
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenaelectro-catalyzed C–H acyloxylation for late-stage tyrosine and oligopeptide diversification†
Xiaoyan
Hou‡
a,
Nikolaos
Kaplaneris‡
a,
Binbin
Yuan
a,
Johanna
Frey
 a,
Tsuyoshi
Ohyama
a,
Antonis M.
Messinis
a and
Lutz
Ackermann
*ab
a,
Tsuyoshi
Ohyama
a,
Antonis M.
Messinis
a and
Lutz
Ackermann
*ab
aInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
bGerman Center for Cardiovascular Research (DZHK), Potsdamer Straße 58, 10785 Berlin, Germany
First published on 25th February 2022
Abstract
Ruthenaelectro(II/IV)-catalyzed intermolecular C–H acyloxylations of phenols have been developed by guidance of experimental, CV and computational insights. The use of electricity bypassed the need for stoichiometric chemical oxidants. The sustainable electrocatalysis strategy was characterized by ample scope, and its unique robustness enabled the late-stage C–H diversification of tyrosine-derived peptides.
Introduction
During the last decade C–H activation has been recognised as a transformative platform in molecular syntheses1 with enabling applications to inter alia natural products syntheses,2 material sciences3 and medicinal chemistry.4 Among the transition metals that have been utilized for direct C–H activations ruthenium complexes,5 due to their robustness and cost-effectiveness, offer an appealing opportunity for sustainable molecular modifications. Due to the widespread occurrence of phenols in a plethora of bioactive compounds related to crop protection,6 molecular syntheses7 and drug development,8 phenol derivatives are uniquely appealing target motifs.9 As a consequence, direct C–H functionalizations on phenol derivatives represent a desirable approach towards value-added compounds.10 Recently, C–H activation has been identified as a powerful tool for late-stage diversification of complex biomolecules.11,12 C–H acyloxylation has attracted major attention in recent years.13 Over the past decade, tremendous efforts have been made to promote C–H acyloxylations, utilizing palladium,14 rhodium15 and ruthenium complexes,16 among others.17 Despite this indisputable progress, stoichiometric amounts of chemical oxidants are generally required, leading to undesired waste-products, jeopardizing the overall atom-economical nature of the C–H activation approach.Electroorganic synthesis has surfaced as an increasingly viable tool for sustainable syntheses.18 In this context, the merger of C–H activation and electrooxidation was recently recognized as a particularly powerful tool for molecular catalysis, avoiding stoichiometric amounts of chemical oxidants.19
Despite of indisputable advances in electrocatalysis,20 late-stage amino acid or peptide diversifications are unprecedented. In stark contrast, we, herein, disclosed the first electrochemical selective oxygenations of sensitive phenols for late-stage tyrosine diversification (Fig. 1). Salient features of our findings include: (a) resource-economical C–H acyloxylations with electricity as sustainable oxidant in lieu of stoichiometric chemical oxidants; (b) effective ruthenium catalysis in an user-friendly undivided cell setup; (c) mechanistic insights into the electrooxidative ruthenium catalysis via the isolation of key intermediates and (d) bioorthogonal C–H acyloxylation for late-stage diversifications of tyrosine-derived peptides.
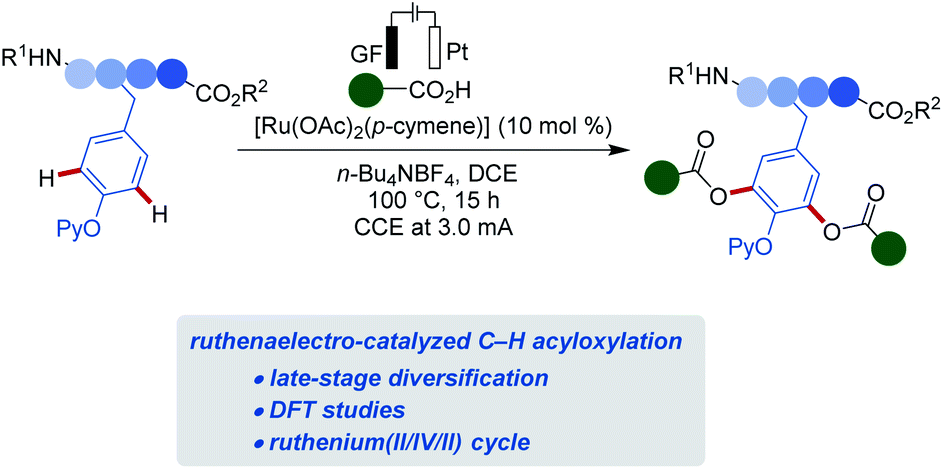 | ||
| Fig. 1 Electrocatalytic C–H acyloxylation of tyrosine containing peptides. | ||
Results and discussion
We initiated our studies by exploring reaction conditions for the envisioned electrochemical C–H acyloxylations of phenol 1a with a removable pyridine21 in an undivided cell setup equipped with graphite felt (GF) and platinum plate (Pt) as anode and cathode, respectively (Table 1). After considerable experimentation, the desired product 3aa was isolated in 87% yield with [Ru(OAc)2(p-cymene)] (10 mol%), and n-Bu4NBF4 (1.0 equiv.) in DCE at 100 °C for 15 h (Table 1, entry 1). TFE in lieu of DCE at 80 °C afforded a sharp decrease of efficacy (entry 2). With an increase of the temperature, the performance could be improved (entry 3). The direct acyloxylation was not viable in either EtOH or t-AmOH/H2O (3/1) (entries 4 and 5). Other ruthenium catalysts proved less effective for the C–H acyloxylation (entries 6–8). Remarkably, ruthenium catalyst outperformed rhodium(III), iridium(III) and cobalt(III) complexes under otherwise identical reaction conditions (entries 9–12). Control experiments verified the essential role of the ruthenium catalyst and the electricity (entries 13 and 14).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield |
| a Reaction conditions: 1a (0.25 mmol), 2a (0.40 mmol), [Ru(OAc)2(p-cymene)] (10 mol%), n-Bu4NBF4 (0.25 mmol), DCE (4.0 mL), 100 °C 15 h, undivided cell, GF anode, Pt cathode, under air, constant current = 3.0 mA. Py = pyridyl, TFE = 2,2,2-trifluoroethanol, DCE = 1,2-dichloroethane, [Ru] = [Ru(OAc)2(p-cymene)]. | ||
| 1 | No change | 87% |
| 2 | TFE as solvent, 80 °C | 36% |
| 3 | TFE as solvent | 65% |
| 4 | EtOH as solvent | — |
| 5 | t-AmOH/H2O (3/1) as solvent | — |
| 6 | [RuCl2(p-cymene)]2 (5 mol%) in place of [Ru] | 78% |
| 7 | [Ru2(OAc)4Cl] (5 mol%) in place of [Ru] | 86% |
| 8 | [RuCl3·3H2O] in place of [Ru] | 66% |
| 9 | [RhCl3·3H2O] in place of [Ru] | — |
| 10 | [Cp*RhCl2]2 (5 mol%) in place of [Ru] | 15% |
| 11 | [Cp*IrCl2]2 (5 mol%) in place of [Ru] | <5% |
| 12 | [Cp*CoI2(CO)] in place of [Ru] | — |
| 13 | Without [Ru] | — |
| 14 | No electricity | — |

With the optimised reaction conditions in hand, we next explored alternative N-heterocycles, such as pyrimidine 1b and benzo[d]oxazole 1c, which also set the stage for effective electro-oxygenations (Scheme 1). The substrate scope of the ruthenaelectro-catalyzed C–H acyloxylation was subsequently examined with a representative set of phenols 1 (Scheme 1). Thus, decorated phenols 1 chemo-selectively afforded the desired acyloxylated products 3. Electron-rich as well as electron-deficient substrates were amenable to the ruthenaelectro-catalyzed acyloxylations. Remarkably, the C–H acyloxylation of naphthalene 1f occurred site-selectively in the β-position, bypassing a potential peri-functionalization regime.
 | ||
| Scheme 1 Substrate scope of acyloxylation with phenols 1 and carboxylic acids 2. a RuCl3·3H2O (10 mol%) as the catalyst. | ||
Our attention was then shifted to explore viable carboxylic acids 2 for the ruthenaelectro-catalyzed acyloxylation (Scheme 1). Both electron-rich and electron-deficient acids delivered good to excellent yields, while electron-deficient acids exhibiting slightly better reactivity. Substituted benzoic acids were efficiently converted to the desired products 3ab–3av. Notably, various sensitive functional groups, including halogens, ester and free hydroxy groups, were fully tolerated under our electrocatalysis. The arene-ligand free RuCl3·3H2O catalyst was also efficient for the ruthenaelectro-catalyzed C–H acyloxylation, suggesting a p-cymene-free mode of action (vide infra). Heteroaromatic acid, such as the thiophene derivative 2r, was also a viable substrate. The power of the ruthenium-catalyzed oxygenations was highlighted by the successful use of alkyl carboxylic acids, such as substrates 2s (acetic acid), 2t (isobutyric acid), 2u (cyclopentanoic acid), and 2v (1-adamantanecarboxylic acid).
The ruthena-electrocatalysis was not limited to the conversion of simple phenols. Next, we devised the envisioned ruthenaelectro-catalyzed late-stage functionalization of tyrosine-containing peptides (Scheme 2). The electrocatalysis avoided the use of stoichiometric oxidants that severely jeopardizes the sustainability and robustness of the late-stage functionalization.22 To our delight, when amino acid 4a was utilized, the desired acyloxylated tyrosine derivative 5aa was selectively obtained. When chemical oxidants were used instead of electricity, the desired oxygenated product was not obtained, providing a strong testament to the robust nature of our electrochemical approach. Moreover, the traceless removal of the pyridyl group was accomplished by a selective methylation/hydrogenation protocol giving rise to NH-free tyrosine-containing acyloxylated amino acid 5aa[OH]. Furthermore, various di-, tri- and tetrapeptides 4b–4i were efficiently functionalized with various (hetero)aromatic acids 2 without signs of epimerization of the sensitive peptides. Remarkably, our mild electroxidative regime bypassed Shono-type manifolds, even when employing proline-containing di- and tetrapeptides 4g and 4i. The broad scope showcased the remarkable efficiency and functional group tolerance of our ruthenaelectro-catalyzed acyloxylations.
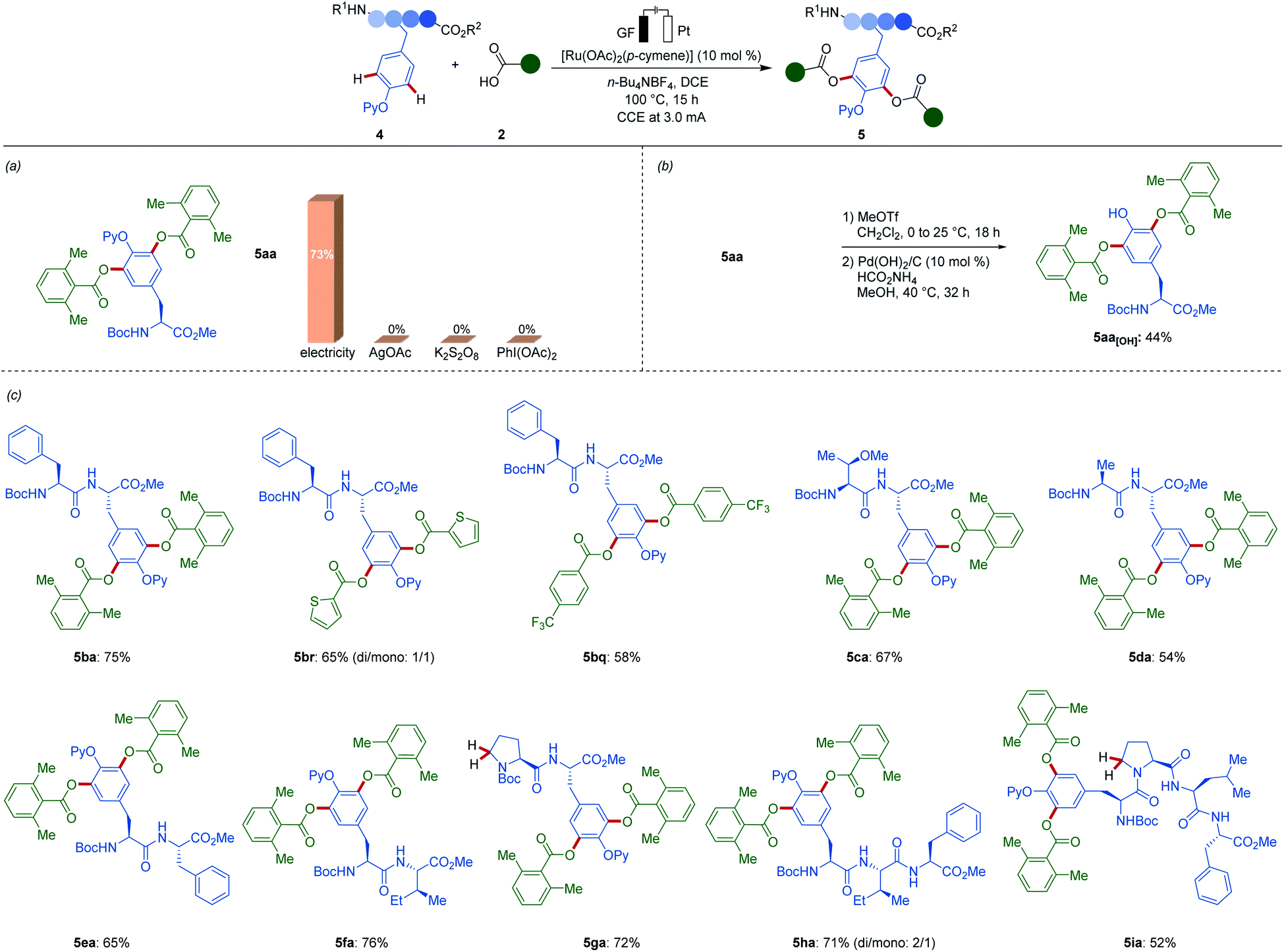 | ||
| Scheme 2 Late-stage tyrosine and peptide diversification: (a) evaluation of oxidants, (b) removal of pyridyl group, (c) scope of viable peptides 4. | ||
Intrigued by the versatility of the electrochemical C–H acyloxylation, we became interested to probe its mode of action. To this end, an intermolecular competition experiment between electronically differentiated phenols 1 was conducted (Scheme 3a). Electron-rich substrate 1a showed an inherently higher reactivity as compared to electron-deficient substrate 1g, showing that nucleophilicity rather than kinetic C–H acidity leads to faster reactions. This observation is not in line with a concerted protonation-deprotonation (CMD) mechanism. Instead, a base-assisted internal electrophilic-type substitution (BIES) mechanism is likely operative. Competition experiments, which were designed to investigate the relative reactivities of electronically-differentiated carboxylic acids 2, showed that the electron-deficient aromatic carboxylic acids 2i reacted faster (Scheme 3a). Furthermore, a competition experiment between aryl and alkyl carboxylic acids 2a and 2s (Scheme 3a) illustrated the superiority of aromatic derivatives. Moreover, a ruthenaelectro-catalysis in the presence of isotopically labelled D2O led to a significant H/D-exchange (Scheme 3b). The H/D scrambling occurred solely at the ortho-position, as judged by the re-isolated substrate [Dn]-1a, being indicative of a fast C–H activation. To further elucidate the key role of the electricity, an on/off experiment was conducted (Scheme 3c). The formation of the acyloxylated product was fully suppressed in the absence of the electric current, thereby ruling a radical-chain process out.
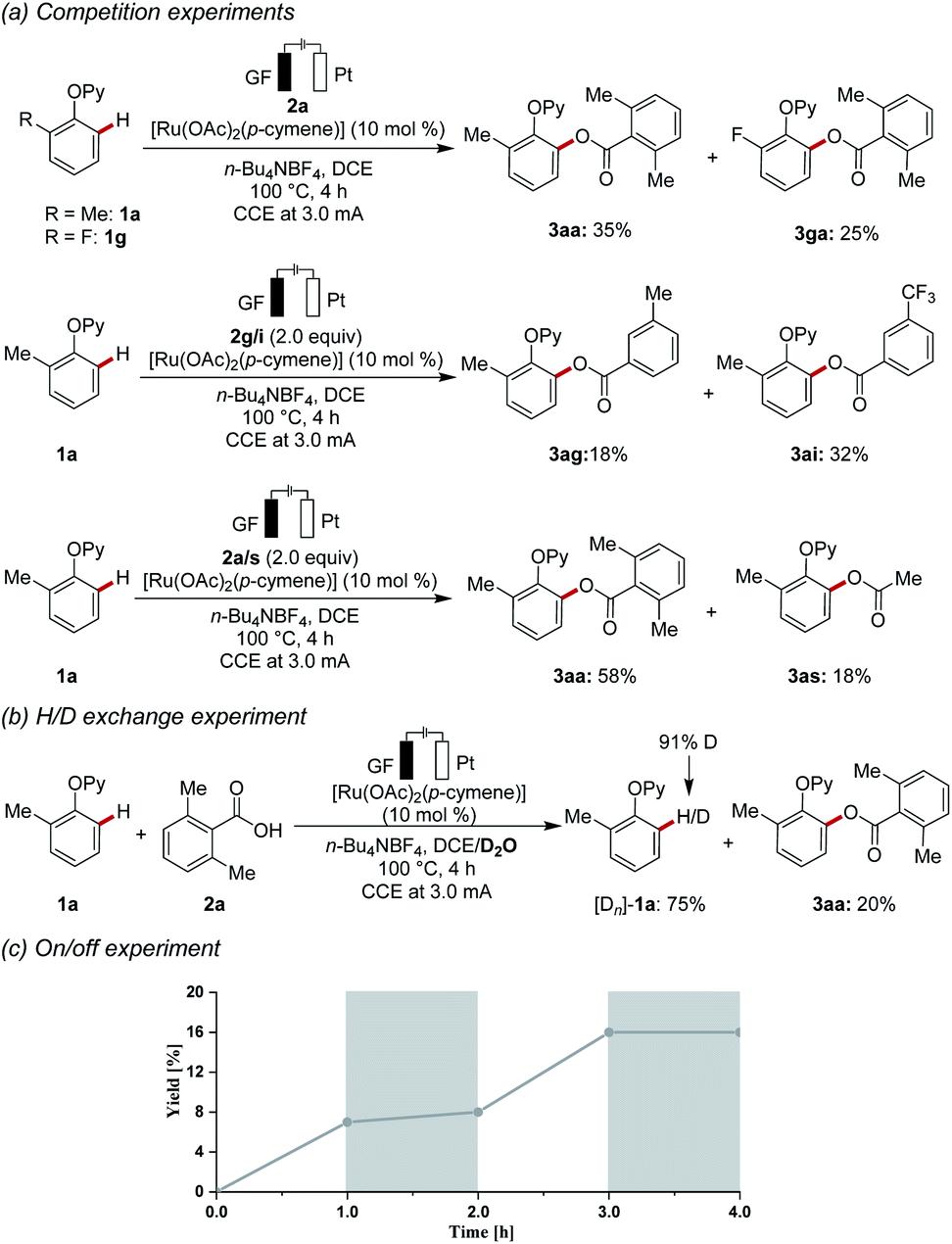 | ||
| Scheme 3 Key mechanistic findings: (a) competition experiments, (b) H/D scrambling experiment, (c) on/off experiment. | ||
During the course of the electrocatalysis, a considerable amount of free p-cymene from the [Ru(OAc)2(p-cymene)] was observed (Scheme 4a). This finding is indicative of a p-cymene-ligand-free complex being catalytically relevant.23 Next, we prepared the well-defined cationic cyclometalated ruthenium complex 7, which showed high catalytic activity under otherwise identical reaction conditions (Scheme 4b). Kinetic studies revealed comparable reactivities for [Ru(OAc)2(p-cymene)] and ruthenacycle 7, as judged by the corresponding initial rates (Fig. S3†).24 With the proposed key intermediate in hand, we probed its redox properties under electrocatalytic conditions (Scheme 4c). Hence, detailed studies by cyclic voltammetry revealed that the complex 7 underwent reversible oxidation to ruthenium(III) at 0.63 V, being significantly lower than the substrates 1f and 2a. Interestingly, the addition of carboxylic acid (e.g. 10 equiv.) slowly led to the gradual disappearance of the reversible peak at 0.63 V, and a new irreversible oxidation peak appeared at 0.98 V. Combined with the analysis from the high-resolution mass spectrometry monitoring of the mixture solution after cyclic voltammetry, carboxylate ions were found to replace acetonitrile to coordinate with the metal centre (Fig. S8†).24 These findings are suggestive of a carboxylate-ligated, arene-ligand-free ruthenacycle as a key intermediate in the C–O reductive elimination.
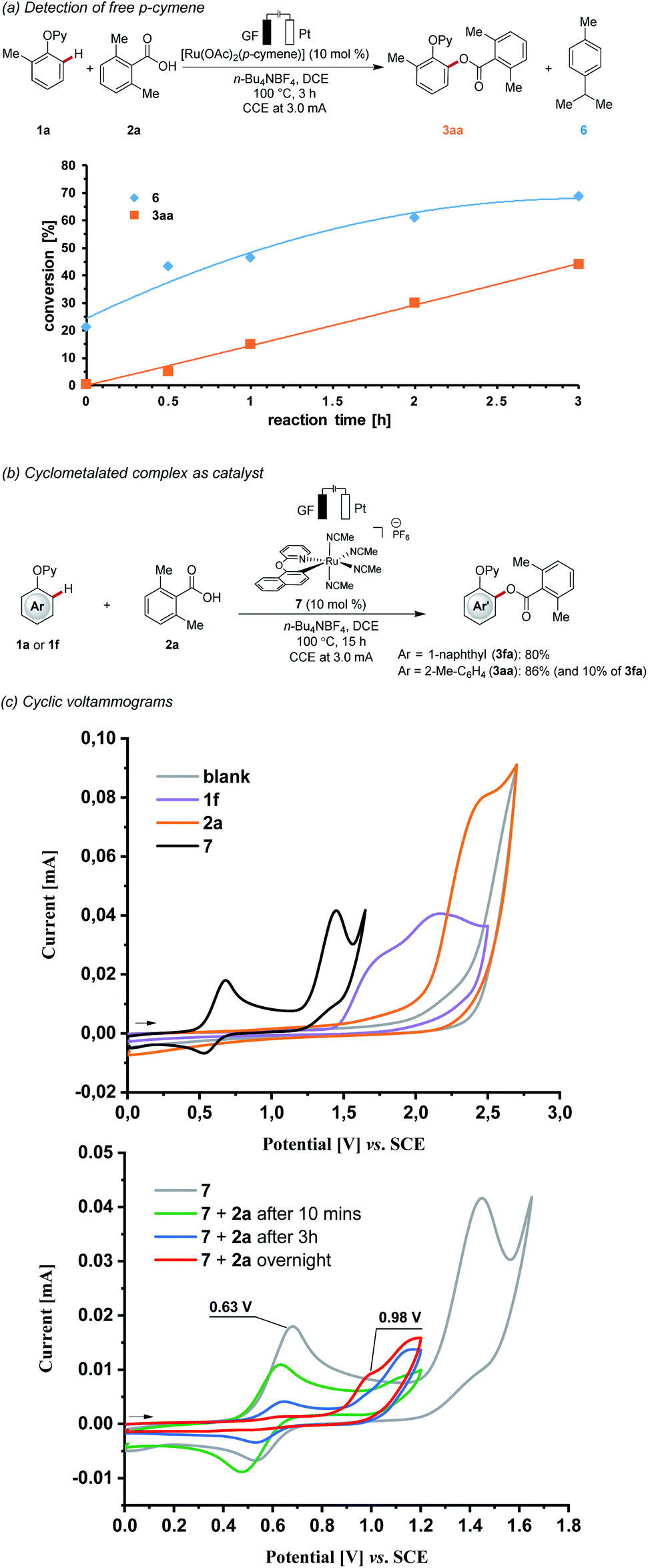 | ||
| Scheme 4 Key mechanistic findings: (a) detection of free p-cymene, (b) cyclometalated complex as catalyst, (c) cyclic voltammograms. | ||
In order to gain better insights into the catalyst mode of action, density-functional theory (DFT) calculations were carried out for the oxidatively induced reductive elimination(OIRE)25 at the PW6B95-D4/def2-TZVP+SMD(DCE)//PBE0-D3BJ/def2-SVP level of theory. In these studies, pathways starting from ruthenium(III) as well as from ruthenium(IV) were considered (Fig. 2). Three computational models were taken into account: (i) p-cymene and 2-phenoxypyridine are coordinated to the ruthenium centre (Fig. S9†),24 (ii) two 2-phenoxypyridines are coordinated, but one of them serves solely as a L-type ligand (Fig. S10†)24 and (iii) both substrates are C–H activated but only one is involved in the reductive elimination (Fig. 2). The first two paths could be ruled out, because they revealed energetically disfavoured due to the high calculated barriers. These results are in good agreement with the experiments, where free p-cymene was detected during the reaction. With regards to potential bis-cyclometalated intermediate,26 the ruthenium(II/IV) manifold pathway was shown to be energetically preferred over the ruthenium(I/III) manifold pathway by 22.2 kcal mol−1. Thus, our computational findings provide strong support for a preferential oxidatively induced reductive elimination through bis-cyclometalated ruthenium(IV) species.
 | ||
| Fig. 2 Computed relative Gibbs free energy profile (ΔG373.15) in kcal mol−1 for two distinct p-cymene free oxidatively induced reductive elimination pathways at the PW6B95-D4/def2-TZVP+SMD(DCE)//PBE0-D3BJ/def2-SVP level of theory. Non-participating hydrogen atoms were omitted for clarity. | ||
On the basis of our findings, a plausible catalytic cycle for the acyloxylation commences with a BIES C–H metalation27 and dissociation of p-cymene, thereby forming the cyclometalated complex I1 (Scheme 5). A second molecule of 1a coordinates to ruthenium complex I1 and undergoes C–H activation to form bis-cyclometalated complex I2, which undergoes anodic oxidation to deliver the ruthenium(IV) intermediate I3. Finally, reductive elimination and ligand exchange deliver the product 3 and regenerate ruthenacycle I1.
 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In summary, experimental and computational mechanistic studies unraveled a C–H acyloxylation manifold through an arene-ligand-free ruthenium complex. Detailed CV studies and DFT computation provided strong support for the formation of a bis-cyclometalated complex as well as an oxidation-induced reductive elimination process within a ruthenium(II/IV/II) manifold. Our strategy provided an expedient access to a plethora of peptides by modular late-stage acyloxylations of tyrosine-derived peptides.Data availability
All experimental data, procedures for data analysis and pertinent data sets are provided in the ESI.†Author contributions
X. H. and L. A. conceived the project. X. H., N. K., J. F. and T. O. performed the experiments. B. Y. performed DFT calculations. A. M. M. assisted X. H. to make the intermediate complexes. X. H. and L. A. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support from the ERC Advanced Grant no. 101021358 and DFG Gottfried-Wilhelm-Leibniz-Preis (L. A.), the China Scholarship Council (fellowship to X. H. and B. Y.) and the European Union's H-2020 Marie Skłodowska-Curie Fellowship grant no. 895404 (fellowship to A. M. M.) is gratefully acknowledged.Notes and references
- (a) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS; (b) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed; (c) Ł. Woźniak, J.-F. Tan, Q.-H. Nguyen, A. Madron du Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef PubMed; (d) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (e) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord and T. Besset, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (f) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (g) H. M. Davies and D. Morton, ACS Cent. Sci., 2017, 3, 936–943 CrossRef CAS PubMed; (h) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (i) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed.
- (a) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- (a) J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed; (b) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed; (c) D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594–1600 CrossRef CAS.
- (a) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS; (b) J. Börgel and T. Ritter, Chem, 2020, 6, 1877–1887 CrossRef.
- (a) P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721–5745 CrossRef CAS; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (c) B. Li and P. H. Dixneuf, in Ruthenium in Catalysis, ed. P. H. Dixneuf and C. Bruneau, Springer, Cham, 2014, pp. 119–193 Search PubMed; (d) C.-J. Li, J. Huang, X.-J. Dai, H. Wang, N. Chen, W. Wei, H. Zeng, J. Tang, C. Li, D. Zhu and L. Lv, Synlett, 2019, 30, 1508–1524 CrossRef CAS.
- (a) A. K. Gautam, P. K. Singh and M. Aravind, in Plant Phenolics in Sustainable Agriculture, ed. R. Lone, R. Shuab and A. N. Kamili, Springer, Singapore, 2020, vol. 1, pp. 579–594 Search PubMed; (b) M. E. Cartea, M. Francisco, P. Soengas and P. Velasco, Molecules, 2011, 16, 251–280 CrossRef CAS PubMed.
- (a) S. W. Youn and C.-G. Cho, Org. Biomol. Chem., 2021, 19, 5028–5047 RSC; (b) Z. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed; (c) R. Mei, J. Koeller and L. Ackermann, Chem. Commun., 2018, 54, 12879–12882 RSC; (d) Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, 2004 Search PubMed.
- (a) J. Nešvera, L. Rucká and M. Pátek, Adv. Appl. Microbiol., 2015, 93, 107–160 CrossRef PubMed; (b) D. Ekinci, M. Şentürk and Ö. İ. Küfrevioğlu, Expert Opin. Ther. Pat., 2011, 21, 1831–1841 CrossRef CAS PubMed; (c) S. Jagani, R. Chelikani and D.-S. Kim, Biofouling, 2009, 25, 321–324 CrossRef CAS PubMed.
- (a) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461–1479 CrossRef CAS; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- (a) Z. Huang and J.-P. Lumb, ACS Catal., 2019, 9, 521–555 CrossRef CAS; (b) F. Yang, H. Zhang, X. Liu, B. Wang and L. Ackermann, Chin. J. Org. Chem., 2019, 39, 59–73 CrossRef CAS.
- For selected recent reviews, see: (a) H.-R. Tong, B. Li, G. Li, G. He and G. Chen, CCS Chem., 2021, 3, 1797–1820 CrossRef CAS; (b) A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed.
- For selected recent examples, see: (a) M. Stangier, A. M. Messinis, J. C. A. Oliveira, H. Yu and L. Ackermann, Nat. Commun., 2021, 12, 4736 CrossRef CAS PubMed; (b) B. Li, X. Li, B. Han, Z. Chen, X. Zhang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 9401–9407 CrossRef PubMed; (c) B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858–5862 CrossRef CAS PubMed; (d) A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576–1580 CrossRef CAS PubMed; (e) L. Mendive-Tapia, S. Preciado, J. García, R. Ramón, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed; (f) W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS PubMed.
- Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS PubMed.
- (a) I. Urruzuno, P. Andrade-Sampedro and A. Correa, Org. Lett., 2021, 23, 7279–7284 CrossRef CAS PubMed; (b) Z. Zhuang, A. N. Herron, Z. Fan and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 6769–6776 CrossRef CAS PubMed; (c) A. K. Cook, M. H. Emmert and M. S. Sanford, Org. Lett., 2013, 15, 5428–5431 CrossRef CAS PubMed; (d) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2012, 14, 3724–3727 CrossRef CAS PubMed; (e) S.-i. Niwa, M. Eswaramoorthy, J. Nair, A. Raj, N. Itoh, H. Shoji, T. Namba and F. Mizukami, Science, 2002, 295, 105–107 CrossRef CAS PubMed.
- (a) S. Jin, J. Kim, D. Kim, J.-W. Park and S. Chang, ACS Catal., 2021, 11, 6590–6595 CrossRef CAS; (b) C. Chen, Y. Pan, H. Zhao, X. Xu, J. Xu, Z. Zhang, S. Xi, L. Xu and H. Li, Org. Chem. Front., 2018, 5, 415–422 RSC; (c) A. Mishra, T. K. Vats, M. P. Nair, A. Das and I. Deb, J. Org. Chem., 2017, 82, 12406–12415 CrossRef CAS PubMed; (d) C. R. Turlington, J. Morris, P. S. White, W. W. Brennessel, W. D. Jones, M. Brookhart and J. L. Templeton, Organometallics, 2014, 33, 4442–4448 CrossRef CAS.
- (a) K. Raghuvanshi, K. Rauch and L. Ackermann, Chem.–Eur. J., 2015, 21, 1790–1794 CrossRef CAS PubMed; (b) V. S. Thirunavukkarasu and L. Ackermann, Org. Lett., 2012, 14, 6206–6209 CrossRef CAS PubMed.
- (a) K. Zhu, K. Xu, Q. Fang, Y. Wang, B. Tang and F. Zhang, ACS Catal., 2019, 9, 4951–4957 CrossRef CAS; (b) W. Wang, C. Pan, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 3978–3980 RSC.
- For selected recent reviews, see: (a) N. Amri and T. Wirth, Chem. Rec., 2021, 21, 2526–2537 CrossRef CAS PubMed; (b) R. C. D. Brown, Chem. Rec., 2021, 21, 2472–2487 CrossRef CAS PubMed; (c) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (d) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed; (e) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS; (f) F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed; (g) H. an, Z.-W. Hou and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592–4595 CrossRef PubMed; (h) Y. Liang, F. Lin, Y. Adeli, R. Jin and N. Jiao, Angew. Chem., Int. Ed., 2019, 58, 4566–4570 CrossRef CAS PubMed; (i) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (j) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (k) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (l) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (m) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- (a) Y. Qiu, C. Zhu, M. Stangier, J. Struwe and L. Ackermann, CCS Chem., 2021, 3, 1529–1552 CrossRef CAS; (b) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (c) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338–352 CrossRef CAS; (d) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS; (e) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- (a) X. Tan, L. Massignan, X. Hou, J. Frey, J. C. A. Oliveira, M. N. Hussain and L. Ackermann, Angew. Chem., Int. Ed., 2021, 60, 13264–13270 CrossRef CAS PubMed; (b) H. Shen, D. Cheng, Y. Li, T. Liu, X. Yi, L. Liu, F. Ling and W. Zhong, Green Synth. Catal., 2020, 1, 175–179 CrossRef; (c) C. Tian, U. Dhawa, J. Struwe and L. Ackermann, Chin. J. Chem., 2019, 37, 552–556 CrossRef CAS; (d) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS PubMed.
- (a) M. Tobisu, J. Zhao, H. Kinuta, T. Furukawa, T. Igarashi and N. Chatani, Adv. Synth. Catal., 2016, 358, 2417–2421 CrossRef CAS; (b) S.-J. Lou, Q. Chen, Y.-F. Wang, D.-Q. Xu, X.-H. Du, J.-Q. He, Y.-J. Mao and Z.-Y. Xu, ACS Catal., 2015, 5, 2846–2849 CrossRef CAS; (c) B. Liu, H.-Z. Jiang and B.-F. Shi, J. Org. Chem., 2014, 79, 1521–1526 CrossRef CAS PubMed; (d) C. Zhang and P. Sun, J. Org. Chem., 2014, 79, 8457–8461 CrossRef CAS PubMed; (e) W. Ma and L. Ackermann, Chem.–Eur. J., 2013, 19, 13925–13928 CrossRef CAS PubMed; (f) L. Ackermann, E. Diers and A. Manvar, Org. Lett., 2012, 14, 1154–1157 CrossRef CAS PubMed.
- (a) M. San Segundo and A. Correa, Chem. Sci., 2020, 11, 11531–11538 RSC; (b) R. B. Bedford, M. F. Haddow, R. L. Webster and C. J. Mitchell, Org. Biomol. Chem., 2009, 7, 3119–3127 RSC.
- (a) T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 15640–15645 CrossRef CAS PubMed; (b) M. Simonetti, D. M. Cannas, X. Just-Baringo, I. J. Vitorica-Yrezabal and I. Larrosa, Nat. Chem., 2018, 10, 724–731 CrossRef CAS PubMed; (c) L. Ackermann, R. Born and P. Álvarez-Bercedo, Angew. Chem., Int. Ed., 2007, 46, 6364–6367 CrossRef CAS PubMed.
- For detailed information, see the ESI.†.
- (a) S.-K. Zhang, A. Del Vecchio, R. Kuniyil, A. M. Messinis, Z. Lin and L. Ackermann, Chem, 2021, 7, 1379–1392 CrossRef CAS; (b) C. Zhu, M. Stangier, J. C. A. Oliveira, L. Massignan and L. Ackermann, Chem.–Eur. J., 2019, 25, 16382–16389 CrossRef CAS PubMed; (c) J. Kim, K. Shin, S. Jin, D. Kim and S. Chang, J. Am. Chem. Soc., 2019, 141, 4137–4146 CrossRef CAS PubMed; (d) L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414–12419 CrossRef CAS PubMed.
- J. M. Zakis, T. Smejkal and J. Wencel-Delord, Chem. Commun., 2022, 58, 483–490 RSC.
- (a) T. Rogge, J. C. A. Oliveira, R. Kuniyil, L. Hu and L. Ackermann, ACS Catal., 2020, 10, 10551–10558 CrossRef CAS; (b) W. Ma, R. Mei, G. Tenti and L. Ackermann, Chem.–Eur. J., 2014, 20, 15248–15251 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc07267f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |