents. | ||
Ligand-to-metal charge transfer (LMCT) of high-valent metal alkoxide complexes is another powerful means to generate oxy radicals from alcohols, as demonstrated by Zuo and his co-workers (Scheme 6B).29 In 2018, they conceived a cerium/alcohol co-catalysed alkane C–H functionalisation reaction and successfully upgraded the light hydrocarbons into value-added products. Albeit the mechanism was still under debate,30 Zuo proposed that through the ligand exchange with methanol or 2,2,2-trichloroethanol, the photoreactive cerium alkoxide complex was formed in situ, which was excited and homolysed to give the alkoxy radical (RO˙).
Methane, ethane, propane, butane, and cyclohexane were all amenable HAT substrates and functionalised by the di-tert-butyl azodicarboxylate (DBAD) to furnish hydrazide products. Such an amination was scalable with continuous-flow reactors. Other than the DBAD, electron-deficient alkene and heteroarene were also applicable alkyl radical acceptors.
Similar to alcohols, N-hydroxy compounds that could result in N-oxy radicals are also viable HAT agents. In 2011, Li and co-workers exploited N-hydroxyphthalimide (NHPI) for C(sp3)–H oxygenation with graphitic carbon nitride (g-C3N4) under photoirradiation (Scheme 7).31 In the presence of g-C3N4 catalysts, O2 mediated the N-oxy radical generation from NHPI, which was proposed as the key radical species to transform allylic or benzylic C–H bonds into C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. Likewise, Gong et al. utilised N-hydroxysuccinimide (NHS) as the HAT agent for formylation of fluoroalkyl imines through the HAT of 1,3-dioxolane, wherein diacetyl was responsible for the oxidation of NHS to generate the N-oxy radical.32
O. Likewise, Gong et al. utilised N-hydroxysuccinimide (NHS) as the HAT agent for formylation of fluoroalkyl imines through the HAT of 1,3-dioxolane, wherein diacetyl was responsible for the oxidation of NHS to generate the N-oxy radical.32
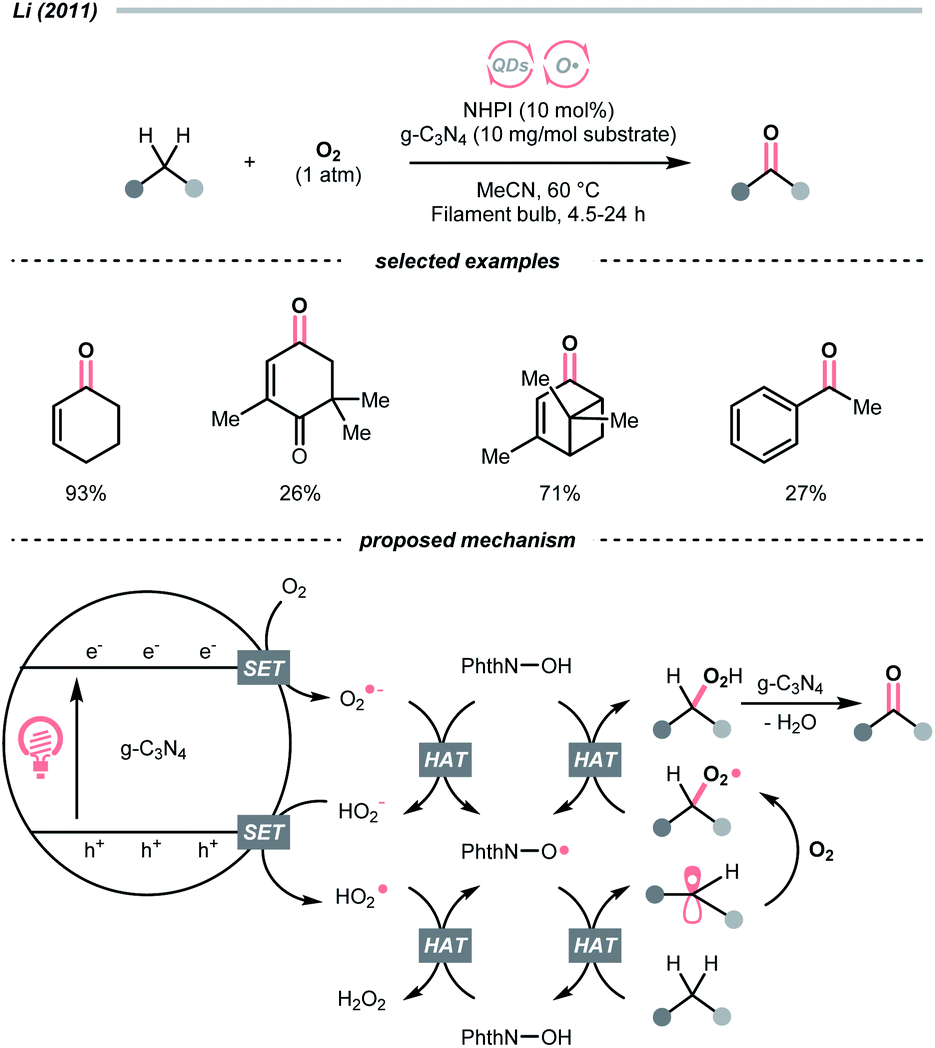 | ||
| Scheme 7 NHPI as HAT agent for C(sp3)–H oxygenation. | ||
Interestingly, some common inorganic salts could produce oxy radicals, however, under strongly oxidizing conditions. As such, these classes of oxy radical precursors remained underutilised thus far. Organophotoredox catalysts (OPCs) often possess broad redox windows, thereby representing ideal chaperones of the inorganic oxides to give oxy radicals under visible light irradiation.
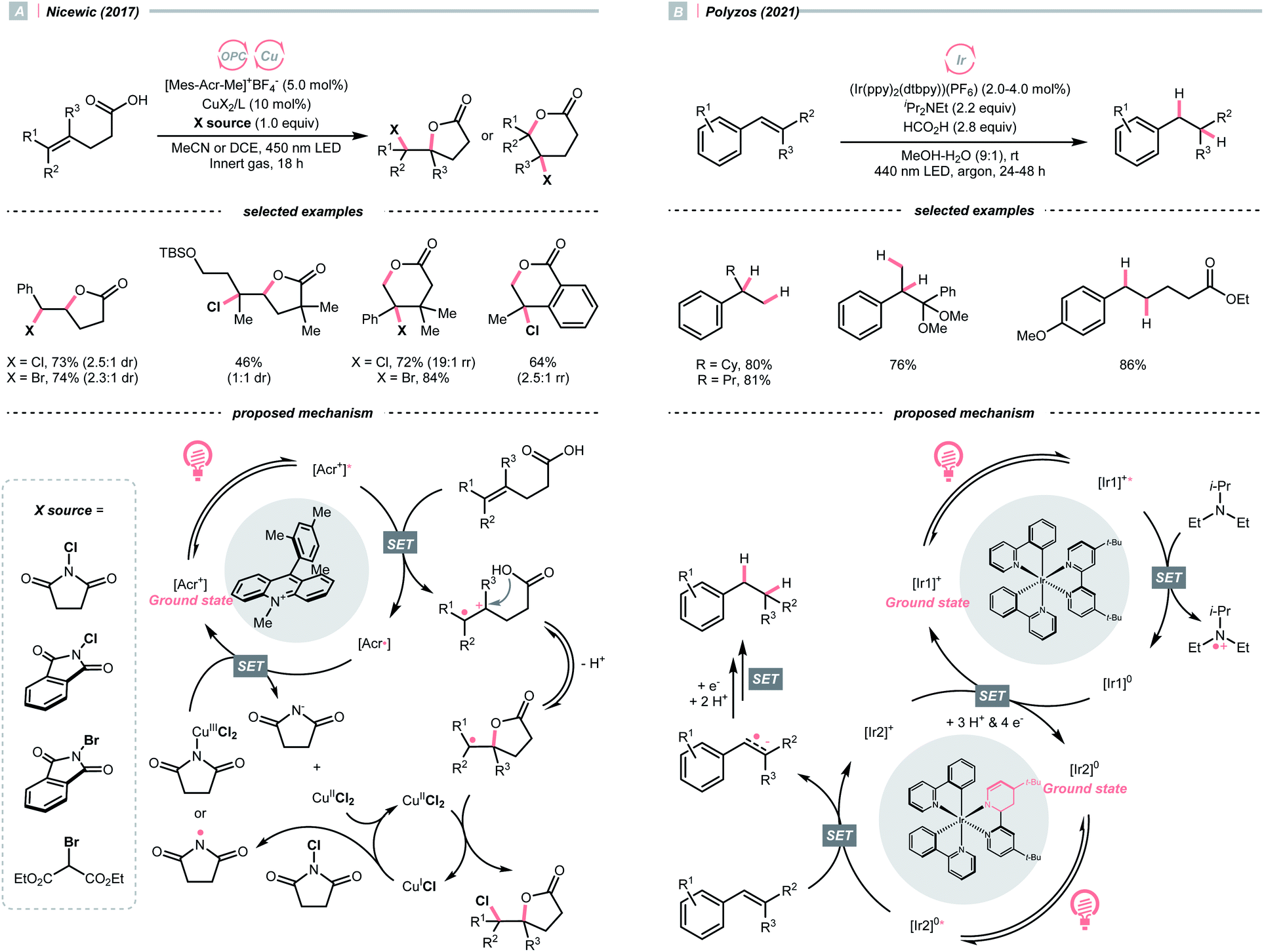
In 2018, the collaboration between Nicewicz's and Alexanian's group showed that the strongly oxidising excited acridinium OPC could catalyse the azidation of non-activated C(sp3)–H bond using K3PO4 as the HAT agent (Scheme 8).33 Taking advantage of the photoexcited acridinium [Mes-Acr+-Ph]* (Nicewicz's catalyst, 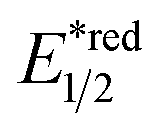 = +2.08 V vs. SCE in MeCN), K3PO4 was directly turned into the phosphate radical and afforded the R˙ via HAT with R–H. Aside from azidation, platform reactions, including fluorination, chlorination, bromination, trifluoromethylation, and alkylation, were successful with the corresponding functionalising reagents to trap the R˙.
= +2.08 V vs. SCE in MeCN), K3PO4 was directly turned into the phosphate radical and afforded the R˙ via HAT with R–H. Aside from azidation, platform reactions, including fluorination, chlorination, bromination, trifluoromethylation, and alkylation, were successful with the corresponding functionalising reagents to trap the R˙.
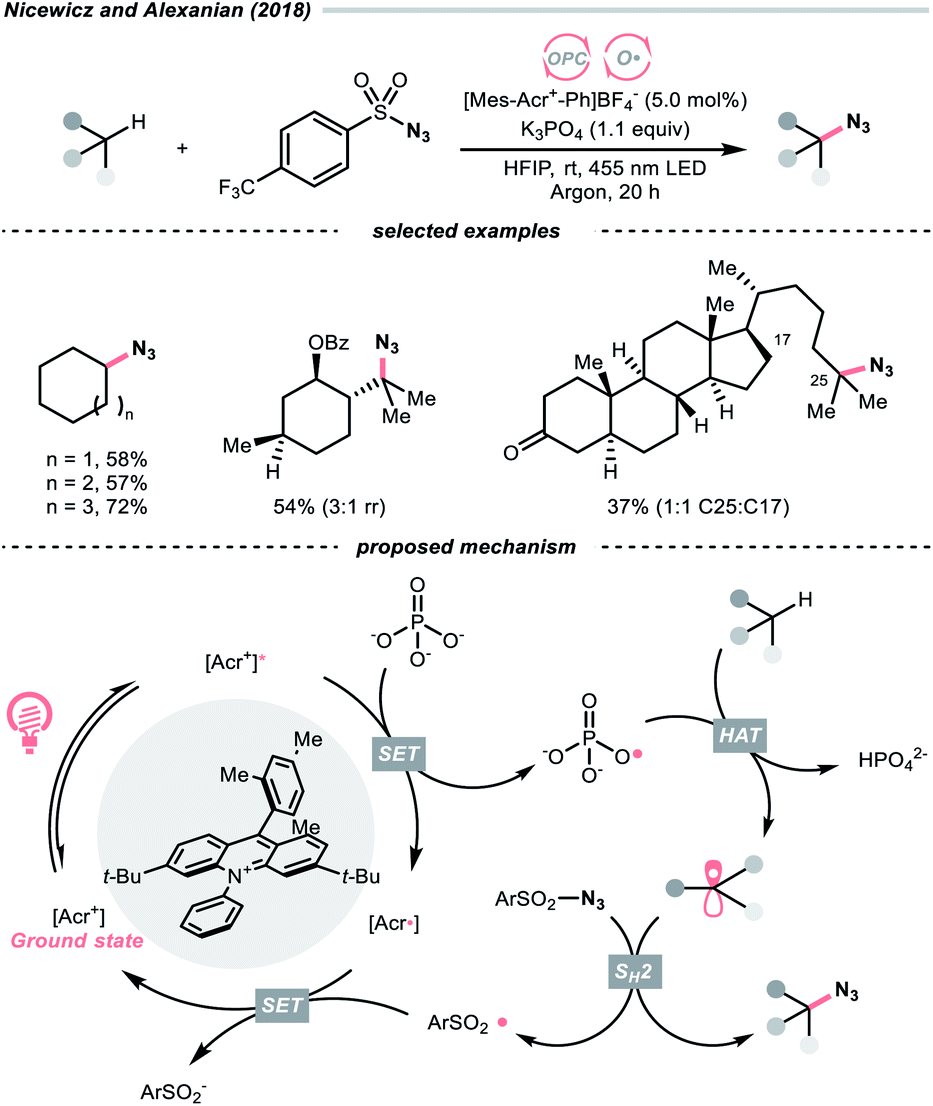 | ||
| Scheme 8 Phosphate as HAT agent. | ||
Another inorganic oxide, nitrate, has also been employed for HAT by Nicewicz's group with a similar acridinium OPC (Scheme 9).34 In general, benzylic C–H bonds are more prone to oxidation relative to their adjacent analogues due in part to the weaker bonding of the former. In Nicewicz's homobenzylic oxygenation reaction of alkylarenes, an OPC/Co dual catalytic system was employed to tackle the challenging C–H oxygenation at homobenzylic positions in favour of benzylic ones. In the plausible mechanism, the nitrate was first oxidised by the photoexcited acridinium, generating a nitrate radical for benzylic HAT. The benzylic radical (R˙) was intercepted by the cobaloxime catalyst to yield a styrene intermediate, which was further subjected into anti-Markovnikov Wacker-type olefin hydration, granting the benzyl ketone product. Interestingly, homobenzylic oxidation could still occur without nitrate for electron-rich substrates.
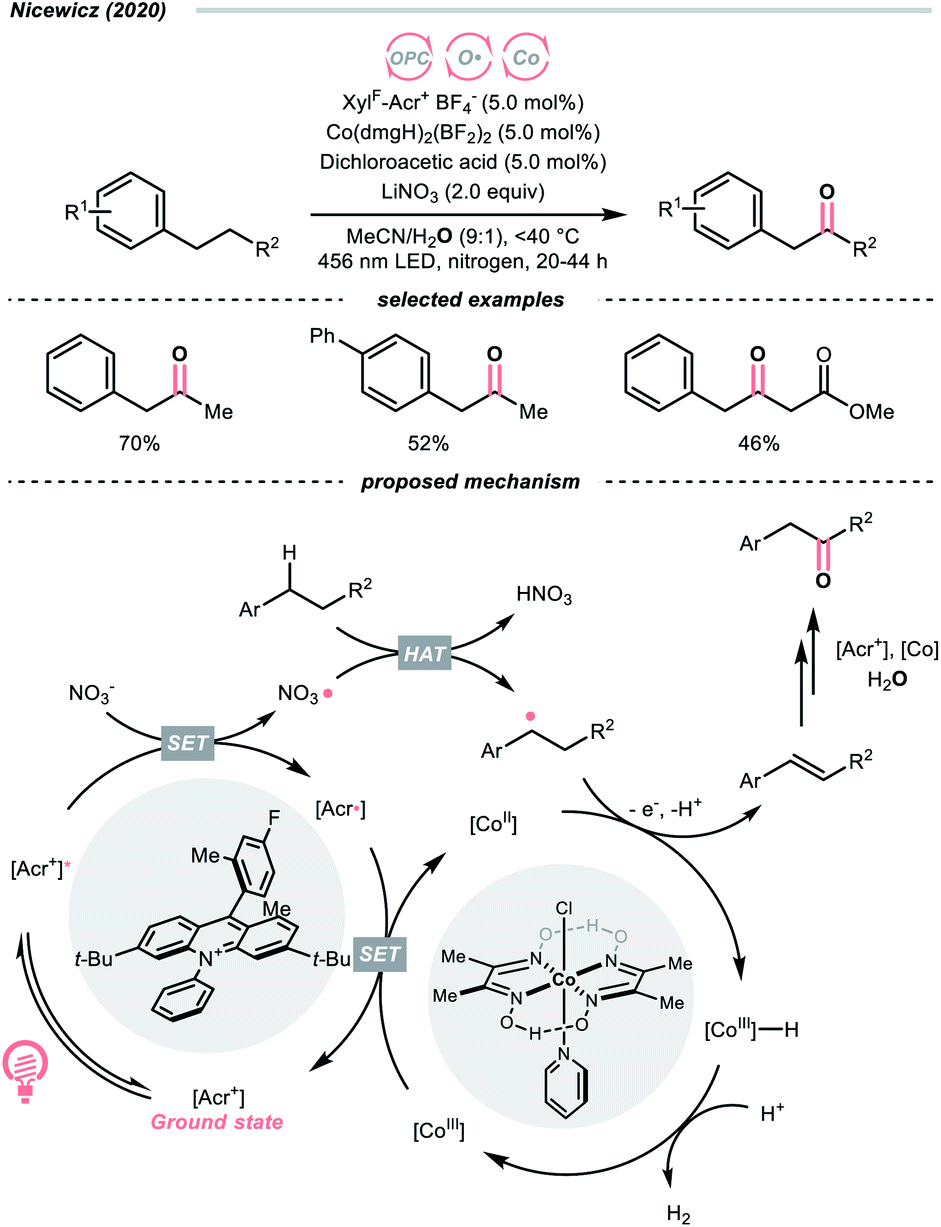 | ||
| Scheme 9 Employing nitrate as HAT agents. | ||
Complementary to the examples above, which accommodated separate photocatalyst and oxy radical precursor in their conditions, some photocatalysts themselves could behave as oxy radicals after light excitation. Ketone is a well-known representative in this class, considering the long and prosperous history of Norrish chemistry. In 2013, Chen's group reported a photocatalytic benzylic C–H fluorination using 9-fluorenone as the HAT agent with Selectfluor as the fluorine source (Scheme 10).35 Under compact fluorescent lamp (CFL) irradiation, the triplet diarylketone abstracted hydrogen at the benzylic position and generated the benzyl radical (R˙). The Selectfluor served as both the fluorine atom source to fluorinate the R˙ and the oxidant for photocatalyst turnover. Interestingly, switching to xanthone gave divergent reaction outcomes under the same photo conditions, making benzyl difluorides the major products. Moreover, this photocatalytic C(sp3)–H fluorination was also feasible for cyclic and acyclic alkanes.
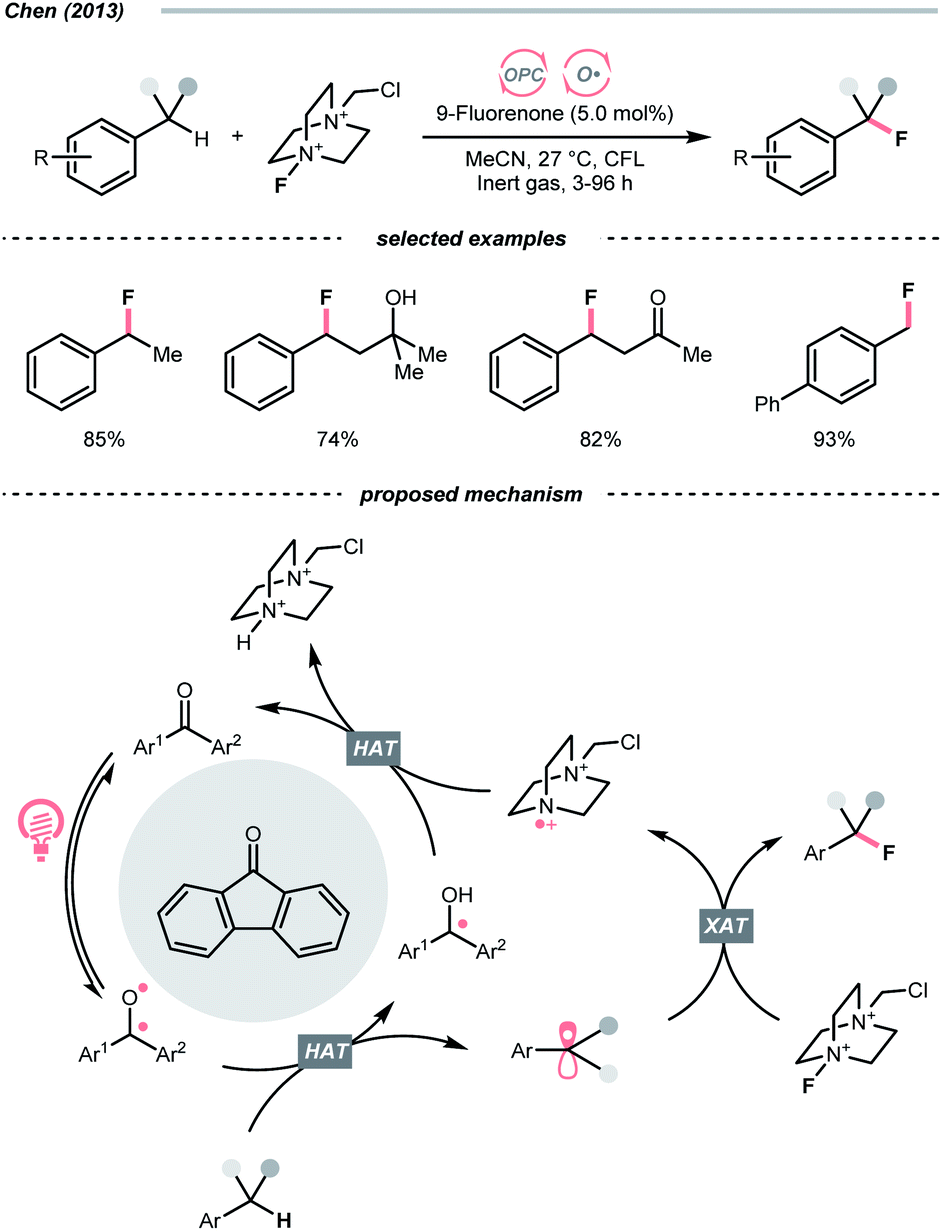 | ||
| Scheme 10 Ketone-catalysed C(sp3)–H fluorination. | ||
Organic dye eosin Y also belongs to this class of photosensitiser. It is visible-light-absorbing and normally used for the single-electron transfer (SET) and energy transfer (EnT) catalysis. In 2020, Wu's group discovered a neutral-eosin Y-catalysed Giese reaction between the ethers and alkenes, in which the underexplored HAT capability of eosin Y was revealed (Scheme 11).36 Mechanistically, the neutral eosin Y, more specifically, its para-quinone methide moiety, was excited to a diradical form and responsible for the HAT with ethereal α-C–H bond. Then, the R˙ was engaged in the Giese reaction with electron-deficient double bonds to give the R′˙.
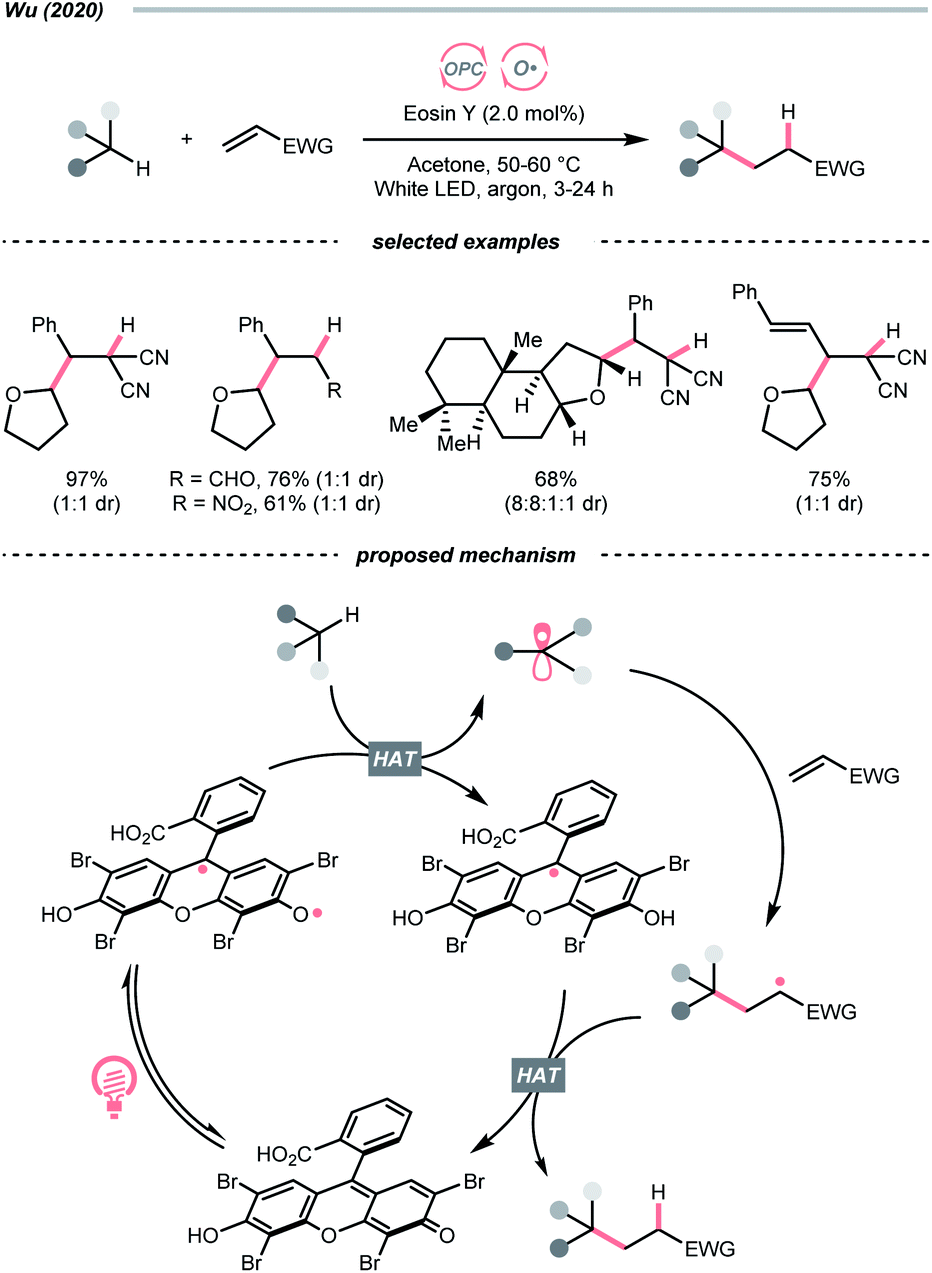 | ||
| Scheme 11 Eosin Y-catalysed C(sp3) radical generation. | ||
Owing to the steric and captodative effect, the reduced eosin Y radical intermediate was relatively stable, which was reformed into the active catalyst by another formal HAT with the R′˙. As such, good to excellent yields of Giese reaction products were also obtained with other C(sp3)–H substrates like alcohols, and amides, while cyclohexane gave only a poor yield. It was worth mentioning that moderate heating was required to increase the reaction efficiency, and eosin Y disodium salt (Na2-eosin Y) was ineffective for this chemistry.
Beyond organophotocatalysis, some metal oxides such as decatungstate (W10O324−, DT), uranyl dication (UO22+),37 and antimony porphyrin complexes (SbTPP)38 could host formal oxy radicals on their periphery under near-ultraviolet (UVA) light irradiation. Capitalizing on this property, Wu et al. designed a CDC reaction19–21 between alkanes and alkenes by merging tungsten and cobalt catalysis (Scheme 12).39 In this dual catalysis system, tungsten was responsible for R˙ generation via HAT, while cobalt was proposed to turn over such a net oxidative coupling via H2 evolution. Specifically, HAT occurred between the photoexcited decatungstate [W10O324−]* and alkane, affording an R˙ and [W10O32]5−H+. While R˙ underwent the Giese addition toward alkenyl CC bond and produced a new stabilised R′˙, the latter (Ered1/2 = −0.97 V vs. SCE in MeCN) reduced the Co(III) (Ered1/2 = −0.16 V vs. SCE in MeCN) into Co(II). Binding of Co(II) and R′˙ followed by β-hydride elimination furnished the alkylated alkene products with a high E/Z ratio and a Co(III)–H. Later, the cobaloxime cycle was closed by quenching the Co(III)–H with proton and releasing H2.
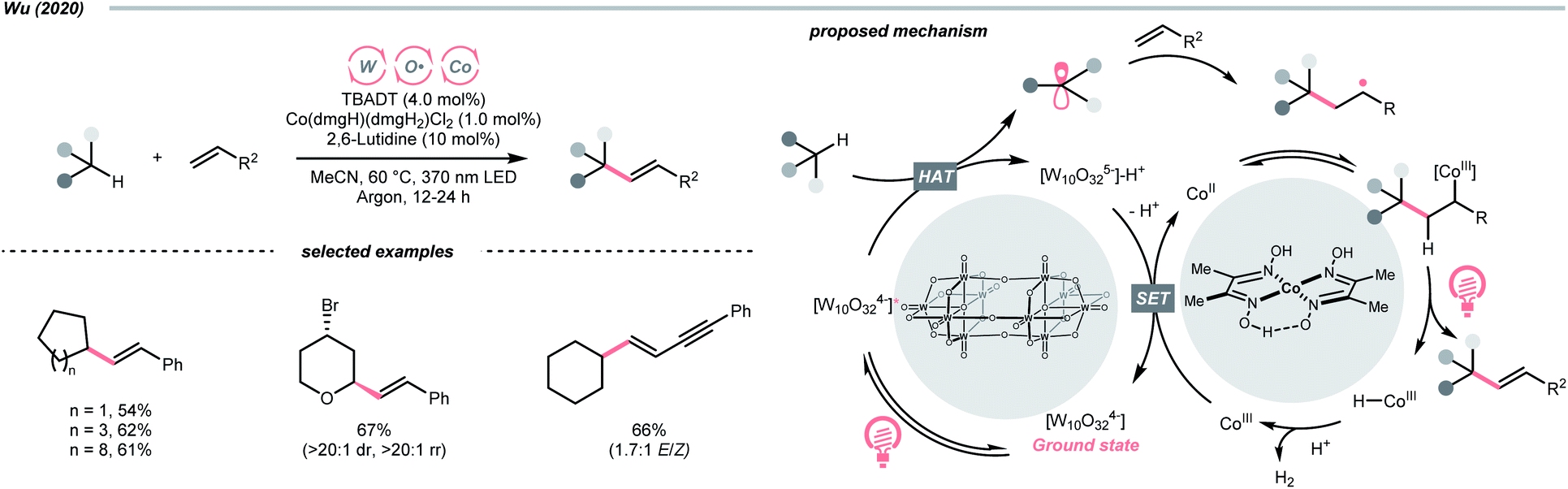 | ||
| Scheme 12 TBADT-catalysed C–H alkenylation of alkanes. | ||
2.2. HAT with nitrogen-centred radicals
Adjacent to oxygen, nitrogen is also a highly electronegative element, and its radicals are suitable for the HAT with C(sp3)–H bonds. Early examples of N-centred radical (NCR) could be traced back to the Hofmann–Löffler–Freytag (HLF) reaction, an intramolecular NCR-mediated HAT reaction.40,41 Although nitrogen is less electronegative than oxygen, NCRs could fine-tune their steric and electronic properties by varying the N-substituents.A photocatalytic example of an HLF reaction to synthesise cyclic amides from N-chloroamides was developed by Yu's group in 2015 (Scheme 13A).42 The photoreduction of the N-chloroalkyl amide by Ir*(III) induced the N–Cl cleavage and formed an NCR, which underwent 1,5-HAT with its pendant alkyl chain to engender a C(sp3) radical (R˙). The R˙ could be oxidised by Ir(IV) following chloride attack, or alternatively, chlorinated by another N-chloroamide, to afford a 5-chloroalkyl amide. With the stepwise addition of a base, intramolecular SN2 amidation occurred, giving the cyclic amide products.
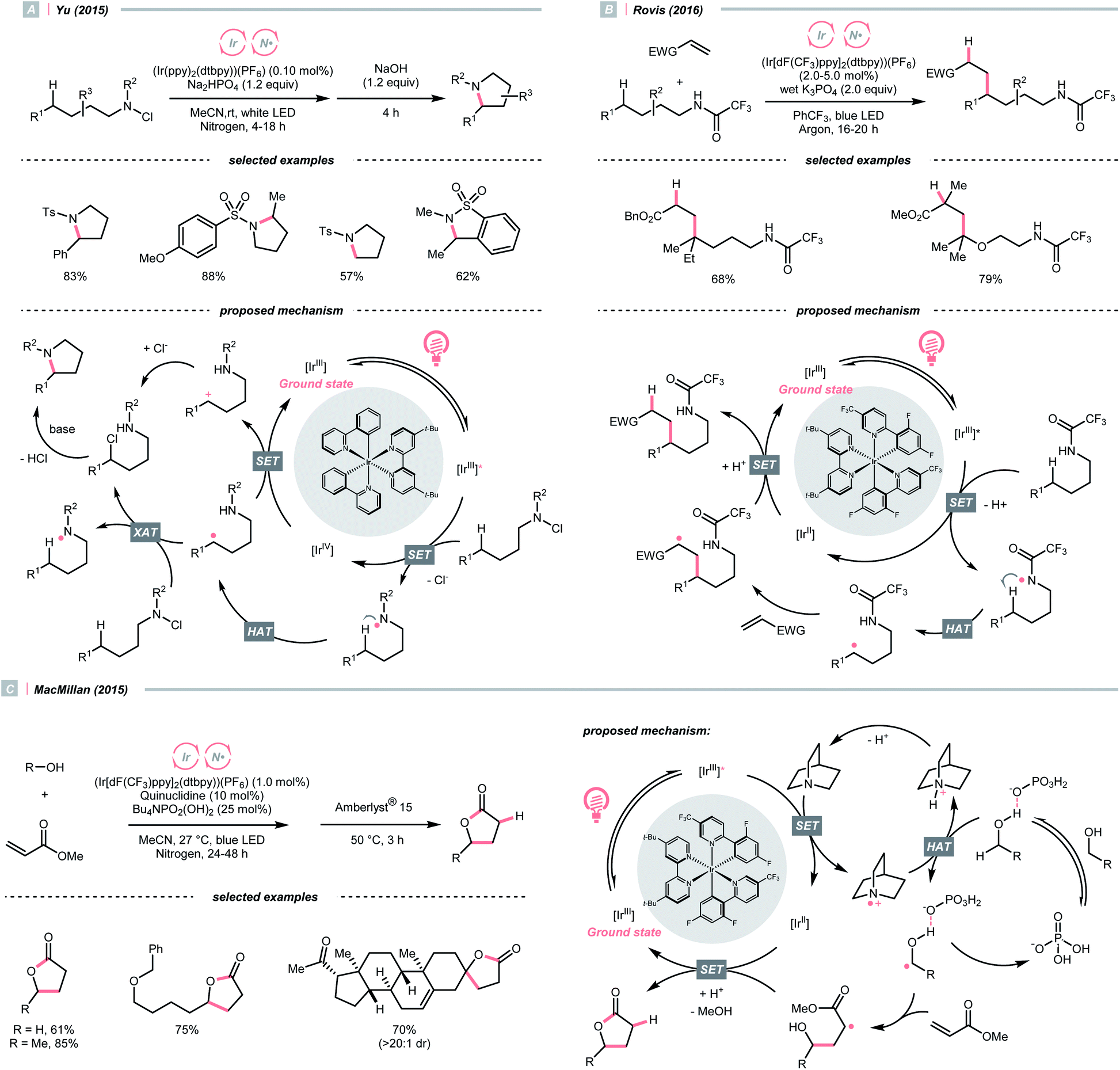 | ||
| Scheme 13 NCR-assisted remote C–H functionalisations. | ||
A similar photocyclisation reaction could also be realised using simple secondary amides as NCR precursors as well as HLF substates, which was revealed by Rovis et al. in 2016 (Scheme 13B).43 Based on their mechanistic studies, acidic amide substrates were crucial, which was partially deprotonated and subjected to oxidation with Ir*(III). Alternatively, PCET might be operative. The NCR triggered the C(sp3) radical generation via 1,5-HAT, which was engaged in the Giese reaction with electron-deficient olefins.
Concurrently, the group of Knowles reported the same photocatalytic remote C–H alkylation but with catalytic Bu4NPO2(OBu)2 as the base.44 Inspired by these elegant chemistries, several amide catalysts were designed to pair with the Ir-photocatalyst, enabling a series of C(sp3)–H alkylation,45 alkenylation46 and arylation.47
In addition to amides and their derivatives, simple amine could accomplish the HAT/Giese reaction sequence under photocatalytic conditions. In 2015, MacMillan et al. reported an iridium/quinuclidine/phosphate triple-catalysed photoredox reaction between alcoholic α-C(sp3)–H bonds and electron-poor alkenes (Scheme 13C).48 In their tentative mechanism, the NCRs was generated from the oxidation of quinuclidine (Ered1/2 = +1.10 V vs. SCE in MeCN) by the photoexcited (Ir[dF(CF3)ppy]2(dtbpy))(PF6) (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN), producing the key R˙ selectively at the alcoholic α-position. The unique regioselectivity, in this case, might stem from hydrogen bonding between alcoholic O–H and phosphate, which weakened the α-C(sp3)–H of alcohol. Such an interaction would allow the selective alkylation with strong C(sp3)–H bond in the presence of weaker ones such as allylic, benzylic, α-ethereal and α-carbonyl C(sp3)–H bonds.
Apart from amines and amides, which formed sp3-hybridised NCRs, sp2 nitrogen radicals are also effective in HAT. Among them, iminyl radicals were often employed in imine remote C(sp3)–H functionalisations, giving ketones after hydrolysis.49,50 Taking Studer's γ-alkylation of ketone as an example (Scheme 14A),51 an α-aminoxy acid auxiliary was condensed with the ketone, of which the carboxylate group could be oxidised by the Ir*(III) to implement decarboxylation and deacetylation, giving an iminyl radical for remote C(sp3)–H abstraction.
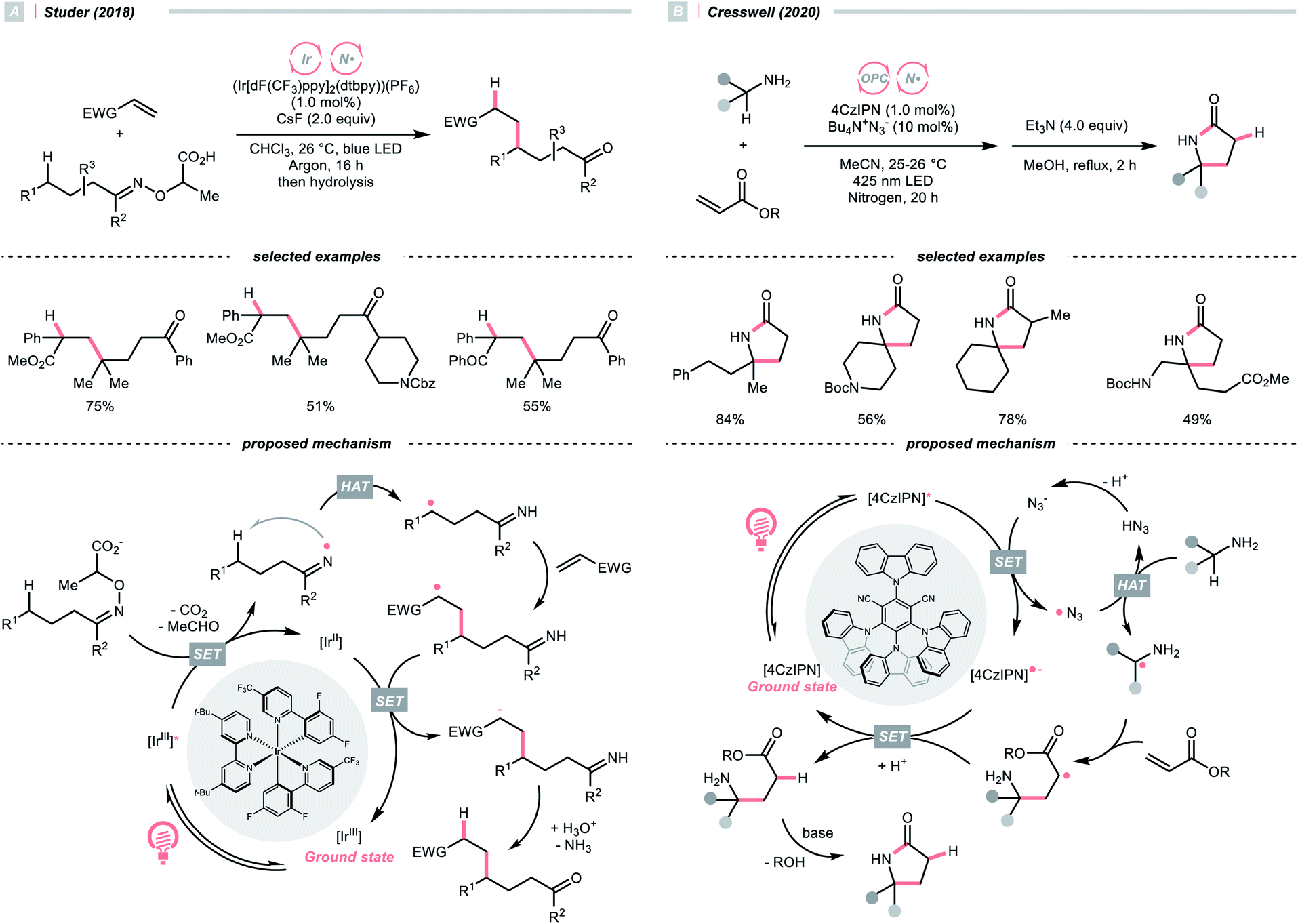 | ||
| Scheme 14 Amine- and azide-catalysed α-C(sp3)–H functionalisations. | ||
In 2020, Cresswell and his team conceived an α-tertiary amine synthesis via azide radical-enabled HAT with unmasked amines (Scheme 14B)52 Similar to MacMillan's quinuclidine radical-mediated HAT (Scheme 13C), the anionic azide was oxidised by photoexcited 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) to provide an azide radical  which performed regioselective C(sp3)–H abstraction from an α-secondary amine and delivered an α-amino R˙. After R˙ addition to a conjugate alkene, the resulting amine could be isolated or subsequently cyclised to afford a lactam.
which performed regioselective C(sp3)–H abstraction from an α-secondary amine and delivered an α-amino R˙. After R˙ addition to a conjugate alkene, the resulting amine could be isolated or subsequently cyclised to afford a lactam.
Similar to oxy radicals, some special photocatalysts could serve as NCR precursors directly, albeit rarely reported in the literature. For instance, trisaminocyclopropenium ion (TAC), which Lambert's group widely applied, was reported to enable site-selective heteroarylation of ether under photoelectrochemical conditions (Scheme 15).53 According to their proposed mechanistic rationale, the TAC was first oxidised into TAC˙+via anodic oxidation. After photoexcitation, it was transformed into an aminyl radical cation, which could abstract the ethereal α-C(sp3)–H to produce R˙ or rearomatise R˙/heteroarene adduct to give the Minisci reaction products. On the other side, cathodic reduction of H+ would release H2. Following the success of this work, C(sp3)–H diamination and amidation were later achieved using the same TAC photocatalyst.54,55
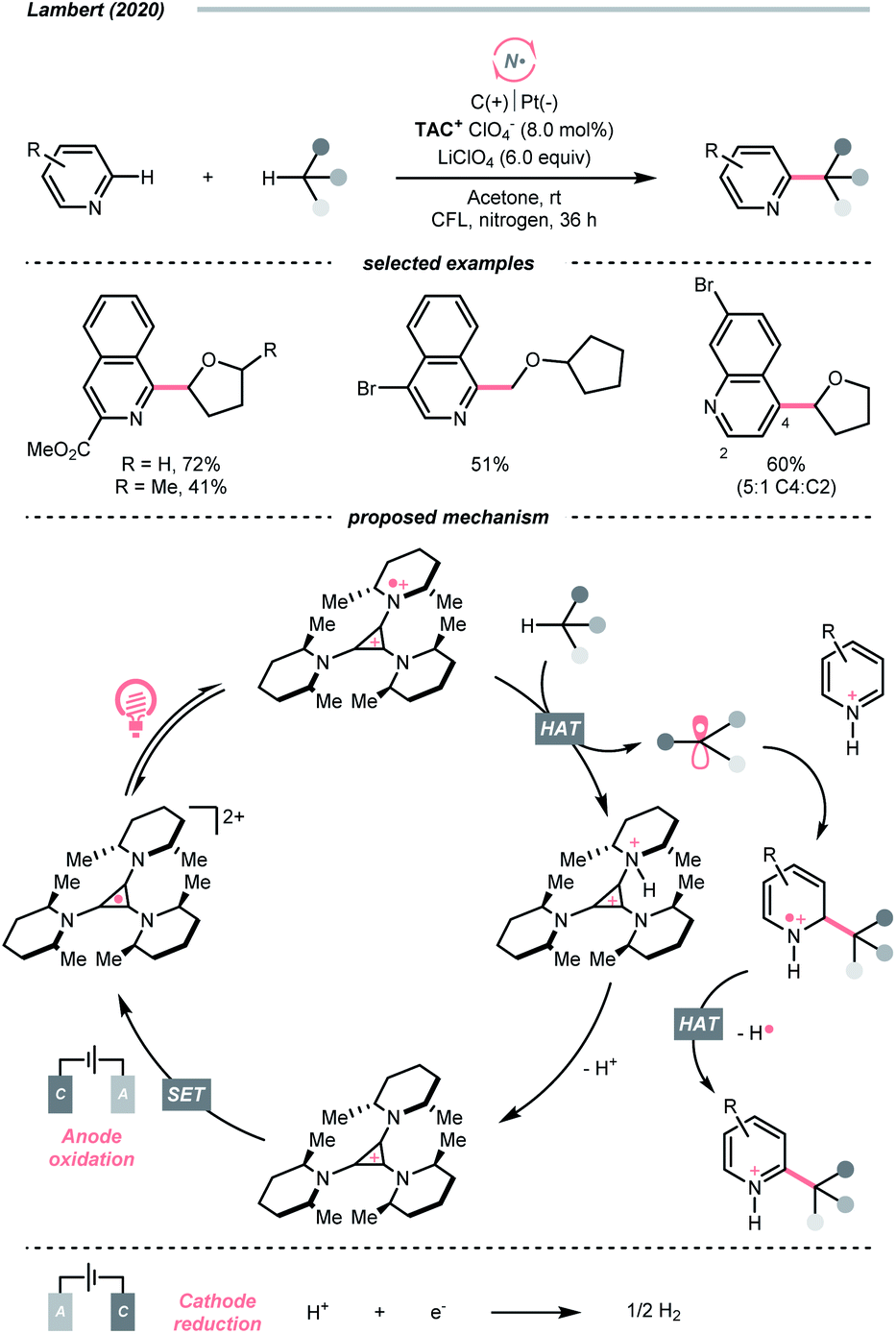 | ||
| Scheme 15 TAC-catalysed HAT under photoelectrochemistry conditions. | ||
2.3. HAT with thiyl radicals
Moving downward in the chalcogen column, S-centred (thiyl) radicals, despite being less electrophilic relative to oxy radicals, could also perform HAT with some C(sp3)–H bonds. In general, thiyl radicals could be generated more easily because of the more polarisable and less electronegative sulfur centres.Like the oxy radical, thiyl radicals can be formed from thiols, thiocarboxylic acids and thiophosphoric acids.56,57 By merging a thiol catalyst and Ir-photocatalyst, the group of MacMillan reported a dehydrative Minisci alkylation using thiol as the HAT agent and alcohols as the alkyl sources (Scheme 16A).56 Mechanistically, the essential thiyl radical came from the SET between [Ir(ppy)2(dtbpy)2]2+ (Ered1/2 Ir(V)/Ir(III) = +1.21 V vs. SCE in MeCN) and mercaptan co-catalyst (Ered1/2 = +0.85 V vs. SCE in MeCN for cysteine). Then, the thiyl radical abstracted the hydrogen atom from alcoholic α-C(sp3)–H assisted by the polar effect; otherwise, such transformation would be thermodynamically unfavourable (BDES–H for thiol ∼87 kcal mol−1; BDEC–H for MeOH = 96 kcal mol−1). The nucleophilic addition of R˙ to the protonated heteroarene, which was followed by a spin-centre shift (SCS)-induced dehydration and some proton/electron transfer steps to give the alkylated Minisci products. Notably, the successful application of methanol for aromatic methylation represented a major breakthrough in the field.
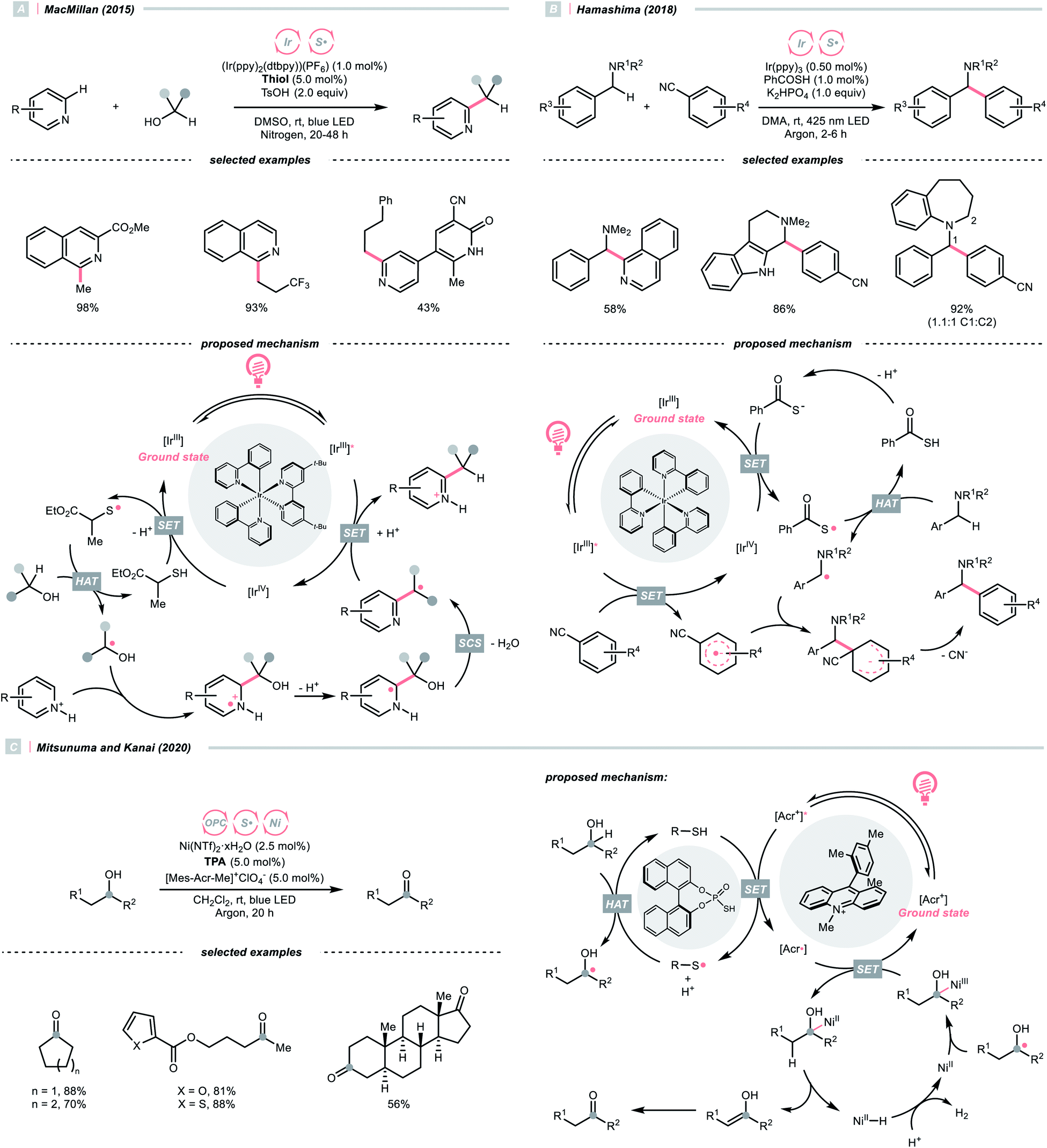 | ||
| Scheme 16 Thiyl radical-enabled C(sp3)–H elaborations. | ||
Using thiobenzoate as a catalytic HAT agent, Hamashima et al. reported a redox-neutral C(sp3)–C(sp2) coupling of benzyl amines and cyanoarenes in aid of Ir-photocatalyst (Scheme 16B).58 The photocatalytic cycle was initiated by reduction of the photoexcited Ir(III) by dicyanoarene to form a radical anion intermediate. Subsequently, electron transfer between Ir(IV) and thiobenzoate oxidised the latter and generated the thiyl radical for benzylic HAT. Due to the persistent radical effect, coupling the α-aminobenzylic radical (R˙) and cyanoaryl radical anion was feasible, which could afford the product after cyanide extrusion.
Switching to another thiyl radical HAT system with acridinium, thiophosphoric acid (TPA) and Ni(II), Mitsunuma and Kanai reported a photocatalytic acceptorless dehydrogenation reaction of alcohols in 2020 (Scheme 16C).59 Based on their seminal results,60 a triple catalytic cycle was designed. The TPA was oxidised by the excited Fukuzumi catalyst to form a thiyl radical for HAT with the alcoholic α-C–H bond. The ensuing R˙ was intercepted by nickel(II), followed by β-hydride elimination and tautomerisation to deliver the ketone product. Terminal oxidant was absent in this reaction because the nickel promoted the H2 evolution and closed the catalytic cycle. Notably, under the same C(sp3) radical generation scenario, intermolecular reactions such as the Giese reaction and oxidative esterification between alcohols and aldehydes were successful.
2.4. HAT with halogen radicals
The application of halogen radicals in organic synthesis could date back to more than 150 years ago when Regnault discovered that dichloromethane could be formed by exposing chloromethane and chloroform to sunlight.61 While the Cl˙-involved process remains a common practice for alkyl chloride synthesis, many novel C(sp3)–H functionalisation reactions have been established by embedding the halogen radical-mediated HAT in visible light photocatalysis.Inorganic chlorides (Cl−) represent a convenient source of Cl˙ for laboratory synthesis. However, oxidation of Cl− to Cl˙ (Ered1/2 = +2.03 V vs. SCE in MeCN) mandates strong oxidants,62 and controlling the reactivity of Cl˙ stays challenging. In 2018, Barriault and his group reported an elegant solution to solve these two problems and realised an (Ir[dF(CF3)ppy]2(dtbpy))Cl-catalysed Giese reaction with alkanes (Scheme 17A).63 Mechanistically, a radical process with Cl˙ and R˙ was proposed. The former was produced from the SET between excited Ir(III) and chloride under gentle heating conditions since the Cl− oxidation was unfavourable in this case (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN). The latter was derived from the HAT between Cl˙ and alkanes and was subjected to the Giese pathway. Interestingly, the reactivity of Cl˙ could be tamed at low concentration with pyridine as the solvent, wherein it exhibited enhanced selectivity toward tertiary C(sp3)–H bonds than others in cyclopentyl methyl ether.
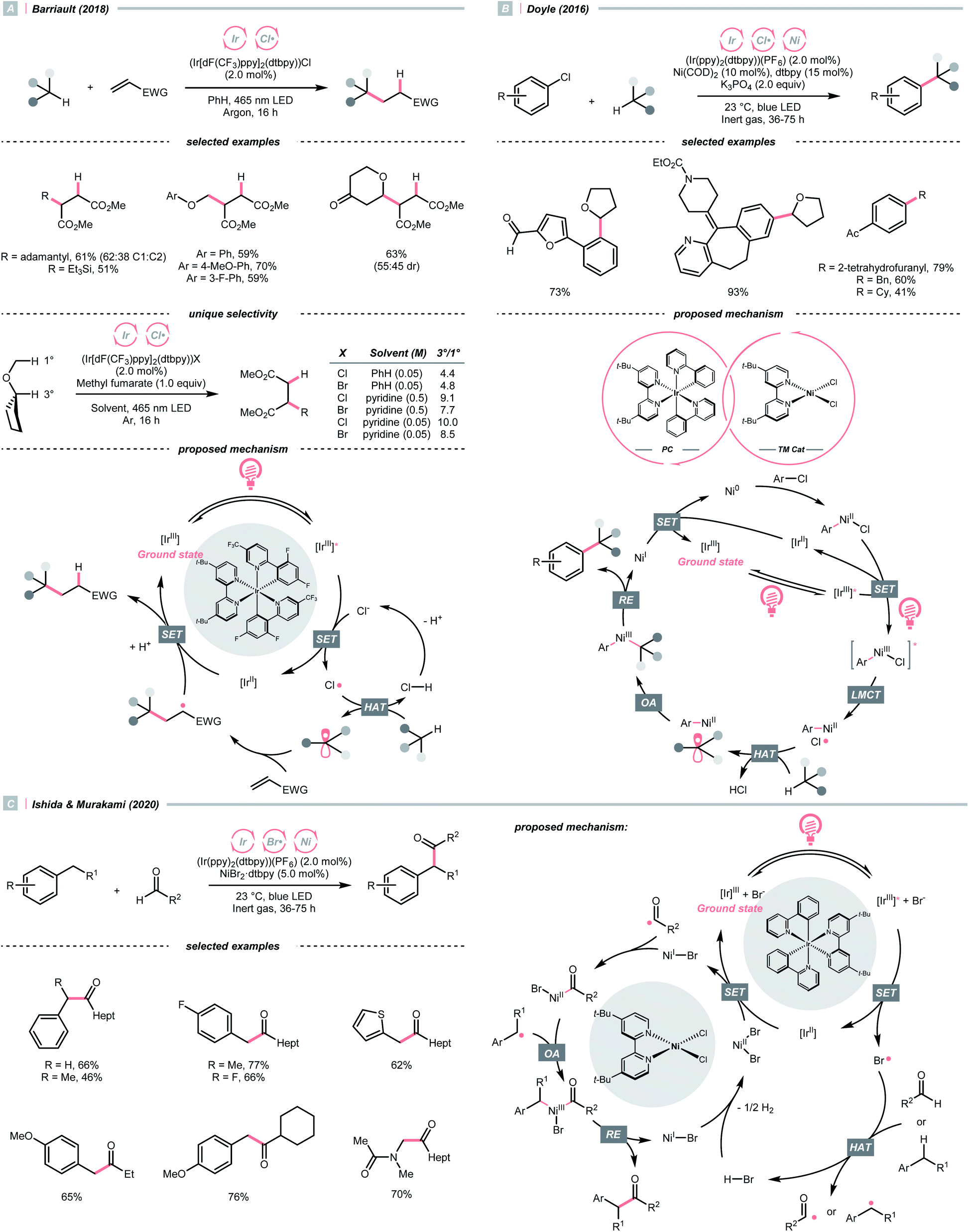 | ||
| Scheme 17 Halogen radical-enabled C(sp3)–H activations. | ||
Complementary to SET, LMCT is also effective in generating Cl˙.62,64 Inspired by Nocera's observation on photoinduced LMCT of nickel(III) trichloride,65 in 2016, Doyle and colleagues designed a dual metallaphotoredox catalysis reaction for a redox-neutral coupling of aryl chlorides and ethers (Scheme 17B).62 By merging iridium photocatalysis and nickel catalysis under visible light, the key Cl˙ could result from an LMCT process of the excited Ni(III)(Ni–Cl. This strategy could bypass the unfavourable outer-sphere oxidation of Cl− by photocatalyst, and external Cl− was absent in Doyle's conditions since oxidative addition of Ni(0) could gain Cl− from a broad range of aryl chlorides. Noticeably, C(sp3)–H arylations with toluene and cyclohexane were also feasible, albeit in lower yields.
Consistent with the trend of the oxygen-to-sulfur switch, compared to Cl−, bromide (Br−) has a less positive reduction potential (Ered1/2 = +1.60 V vs. SCE in MeCN), weaker hydrogen–halide bond (BDE for H–Br = 87 kcal mol−1) and lower electronegativity; therefore, bromine radical (Br˙) could be a theoretically more selective HAT agent that is easier to obtain.66 Based on these properties, Ishida and Murakami et al. utilised the nickel/iridium dual metallaphotocatalytic system for the CDC between toluene derivatives and benzaldehydes (Scheme 17C).67 The direct Br−-to-Ir*(III) electron transfer of the in situ formed [Ir(ppy)2(dtbpy)]Br led to the Br˙ formation. Impressively, the yield of cross-coupling products could be optimised by fine-tuning the molar ratio of toluenes and aldehydes.
As demonstrated by Wu's laboratory, the same Br˙ could also be derived from the CH2Br2 oxidation by photoexcited acridinium catalyst, which was submitted to achieve alkyl C–H abstraction for Giese reaction.68 To be noted, the HAT byproduct, HBr, could serve as additional Br˙ sources.
2.5. HAT with carbon-centred radicals
Unlike heteroatom-based radicals, most non-functionalised C-centred radicals are nucleophilic. Since the components on both sides of the HAT equation are very similar in terms of the C–H bond strength and C-centred radical polarity, low kinetics of the HAT step, premature coupling process and other side reactions are major concerns of this HAT protocol. This explained its rare application in the intermolecular process.However, C-centred radical-mediated HAT enjoyed rapid development in recent years owing to the renaissance of photocatalysis. Along this line, Gevorgyan, Reiser, Zhu and other research groups have devoted themselves to advancing HAT chemistry with C-centred hydrogen abstractors in transition metal-catalysed or metal-free reactions.69–71
In 2020, Gevorgyan's group described a photoinduced intramolecular atom-transfer radical cyclisation (ATRC) reaction of vinyl iodides to synthesise 3-iodomethyl dihydrobenzofurans under palladium photocatalysis (Scheme 18).72 An unprecedented hybrid vinyl/Pd(I) radical pair intermediate was proposed as a consequence of SET between the photoexcited Pd(0) catalyst and vinyl iodide. A 1,5-HAT process between the vinyl radical and tertiary C(sp3)–H bonds then proceeded, generating an R˙ for the iodocyclisation.
 | ||
| Scheme 18 Vinyl radical as HAT agent. | ||
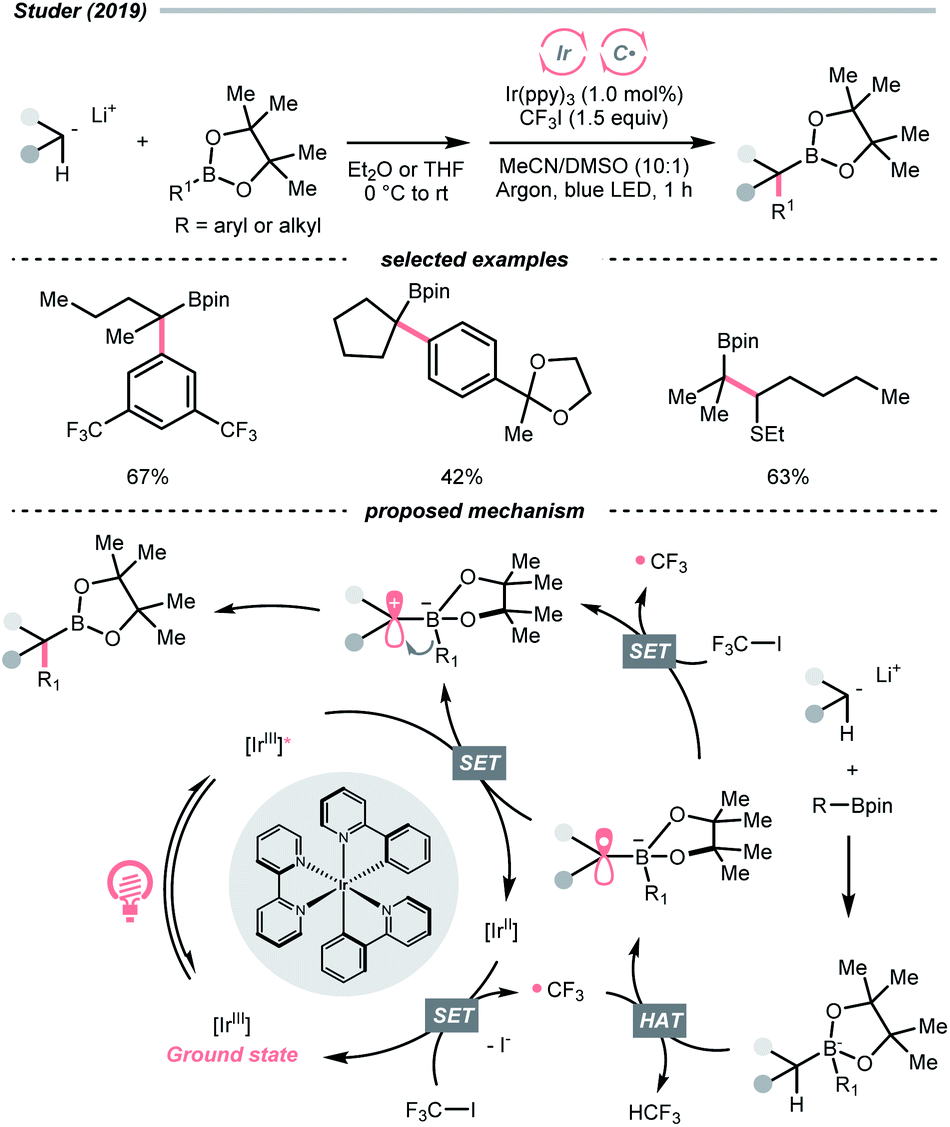
Other than C(sp2)-centred radicals, C(sp3)-centred radicals were also versatile HAT agents. In 2019, Studer's group reported photocatalysed α-C–H alkylation and arylation of alkylboronic esters, in which trifluoromethyl iodide mediated the HAT under photocatalytic conditions (Scheme 19).73 In their original mechanistic proposal, the trifluoromethyl radical 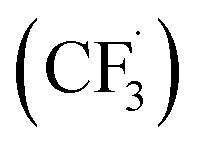 was generated through SET between the CF3I (Ered1/2 = −1.52 V vs. SCE in DMF) and Ir*(III) (Ered1/2 Ir(IV)/Ir*(III) = −1.73 V vs. SCE in MeCN). The
was generated through SET between the CF3I (Ered1/2 = −1.52 V vs. SCE in DMF) and Ir*(III) (Ered1/2 Ir(IV)/Ir*(III) = −1.73 V vs. SCE in MeCN). The 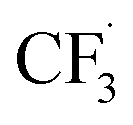 could execute HAT with the pre-synthesised boronate complex and form a radical anion intermediate (R˙), which was further oxidised by Ir(IV) or another trifluoromethyl iodide and underwent 1,2-alkyl or aryl migration to afford the α-substituted boronates. It should be noted that the radical chain process was supported by the reaction quantum yield measurement (ϕ = 8.8). Also, the fluorescence quenching of the Ir-photocatalyst with the boronate complex indicated another plausible catalytic cycle initiated by the borate oxidation.
could execute HAT with the pre-synthesised boronate complex and form a radical anion intermediate (R˙), which was further oxidised by Ir(IV) or another trifluoromethyl iodide and underwent 1,2-alkyl or aryl migration to afford the α-substituted boronates. It should be noted that the radical chain process was supported by the reaction quantum yield measurement (ϕ = 8.8). Also, the fluorescence quenching of the Ir-photocatalyst with the boronate complex indicated another plausible catalytic cycle initiated by the borate oxidation.
 | ||
| Scheme 19 Trifluoromethyl radical-mediated C(sp3)–H arylation. | ||
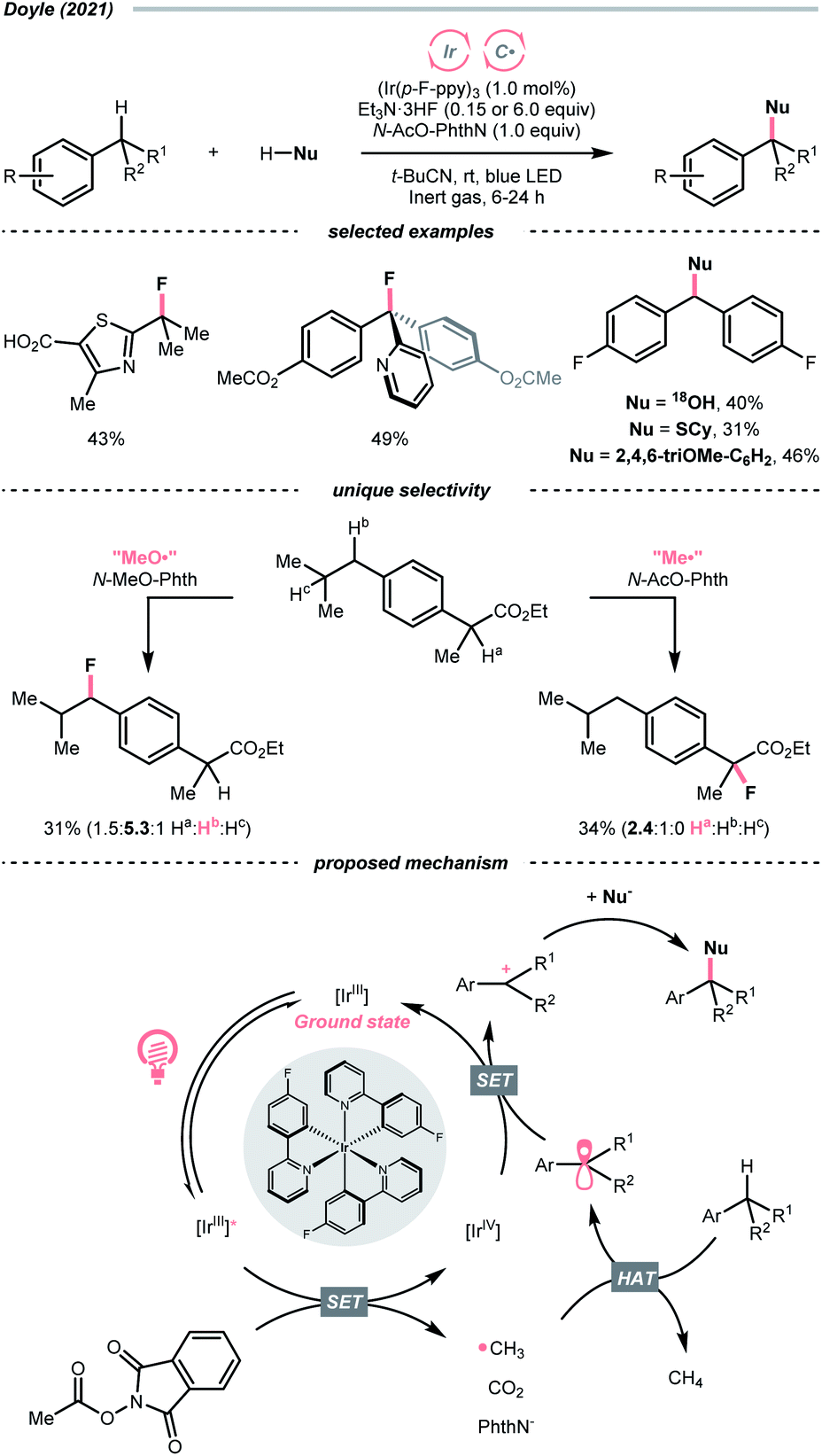
Very recently, Doyle's group documented a methyl radical (Me˙)-mediated C(sp3)–H fluorination using N-acetyloxyphthalimide as Me˙ precursor and triethylamine trihydrofluoride (Et3N·3HF) as the fluoride (F−) source under radical-polar crossover mechanism (Scheme 20).74 In their tentative catalytic cycle, N-acetyloxyphthalimide was reduced to Me˙ by excited Ir(III), with the concurrent release of CO2 and phthalimide. The alkane substrate underwent HAT with the Me˙. Afterwards, the generated R˙ was transformed into a carbocation, which was intercepted by the F− to give the fluorinated product. Distinct HAT selectivity toward electron-deficient C(sp3)–H bonds was observed with the nucleophilic Me˙, which was complementary to the electrophilic radicals. Besides fluorination, C(sp3)–H functionalisations with different nucleophiles such as water, alcohol, chloride, azide, thiol, and electron-rich arene were also demonstrated under slightly modified conditions.
 | ||
| Scheme 20 Methyl radical-mediated C(sp3)–H functionalisation. | ||
2.6. Miscellaneous examples
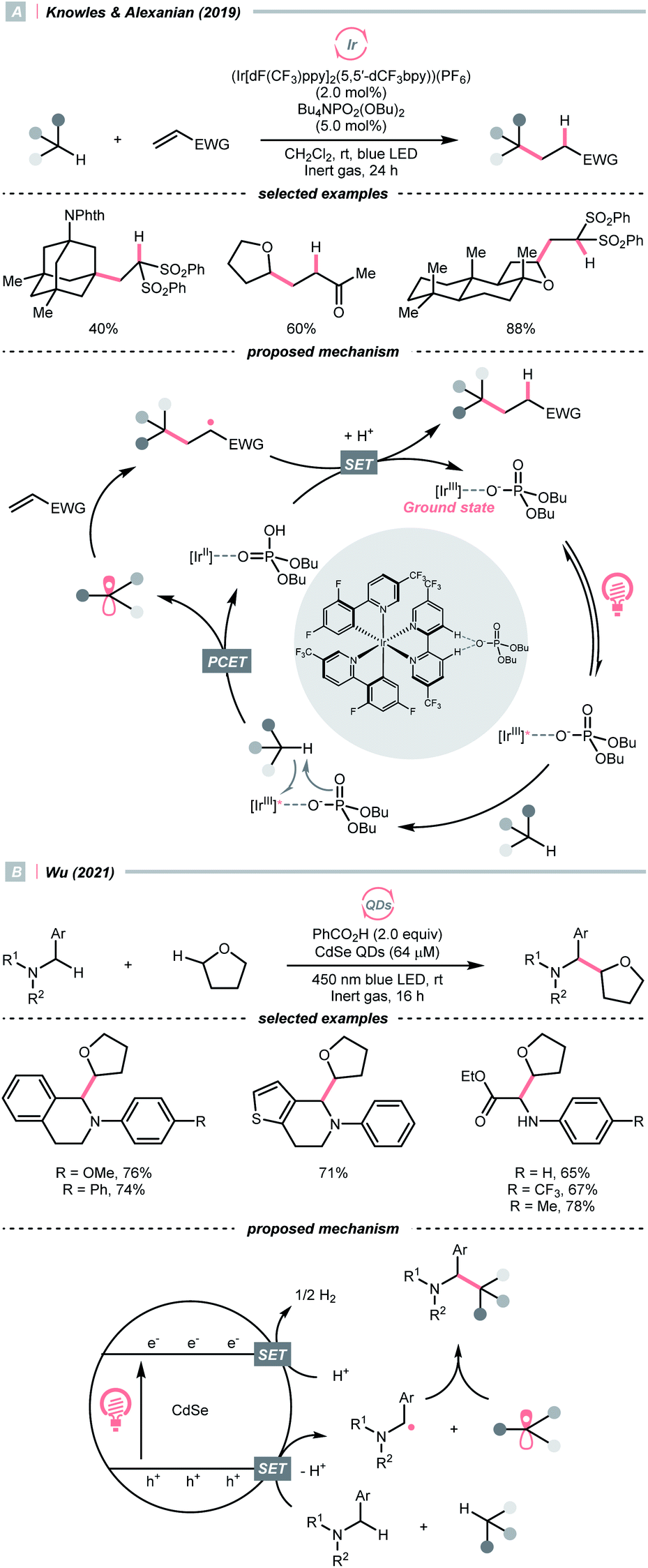
C–H bond oxidation followed by deprotonation is another R˙ generation pathway that resembles the HAT process. Unlike conventional HAT, wherein HAT agent was required, this kind of formal HAT process mandated strong oxidants or unique mechanisms to realise the stepwise R˙ generation.In 2019, the team of Knowles and Alexanian disclosed an unprecedented, intermolecular multisite-PCET (MS-PCET) interaction of the C(sp3)–H bond in the unactivated alkane with a noncovalent complex assembled from the Ir(III) polypyridyl catalyst and an organic phosphate base (PO2(OBu)2−) (Scheme 21A).75
 | ||
| Scheme 21 Forming R˙ through C(sp3)–H oxidations. | ||
Detailed spectroscopic studies supported the ion-pairing between the Ir*(III) (Ered1/2 Ir*(III)/Ir(II) = +1.72 V vs. SCE in MeCN), and PO2(OBu)2− (Ered1/2 ≥ +2.02 V vs. SCE in MeCN) and precluded the possibility of the direct electron-transfer mechanism. Such an association decreased the molecularity of the elementary C–H cleavage step, which facilitated the concerted transfers of proton and electron. Various C(sp3)–H bonds were transformed into R˙ for the Giese reaction, with observed selectivity consistent with their bond dissociation free energies (BDFEs).
When exploiting the heterogeneous cadmium selenide quantum dots (CdSe QDs), Wu's group disclosed another new mechanism that directly activates C(sp3)–H bonds under blue light irradiation (Scheme 21B).76 Due to the large surface area to accommodate reactive species, two types of electron-rich α-heteroatomic C(sp3)–H bonds, in ether and 2-aryl tetrahydroisoquinoline (THIQ), respectively, were simultaneously oxidised by the holes (h+s) on QDs, forming two different R˙ and setting the stage for subsequent radical–radical cross-couplings. Synergistically, the electrons distributed on CdSe were consumed via H2 evolution, accomplishing the CDC reaction. This powerful QD material was also used in other radical transformations by the same group, which showed fruitful reactivities in Minisci alkylation77 and radical thiolation.78
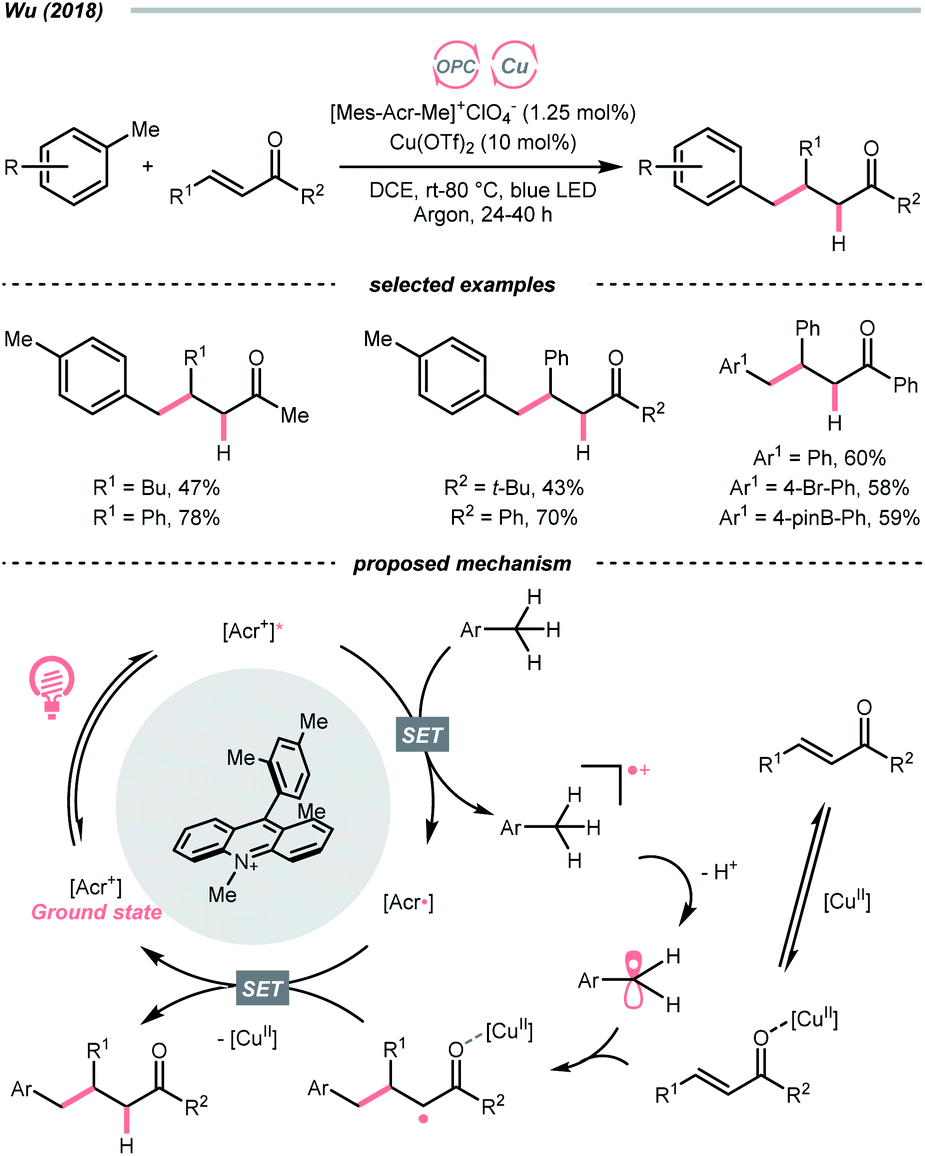
The R˙ production via the SET/deprotonation pathway of benzylic C–H bonds in toluene derivatives could be facilitated by the initial oxidation of their arene moieties. Along this line, Chen and Wu et al. documented a Giese reaction between toluene and chalcone derivatives with an acridinium photocatalyst and a Cu(II) Lewis acid catalyst (Scheme 22).79 In the tentative mechanism, the excited acridinium (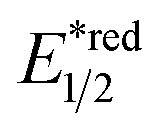 = +2.06 V vs. SCE in MeCN) oxidised the toluene (Ered1/2 = +2.36 V vs. SCE in MeCN) to a radical cation, in which the spin centre might initially reside in the arene, then shifted to the benzylic position after deprotonation. Despite the uphill SET process, which was crucial for breaking the benzylic C–H bond since its acidity was pronouncedly enhanced after arene oxidation, some innovative techniques (e.g., “stop-flow” micro-tubing (SFMT) reactors) could expedite the electron transfer.
= +2.06 V vs. SCE in MeCN) oxidised the toluene (Ered1/2 = +2.36 V vs. SCE in MeCN) to a radical cation, in which the spin centre might initially reside in the arene, then shifted to the benzylic position after deprotonation. Despite the uphill SET process, which was crucial for breaking the benzylic C–H bond since its acidity was pronouncedly enhanced after arene oxidation, some innovative techniques (e.g., “stop-flow” micro-tubing (SFMT) reactors) could expedite the electron transfer.
 | ||
| Scheme 22 Photocatalytic benzylic C(sp3)–H oxidation. | ||
3. C–C cleavage
C–C bonds are common skeletons in organic molecules, which partially explains the high diversity of types of R˙ precursors that involve C–C cleavage to form R˙. Since C–C bonds are relatively inert, driving forces such as small molecule extrusion (i.e., CO2, CO, acetone), strain release, and aromatisation are frequently considered to facilitate the C–C bond cleavage. In this regard, oxygen-containing molecules, such as carboxylic acids, aldehydes and alcohols, and some dearomatised compounds, are versatile R˙ precursors that undergo photoscission of C–C bonds.3.1. C–C cleavage of carboxylic acid derivatives
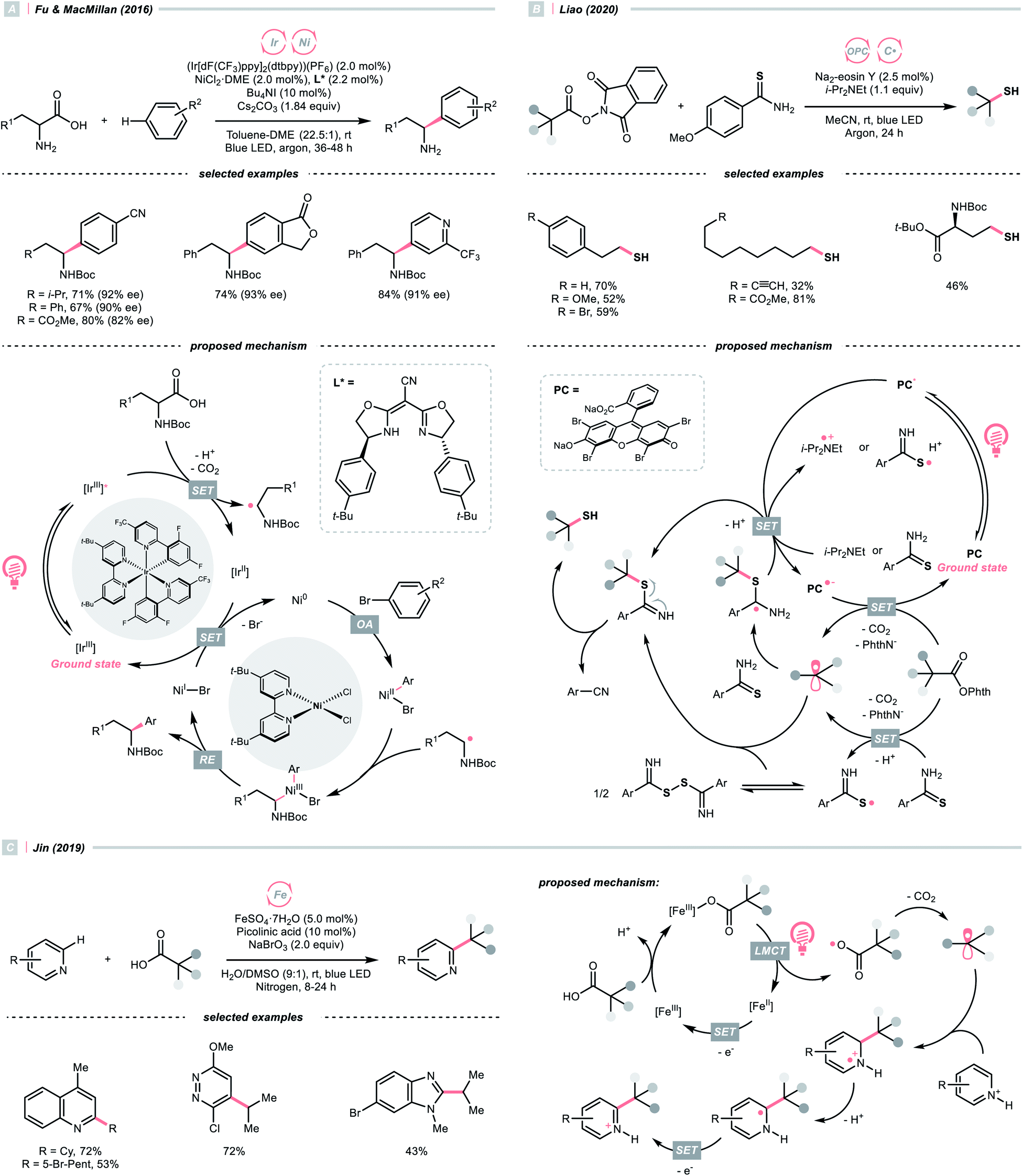
Alkyl carboxylic acids are naturally abundant and bench-stable.80 Although CO2 extrusion offers the enthalpic advantage to elicit R˙ from carboxylic acids, high temperatures and strong oxidants, and sometimes the presence of transition metals, were required in traditional decarboxylation methods. Fortunately, advancements in photocatalysis have allowed decarboxylative R˙ generation to procced under mild conditions.81In 2014, Doyle, MacMillan and their co-workers reported the seminal work of metallaphotoredox catalysis by combining photoredox catalysis with nickel catalysis for decarboxylative arylation of α-amino acids with aryl halides,82 which revolutionised the conventional design for transition metal-catalysed cross-couplings.5,83 Later in 2016, the team of Fu and MacMillan upgraded this dual catalysis protocol to an asymmetric version using a chiral bisoxazoline ligand (Scheme 23A).84 In this metallaphotoredox mechanism, single-electron oxidation of the carboxylate (for tert-butyl carbamoyl Boc-Pro-OCs, Ered1/2 = +0.95 V vs. SCE in MeCN) with the excited Ir(III) (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN) generated a carboxyl radical, which decomposed into an R˙ via releasing CO2. Meanwhile, the chiral aryl nickel(II) bromide generated from the oxidative addition of nickel(0) to aryl bromide trapped the R˙, followed by the reductive elimination to afford the α-arylated amines in moderate to good yields with good to excellent enantioselectivities.
 | ||
| Scheme 23 R˙ formation via carboxyl radical intermediates. | ||
By strategically designing some carboxylic acid derivatives, the decarboxylation could also be put into a reduction-initiated manifold. For example, alkyl diacyl peroxides ((RCOO)2) that were synthesised from aliphatic acids could reverse the redox properties of their parent acids and favour the reductive decarboxylation.85 However, they are often chemically labile and potentially explosive, which cause safety concerns. Pioneered by Okada, and later revisited by Baran, Glorius and others, the carboxylic acid was converted into the relatively stable N-(acyloxy)phthalimides, which were repurposed radical alkylating reagents and so-called redox-active esters (RAEs).86–92 Since these pioneering research, a diverse range of radical alkylation reactions have been published. One recent photocatalytic example was Liao's report of decarboxylative thiolation of N-(acyloxy)phthalimides utilizing Na2-eosin Y in the presence of sulfur donor and amine reductant (Scheme 23B).93 Mindful of the sensitivity of thiol products toward oxidative conditions, reductive generation of R˙ from photocatalytic decarboxylation could effectively avoid the undesired sulfur oxidation. Under the photoreduction conditions, RAE underwent a SET/fragmentation sequence to release CO2 and phthalimide, giving the R˙ to be thiolated subsequently. To be noticed, other than these carboxylic acid derivatives, using anhydride94 and pyridine N-oxide95 were also reported for C(sp3) radical generation with photoredox catalysis.
Apart from undergoing the SET pathway, aliphatic acid could be oxidised to form R˙ via LMCT of the photoexcitable transition metal carboxylate. In 2019, Inspired by the photoinduced iron(III)-mediated decarboxylative Minisci alkylation by Sugimori,96 Jin's group advanced an iron-catalysed version with the picolinic acid ligand under visible light (Scheme 23C).97 In their proposed reaction mechanism, iron(II) carboxylate was oxidised by BrO3− to iron(III), which, upon photoexcitation, was susceptible to Fe–O bond homolysis. Decarboxylation ensued, producing the R˙ for the sequential Minisci alkylation.
The picolinic acid ligand was crucial in altering the photophysical properties of the iron catalyst since the desired reactivity was inhibited in its absence. To be noticed, with the same iron/ligand set, Jin's group also applied this decarboxylative R˙ generation strategy for conjugate addition to construct C–C and C–N bonds under redox-neutral conditions.98
3.2. C–C cleavage of carbonyl derivatives
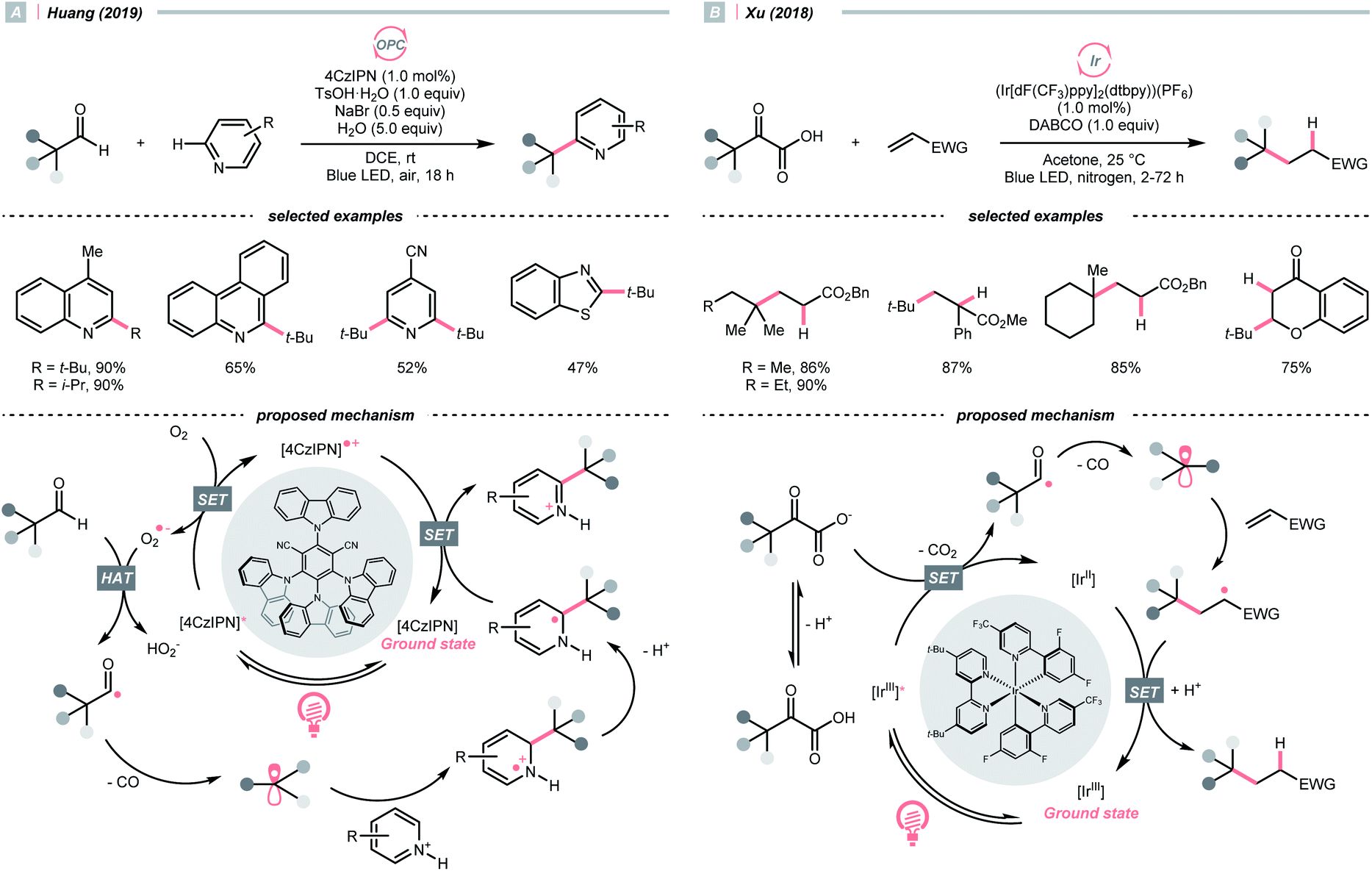
The decarbonylation of carbonyl compounds is another common way to generate an R˙, wherein the R˙ precursor, acyl radical, is frequently obtained from aldehydes through HAT.99,100In 2019, Huang and his co-workers developed a photocatalysed decarbonylative Minisci alkylation with aldehydes under air (Scheme 24A).101 In their critical R˙-generating steps, the O2 in the atmosphere was reduced by photoexcited 4CzIPN, generating a superoxide radical anion (O2˙−). This oxy radical could perform HAT at the formyl C–H to give an acyl radical, followed by a radical decarbonylation to release CO and give an R˙.
 | ||
| Scheme 24 R˙ formation from acyl radical intermediates. | ||
Alternatively, acyl radicals could derive from an oxidative decarboxylation reaction of α-ketoacids, followed by the decarbonylation to give the R˙. Capitalizing on this special class of oxocarboxylic acids, in 2018, Xu and co-workers described a Giese reaction with α-ketoacids under photoredox conditions (Scheme 24B).102 In the presence of a base, the α-oxocarboxylate (for pyruvic acid, Ered1/2 = +1.03 V vs. SCE in DMSO) was oxidised by the excited (Ir[dF(CF3)ppy]2(dtbpy))(PF6) (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN) into carboxyl radical, delivering the R˙ with concurrent evolution of CO2 and CO. However, the acylation product dominated when targeting the less stabilised 2° and 1° radical, or the bulky adamantly one. Fortunately, alkylation with these R˙ precursors could be more efficient toward alkylation with refluxing.
3.3. C–C cleavage of alcohol derivatives
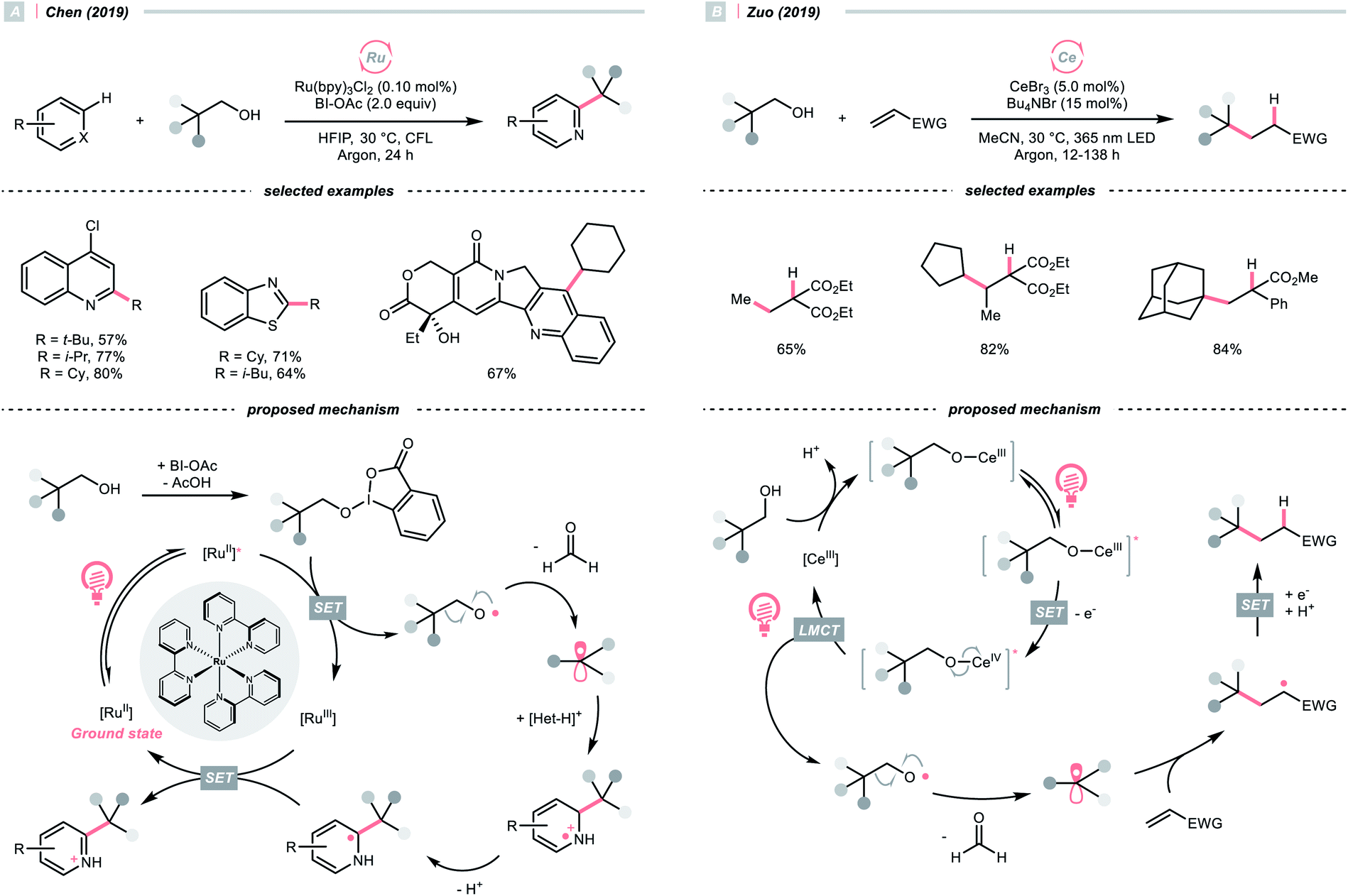
SET of aliphatic alcohols could also effect the R˙ generation via different C–C bond-breaking pathways. Among them, dehydroxymethylation of β-substituted alcohols could give the R˙ after decarbonylation.To sidestep the sluggish oxidation of the –OH group, Chen's group converted the alcohol into alkoxybenziodoxolone (BI–OCH2R) through condensation with 1-acetoxy-1,2-benziodoxol-3-(1H)-one (BI-OAc). The in situ assembled BI-OCH2R underwent reductive SET by the excited Ru(II)-photocatalyst and produced an RCH2O˙, which delivered an R˙ for Minisci alkylation after β-scission and removal of formaldehyde (Scheme 25A).103
 | ||
| Scheme 25 R˙ generation from alcohol and its derivatives. | ||
Alternatively, Zuo and his co-workers demonstrated a Ce-catalysed dehydroxymethylative functionalisation reaction with β-substituted alcohols, wherein the key alkoxy radicals were generated by the LMCT of photoexcited Ce(IV) alkoxide (Scheme 25B).104,105 Similar to Chen's case, β-cleavage of the C–C bonds in alkoxy radical give the R˙, which could engage in alkylation, alkenylation, oxygenation, and hydrogenation cross-couplings with alkyl halides. The catalyst turnover mechanism varied with the light sources as cerium could catalyse the dehydroxymethylative Giese reaction alone under 365 nm. However, it dictated the presence of 9,10-diphenylanthracene as co-catalyst to accomplish the same reaction with 400 nm irradiation.
3.4. C–C cleavage via strain release
Strain release of congested cyclic compounds could be induced by ring-opening radical translocation or radical addition, which were two common pathways to generate the R˙ from strained structures.NCR resulting from SET could enable R˙ generation via breaking neighbouring C–C bonds. Especially, ring-opening of cyclic iminyl radical intermediates, which are commonly derived from oxime derivatives, are useful in synthesizing alkyl nitriles.
In 2018, Leonori's group showcased a photoredox synthesis of remotely functionalised alkyl nitriles using O-alkylated oximes derived from carboxylic acid (Scheme 26A).106 With the equimolar carbonate base, the carboxylate was oxidised by Fukuzumi's catalyst. Following CO2 and acetone extrusion, the cyclic iminyl radical quickly fragmented into the R˙ with a terminal nitrile group for the subsequent fluorination, chlorination, or azidation. Ring-opening of four-membered rings was facile, while the unstrained five to seven-membered ones required two α-methyl groups or one α-phenyl substituent to facilitate C–C cleavage. By intercepting the cyanated R˙ with nickel catalysis, radical ring-opening arylation, vinylation and alkylation cascades were achieved by the same group with corresponding carbon electrophiles.107
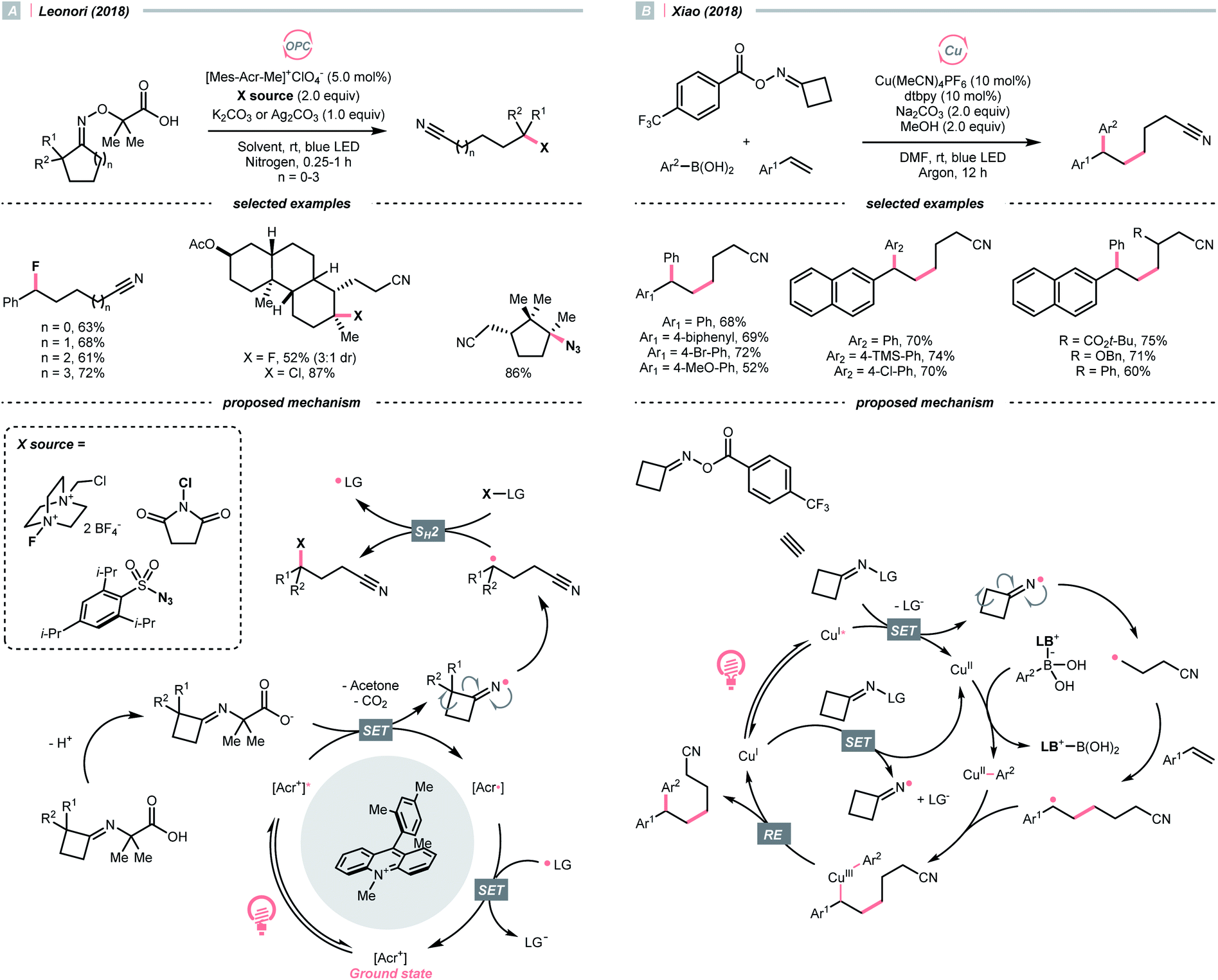 | ||
| Scheme 26 R˙ formation via iminyl radical-induced ring-openings. | ||
Reductive SET of engineered oximes could also furnish the iminyl radicals for R˙ generation via ring-opening. In the same year, Xiao et al. exemplified a visible-light-induced copper-catalysed styrene difunctionalisation reaction with oxime esters and aryl boronic acids (Scheme 26B).108 In their proposed mechanism, the excited copper(I) reduced the O-benzoyl oxime ester and triggered its N–O cleavage and β-scission to give the cyanated R˙. Meanwhile, copper(II) exchanged its ligand with boronic acid to form an aryl Cu(II) species. The R˙ was trapped by the styrene and subsequently by the aryl copper(II) intermediate, which gave the 1,2-difunctionalised alkane after a facile Cu(III) reductive elimination and closed the copper catalytic cycle.
Aligned with iminyl radical, amidyl radical is another NCR that could generate R˙ by breaking the C–C bond in cyclic structures. In 2020, Waser's laboratory developed a ring-opening strategy to achieve oxidative difunctionalisations of N-cyclopropyl and N-cyclobutyl amides with Selectfluor as the fluorine source and benzophenone as the photocatalyst (Scheme 27).109 Mechanistically speaking, a strongly oxidising benzophenone radical cation (Ered1/2 (BP+/BP) = +2.37 V vs. SCE in MeCN) was formed by photooxidation with Selectfluor or its amino radical cation (for benzophenone, Ered1/2 (BP+/BP*) = −0.62 V vs. SCE in MeCN; for Selectfluor, Ered1/2 = +0.33 V vs. SCE in MeCN and for Selectfluor-derived N-radical cation, Ered1/2 = +0.79 V vs. SCE in MeCN). With this potent oxidising species, the amide (for cyclopropyl amide, Ered1/2 = +1.67 V vs. SCE in MeCN) was turned into an amido radical cation, then an iminium-containing R˙ after ring-opening. Selectfluor serves as both fluorine source and oxidant for benzophenone regeneration. Stepwise addition of a nucleophile, including alcohol, hydroperoxide, thiol, trifluoroborate, and electron-rich arene to the fluorinated imine intermediates, could accomplish the corresponding difunctionalisations.
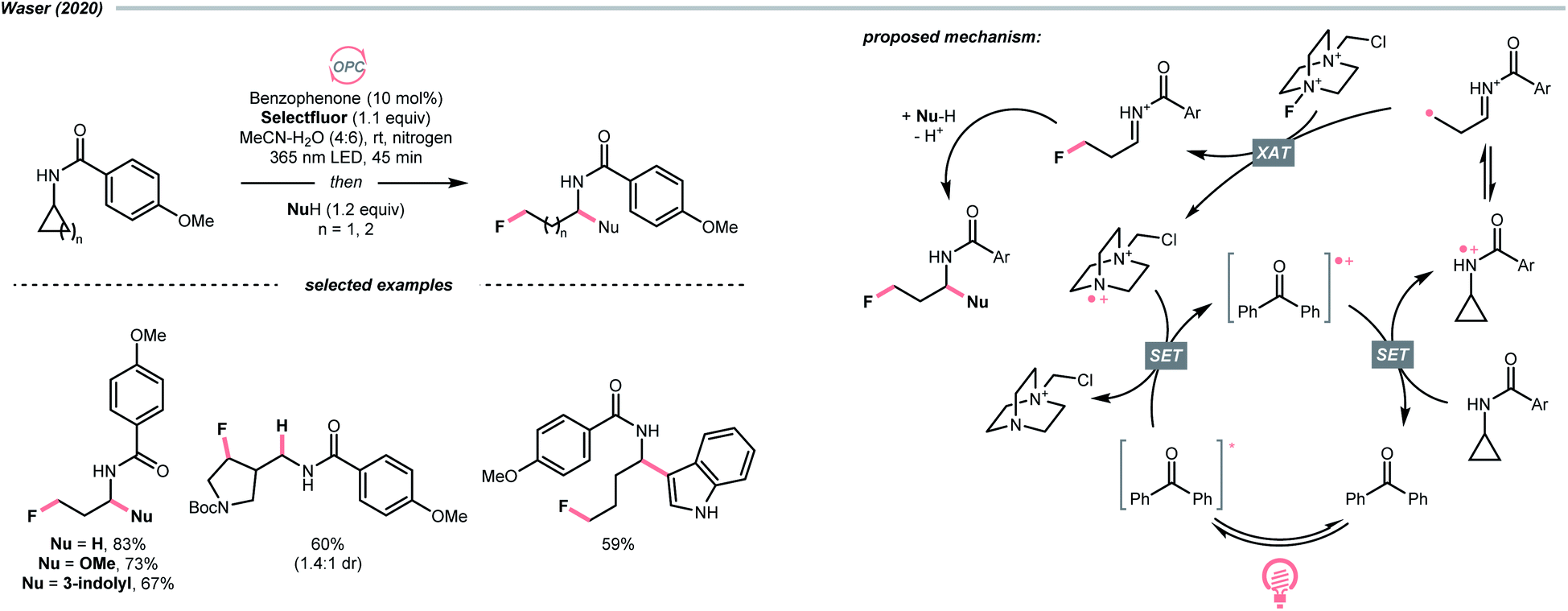 | ||
| Scheme 27 Generating R˙ from oxidative ring-opening of amide. | ||
Besides C–C cleavage of cyclic oxime and amides [1.1.1]propellane, a simple and strained bicyclic molecule, could directly interact with a radical species to cleave its central C–C bond and afford a C(sp3) radical. Since [1.1.1]propellane has been demonstrated as a useful synthon to access disubstituted bicyclo[1.1.1]pentane (BCP),110–115 a high-value three-dimensional bioisostere for phenyl, alkynyl, and tert-butyl groups in drug designs,116,117 its synthetic application has received attention in photocatalysis.
Driven by its great utility, MacMillan's group reported a difunctionalisation reaction of [1.1.1]propellane by merging copper catalysis and iridium photocatalysis (Scheme 28).114 In their tentative mechanism, the excited Ir(III)-photocatalyst (Ered1/2 Ir(IV)/Ir*(III) = −1.73 V vs. SCE in MeCN) reduced the iodonium dicarboxylate (for the 4-tetrahydropyranyl one, Ered1/2 = −0.82 V vs. SCE in MeCN), alkyl bromide, or sulfonothioate via SET to generate a C- or S-centred radicals, respectively, which could readily combine with [1.1.1]propellane to give a bicyclo[1.1.1]pentyl radical (R˙) through strain release. By leveraging copper catalysis, the R˙ was then coupled with various nucleophiles such as electron-rich arenes, anilines, and sulfonamides, offering 1,3-disubstituted BCPs through the facile R–Cu(III)–Nu reductive elimination.
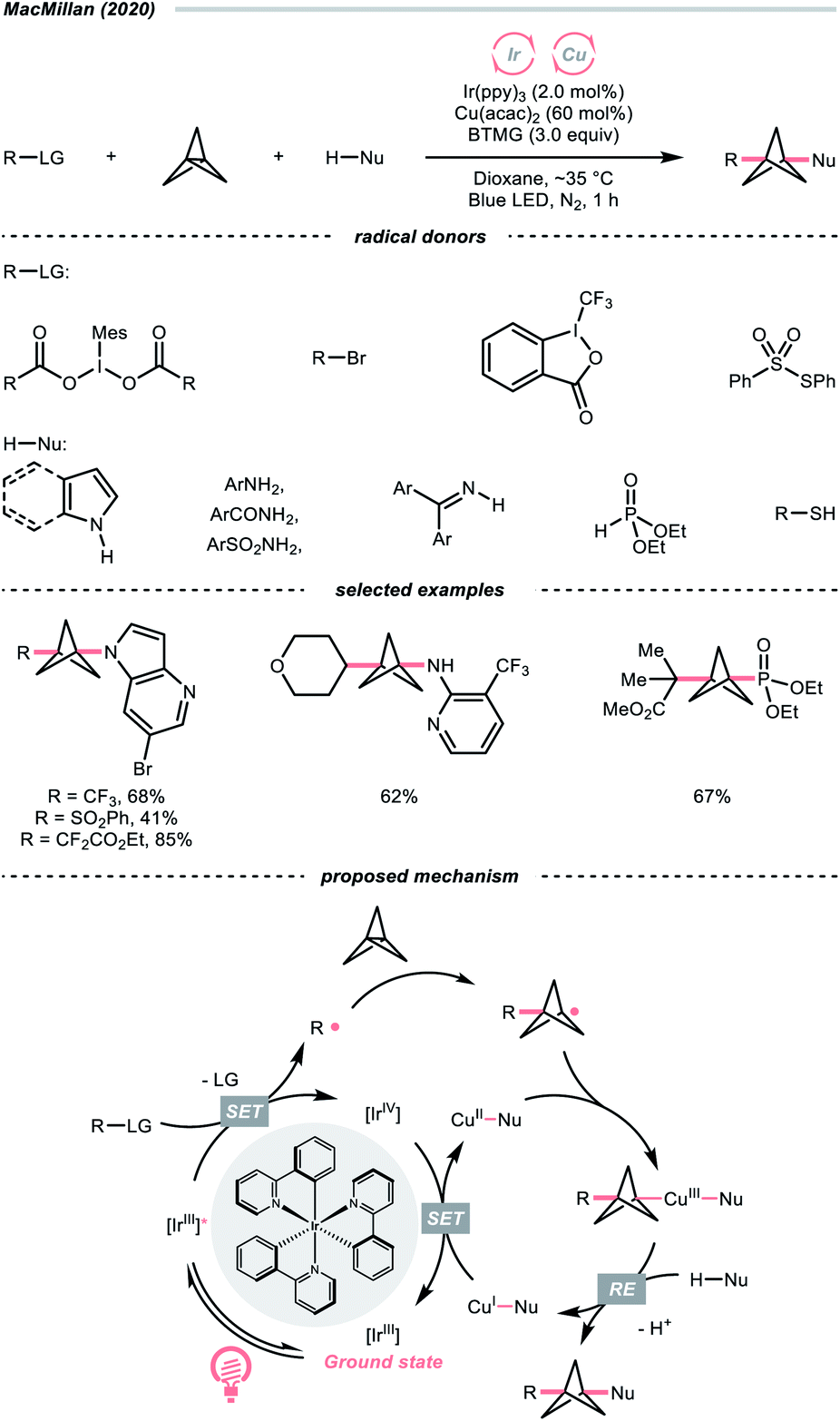 | ||
| Scheme 28 Radical addition to [1.1.1]propellane. | ||
3.5. C–C cleavage via aromatisation
Dearomatised arenes and heteroarenes are reducing and tend to rearomatise. With such a strong driving force, various alkylating reagents in this class are designed to release the R˙ via rearomative C–C cleavage under photocatalytic conditions.Aza-cyclohexadienyl radical is a special amino radical that mostly stems from the oxidative SET with dihydropyridines (DHPs). Bearing a strong tendency toward aromatisation via C–C cleavage at its para-position, 4-alkylated DHPs are viable R˙ sources. Taking a C4-iminoalkylated Hantzsch ester (Ered1/2 = +1.01–1.23 V vs. SCE in MeCN) as the starting material, Romanov-Michailid et al. elaborated the synthesis of several 3-aryl morpholines via R˙ cyclisation (Scheme 29A).118 Mechanistically, PCET between the (Ir[dF(CF3)ppy]2(bpy))(PF6) (Ered1/2 Ir*(III)/Ir(II) = +1.32 V vs. SCE in MeCN) and a Lewis base was believed to be a key step in this O2-mediated cyclisation protocol. Ring-closure of the R˙ with its imine tail and the subsequent reductive quenching will give the morpholine as the desired product. Interestingly, pioneered by Melchiorre's group, several photocatalyst-free alkylation protocols with direct excitation of Hantzsch esters were also documented.119,120
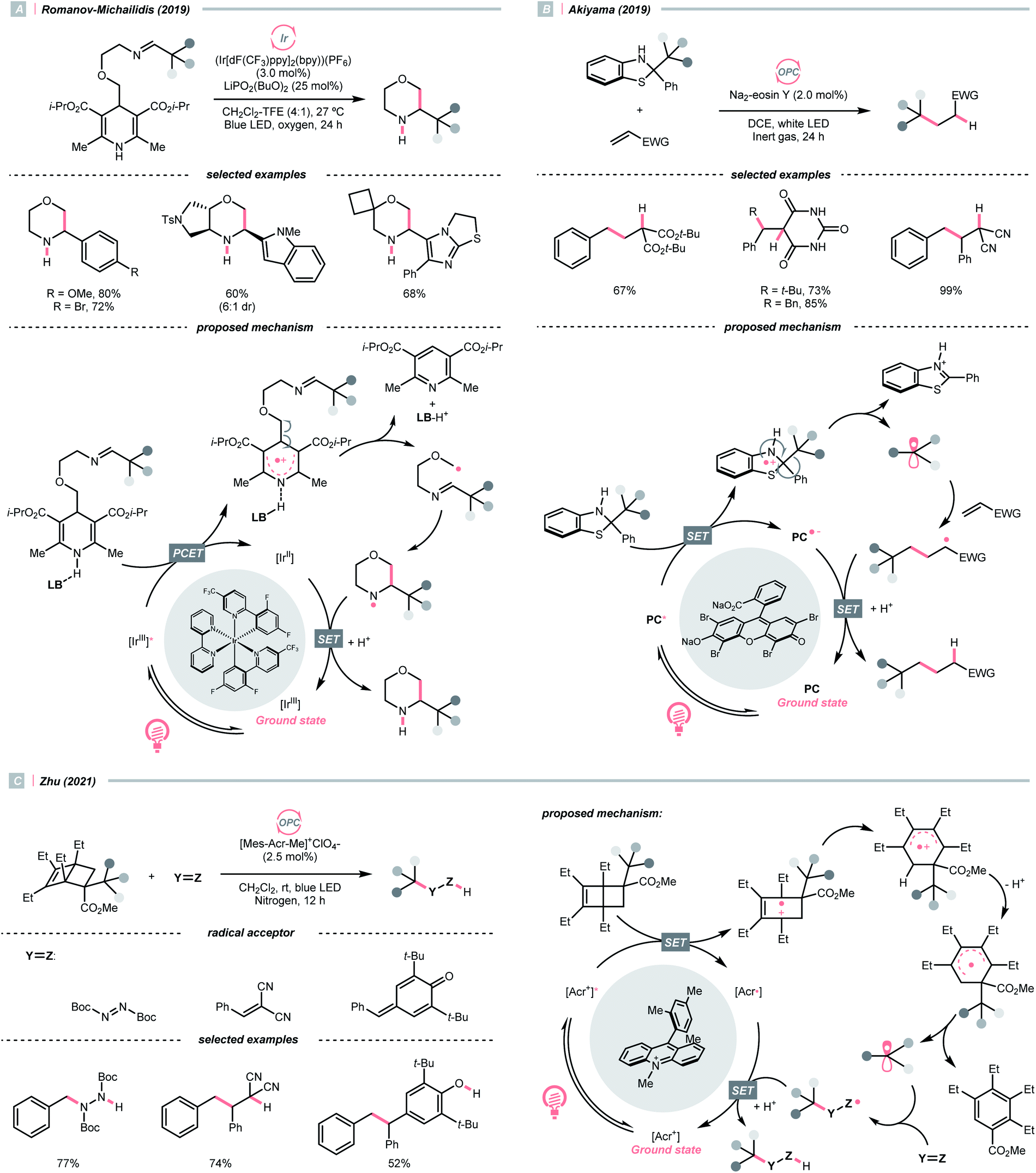 | ||
| Scheme 29 Aromatisation-induced R˙ extrusions. | ||
Aside from DHPs, other heterocycles and carbocycles that could enforce the rearomative C–C cleavage to give the R˙ were reported. For instance, Akiyama's group used C2-alkylated 2-phenyl dihydrobenzothiazoles (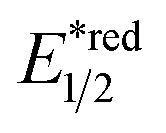 = +0.70 V vs. SCE in MeCN) for photoinduced radical alkylation with electron-deficient alkenes as the acceptors and Na2-eosin Y as the photocatalyst (Scheme 29B, Ered1/2 (eosin Y*/eosin Y˙−) = +0.83 V vs. SCE in 1:1 MeCN–H2O).121 Very recently, Zhu's group reported a novel alkylated bicyclo[2.2.0]hexene reagent (for benzylation, Ered1/2 = +1.59 V vs. SCE in MeCN) for 1,2-addition to azodicarboxylate and electron-poor alkene, as well as 1,6-addition to para-quinone methides with [Mes-Acr-Me]+ClO4− (Scheme 29C, E*1/2red = +2.06 V vs. SCE in MeCN).122 In both cases, single-electron oxidation of R˙ precursors afford radical cations, which split into a (hetero)arene and an R˙. The latter was submitted to typical photocatalysed Giese-type pathways to give the alkylated products.
= +0.70 V vs. SCE in MeCN) for photoinduced radical alkylation with electron-deficient alkenes as the acceptors and Na2-eosin Y as the photocatalyst (Scheme 29B, Ered1/2 (eosin Y*/eosin Y˙−) = +0.83 V vs. SCE in 1:1 MeCN–H2O).121 Very recently, Zhu's group reported a novel alkylated bicyclo[2.2.0]hexene reagent (for benzylation, Ered1/2 = +1.59 V vs. SCE in MeCN) for 1,2-addition to azodicarboxylate and electron-poor alkene, as well as 1,6-addition to para-quinone methides with [Mes-Acr-Me]+ClO4− (Scheme 29C, E*1/2red = +2.06 V vs. SCE in MeCN).122 In both cases, single-electron oxidation of R˙ precursors afford radical cations, which split into a (hetero)arene and an R˙. The latter was submitted to typical photocatalysed Giese-type pathways to give the alkylated products.
3.6. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C π-bond cleavage
C π-bond cleavage
Radical addition to the alkenyl π-bond is a method to generate a C(sp3) radical that could increase the structure and functional complexities of incoming radical species. In some cases, such a radical translocation process could expedite the new radical generation since the radical adducts are usually more stable. Furthermore, radical philicity reversal is also a common purpose by introducing these primary radical acceptors, which could be highly useful in organic synthesis and polymer chemistry.123,124
The addition of R˙ to electronically matched double bonds is commonly seen in the literature of alkene hydrofunctionalisations and difunctionalisations, some of which have been reviewed in previous examples. Instead, adding heteroatomic radicals to the olefins and merging the new R˙ in different radical cascade reactions was rarely seen and will be exemplified below.
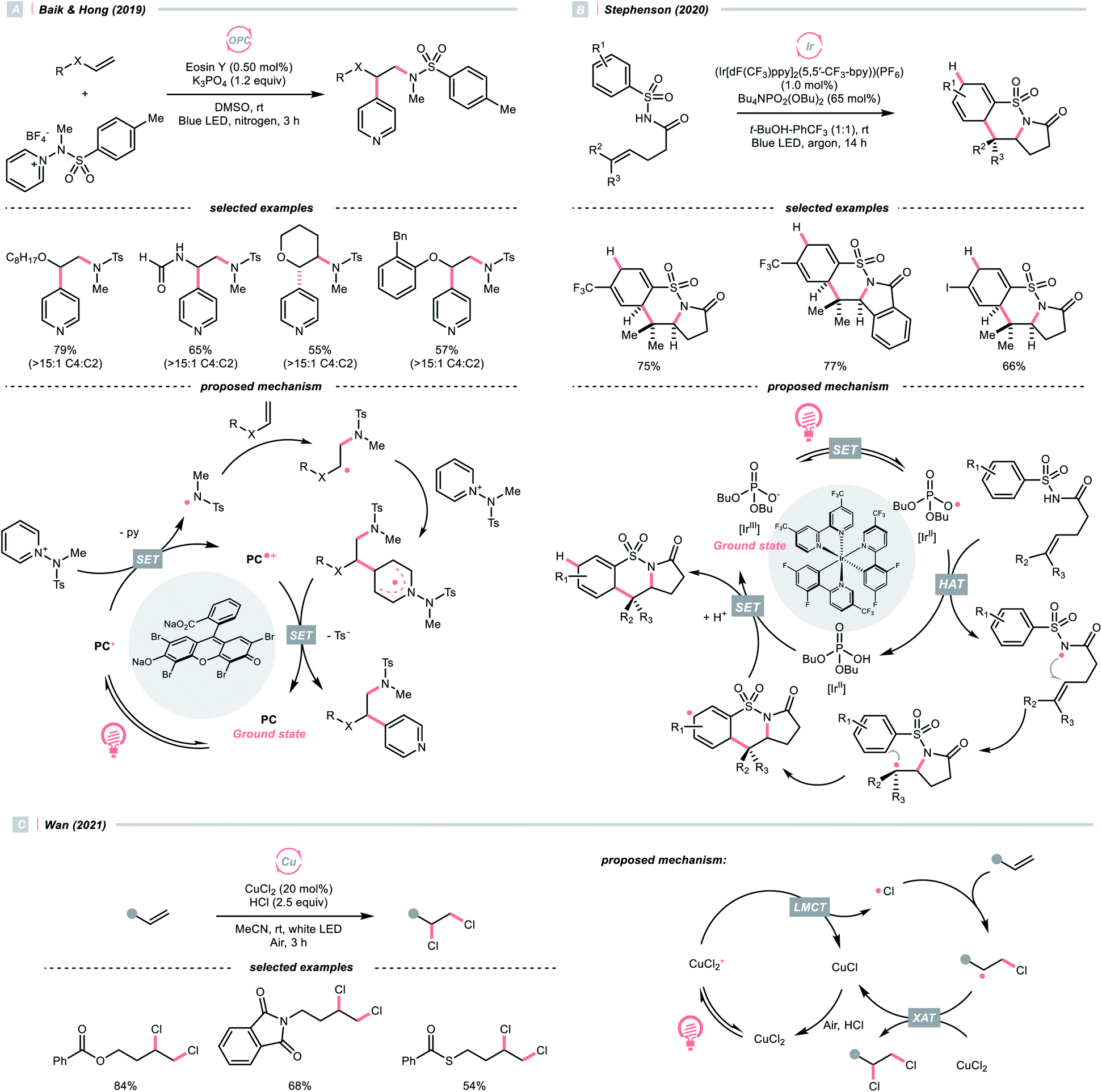
As an intermolecular example, in 2019, Baik and Hong's team made a breakthrough on C4-selective heteroarene alkylation, which is a long-lasting challenge when both the heteroaromatic C2 and C4 sites are non-substituted (Scheme 30A).125 The key for such a unique selectivity was the usage of N-(methyltosyl)aminopyridinium salts (Ered1/2 = −0.70 V vs. SCE in MeCN), which provided the NCR upon single-electron reduction by the excited eosin Y (Ered1/2 (eosin Y*/eosin Y˙+) = −1.11 V vs. SCE in 1:1 MeCN–H2O) and steered the R˙ formed from the NCR and alkene toward its C4 position via steric and some secondary interaction. The observed high regioselectivity complied with the density functional theory (DFT) calculation, suggesting an N–N bond cleavage mechanism of the N-aminopyridinium to give the final product and a new NCR. It was worth mentioning that after the combination of the electrophilic NCR and the electron-rich alkene, a nucleophilic R˙ was generated for the alkylation of electron-poor pyridinium, which demonstrated the significance of matching the polarity and the versatility of alkene in inversing the radical polarity.
 | ||
| Scheme 30 Photocatalysed R˙ formation via radical addition to CC bonds. | ||
A similar reaction sequence in an intramolecular fashion could lead to different reaction outcomes. Stephenson's laboratory reported a photoinduced dearomative cyclisation of alkenyl N-arylsulfonamides with (Ir[dF(CF3)ppy]2(5,5′-CF3-bpy))(PF6) and a HAT agent Bu4NPO2(OBu)2 (Scheme 30B).126 At first, a ground-state Ir(III) and dibutyl phosphate aggregation, as proposed by Alexanian and Knowles (Scheme 21A), was suspected of enabling the NCR generation. Nonetheless, careful spectroscopic studies suggested the MS-PCET and deprotonation/oxidation mechanism to generate the NCR were less likely in this case. Instead, after light-induced SET between the Ir(III) and phosphate, an oxy radical departed and performed the HAT with the sulfonamide N–H. NCR addition to the terminal alkene formed an R˙, which then performed an intramolecular radical cyclisation to the pendent arene. Subsequent SET/protonation steps of the cyclohexadienyl radical delivered the dearomatised product.
In addition to NCRs, the addition of chlorine radical to aliphatic alkene could also effect the alkyl radical generation, as demonstrated by Wan's group (Scheme 30C).127 In this case, a vicinal dichlorination was succeeded, wherein CuCl2 catalyst and HCl supplemented the chlorine sources under ambient conditions. However, styrene 1,2-dichlorination was compromised by some side reactions, which could be suppressed using over-stoichiometric CuCl2.
In contrast to the radical addition mechanism above, some novel photocatalysis that could provide highly oxidising or reducing environments has been used to generate R˙ via direct SET on alkenes, which was trapped by some nucleophiles or electrophiles, respectively. Single-electron oxidation of a CC bond gives a C–C radical cation, simultaneously serving as an R˙ source and an electrophile for alkene difunctionalisations. Under this reaction paradigm, Nicewicz and his co-workers disclosed their seminal discovery on photocatalysed intramolecular hydroetherification of alkenols in 2012.128 Later, several intra- and intermolecular alkene hydrofunctionalisations followed.129–131 In 2017, they advanced this protocol by merging Cu(II) catalyst and electrophilic halogen sources for halolactonisation of unsaturated fatty carboxylic acids (Scheme 31A).132 The strongly oxidising [Mes-Acr+-Me]ClO4− (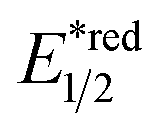 = +2.06 V vs. SCE in MeCN) played a pivotal role in this alkene radical cation chemistry since the reduction potential of alkenes were often beyond the reach of many common photocatalysts.
= +2.06 V vs. SCE in MeCN) played a pivotal role in this alkene radical cation chemistry since the reduction potential of alkenes were often beyond the reach of many common photocatalysts.
 | ||
| Scheme 31 Photocatalysed SET-induced CC cleavages. | ||
On the other hand, single-electron reduction of the alkene will leave a radical anion. In 2021, Polyzos published a photocatalysed styrene reductive hydrofunctionalisation with proton or ketone as electrophiles and amine as the sacrificial reductant (Scheme 31B).133 Interestingly, a multiphoton tandem photoredox cycle of the Ir-photocatalyst [Ir(ppy)2(dtbbpy)]PF6 was proposed. Central to this method was the photocatalytic generation of a new Ir species with a partially reduced bipyridine ligand, which could initiate another photocatalytic cycle and deliver higher-energy electrons to overcome the high reduction barrier of styrene (typically, Ered1/2 = <−2.0 V vs. SCE in MeCN).
4. C–X cleavage
Exploring versatile aliphatic derivatives as R˙ sources can offer more synthetic opportunities for retrosynthetic analysis. Since each type of C–X bond has distinct chemical and photophysical properties, they are often paired with unique activation strategies, which are useful in different synthetic settings. With considerable efforts devoted to the field of photocatalysis, various alkylating reagents, including aliphatic halides, alcohols, amines, boronic acids, and others, have been demonstrated as effective R˙ sources.4.1. C–N cleavage
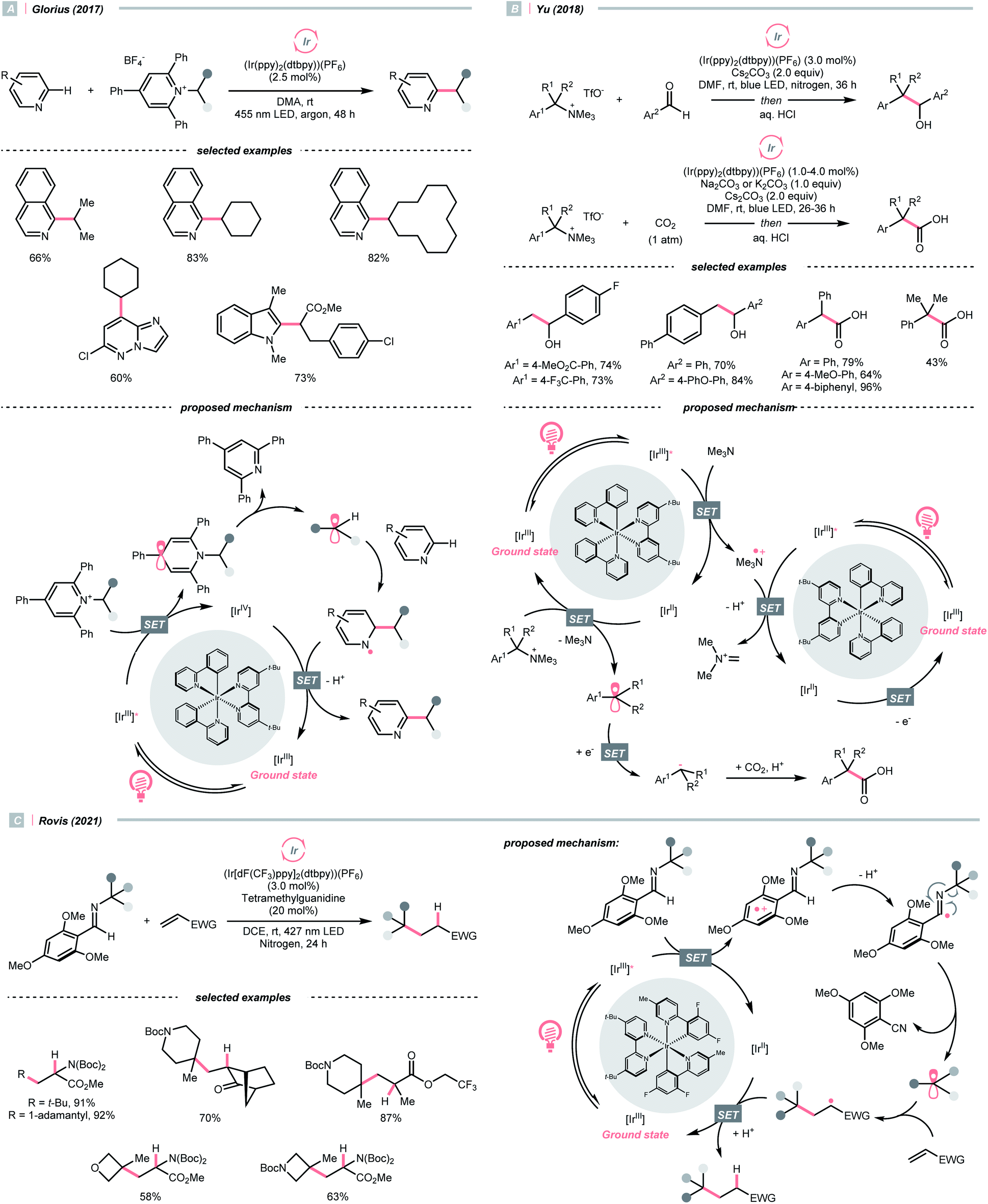
While photocatalytic radical C–N bond formation is of tremendous interest in both academic and industrial settings, C–N bond cleavage typifies an equally important direction, which played an indispensable role in R˙ generation strategies within the SET manifold.Quaternary ammonium or pyridinium salts, featuring highly polarised C–N bonds because of the cationic nitrogen, are widely used in radical alkylations via single-electron reduction. In 2017, Glorius et al. described a novel Minisci alkylation reaction with Katritzsky salts (R-TPP) under iridium photocatalysis (Scheme 32A).134 Mechanistically, the electron-deficient pyridinium (for Et-TPP, E1/2 = −0.93 V vs. SCE in DMF) accepted an electron from the excited Ir(III) (Ered1/2 Ir(IV)/Ir*(III) = −0.96 V vs. SCE in MeCN) and formed a dihydropyridine radical, whose C–N fragmentation will engender the R˙ for addition to the neutral heteroarene. Notably, Brønsted acids, typical additives in Minisci alkylation conditions, were absent in Glorius's case. Moreover, electron-rich heteroarenes were accommodated in this photocatalytic approach, in which various proteinogenic amino acids were masked as R˙ sources via radical deamination.
 | ||
| Scheme 32 R˙ generation via C–N cleavage of quaternary nitrogen salt. | ||
For the Katritzsky salt-based method above, the pyridinium-forming process is limited to the condensation with less hindered primary amines.135–138 In contrast, quaternary ammonium salts could originate from nearly all types of amines via exhaustive alkylation, typically with methyl iodide or triflate. Using these bench-stable R˙ sources, Yu's group achieved the photocatalysed reductive cross-electrophile coupling reactions with alkyl ammonium salts and benzaldehydes or CO2 (Scheme 32B).139 Resembling the Katritzsky salts, a single-electron reduction of the ammonium salt (for BnNMe3OTf, Ered1/2 = −1.58 V vs. SCE in DMF) with the in situ generated Ir(II) (Ered1/2 Ir(III)/Ir(II) = −1.49 V vs. SCE in DMF) could break the C–N bond and produce the R˙. The NMe3 released from ammonium decomposition served as a reductant to reduce Ir(III) and regenerate the Ir(II), which further reduced R˙ into a carbanion to react with benzaldehyde or CO2, giving benzyl alcohols and carboxylic acids, respectively.
Besides quaternary salts, Rovis's group recently demonstrated that N-alkylated redox-active imines could generate R˙ under photoredox conditions (Scheme 32C).140 Unlike the previous case, due to the electron richness of (2,4,6-trimethoxyphenyl)methanimine (for N-tert-butyl one, Ered1/2 = +1.40 V vs. SCE in MeCN), it was oxidised into a radical cation by the Ir*(III) (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN). The generated radical cation underwent deprotonation/SCS to form an imidoyl radical, followed by the β-scission to give the R˙ with the loss of 2,4,6-trimethoxybenzonitrile. However, the concerted or stepwise mechanism of the above PCET process could not be ascertained. With careful experimentations, the authors suggested the HAT with the iminyl C–H was less likely to contribute to the R˙ formation. Additionally, it is worth noting that owing to the facile condensation of the aldehyde with sterically encumbered amine, Rovis's method could generate tertiary R˙ easily, which was difficult with the quaternary nitrogen salts.
4.2. CN cleavage
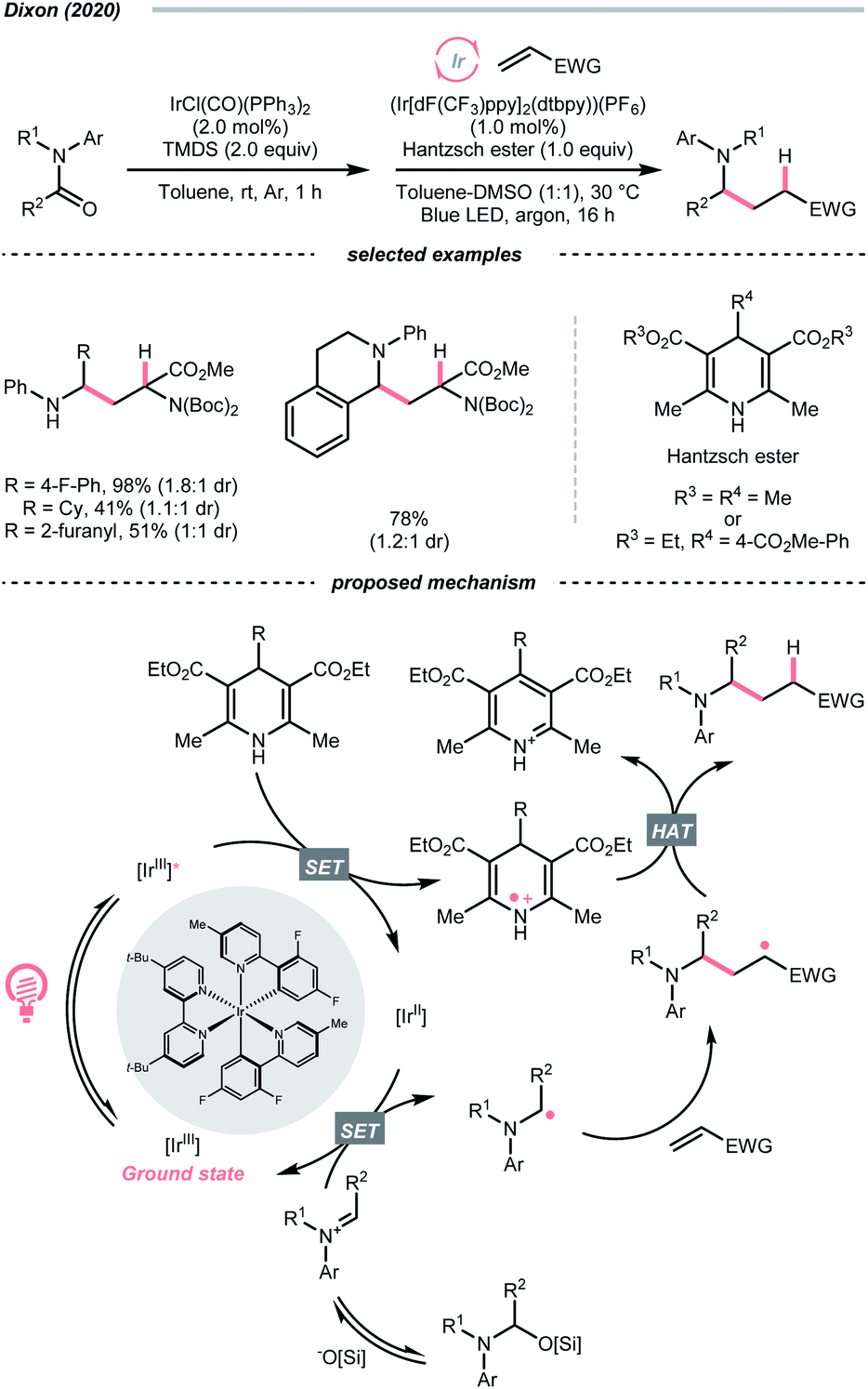
Instead of breaking C–N σ-bond, cleaving the π-bond in some CN-containing compounds could afford R˙ as well. In the case of iminiums, the group of Duarte and Dixon detailed a one-pot-two-step deoxygenative alkylation reaction of tertiary amides in 2020, with iminium ions as the key intermediates to generate α-amino radicals for Giese addition (Scheme 33).141 In the first step, hydrosilylation of the amide to furnish an iminium was executed with 1,1,3,3-tetramethyl disiloxane (TMDS) and Vaska's catalyst (IrCl(CO)(PPh3)2). Then, the iminium (Ered1/2 = −0.96 V vs. SCE in DMSO) was subject to the reductive coupling with Ir-photocatalyst (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN; Ered1/2 Ir(III)/Ir(II) = −1.42 V vs. SCE in DMSO), Hantzsch ester (for 4-phenyl ethyl ester, calculated Ered1/2 = +0.75 V vs. SCE in DMSO) and Michael acceptors under photo-irradiation, which shares a similar mechanism to their developed enol ether alkylations (Scheme 40C).142
 | ||
| Scheme 33 Reductive CN π-bond cleavage of iminium salt. | ||
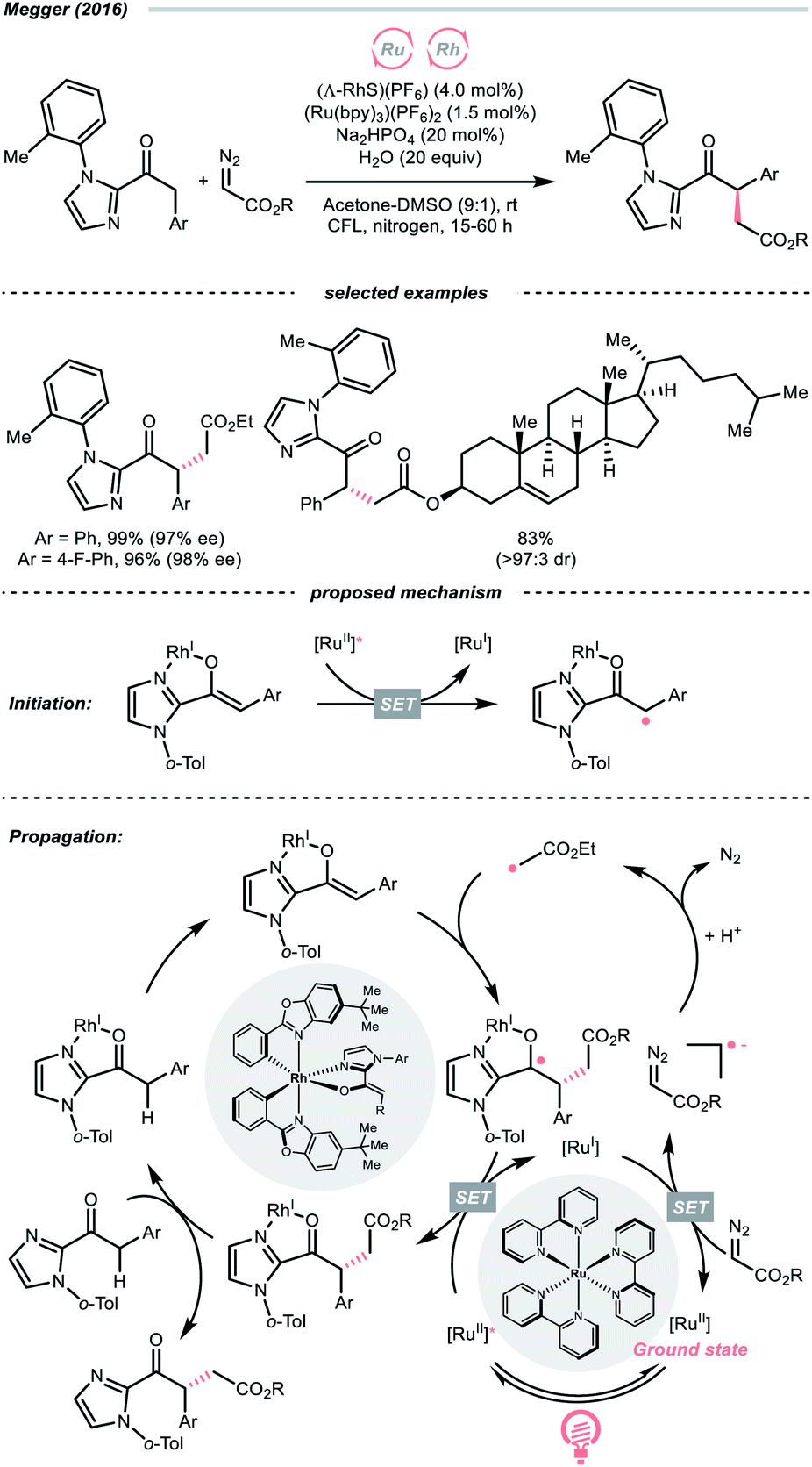
The R˙ generation could also proceed with complete CN cleavage of diazo compounds, which has been explored by many laboratories.143–146 In 2016, the group of Megger merged the Ru-photoredox catalysis with a chiral Rh(I) catalysis for asymmetric α-C(sp3)–H functionalisation of arylacetyl imidazoles with diazoacetate as the R˙ source (Scheme 34).147 In the early stage, the Rh(I) chelated the carbonyl and imidazolyl groups of the substrate and formed an enolate complex (Rh-enolate). Based on the fluorescence quenching studies, the reaction was believed to start from the SET between the Rh-enolate (with 2-acyl imidazole, Ered1/2 = +0.50 V vs. SCE in MeCN) and the Ru(II)-photocatalyst (Ered1/2 Ru*(II)/Ru(I) = +0.77 V vs. SCE in MeCN). After the initiation, albeit slightly endergonic, the single-electron reduction of the diazo substrate (for ethyl diazoacetate, Ered1/2 = −1.55 V vs. SCE in MeCN) by the reducing Ru(I) (Ered1/2 Ru(II)/Ru(I) = −1.33 V vs. SCE in MeCN) will give the R˙ after protonation and N2 extrusion. The stereo-determining R˙ addition to the chiral Rh-enolate and the SET with Ru*(II) closed both catalytic cycles and propagated the radical chain.
 | ||
| Scheme 34 Generating R˙ from aliphatic diazo compounds. | ||
Notably, despite the proposed radical chain mechanism, a low quantum yield (ϕ = 0.10) was determined for the model reaction, which could attribute to some sophisticated deactivation pathways like competing light absorption and quenching effects. However, based on this dual photocatalytic protocol, a wide range of similar enantioselective reactions were achieved with various radical sources, such as trifluoroborate,148 silanes149 and redox-active esters.150
4.3. C–O cleavage
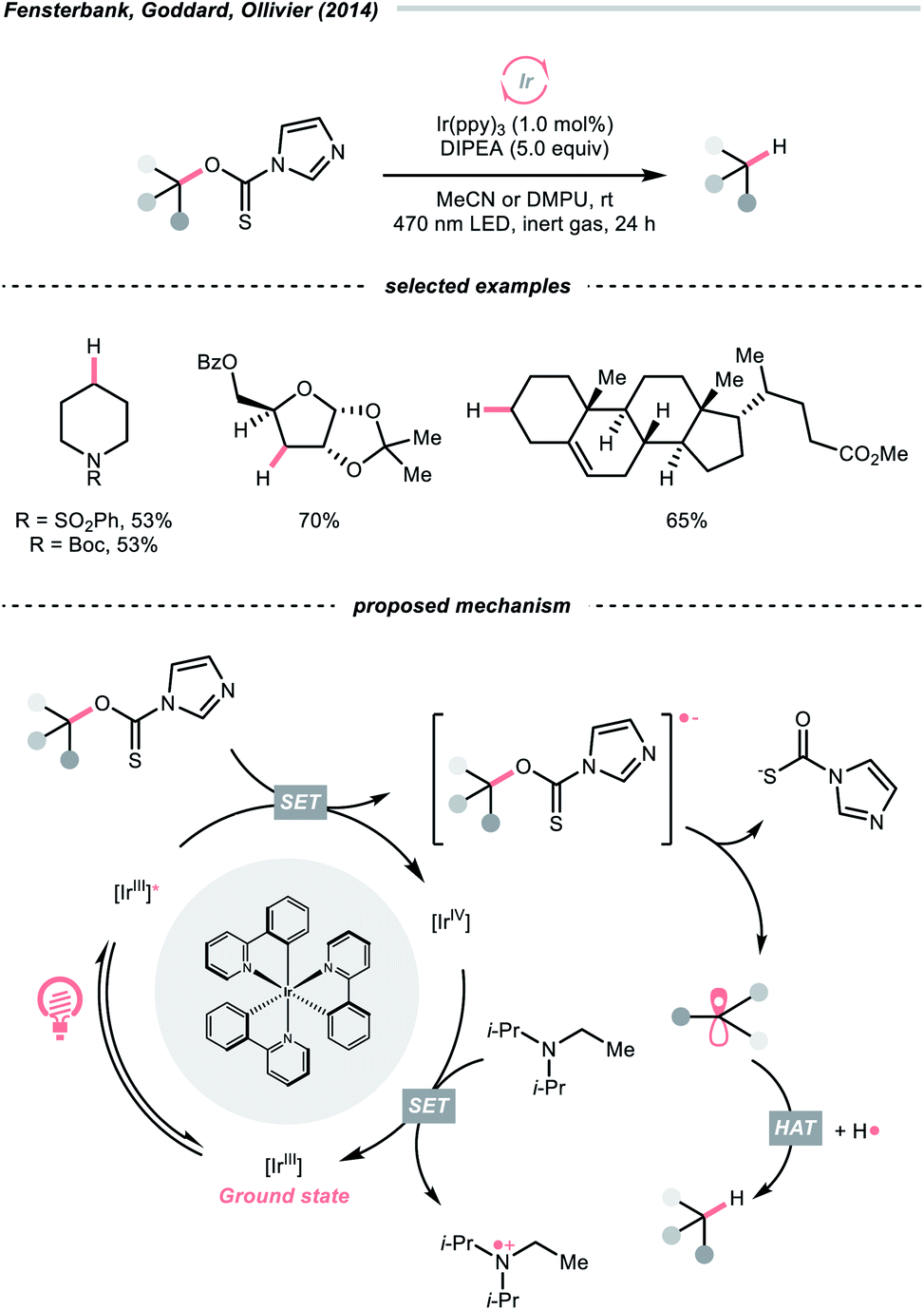
C–O cleavage exemplified a typical reaction outcome of SET to generate R˙ from alcohol and its activated derivatives. Enlightened by the well-known Barton–McCombie deoxygenation with xanthate and its later generations,151 recent reports revealed various benign reaction conditions using photocatalysis.In 2014, a photocatalysed tin-free Barton–McCombie-type deoxygenation reaction was demonstrated by the collaboration between Fensterbank's, Goddard's and Ollivier's group (Scheme 35).152 In the plausible mechanism, the O-thiocarbamate, (Ered1/2 −1.56 to −1.73 V vs. SCE in MeCN) was reduced by the photoexcited Ir(ppy)3 (Ered1/2 Ir(IV)/Ir*(III) = −1.73 V vs. SCE in MeCN), followed by C–O cleavage to afford an R˙. The N,N-diisopropylethylamine (DIPEA) served not only as a reductant to reduce the Ir(IV) back to Ir(III) but also as a hydrogen atom source to deliver the deoxygenated product.
 | ||
| Scheme 35 Photocatalytic deoxygenation of O-thiocarbamates. | ||
Pertinent to the RAEs derived from carboxylic acids, Overman's group unveiled a similar N-phthalimidoyl oxalate as the activated alcohol and successfully applied it in the photocatalytic Giese-type approach to construct quaternary carbon centres.153 Due to the lability of such oxalate starting materials that led to purification issues, in 2015, the groups of Overman and MacMillan collaboratively advanced a novel protocol using alkali oxalate as the activating group, which possessed opposite redox property relative to the N-phthalimidoyl ones (Scheme 36A).154 In their new redox-neutral Giese reaction, reductive quenching of the Ir*(III) (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN) by tertiary alcohol-derived cesium oxalate (for tert-BuOCOCO2Cs, Ered1/2 = +1.28 V vs. SCE in MeCN) was conceived feasible due to the close reduction potential and the facile evolution of two CO2 molecules. The R˙ obtained from the C–O cleavage was added toward electron-deprived alkene, furnishing the alkylated product through electron and proton transfer with the rest of the photocatalytic cycle. To be noticed, the double decarboxylation of secondary alkali metal oxalates was less efficient, which slightly limited the scope of applicable alcohols. However, such an oxalate-based approach opened chemical space for various radical transformations soon after, as the same group published a metallaphotoredox cross-coupling reaction with the oxalate salts and aryl halides.155 Enlightened by these elegant examples, Wu's group disclosed a sodium xanthate-based photocatalysed Giese reaction, in which stoichiometric phosphine behaved as sacrificial reductant and sulfur transfer reagent.156
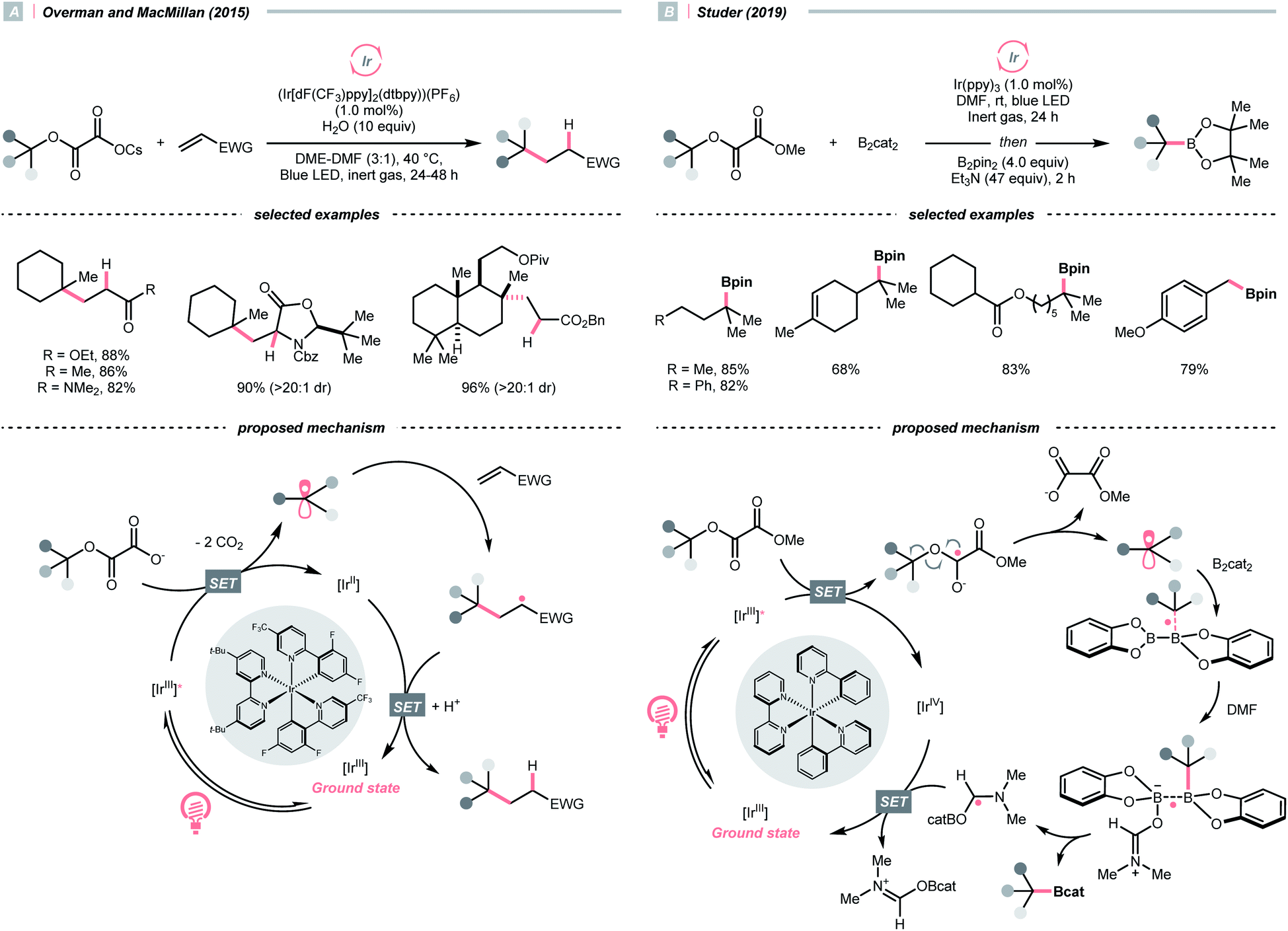 | ||
| Scheme 36 R˙ generation via C–O cleavage of oxalates. | ||
In contrast to the anionic oxalates, neutral dialkyl oxalate esters feature different redox properties, which could oxidatively quench the excited Ir(III)-photocatalyst. In 2019, Studer and his co-workers successfully applied such a reaction paradigm in a radical borylation, wherein the tertiary alcohols were converted to methyl oxalates and subjected to single-electron reduction-initiated C–O cleavage to give the R˙ (Scheme 36B).157 In the same work, the substrate scope was extended to secondary alcohols via the xanthates and under thermal-induced radical conditions with tris(trimethylsilyl)silane (TTMSS) and 2,2′-azobis(2-methylpropionitrile) (AIBN).
While the abovementioned C–C cleavages with CO2 extrusion as the driving force, the group of DiRocco demonstrated a photocatalysed alkylation of bioactive heteroarenes by decomposing peroxides into R˙, with the formation of acetone as the driving force (Scheme 37).158 Using photoexcited Ir(III) to reduce tert-butyl or tert-pentyl peracetate, tert-butoxy and tert-pentoxy radicals could be generated, respectively, which could undergo β-scission to deliver methyl and ethyl radicals for Minisci alkylations.
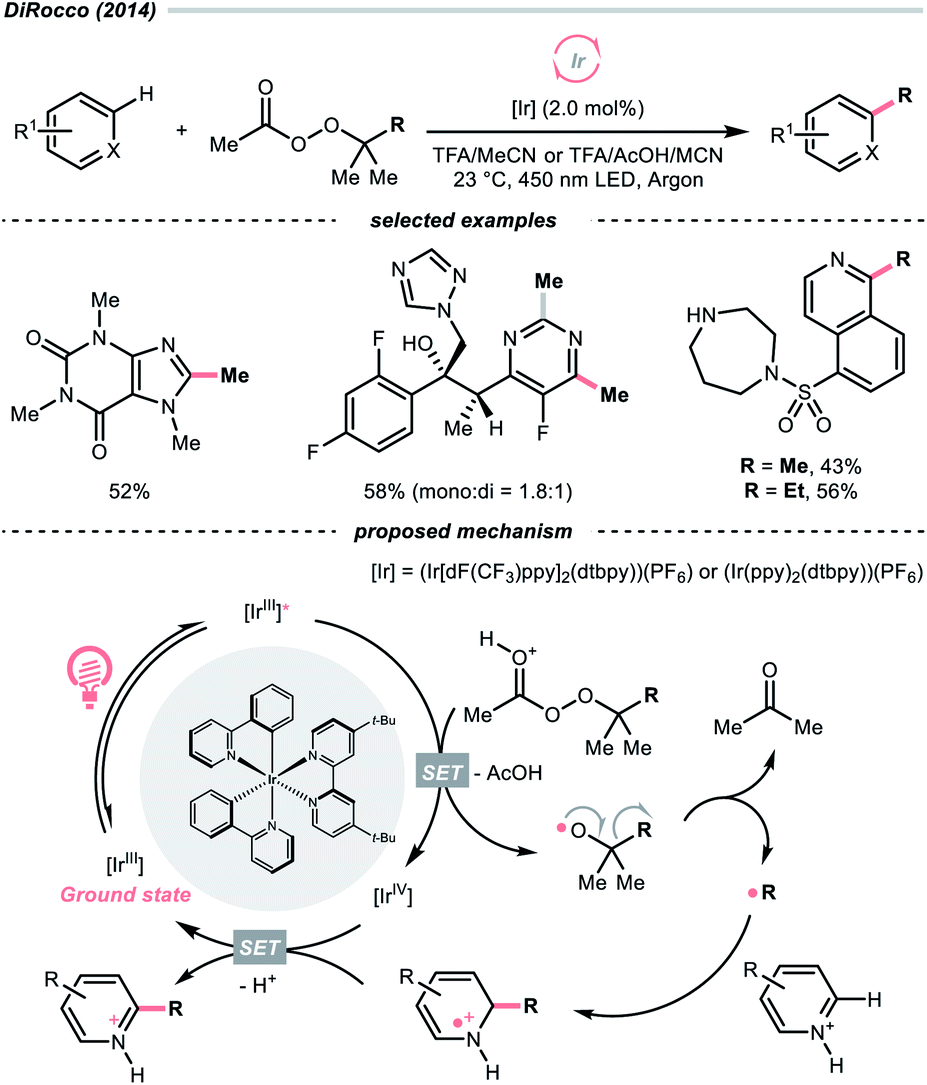 | ||
| Scheme 37 R˙ generation from peroxides. | ||
Unlike the SET-based strategies used above, Xie et al. used dialkyl acetals as masked alcohols and conceived an HAT-enabled C–O trifluoromethylation reaction under a dual-photocatalytic manifold (Scheme 38).159 Mechanistically, the thiophosphate co-catalyst was firstly oxidised by the excited 4-CzIPN to form a thiyl radical, which performed a highly regioselective HAT at the acetal α-C–H position. The ensuing dialkoxy radical underwent β-C–O cleavage and turned into an R˙. The R˙ was then intercepted by the N-(trifluoromethylthio)phthalimide to deliver the aliphatic trifluoromethylthioether product. Although only tertiary alcohols were demonstrated in the scope, such an R˙-generation protocol were shown applicable in C–O fluorination in their later publication.160
 | ||
| Scheme 38 Using masked alcohol for R˙ generation. | ||
Interestingly, free alcohols could be directly utilised as alkylating reagents via deoxygenation. As shown by Doyle and Rovis et al., under the iridium photocatalysis, phosphoranyl radical could be generated catalytically from the corresponding phosphine, which deoxygenated the free alcohol and released the R˙ (Scheme 39).161 Although this hydrodeoxygenative condition could only accommodate benzyl alcohols, it opened up new chemical space to efficiently extract alkyl radicals from alcohols.
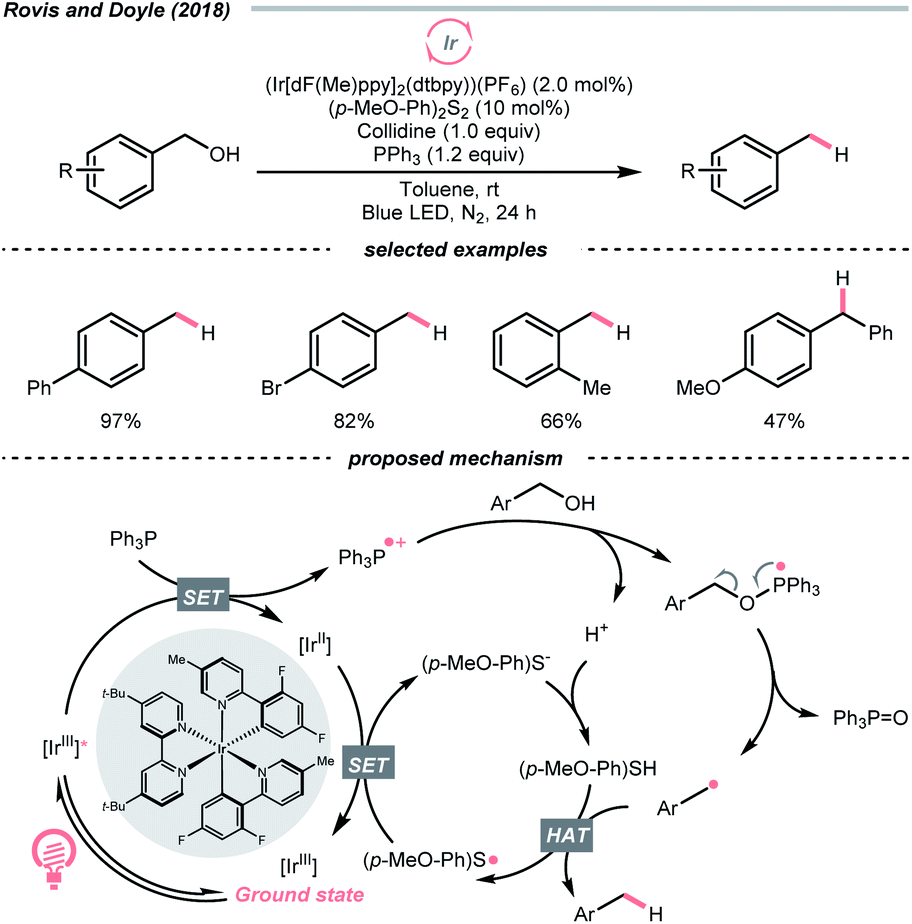 | ||
| Scheme 39 Direct hydrodeoxygenation of alcohols via R˙. | ||
4.4. CO cleavage
With photoredox catalysis, CO could also act as an R˙ precursor via SET-enabled CO cleavage. Combining [Ir(ppy)2(dtbbpy)]PF6, TTMSS, and trifluoroacetic acid (TFA), Wang's group realised a photocatalysed deoxygenative hydroheteroarylation of ketone (Scheme 40A).162 A stepwise C–O cleavage mechanism involving ketyl radical (R˙) was proposed. Hypothetically, the protonated ketone obtained an electron via a PCET mechanism, which derived from the photoreduction of [Ir(III)]* (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN) by TTMSS (Ered1/2 = +0.73 V vs. SCE in MeCN). The nucleophilic ketyl radical was added to the protonated heteroarene, which gave an α-aminoradical after proton transfer. Driven by the rearomatisation, an SCS process proceeded with the removal of H2O, providing the alkylated heteroarene via the HAT with solvent. Concurrently, Huang's group reported a similar deoxygenative Minisci alkylation with aldehydes, which was proposed as a photocatalytic Br˙-mediated process.163
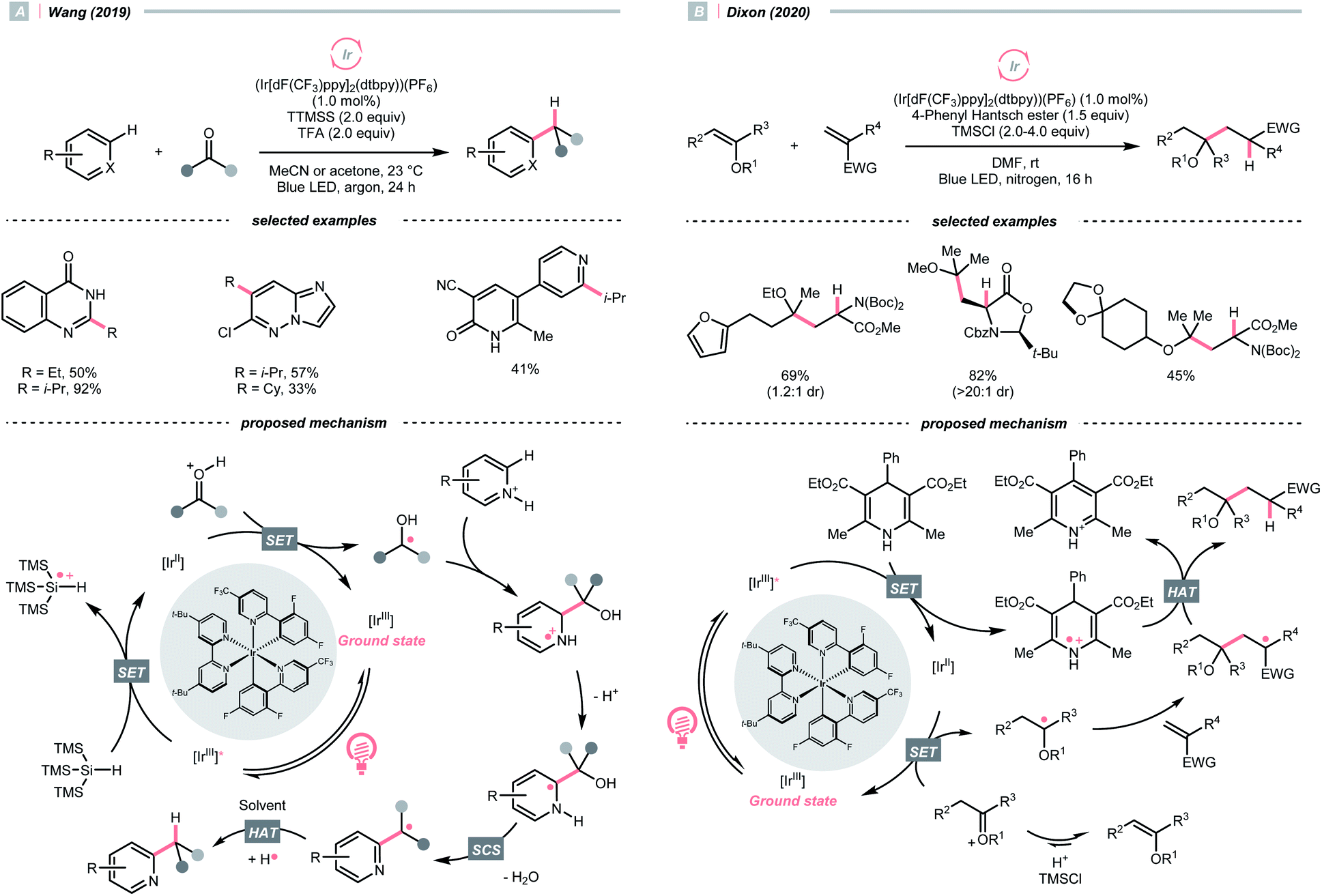 | ||
| Scheme 40 R˙ generation via carbonyl group reductions. | ||
Carbonyl derivative or in situ generated carbonyl analogue are also viable precursors of R˙, while preserving the C–O bonds in products were typical reaction outcomes. In 2020, Dixon et al. utilised alkyl enol ethers under acidic conditions and generated oxonium tautomers in situ as activated carbonyl and R˙ precursor (Scheme 40B).142 In this work, the oxonium ion (calculated Ered1/2 = −1.12 V vs. SCE in MeCN) was reduced by the Ir(II) (Ered1/2 Ir(III)/Ir(II) = −1.37 V vs. SCE in MeCN) derived from the SET between excited Ir(III)-photocatalyst (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN) and 4-phenyl Hantzsch ester (calculated Ered1/2 = +0.75 V vs. SCE in MeCN), forming an α-ethereal radical (R˙). Adding such an R˙ to the conjugated alkenes followed by HAT with the Hantzsch ester brought the α-tertiary dialkyl ethers as desired products.
4.5. C–S cleavage
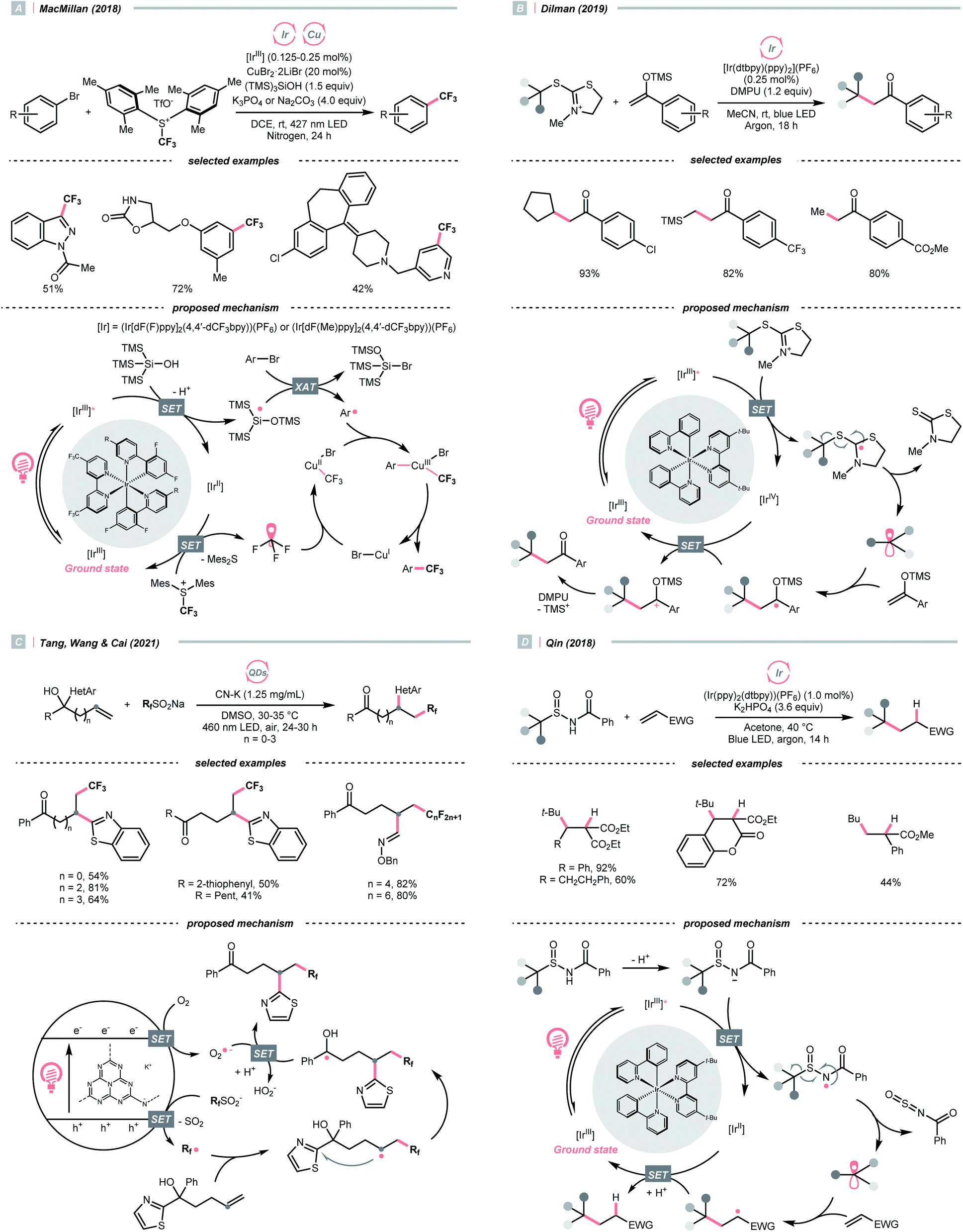
Due to the flexible oxidation state of sulfur, various S-based radical alkylating reagents were developed, accompanied by their photocatalytic systems.Alkyl sulfonium salt can be easily synthesised from alkyl halides, thiols and alcohols.164–168 Upon receiving an electron, such a trivalent species undergoes R–S cleavage to release an R˙ and a thioether. In 2018, MacMillan et al. disclosed a Cu(II)/Ir(III) dual photocatalytic system to couple aryl bromides and trifluoromethylsulfonium salt in the presence of a supersilanol (Scheme 41A).169 The photoredox cycle began with the generation of Si˙ via SET (Ered1/2 Ir*(III)/Ir(II) = +1.55 V vs. SCE in MeCN; for (TMS)3SiOH, Ered1/2 = +1.54 V vs. SCE in MeCN). The aryl bromide was subjected to Si˙-mediated halogen atom transfer (XAT, see Section 4.6), giving the aryl radical (Ar˙). On the other hand, interaction between modified Umemoto's reagent, dimesityl(trifluoromethyl)sulfonium (dMesSCF3 or MacMillan's trifluoromethylation reagent, Ered1/2 = −0.52 V vs. SCE in MeCN) and Ir(II) (Ered1/2 Ir(III)/Ir(II) = −0.83 V vs. SCE in MeCN) gave 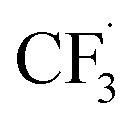 through reductive C–S cleavage. On the copper side, the interception of
through reductive C–S cleavage. On the copper side, the interception of  by Cu(I) preceded the Ar˙ trapping, and the facile Cu(III) reductive elimination rendered the trifluorotoluenes as desired products. The same tactic could apply to alkyl bromides, as shown in their later report.170
by Cu(I) preceded the Ar˙ trapping, and the facile Cu(III) reductive elimination rendered the trifluorotoluenes as desired products. The same tactic could apply to alkyl bromides, as shown in their later report.170
 | ||
| Scheme 41 R˙ from sulfoniums, thiazoliniums, sulfinates and sulfinamides. | ||
With the same Ir-photocatalyst for SET reduction, Dilman et al. exploited the ketone synthesis using dihydrothiazolinium salts as R˙ precursors, which could be simply prepared from alkyl bromides (Scheme 41B).171 Upon being reduced by the photoexcited Ir(III), dihydrothiazoline radical fragmented into an R˙ along with a thione byproduct. Silyl enol ether trapped the R˙, followed by oxidation and silyl group removal to give the ketone product.
Instead of SET reduction, sulfinate and sulfinamide are other versatile S-containing radical alkylating reagents that give R˙ upon oxidative decomposition. In 2021, an application of perfluoroalkyl sulfinates (NaSO2Rf) in migratory alkene difunctionalisations with intramolecular heteroaryl groups was presented by the research team of Tang, Wang, and Cai (Scheme 41C).172 Using potassium-modified carbon nitride (CN–K) as a recyclable photosensitive material, the electron–hole pair generated under light irradiation and ambient atmosphere could oxidise NaSO2Rf into a perfluoroalkyl radical 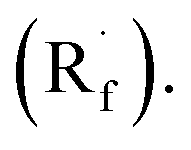 The
The  addition to the olefin tail triggered the heteroaryl group migration, which furnished the terminally perfluoroalkylated ketone as the expected product after losing a proton and electron. Interestingly, formyl and benzyl oxime ether groups can undertake the same type of functional group translocation.
addition to the olefin tail triggered the heteroaryl group migration, which furnished the terminally perfluoroalkylated ketone as the expected product after losing a proton and electron. Interestingly, formyl and benzyl oxime ether groups can undertake the same type of functional group translocation.
In 2018, Qin et al. documented a desulfurative R˙ generation strategy with N-benzoyl alkylsulfinamide using Ir-photoredox catalysis (Scheme 41D).173 As in typical oxidation-initiated Giese reaction, the R˙ was produced by the SET between Ir*(III) (Ered1/2 Ir*(III)/Ir(II) = +1.21 V vs. SCE in MeCN) and deprotonated sulfinamide (for neutral tert-butyl sulfinamide, Ered1/2 = +0.66 V vs. SCE in MeCN), which was accompanied by a C–S bond cleavage to give the R˙ and N-sulfinylbenzamide byproduct. By performing the R˙ conjugate addition to electron-deficient alkenes, reactions with secondary and tertiary alkylsulfinamides provided alkylated products with generally good to excellent yields, while primary ones were less efficient.
Beyond the SET territory, homolytic cleavage of C–S bonds to give R˙ could be realised under visible light irradiation. However, this kind of bond-breaking pattern remained rare in practical synthesis, especially for intermolecular transformations, since controlling the reaction outcome with a radical pair could be challenging. Also, these twin radicals have more tendency to recombine or undergo side reactions, which could undermine the effective concentration of desired R˙. In light of these difficulties, the strategic introduction of a photocatalyst that could mediate the reactivities of both radicals was crucial to the success of two-component or even multi-component radical reactions.
In 2019, the group of Melchiorre designed an elegant indole-based dithiocarbamate organocatalyst to tackle this challenge (Scheme 42).174 In their proposed mechanism, the nucleophilic attack of the dithiocarbamate to alkyl halides/pseudohalides could form a visible light-absorbing species, which could undergo R–S bond homolysis to afford a thiyl radical and the desired R˙. With a substoichiometric quantity of dithiocarbamate as a photochemical trigger, the σ-bond homolysis to give R˙ becomes much more efficient and controllable. To this end, R˙ could be engaged in the conjugate addition with electron-deficient alkenes smoothly (Giese addition), wherein the γ-terpinene was added as the terminal reductant, serving as the product hydrogen atom source and for catalyst turnover. It was noteworthy that the side reactions with thiyl radical were inconsequential since the so-formed adduct could be resubjected to the light-enabled homolysis and liberate the thiyl radical for its catalytic cycle. Soon after, this organocatalytic photochemical R˙ generation method was embedded in many other alkylative transformations, including alkene difunctionalisations, two- and three-component aromatic alkylation,175 and borylation.176
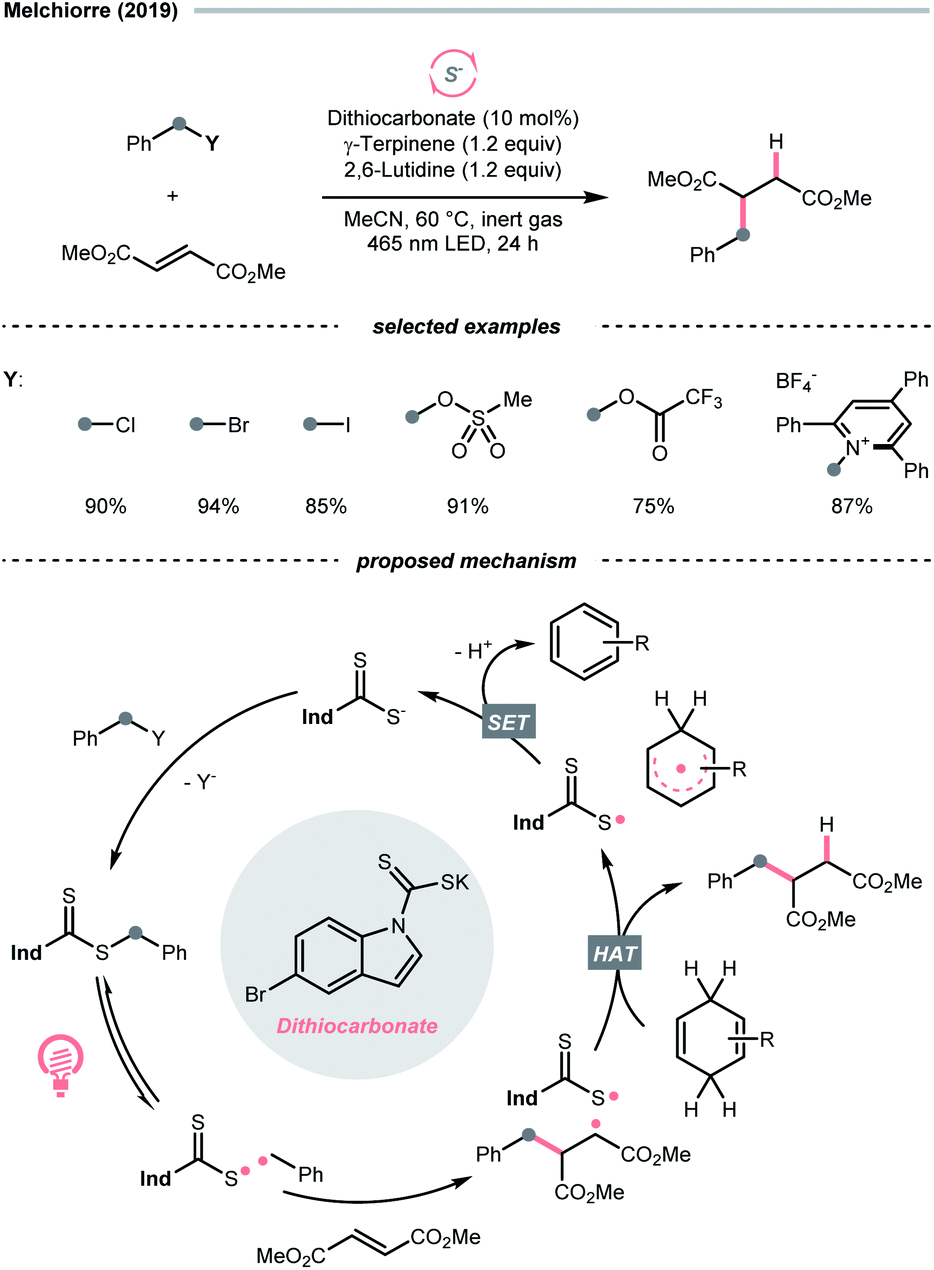 | ||
| Scheme 42 Photocatalytic R˙ generation via R–S homolysis. | ||
4.6. C–Br and C–I cleavages
Alkyl halides are among the earliest employed R˙ precursors and still widely used in modern synthesis because of their structural diversity and commercial availability. Among them, iodides and bromides, which possess relatively weak C–Br and C–I bonds, are more popular choices than the corresponding chlorides and fluorides.177–181 Although many photochemical alkylation reactions via homolytic cleavage of weak C–I bond were precedented, they often involve direct irradiation with UV light, leading to narrow substrate scope and less controllable radical process. Gratifyingly, recent advancements have demonstrated that this type of R˙ generation conditions could be significantly improved under visible light irradiation with the aid of photocatalysts.182The rapid progress of photoredox chemistry endows alkyl halides with new activation modes, among which SET represents the mostly seen ways to streamline the generation of R˙.183 In 2018, Jiang et al. disclosed an elegant enantioselective cross-coupling between α-bromoketones and α-amino acids with the aid of chiral phosphoric acid (SPINOL-CPA) and dicyanopyrazine-derived chromophore (DPZ) under visible light irradiation (Scheme 43).184 The photoexcited DPZ functioned as an electron transfer catalyst, which mediated the generation of two electronically distinct R˙ from the alkyl bromide and carboxylic acid via reductive and oxidative SET, respectively. The CPA then managed the radical–radical cross-coupling with these two R˙ in an enantioselective fashion, synthesizing numerous β-amino ketones in good to excellent enantioselectivities.
 | ||
| Scheme 43 Employing alkyl bromides as R˙ precursors. | ||
For iodides, in 2020, Studer's laboratory showcased a synthetic approach toward perfluoroalkylated diborylalkanes using perfluoroalkyl iodides and anionic diboron complexes, which were formed by in situ mixing Grignard reagent and bis(pinacolato)diboron (B2pin2) (Scheme 44).185 Similar to the alkyl bromides above, the perfluoroalkyl iodide (Ered1/2 = −1.52 V vs. SCE in DMF for CF3I) received an electron from the excited Rhodamine B, forming an  which combined with an alkene to give a new R′˙. Distal boron migration with the intramolecular Bpin group following single-electron oxidation delivered the desired 1,n-bisborylalkane products (n = 3 and 4).
which combined with an alkene to give a new R′˙. Distal boron migration with the intramolecular Bpin group following single-electron oxidation delivered the desired 1,n-bisborylalkane products (n = 3 and 4).
 | ||
| Scheme 44 R˙ formation from alkyl iodide. | ||
Besides the SET pathway to form R˙ from alkyl halides, halogen atom transfer (XAT) has demonstrated its synthetic utility for decades. Historically, XAT was enabled by some metallic radicals (e.g., tin, chromium, cerium, gold) that can abstract halogen atoms from alkyl halides and give R˙.186–190 With the surging development of photocatalysis, various new XAT agents could be accessed efficiently under mild photocatalytic conditions, especially with those non-metallic ones such as silyl and C-centred radicals, which significantly broaden the application of such an R˙-generating strategy.190
In this context, MacMillan and Houk's team developed a photoinduced reductive fluorination reaction of alkyl bromide via the XAT process (Scheme 45).191 Based on the supersilanol-mediated bromine atom transfer strategy developed in MacMillan's laboratory,192–194 this reaction was optimised with catalytic benzophenone and stoichiometric quantity of silyl radical source, (tris(trimethylsilyl)silanol (TMS)3SiOH) and electrophilic fluorinating reagent, N-fluorobenzenesulfonimide (NFSI). Initially, under blue LED irradiation, the excited benzophenone catalysed the generation of silyl radical (Si˙), in which the supersilanol might experience an HAT or SET process followed by the Brook rearrangement. Then, the Si˙ was subjected to the XAT with the alkyl bromide to give R˙. The R˙ was fluorinated by NFSI to produce an alkyl fluoride and an NCR (sulfonamido radical), which could regenerate the Si˙ and propagate the radical chain process.
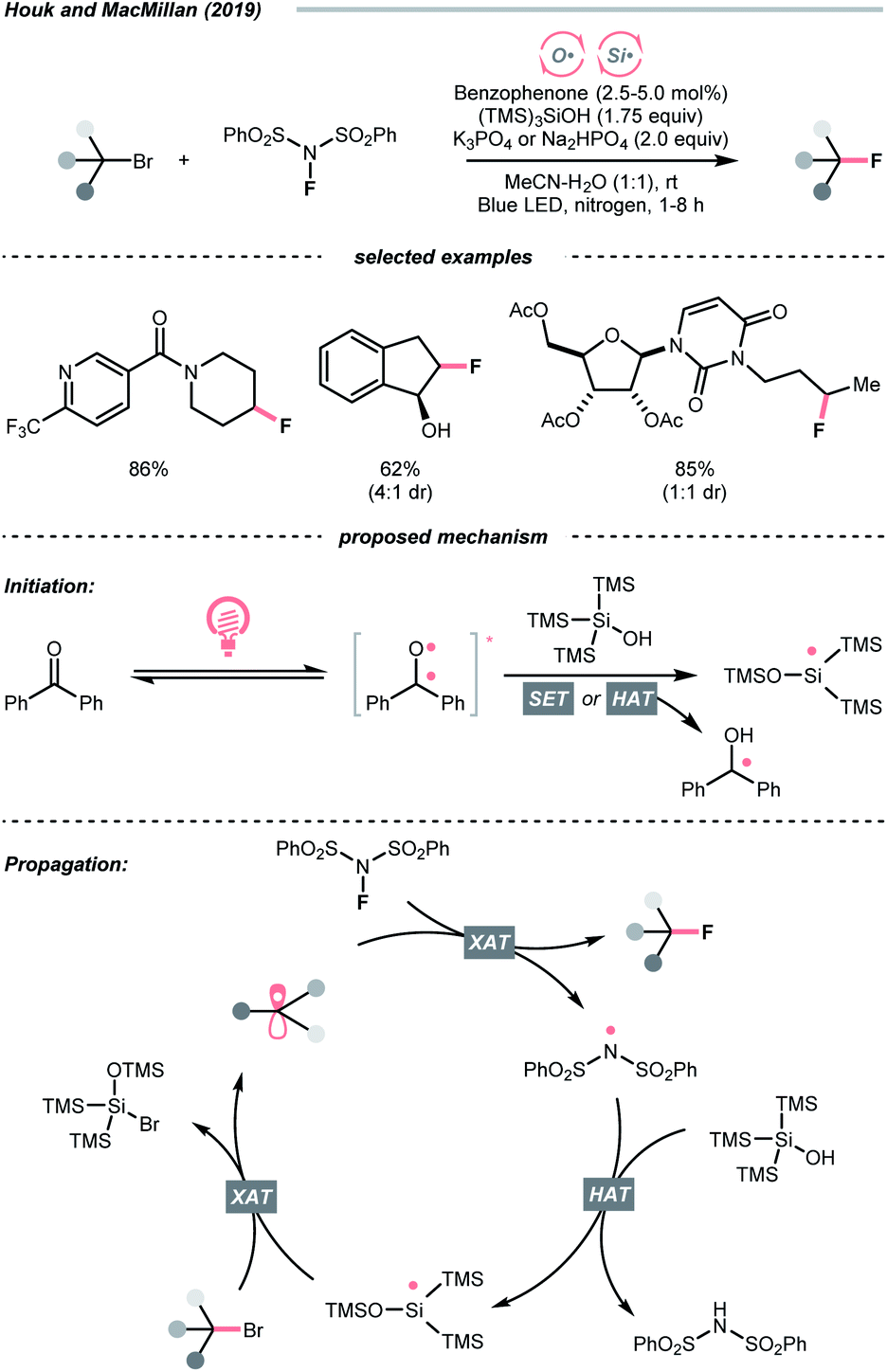 | ||
| Scheme 45 Silyl radical-mediated XAT for R˙ generation. | ||
In addition to Si˙, using C-centred radicals for XAT with alkyl halides was recently demonstrated by Juliá, Leonori and their co-workers. In this contribution, a C(sp3)–C(sp3) bond formation reaction between alkyl halides and alkenes was disclosed, which was mediated by organophotoredox catalyst 4CzIPN under visible light irradiation (Scheme 46).195 Initially, an α-amino radical resulted from the single-electron oxidation of tertiary amine by excited 4CzIPN. The key XAT benefited from the strong nucleophilicity of α-aminoalkyl radicals, which stabilised the polar XAT transition state and facilitated the R˙ generation. Then, the R˙ followed the typical reaction pathway of Giese-type radical addition to alkenes. A judicious choice of XAT reagent is essential in this design since the XAT byproduct, α-iodoamine, could degrade into an iminium iodide, therefore, combating the back halogen atom transfer.
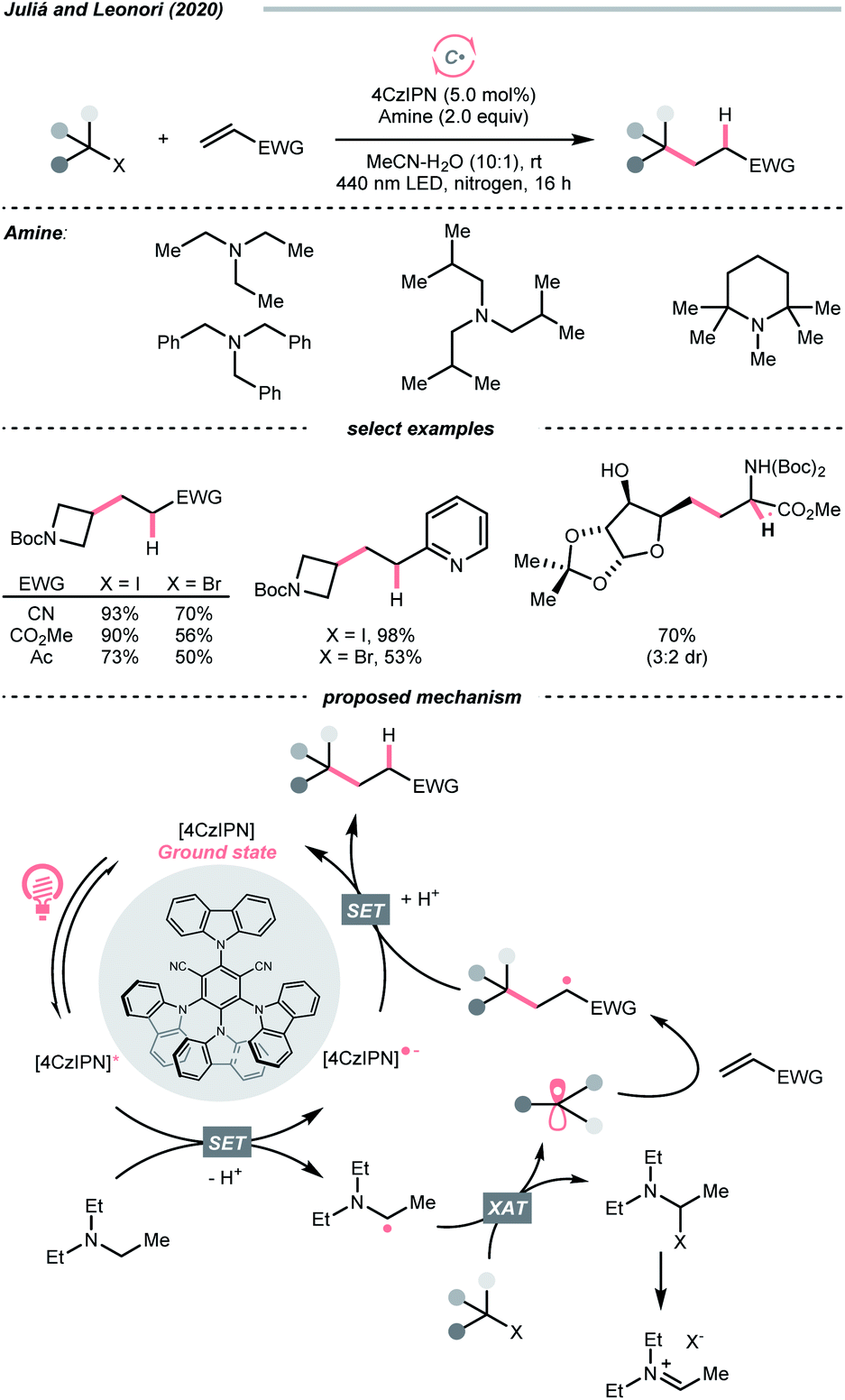 | ||
| Scheme 46 Tertiary amine-mediated XAT for R˙ formation. | ||
4.7. C–B cleavage
Like the alkyl halides, boronic acid and its derivatives are highly enabling R˙ precursors, however, featuring opposite electronic demand during the SET with photoredox catalysts (Scheme 47). Among them, potassium trifluoroborate, which was intensively studied by Molander's and other groups, is a common option due to its high shelf stability and easily accessible redox potential.196–206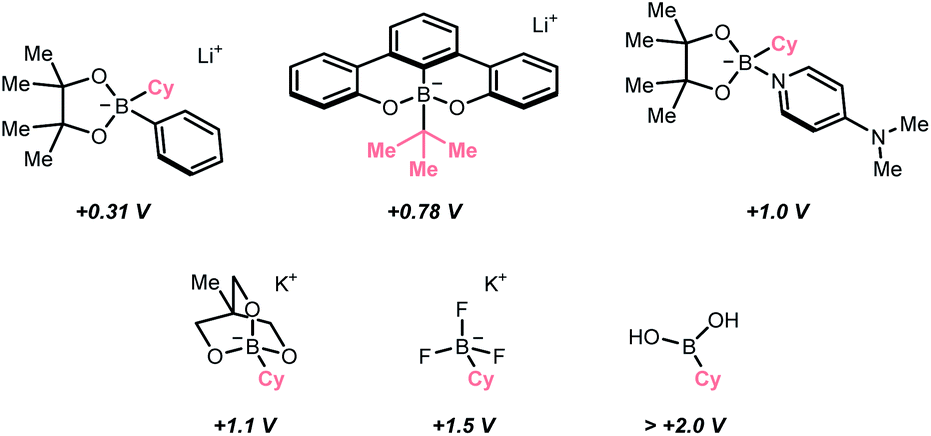 | ||
| Scheme 47 Common boron-based R˙ precursors. | ||
By merging photocatalysis and nickel catalysis, the challenging C(sp2)–C(sp3) Suzuki–Miyaura coupling with aryl bromides and benzyltrifluoroborates was realised by Molander's group in 2014 (Scheme 48A).207 Soon after, secondary,208 tertiary209 trifluoroborates and other variants are shown as effective in their later publications. The SET between Ir*(III) (Ered1/2 Ir*(III)/Ir(II) = +1.32 V vs. SCE in MeCN) and trifluoroborate (for the potentials of R–BF3K vs. SCE in MeCN, R = Bn, Ered1/2 = +1.10 V; R = Cy, Ered1/2 = +1.50 V; R = tert-Bu, Ered1/2 = +1.26 V) and the trapping of corresponding R˙ by Ar–Ni(II) to promote the product-forming reductive elimination were two common mechanistic traits for all these dual catalysed cross-couplings. Unlike the conventional polar transmetalation pathways in which alkyl nucleophiles were often problematic organometallic partners, such a single-electron scenario with alkyl trifluoroborates grants unique and complementary reactivities under the radical mechanism.
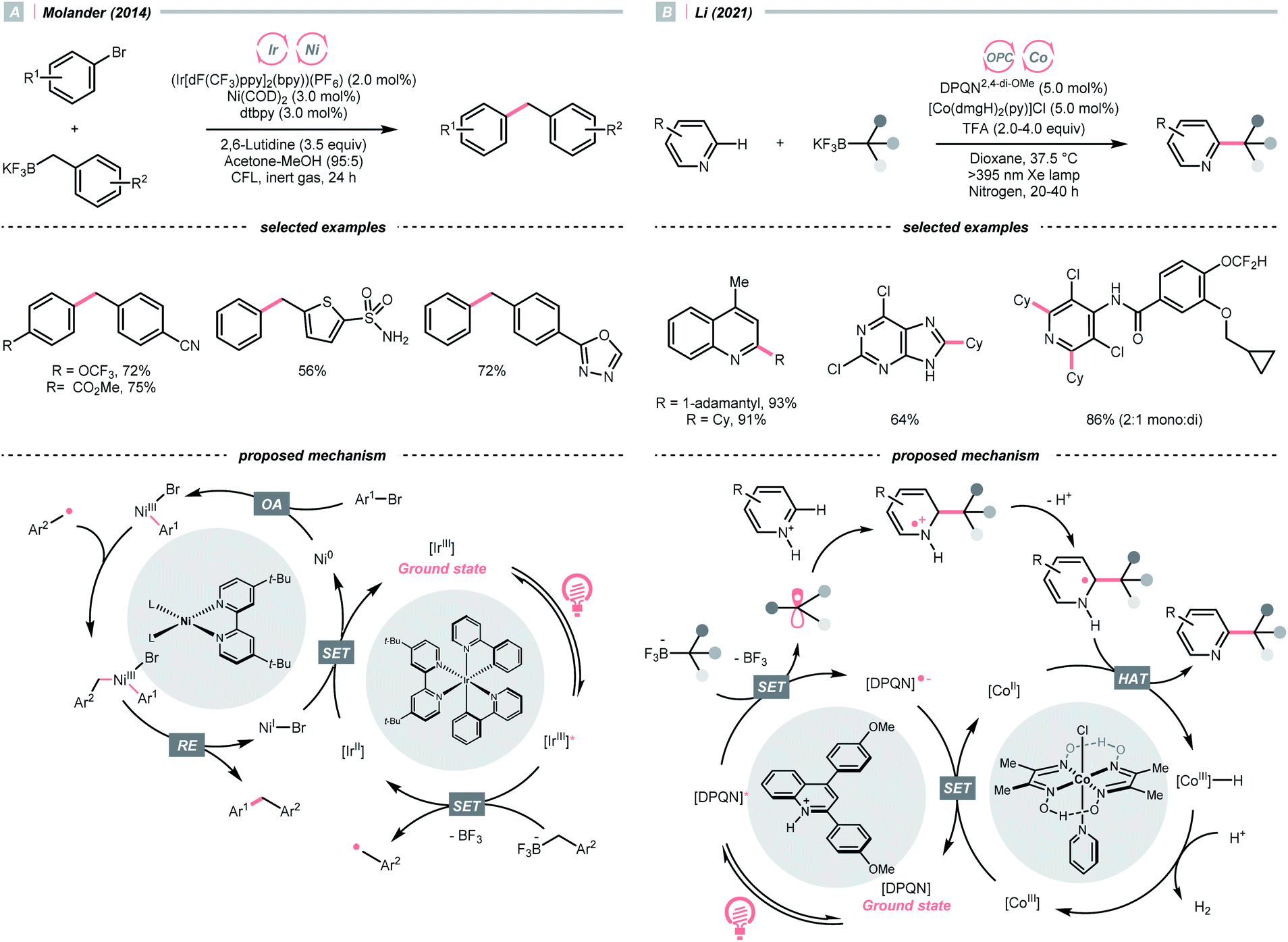 | ||
| Scheme 48 Alkyl trifluoroborate as R˙ source. | ||
Instead of metallaphotocatalysis, organophotoredox catalysts that were highly oxidising at the excited state could also pair with the alkyl trifluoroborate to effect R˙ generation. In 2021, Li's group reported a quinolinium/cobaloxime co-catalysed Minisci alkylation of alkyl trifluoroborates without external chemical oxidant (Scheme 48B).210 With detailed mechanistic studies, they conceptualised a proton-activation mode of N-heteroaromatics and developed a novel 2,4-bis(4-methoxyphenyl)quinoline organophotocatalyst (DPQN2,4-di-OMe) with an extensive oxidation window (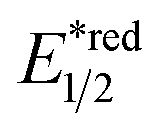 = +1.96 V vs. SCE in MeCN).62 SET with different alkyl trifluoroborates broke the C–B bonds and turned them into R˙, which was enrolled in the typical Minisci alkylation mechanism. Strategically, a cobalt(III) catalyst was introduced to drive this transformation and balance its redox equation via H2 evolution. This novel organophotocatalyst could work with many other R˙ precursors, e.g., Hantzsch ester, sulfinate, hydrazide, dihydrobenzothiazoles, bicyclo[2.2.0]hexene as well as oxalic acid. The same group also showed that other types of oxidative alkylations with alkenes and alkyne could be achieved, and the immobilised quinolinium photocatalyst on polymeric support could be recycled multiple times.
= +1.96 V vs. SCE in MeCN).62 SET with different alkyl trifluoroborates broke the C–B bonds and turned them into R˙, which was enrolled in the typical Minisci alkylation mechanism. Strategically, a cobalt(III) catalyst was introduced to drive this transformation and balance its redox equation via H2 evolution. This novel organophotocatalyst could work with many other R˙ precursors, e.g., Hantzsch ester, sulfinate, hydrazide, dihydrobenzothiazoles, bicyclo[2.2.0]hexene as well as oxalic acid. The same group also showed that other types of oxidative alkylations with alkenes and alkyne could be achieved, and the immobilised quinolinium photocatalyst on polymeric support could be recycled multiple times.
Interestingly, capitalizing on the unique solvent effect of N,N-dimethylacetamide (DMA), Sharma's group tackled the intractable one-electron activation of free boronic acids, realizing various R˙-involving processes via C–B bond cleavage.211
In addition to the single-electron oxidation of boronic acids or boronates, SH2 of aliphatic boronic acids to extrude R˙ was also operative. In 2016, Liu and Chen's team reported a photocatalytic Minisci alkylation with alkyl boronic acids in the presence of BI-OAc oxidant and ruthenium(II) photocatalyst (Scheme 49).212 The photoexcited Ru(II) firstly reduced BI-OAc to 2-iodobenzoyloxy radical. Counting on the B–O affinity, the oxy radical addition to boronic acid would induce the C–B cleavage and release an R˙, which was supported by the DFT calculations. The R˙ was then subjected to the heteroarene alkylation.
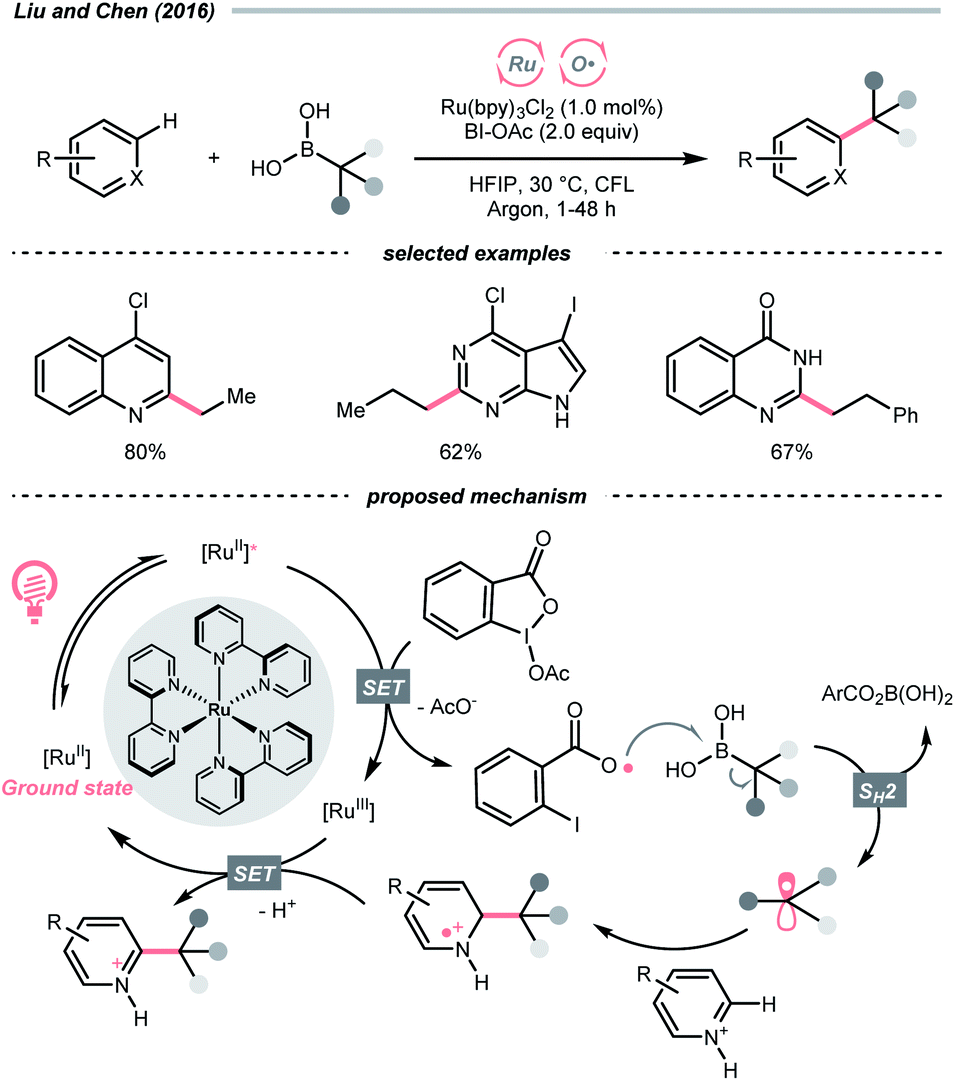 | ||
| Scheme 49 R˙ formation via SH2 of alkyl boronic acid. | ||
4.8. C–Si cleavage
Analogous to boronic acids, silane and other silica-based compounds are organometallic reagents that effect the R˙ generation by C–Si bond scission under mildly photoredox conditions. One application of alkylsilane in asymmetric addition to α,β-unsaturated ketones was demonstrated by Melchiorre's laboratory in 2018 (Scheme 50).213 Combining the elegant iminium-catalysed photooxidation214 and electron-donor acceptor (EDA) chemistry established in his group,215 a chiral carbazole-tethered amine organocatalyst was developed. The iminium EDA intermediate formed by condensing the amine catalyst and conjugated ketone absorbed visible light and enabled an intramolecular electron transfer to create a long-lived carbazole radical cation (Ered1/2 = +1.11 V vs. SCE in MeCN). Silanes within this redox window, typically those α-nitrogenated, were applicable R˙ precursors, which would undergo an oxidative fragmentation to break the C–Si bonds. Importantly, the MeCN coordination could facilitate the desilylation by forming a [MeCN-SiR3]+ complex and inhibiting back-electron transfer.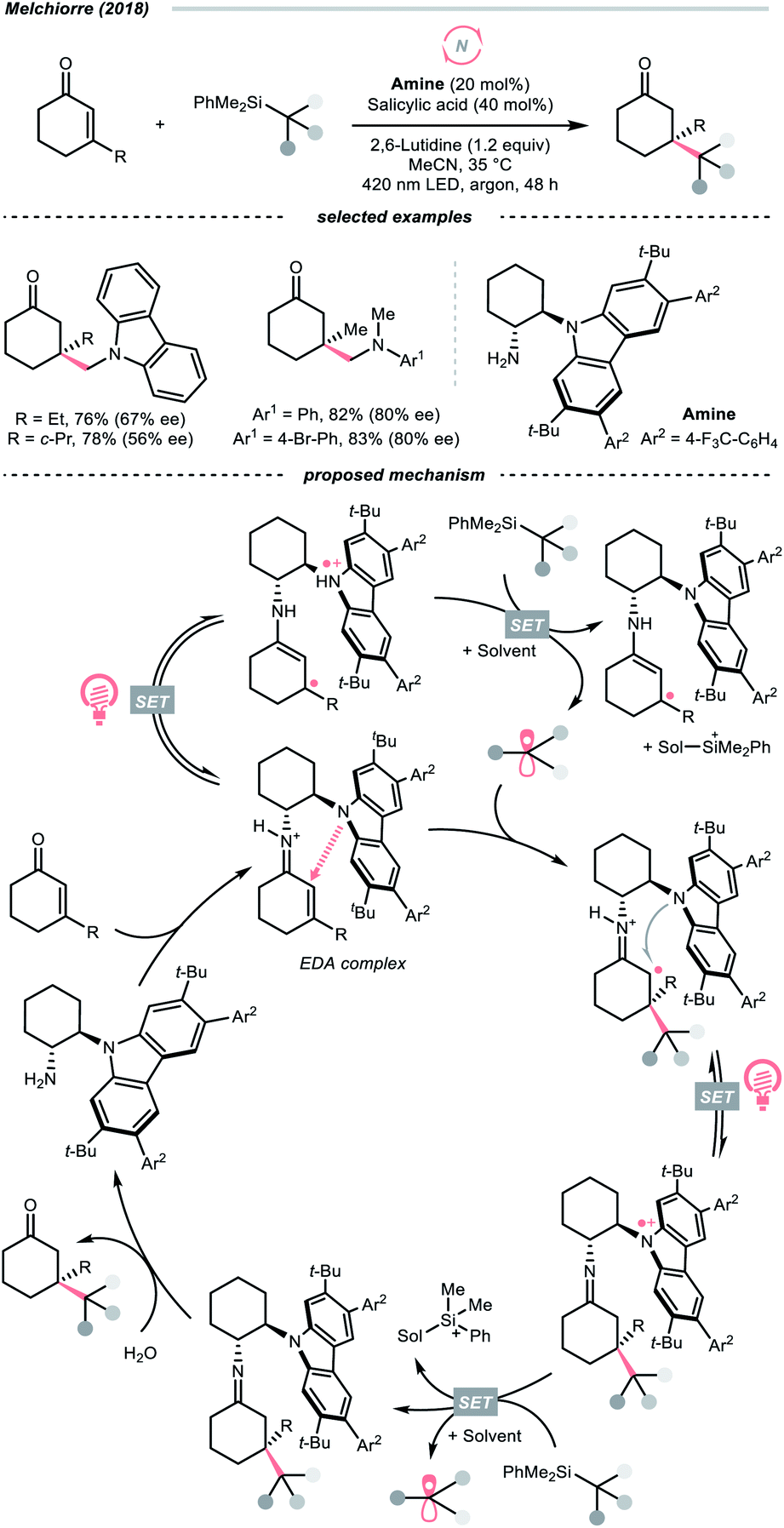 | ||
| Scheme 50 Oxidation of alkyl silane for R˙ formation. | ||
The carbazole handle was bifunctional. On the one hand, guided by such a bulky shield, R˙ will attack the conjugated iminium preferentially at the less hindered face, giving an α-iminyl radical. On the other hand, the electron-rich carbazole could be an electron sink; therefore, another intramolecular SET will give a new carbazole radical cation, which substantiated a radical chain mechanism. Subsequent hydrolysis of the β-functionalised imine led to product formation and catalyst regeneration. It is notable that recently, the same group reported an asymmetric C–C cross-coupling between aliphatic silanes and allylic alcohols, in which the photoexcitable allylic Ir(III) complex mediated the R˙ formation and the chiral induction for the C–C bond-forming step.216
As explored in Fensterbank's and Kano's studies and others, anionic hypervalent silicate could generate the R˙ under comparably milder conditions due to its anionic character. On this basis, two different types of silicates were applied in photoredox chemistry.
In 2017, Fensterbank, Ollivier, Goddard and their co-workers collaboratively disclosed a photocatalysed radical alkylation reaction via oxidation of biscatecholato silicates (Scheme 51A).217 A series of silicates were prepared via modular synthesis, which showed accessible reduction potentials (for [CySi−(cat)2][K+(18-C-6)], Ered1/2 = +0.69 V vs. SCE in DMF) by the commercially available Ir(III)-photocatalyst (Ered1/2 Ir*(III)/Ir(II) = +1.32 V vs. SCE in MeCN). The R˙ resulting from the C–Si cleavage could be employed in Giese-type addition or allylation reaction via the SH2 mechanism. Additionally, the versatility of this type of silicate reagent could be extended to cross-couplings with nickel catalysis.218,219
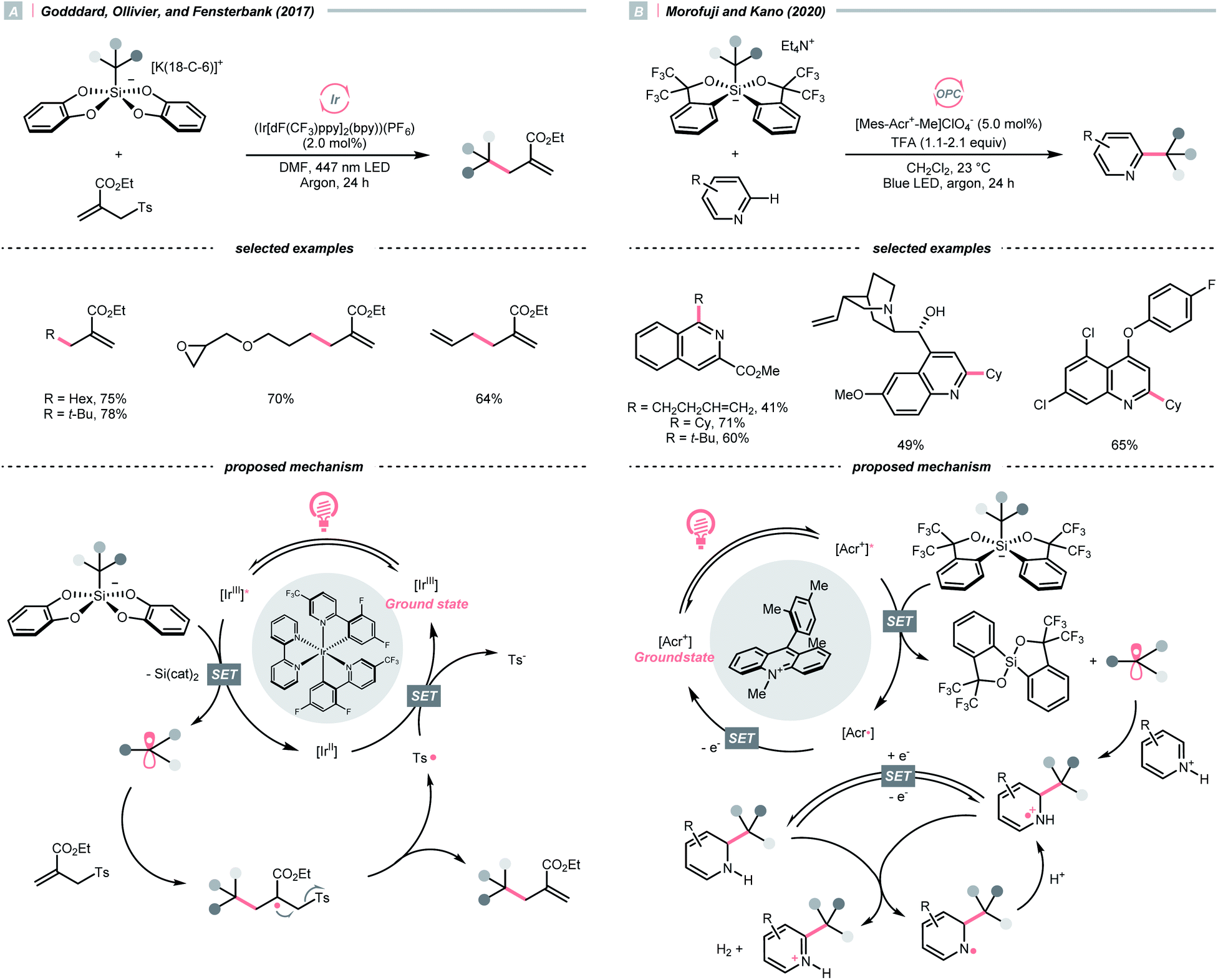 | ||
| Scheme 51 Silicate-based reagents as R˙ precursors. | ||
However, when biscatecholato silicates were applied to Minisci-type alkylation, the reaction efficiency was largely compromised by their decomposition under acidic conditions. Stepping forward, Kano et al. devised a new acid-stable pentavalent silicate reagent (for cyclohexyl one, Ered1/2 = +1.47 V vs. SCE in MeCN), which could generate the desired R˙ for heteroarene alkylation after being oxidised by [Mes-Acr-Me]+ClO4− (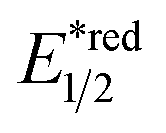 = +2.06 V vs. SCE in MeCN, Scheme 51B).220 Interestingly, with the [Mes-Acr+-Ph]BF4− photocatalyst (
= +2.06 V vs. SCE in MeCN, Scheme 51B).220 Interestingly, with the [Mes-Acr+-Ph]BF4− photocatalyst (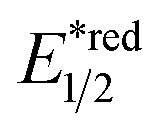 = +2.08 V vs. SCE in MeCN), radical addition to electron-deficient alkenes was also achieved by the same group, including methylation.221
= +2.08 V vs. SCE in MeCN), radical addition to electron-deficient alkenes was also achieved by the same group, including methylation.221
5. Conclusions
C(sp3) radicals have been increasingly serving as aliphatic functionalising species in organic synthesis, and photocatalysis represents one of the most advanced approaches to enable the C(sp3) radical generation and the subsequent radical functionalisations in recent years.This review listed some typical contributions in this field and classified them based on types of bond cleavage and the corresponding R˙ generation strategies. Along this line, distinguished mechanistic traits of these radical C–H, C–C and C–X functionalisation examples were analysed, discussed and compared in detail. By summarising these works, we wish to offer the readers a systematic overview of this exciting area and, more importantly, inspire future research endeavours.
We are optimistic that photocatalytic examples that provide more and more bond-breaking and -forming opportunities will appear in the literature. Especially, those incorporating some interdisciplinary techniques, e.g., photoelectrochemistry and biocatalysis, would pronouncedly enrich the chemist's toolkit for C(sp3)-based skeleton synthesis. Moreover, we believe that these established C(sp3) radical logic could be extended, effecting the generation of other C-centred radicals such as aryl, vinyl and alkynyl radicals.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Canada Research Chair Foundation (to C.-J. L.), the Canada Foundation for Innovation, the FQRNT Centre in Green Chemistry and Catalysis, the Natural Sciences and Engineering Research Council of Canada, and McGill University for supporting our research.References
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- S. G. E. Amos, M. Garreau, L. Buzzetti and J. Waser, Beilstein J. Org. Chem., 2020, 16, 1163–1187 CrossRef CAS PubMed.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 90, 9540–9685 RSC.
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed.
- C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC.
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- A. L. Gant Kanegusuku and J. L. Roizen, Angew. Chem., Int. Ed., 2021, 60, 21116–21149 CrossRef CAS PubMed.
- M. A. Emmanuel, N. R. Greenberg, D. G. Oblinsky and T. K. Hyster, Nature, 2016, 540, 414–417 CrossRef CAS.
- K. F. Biegasiewicz, S. J. Cooper, X. Gao, D. G. Oblinsky, J. H. Kim, S. E. Garfinkle, L. A. Joyce, B. A. Sandoval, G. D. Scholes and T. K. Hyster, Science, 2019, 364, 1166–1169 CrossRef CAS PubMed.
- J. S. DeHovitz, Y. Y. Loh, J. A. Kautzky, K. Nagao, A. J. Meichan, M. Yamauchi, D. W. C. MacMillan and T. K. Hyster, Science, 2020, 369, 1113–1118 CrossRef CAS PubMed.
- M. J. Black, K. F. Biegasiewicz, A. J. Meichan, D. G. Oblinsky, B. Kudisch, G. D. Scholes and T. K. Hyster, Nat. Chem., 2020, 12, 71–75 CrossRef PubMed.
- X. Huang, B. Wang, Y. Wang, G. Jiang, J. Feng and H. Zhao, Nature, 2020, 584, 69–74 CrossRef CAS PubMed.
- H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catalysis, 2021, 1, 523–598 CrossRef.
- Z. Ye, Y. M. Lin and L. Gong, Eur. J. Org. Chem., 2021, 2021, 5545–5556 CrossRef CAS.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- M. Bhakat, P. Biswas, J. Dey and J. Guin, Org. Lett., 2021, 23, 6886–6890 CrossRef CAS.
- J. Jin and D. W. C. Macmillan, Angew. Chem., Int. Ed., 2015, 54, 1565–1569 CrossRef CAS PubMed.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- C.-Y. Huang, H. Kang, J. Li and C.-J. Li, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS PubMed.
- S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200–16203 CrossRef CAS PubMed.
- Y. Wang, G.-X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679–2683 RSC.
- X. Wu, M. Wang, L. Huan, D. Wang, J. Wang and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 1640–1644 CrossRef CAS.
- X. Tang and A. Studer, Chem. Sci., 2017, 8, 6888–6892 RSC.
- X. Tang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 814–817 CrossRef CAS PubMed.
- M. Wang, L. Huan and C. Zhu, Org. Lett., 2019, 21, 821–825 CrossRef CAS PubMed.
- X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847–852 CrossRef CAS PubMed.
- P. Zhang, Y. Wang, J. Yao, C. Wang, C. Yan, M. Antonietti and H. Li, Adv. Synth. Catal., 2011, 353, 1447–1451 CrossRef CAS.
- S. Yang, S. Zhu, D. Lu and Y. Gong, Org. Lett., 2019, 21, 2019–2024 CrossRef CAS PubMed.
- K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef CAS PubMed.
- J. B. McManus, J. D. Griffin, A. R. White and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 10325–10330 CrossRef CAS PubMed.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed.
- X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed.
- Y. Li, J. Su, E. Mitchell, G. Zhang and J. Li, Sci. China Chem., 2013, 56, 1671–1681 CrossRef CAS.
- T. Shiragami, J. Matsumoto, H. Inoue, M. Yasuda, J. Photochem and C. Photobiol, Photochem. Rev., 2005, 6, 227–248 CrossRef CAS.
- H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Nat. Commun., 2020, 11, 1956 CrossRef CAS PubMed.
- H. Chen and S. Yu, Org. Biomol. Chem., 2020, 18, 4519–4532 RSC.
- X. Wu and C. Zhu, J. Chin. Chem. Soc., 2020, 2, 813–828 CAS.
- Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894–1897 CrossRef CAS PubMed.
- J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- K. Ohmatsu, R. Suzuki, Y. Furukawa, M. Sato and T. Ooi, ACS Catal., 2020, 10, 2627–2632 CrossRef CAS.
- K. Minami, K. Ohmatsu and T. Ooi, ACS Catal., 2022, 1971–1976, DOI:10.1021/acscatal.1c05604.
- H. Tanaka, K. Sakai, A. Kawamura, K. Oisaki and M. Kanai, Chem. Commun., 2018, 54, 3215–3218 RSC.
- J. L. Jeffrey, J. A. Terrett and D. W. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS PubMed.
- W. Shu and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1881–1884 CrossRef CAS PubMed.
- I. B. Krylov, O. O. Segida, A. S. Budnikov and A. O. Terent'Ev, Adv. Synth. Catal., 2021, 363, 2502–2528 CrossRef CAS.
- H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692–1696 CrossRef CAS PubMed.
- A. S. H. Ryder, W. B. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay, M. N. Grayson and A. J. Cresswell, Angew. Chem., Int. Ed., 2020, 59, 14986–14991 CrossRef CAS PubMed.
- H. Huang, Z. M. Strater and T. H. Lambert, J. Am. Chem. Soc., 2020, 142, 1698–1703 CrossRef CAS PubMed.
- T. Shen and T. H. Lambert, Science, 2021, 371, 620–626 CrossRef CAS PubMed.
- T. Shen and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 8597–8602 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed.
- Y. Shen, I. Funez-Ardoiz, F. Schoenebeck and T. Rovis, J. Am. Chem. Soc., 2021, 143, 18952–18959 CrossRef CAS PubMed.
- T. Ide, J. P. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Chem. Sci., 2018, 9, 8453–8460 RSC.
- H. Fuse, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 4493–4499 CrossRef CAS PubMed.
- S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, J. Am. Chem. Soc., 2017, 139, 2204–2207 CrossRef CAS PubMed.
- V. Regnault, J. Prakt. Chem., 1840, 19, 193–218 CrossRef.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS PubMed.
- M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191–7194 CrossRef CAS PubMed.
- S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S. L. Zheng, Y. S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 6472–6475 CrossRef CAS PubMed.
- L. Troian-Gautier, M. D. Turlington, S. A. M. Wehlin, A. B. Maurer, M. D. Brady, W. B. Swords and G. J. Meyer, Chem. Rev., 2019, 119, 4628–4683 CrossRef CAS PubMed.
- T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef CAS PubMed.
- P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Chem, 2020, 6, 1766–1776 CAS.
- D. Nagib, L. Stateman and K. Nakafuku, Synthesis, 2018, 50, 1569–1586 CrossRef PubMed.
- P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed.
- S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974–12993 RSC.
- M. Ratushnyy, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2018, 57, 2712–2715 CrossRef CAS PubMed.
- D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2019, 141, 14126–14130 CrossRef CAS PubMed.
- I. N.-M. Leibler, M. A. Tekle-Smith and A. G. Doyle, Nat. Commun., 2021, 12, 6950 CrossRef CAS PubMed.
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253–13260 CrossRef CAS PubMed.
- L.-Z. Wu, J. Qiao, Z.-Q. Song, C. Huang, R.-N. Ci, Z. Liu, B. Chen and C.-H. Tung, Angew. Chem., Int. Ed., 2021, 60, 27201–27205 CrossRef PubMed.
- C. Huang, J. Qiao, R.-N. Ci, X.-Z. Wang, Y. Wang, J.-H. Wang, B. Chen, C.-H. Tung and L.-Z. Wu, Chem, 2021, 7, 1244–1257 CAS.
- C. Huang, R. N. Ci, J. Qiao, X. Z. Wang, K. Feng, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 11779–11783 CrossRef CAS PubMed.
- H. Liu, L. Ma, R. Zhou, X. Chen, W. Fang and J. Wu, ACS Catal., 2018, 8, 6224–6229 CrossRef CAS.
- J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed.
- L. Ge, Y. Li, W. Jian and H. Bao, Chem. –Eur. J., 2017, 23, 11767–11770 CrossRef CAS PubMed.
- K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS.
- L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed.
- J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213–218 CrossRef CAS PubMed.
- J. M. Smith, T. Qin, R. R. Merchant, J. T. Edwards, L. R. Malins, Z. Liu, G. Che, Z. Shen, S. A. Shaw, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 11906–11910 CrossRef CAS PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin, D. G. Blackmond and P. S. Baran, ACS Catal., 2018, 8, 9537–9542 CrossRef CAS PubMed.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed.
- T. Cao, T. Xu, R. Xu, X. Shu and S. Liao, Nat. Commun., 2020, 11, 5340 CrossRef CAS PubMed.
- J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed.
- K. Zhang, D. Rombach, N. Y. Nötel, G. Jeschke and D. Katayev, Angew. Chem., Int. Ed., 2021, 60, 22487–22495 CrossRef CAS PubMed.
- A. Sugimori and T. Yamada, Chem. Lett., 1986, 15, 409–412 CrossRef.
- Z. Li, X. Wang, S. Xia and J. Jin, Org. Lett., 2019, 21, 4259–4265 CrossRef CAS PubMed.
- G. Feng, X. Wang and J. Jin, Eur. J. Org. Chem., 2019, 2019, 6728–6732 CrossRef CAS.
- R.-J. Tang, L. Kang and L. Yang, Adv. Synth. Catal., 2015, 357, 2055–2060 CrossRef CAS.
- R. X. Liu, F. Zhang, Y. Peng and L. Yang, Chem. Commun., 2019, 55, 12080–12083 RSC.
- Z. Wang, X. Ji, J. Zhao and H. Huang, Green Chem., 2019, 21, 5512–5516 RSC.
- J.-Q. Chen, R. Chang, Y.-L. Wei, J.-N. Mo, Z.-Y. Wang and P.-F. Xu, J. Org. Chem., 2018, 83, 253–259 CrossRef CAS PubMed.
- X. Hu, G.-X. Li, G. He and G. Chen, Org. Chem. Front., 2019, 6, 3205–3209 RSC.
- K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef CAS PubMed.
- Y. Chen, X. Wang, X. He, Q. An and Z. Zuo, J. Am. Chem. Soc., 2021, 143, 4896–4902 CrossRef CAS PubMed.
- E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744–748 CrossRef CAS PubMed.
- E. M. Dauncey, S. U. Dighe, J. J. Douglas and D. Leonori, Chem. Sci., 2019, 10, 7728–7733 RSC.
- X. Y. Yu, Q. Q. Zhao, J. Chen, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505–15509 CrossRef CAS PubMed.
- M.-M. Wang and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 16420–16424 CrossRef CAS PubMed.
- J. Kanazawa, K. Maeda and M. Uchiyama, J. Am. Chem. Soc., 2017, 139, 17791–17794 CrossRef CAS PubMed.
- S. K. Rout, G. Marghem, J. Lan, T. Leyssens and O. Riant, Chem. Commun., 2019, 55, 14976–14979 RSC.
- J. Nugent, C. Arroniz, B. R. Shire, A. J. Sterling, H. D. Pickford, M. L. J. Wong, S. J. Mansfield, D. F. J. Caputo, B. Owen, J. J. Mousseau, F. Duarte and E. A. Anderson, ACS Catal., 2019, 9, 9568–9574 CrossRef CAS.
- M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef CAS PubMed.
- X. Zhang, R. T. Smith, C. Le, S. J. McCarver, B. T. Shireman, N. I. Carruthers and D. W. C. MacMillan, Nature, 2020, 580, 220–226 CrossRef CAS PubMed.
- M. Ociepa, A. J. Wierzba, J. Turkowska and D. Gryko, J. Am. Chem. Soc., 2020, 142, 5355–5361 CrossRef CAS PubMed.
- J. Kanazawa and M. Uchiyama, Synlett, 2019, 30, 1–11 CrossRef CAS.
- J. Turkowska, J. Durka and D. Gryko, Chem. Commun., 2020, 56, 5718–5734 RSC.
- N. B. Bissonnette, J. M. Ellis, L. G. Hamann and F. Romanov-Michailidis, Chem. Sci., 2019, 10, 9591–9596 RSC.
- E. Gandolfo, X. Tang, S. Raha Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16854–16858 CrossRef CAS PubMed.
- T. Van Leeuwen, L. Buzzetti, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 4953–4957 CrossRef CAS PubMed.
- T. Uchikura, K. Moriyama, M. Toda, T. Mouri, I. Ibanez and T. Akiyama, Chem. Commun., 2019, 55, 11171–11174 RSC.
- B. Wu, J. Wang, X. Liu and R. Zhu, Nat. Commun., 2021, 12, 3680 CrossRef CAS PubMed.
- H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC.
- W.-J. Xiao, F.-D. Lu, G.-F. He and L.-Q. Lu, Green Chem., 2021, 23, 5379–5393 RSC.
- Y. Moon, B. Park, I. Kim, G. Kang, S. Shin, D. Kang, M.-H. Baik and S. Hong, Nat. Commun., 2019, 10, 4117 CrossRef PubMed.
- R. C. McAtee, E. A. Noten and C. R. J. Stephenson, Nat. Commun., 2020, 11, 2528 CrossRef CAS PubMed.
- P. Lian, W. Long, J. Li, Y. Zheng and X. Wan, Angew. Chem., Int. Ed., 2020, 59, 23603–23608 CrossRef CAS PubMed.
- D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed.
- A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334–10337 CrossRef CAS PubMed.
- T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed.
- T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 CrossRef CAS PubMed.
- J. D. Griffin, C. L. Cavanaugh and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 2097–2100 CrossRef CAS PubMed.
- M. L. Czyz, M. S. Taylor, T. H. Horngren and A. Polyzos, ACS Catal., 2021, 11, 5472–5480 CrossRef CAS.
- F. J. R. Klauck, M. J. James and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12336–12339 CrossRef CAS PubMed.
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed.
- S. L. Rössler, B. J. Jelier, E. Magnier, G. Dagousset, E. M. Carreira and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 9264–9280 CrossRef PubMed.
- J. T. M. Correia, V. A. Fernandes, B. T. Matsuo, J. A. C. Delgado, W. C. De Souza and M. W. Paixão, Chem. Commun., 2020, 56, 503–514 RSC.
- S. L. Rössler, B. J. Jelier, E. Magnier, G. Dagousset, E. M. Carreira and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 9264–9280 CrossRef PubMed.
- L. L. Liao, G. M. Cao, J. H. Ye, G. Q. Sun, W. J. Zhou, Y. Y. Gui, S. S. Yan, G. Shen and D. G. Yu, J. Am. Chem. Soc., 2018, 140, 17338–17342 CrossRef CAS PubMed.
- M. A. Ashley and T. Rovis, J. Am. Chem. Soc., 2020, 142, 18310–18316 CrossRef CAS PubMed.
- T. Rogova, P. Gabriel, S. Zavitsanou, J. A. Leitch, F. Duarte and D. J. Dixon, ACS Catal., 2020, 10, 11438–11447 CrossRef CAS.
- J. A. Leitch, T. Rossolini, T. Rogova and D. J. Dixon, ACS Catal., 2020, 10, 11430–11437 CrossRef CAS.
- Ł. W. Ciszewski, J. Durka and D. Gryko, Org. Lett., 2019, 21, 7028–7032 CrossRef PubMed.
- Y.-L. Su, G.-X. Liu, J.-W. Liu, L. Tram, H. Qiu and M. P. Doyle, J. Am. Chem. Soc., 2020, 142, 13846–13855 CrossRef CAS PubMed.
- F. Li, C. Pei and R. M. Koenigs, Angew. Chem., Int. Ed., 2021, 61, e202111892 Search PubMed.
- Z. Zhang, N. Kvasovs, A. Dubrovina and V. Gevorgyan, Angew. Chem., Int. Ed., 2022, 61, e202110924 CAS.
- X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed.
- H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936–6939 CrossRef CAS PubMed.
- J. Ma, K. Harms and E. Meggers, Chem. Commun., 2016, 52, 10183–10186 RSC.
- C. Wang, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 13495–13498 CrossRef CAS PubMed.
- L. Chenneberg and C. Ollivier, Chimia, 2016, 70, 67–76 CrossRef CAS PubMed.
- L. Chenneberg, A. Baralle, M. Daniel, L. Fensterbank, J.-P. Goddard and C. Ollivier, Adv. Synth. Catal., 2014, 356, 2756–2762 CrossRef CAS.
- G. L. Lackner, K. W. Quasdorf and L. E. Overman, J. Am. Chem. Soc., 2013, 135, 15342–15345 CrossRef CAS PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef CAS PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929–7933 CrossRef CAS PubMed.
- H.-M. Guo and X. Wu, Nat. Commun., 2021, 12, 5365 CrossRef CAS PubMed.
- F. W. Friese and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9561–9564 CrossRef CAS PubMed.
- D. A. Dirocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed.
- W. Xu, J. Ma, X.-A. Yuan, J. Dai, J. Xie and C. Zhu, Angew. Chem., Int. Ed., 2018, 130, 10514–10518 CrossRef.
- J. Ma, W. Xu and J. Xie, Sci. China Chem., 2020, 63, 187–191 CrossRef CAS.
- E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed.
- J. Dong, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Sci. Adv., 2019, 5, eaax9955 CrossRef CAS PubMed.
- Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154–159 CrossRef CAS.
- Á. Péter, G. J. P. Perry and D. J. Procter, Adv. Synth. Catal., 2020, 362, 2135–2142 CrossRef.
- J. Gao, J. Feng and D. Du, Chem.–Asian J., 2020, 15, 3637–3659 CrossRef CAS PubMed.
- C. Chen, Z.-J. Wang, H. Lu, Y. Zhao and Z. Shi, Nat. Commun., 2021, 12, 4526 CrossRef CAS PubMed.
- X. Zhu, M. Jiang, X. Li, E. Zhu, Q. Deng, X. Song, J. Lv and D. Yang, Org. Chem. Front., 2022, 9, 347–355 RSC.
- H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed.
- D. J. P. Kornfilt and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 6853–6858 CrossRef CAS PubMed.
- A. A. Zemtsov, S. S. Ashirbaev, V. V. Levin, V. A. Kokorekin, A. A. Korlyukov and A. D. Dilman, J. Org. Chem., 2019, 84, 15745–15753 CrossRef CAS PubMed.
- Y. He, X. Dan, Y. Tang, Q. Yang, W. Wang and Y. Cai, Green Chem., 2021, 23, 9577–9582 RSC.
- F. Xue, F. Wang, J. Liu, J. Di, Q. Liao, H. Lu, M. Zhu, L. He, H. He, D. Zhang, H. Song, X. Y. Liu and Y. Qin, Angew. Chem., Int. Ed., 2018, 57, 6667–6671 CrossRef CAS PubMed.
- B. Schweitzer-Chaput, M. A. Horwitz, E. De Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed.
- S. Cuadros, M. A. Horwitz, B. Schweitzer-Chaput and P. Melchiorre, Chem. Sci., 2019, 10, 5484–5488 RSC.
- D. Mazzarella, G. Magagnano, B. Schweitzer-Chaput and P. Melchiorre, ACS Catal., 2019, 9, 5876–5880 CrossRef CAS.
- K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed.
- H. Wang and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 163–166 CrossRef CAS PubMed.
- D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed.
- J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, M. Medebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noel, C. W. Am Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS PubMed.
- C. Liu, N. Shen and R. Shang, Nat. Commun., 2022, 13, 354 CrossRef CAS PubMed.
- Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- S. Ye, T. Xiang, X. Li and J. Wu, Org. Chem. Front., 2019, 6, 2183–2199 RSC.
- J. Li, M. Kong, B. Qiao, R. Lee, X. Zhao and Z. Jiang, Nat. Commun., 2018, 9, 2445 CrossRef PubMed.
- D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2020, 142, 9119–9123 CrossRef CAS PubMed.
- W. H. Tamblyn, E. A. Vogler and J. K. Kochi, J. Org. Chem., 1980, 45, 3912–3915 CrossRef CAS.
- C. Chatgilialoglu, Acc. Chem. Res., 1992, 25, 188–194 CrossRef CAS.
- F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 2002, 51, 4411–4416 CrossRef.
- E. Tatunashvili and C. S. P. McErlean, Org. Biomol. Chem., 2020, 18, 7818–7821 RSC.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- G. H. Lovett, S. Chen, X.-S. Xue, K. N. Houk and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 20031–20036 CrossRef CAS PubMed.
- P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 CrossRef CAS PubMed.
- T. Q. Chen and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2019, 58, 14584–14588 CrossRef CAS PubMed.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed.
- G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852–1855 CrossRef CAS PubMed.
- M. Presset, N. Fleury-Brégeot, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 4615–4619 CrossRef CAS PubMed.
- Y. Yamashita, J. C. Tellis and G. A. Molander, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 12026–12029 CrossRef CAS PubMed.
- G. Duret, R. Quinlan, P. Bisseret and N. Blanchard, Chem. Sci., 2015, 6, 5366–5382 RSC.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed.
- M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254–258 CrossRef CAS PubMed.
- J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512–3522 RSC.
- D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 9847–9850 CrossRef CAS PubMed.
- D. P. Plasko, C. J. Jordan, B. E. Ciesa, M. A. Merrill and J. M. Hanna, Photochem. Photobiol. Sci., 2018, 17, 534–538 CrossRef CAS PubMed.
- S. Zhou, K. Lv, R. Fu, C. Zhu and X. Bao, ACS Catal., 2021, 11, 5026–5034 CrossRef CAS.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 4345, 433–436 CrossRef PubMed.
- D. N. Primer, I. Karakaya, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2015, 137, 2195–2198 CrossRef CAS PubMed.
- D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 9847–9850 CrossRef CAS PubMed.
- J. Li, C.-Y. Huang, J.-T. Han and C.-J. Li, ACS Catal., 2021, 11, 14148–14158 CrossRef CAS.
- P. Ranjan, S. Pillitteri, G. Coppola, M. Oliva, E. V. Van Der Eycken and U. K. Sharma, ACS Catal., 2021, 11, 10862–10870 CrossRef CAS.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- Z.-Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- G. E. M. Crisenza, A. Faraone, E. Gandolfo, D. Mazzarella and P. Melchiorre, Nat. Chem., 2021, 13, 575–580 CrossRef CAS PubMed.
- V. Corcé, L. M. Chamoreau, E. Derat, J. P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414–11418 CrossRef PubMed.
- C. Lévêque, L. Chenneberg, V. Corcé, J.-P. Goddard, C. Ollivier and L. Fensterbank, Org. Chem. Front., 2016, 3, 462–465 RSC.
- M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef CAS PubMed.
- G. Ikarashi, T. Morofuji and N. Kano, Chem. Commun., 2020, 56, 10006–10009 RSC.
- T. Morofuji, Y. Matsui, M. Ohno, G. Ikarashi and N. Kano, Chem. –Eur. J., 2021, 27, 6713–6718 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |