
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(Fluoro)alkylation of alkenes promoted by photolysis of alkylzirconocenes†
Xiaoxiao
Ren
a,
Xing
Gao
b,
Qiao-Qiao
Min
b,
Shu
Zhang
c and
Xingang
Zhang
 *ab
*ab
aGreen Catalysis Center, and College of Chemistry, Zhengzhou University, Zhengzhou 450001, P. R. China
bKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xgzhang@mail.sioc.ac.cn
cThe Yangtze Delta Region Institute (Huzhou), University of Electronic Science and Technology of China, Huzhou 313001, China
First published on 2nd March 2022
Abstract
Difluoroalkylated compounds have important applications in pharmaceutical, agrochemical, and materials science. However, efficient methods to construct the alkylCF2–alkyl bond are very limited, and the site-selective introduction of a difluoromethylene (CF2) group into an aliphatic chain at the desired position remains challenging. Here, we report an unprecedented example of alkylzirconocene promoted difluoroalkylation of alkyl- and silyl-alkenes with a variety of unactivated difluoroalkyl iodides and bromides under the irradiation of visible light without a catalyst. The resulting difluoroalkylated compounds can serve as versatile synthons in organic synthesis. The reaction can also be applied to activated difluoroalkyl, trifluoromethyl, perfluoroalkyl, monofluoroalkyl, and nonfluorinated alkyl halides, providing a general method to controllably access fluorinated compounds. Preliminary mechanistic studies reveal that a single electron transfer (SET) pathway induced by a Zr(III) species is involved in the reaction, in which the Zr(III) species is generated by the photolysis of alkylzirconocene with blue light.
Introduction
The site-selective introduction of fluorine atom(s) into organic molecules has important applications in medicinal chemistry, chemical biology, and materials science.1 For instance, biologically active molecules containing a difluoromethylene (CF2) moiety at the specific position exhibit improved bioactivities compared with their nonfluorinated counterparts, because the CF2 can change the metabolic stability, conformation, acidity and polarity of the molecules.2 Some important pharmaceuticals containing the CF2 moiety have been discovered for the treatment of tumors and other deceases, such as gemcitabine,3 vinflunine,4 and lubiprostone5 (Fig. 1A). To this end, efforts toward the development of efficient methods to access CF2-containing molecules have been witnessed over the past decade.6 However, most of these developed synthesis methods mainly focus on the preparation of difluoroalkylated arenes (ArCF2R) with CF2 at the benzylic position.6a,7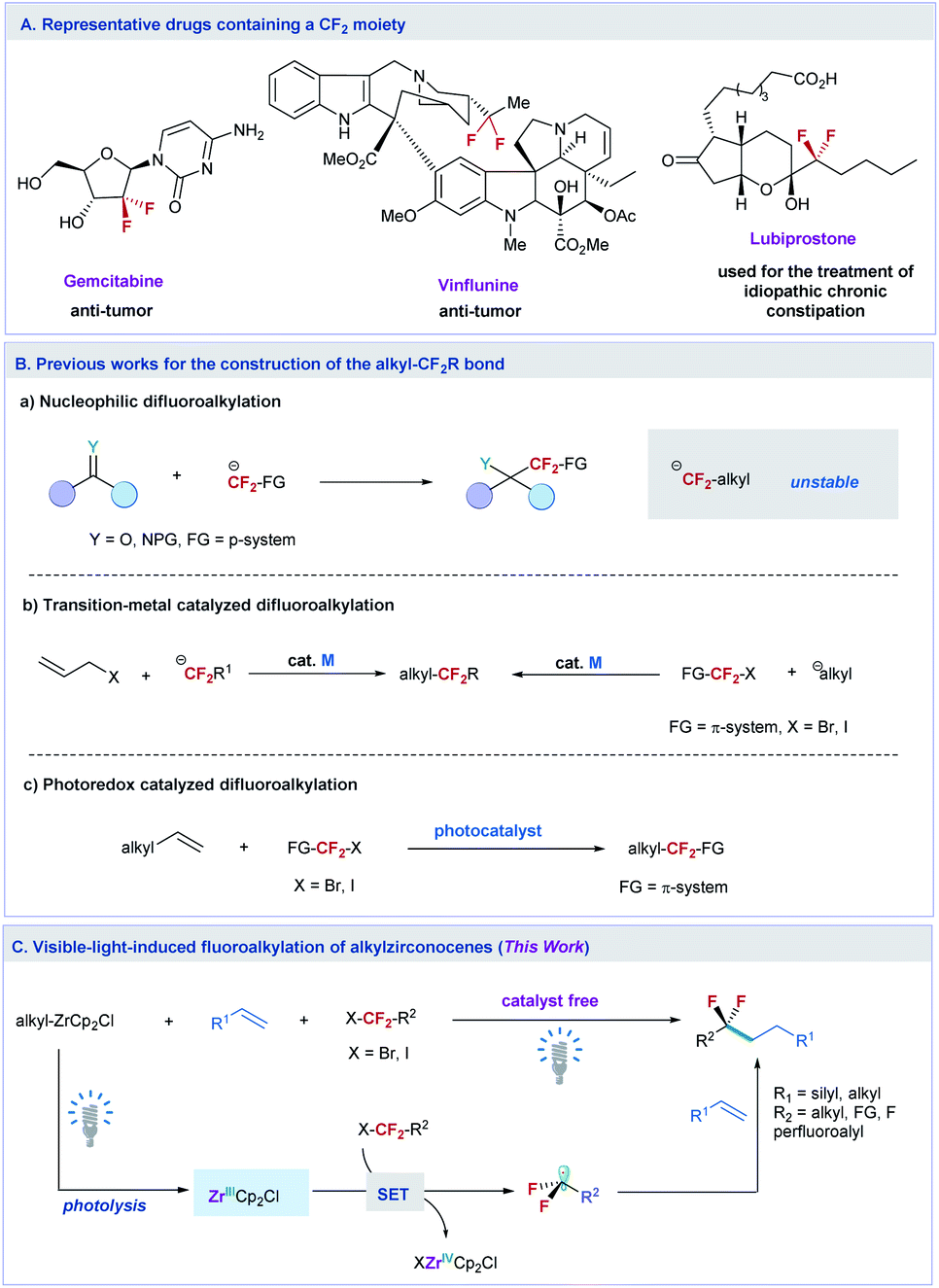 | ||
| Fig. 1 Strategies for the difluoroalkylation and selected examples of pharmaceutical bearing CF2 moieties. (A) Representative drugs containing a CF2 moiety. (B) Previous works for the construction of the alkyl-CF2R bond. (C) This work: visible-light-induced fluoroalkylation of alkylzirconocenes. | ||
To date, efficient methods to site-selectively introduce CF2 into an aliphatic chain remain limited. The traditional method to prepare difluoroalkylated compounds relies on the deoxyfluorination of the carbonyl group with dialkylaminosulfur trifluorides, such as diethylaminosulfur trifluoride (DAST).8 But the modest functional group tolerance of this method restricts its synthetic applications. Another common method to synthesize such a valuable fluorinated structure is based on the nucleophilic addition of difluoroalkylating reagents to aldehydes, ketones, and imines (Fig. 1B(a)).6c,9 However, owing to the instability of nucleophilic difluoroalkylating reagents, usually a π-system adjacent to the CF2 moiety, is needed to stabilize the difluoroalkyl anion,10 making the method difficult in constructing the alkylCF2–alkyl bond. Although nucleophilic substitution of aliphatic electrophiles with carbon nucleophiles is well known, the adoption of a similar strategy to react difluoroalkyl halides with aliphatic nucleophiles remains challenging and has not been reported yet.11 In this context, transition-metal12 or photo-redox13 catalyzed difluoroalkylation reactions to construct the alkylCF2-alkyl bond have been developed (Fig. 1B(b) and B(c)). These methods either need activated coupling partners, such as allylic substrates12a and π system functionalized difluoroalkyl halides12b (XCF2-FG, FG = π system, transition-metal catalyzed process), or are difficult to be used in the reaction of aliphatic alkenes with XCF2–alkyl (e.g. for the photoredox catalyzed process),13c thus restricting their widespread synthetic applications.
To overcome these limitations and meet the increasing demands of life and materials sciences, new methods that can extend the diversity of the difluoroalkylated structure with site-selective introduction of CF2 into the aliphatic chain at the desired position are highly desired. Herein, we report an unprecedented example of catalyst free difluoroalkylation of silyl- and alkyl-alkenes with unactivated difluoroalkyl halides promoted by photolysis of alkylzirconocenes (Fig. 1C). The reaction can also be applied to π-functionalized difluoroalkyl, trifluoromethyl, perfluoroalkyl, monofluoroalkyl, and non-fluoroalkyl halides, providing a general method to access fluoroalkylated alkanes. Preliminary mechanistic studies reveal that a single electron transfer (SET) pathway induced by a Zr(III) species is involved in the reaction, in which the Zr(III) species is generated by the photolysis of alkylzirconocenes with visible light. This protocol paves a new way to construct the alkyl–fluoroalkyl bond by using readily available fluoroalkyl halides and aliphatic alkenes.
Results and discussion
Inspired by our nickel-catalyzed difluoroalkylation cross-coupling,12b,c,14 we chose unactivated difluoroalkyl iodide 1a15 and alkylzirconocene 2a as the model substrates (Table 1), in which alkylzirconocene can be readily prepared from the corresponding alkene 5 and Schwartz reagent (HZrCp2Cl, 6).16 Directly adopting the nickel-catalyzed cross coupling failed to provide desired difluoroalkylated alkane 3a (entry 1). In contrast, the irradiation of the nickel catalyzed reaction with a blue LED (12 W) generated product 3a in 51% yield along with 27% yield of hydrodeiodinated by-product 4a (entry 2).17 The absence of the nickel catalyst could also provide a comparable yield (entry 3), and no reaction occurred without the blue LED (entry 4), demonstrating that blue light is essential in promoting the reaction. These mild conditions are quite different from the challenge SN1 or SN2 substitution of difluoroalkyl halides, because of the difficulty in forming a difluoroalkyl carbocation (SN1) or the repulsion of the lone pairs of fluorine atoms to the carbon nucleophile (SN2).18 This difference implies a novel mechanism, which is discussed below. Encouraged by these results, we tested a series of reaction parameters (for details, see the ESI†), and found that the use of 3.0 equiv. of 2a with NMP as the solvent could provide 3a in 81% yield (62% isolated yield) (entry 5).|
|
||||
|---|---|---|---|---|
| Entry | [Ni] (x) | Solvent | 3a (%) | 4a (%) |
| a Reaction conditions (unless otherwise specified): 1a (1 equiv., 0.3 mmol), 5a (0.72 mmol, 2.4 equiv.), 6 (0.6 mmol, 2.0 equiv.), solvent (2 mL), and 12 h. b The yield was determined by 19F NMR using fluorobenzene as an internal standard, and the number given in parentheses is the isolated yield. c The reaction was conducted without the blue LED. d 5a (1.08 mmol, 3.6 equiv.), 6 (0.9 mmol, 3.0 equiv.), and NMP (3 mL) were used. e nd, not detected. | ||||
| 1c | NiBr2·DME/bpy (10) | THF | 0 | 5 |
| 2 | NiBr2·DME/bpy (10) | THF | 51 | 27 |
| 3 | None | THF | 53 | 33 |
| 4c | None | THF | nde | nde |
| 5d | None | NMP | 81(62) | 15 |
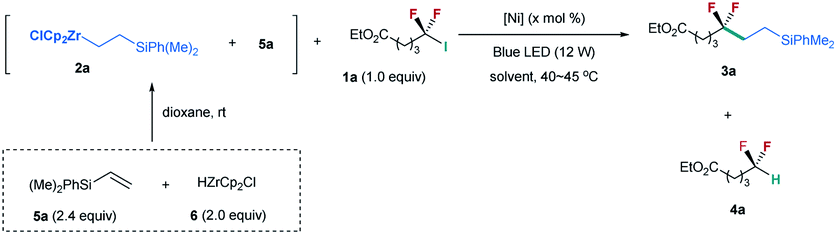
Because 2a was derived from the reaction of 6 with excessive silylalkene 5a (Table 1), this posed a question whether 5a, instead of 2a, is involved in the formation of 3a. To identify the role of 2a, we prepared 2a by reaction of alkylmagnesium bromide 7 with ZrCp2Cl2 (Scheme 1a).19 However, no 3a was observed by reaction of 2a with 1a (Scheme 1a). In contrast, the addition of 5a to the reaction could provide the desired product in 48% yield (Scheme 1b). These findings suggest that excessive silylalkene 5a in the reaction is the substrate responsible for generating 3a with 1a. This deduction was further supported by the addition of another silylalkene 5b to the reaction mixture of 1a, 2a, and 5a under the irradiation of blue light, in which both difluoroalkylated products 3a and 3a′ were formed (Scheme 1c). Since [Zr]-migration from alkylzirconocene to alkene cannot occur (Scheme 1d),20 these results clearly demonstrate that the alkene is the substrate to react with difluoroalkyl halide. In addition, the reaction of alkene 5a with 1a in the absence of 2a under the irradiation of a blue LED did not lead to 3a (Scheme 1e), demonstrating that the alkylzirconocene is essential in promoting the reaction.
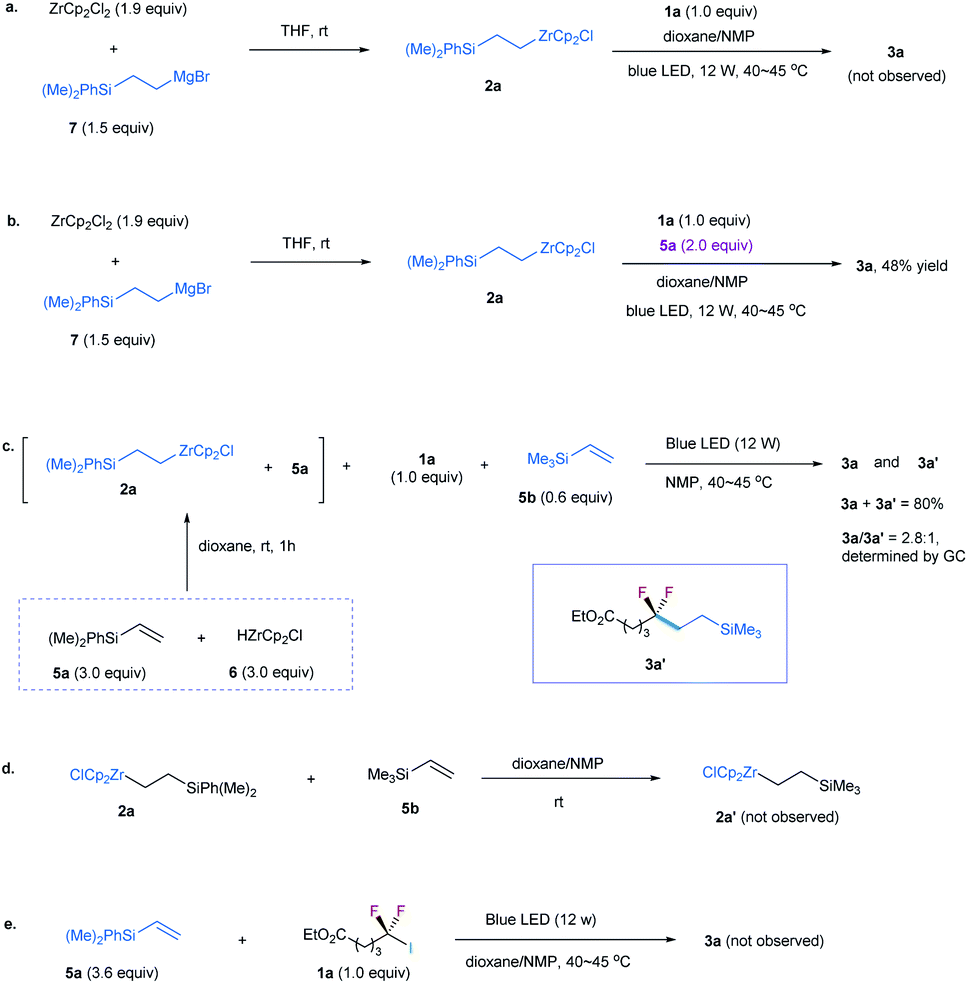 | ||
| Scheme 1 Control experiments for the reaction. (a) Reaction of 2a with 1a. (b) Reaction of 2a with 1a and 5a. (c) Reaction of 2a with 1a and 5a in the presence of 5b. (d) Reaction of 2a with 5b. (e) Reaction of 5a with 1a under irradiation of blue LED. | ||
Based on the above results, we reoptimized the reaction conditions, and found that the use of 2.5 equiv. of 5a, 1.2 equiv. of 6, and 1.0 equiv. of 1a could provide 3a in a comparable yield (80% determined by 19F NMR and 62% isolated yield) (for details, see the ESI†). With the viable reaction conditions in hand, we examined the substrate scope of this process (Scheme 2A). Generally, the reaction of silylalkene 5a with a series of unactivated difluoroalkyl iodides (ICF2–alkyl) 1 furnished the corresponding products 3 efficiently. The chain length of 1 did not affect the reaction efficiency. Substrates 1 bearing a linear aliphatic chain from one to five carbons underwent the coupling smoothly (3a–3k). The reaction exhibited good functional group tolerance. A variety of important functional groups, such as ester, silyl ether, arylchloride, and nitrile, were compatible with the reaction conditions (3a–3g, 3i, 3j, and 3l). Even alkylchloride- and sulphonate-containing substrates (3h and 3k) were competent coupling partners, providing good opportunities for downstream derivatization. The reaction was not restricted to difluoroalkyl iodides, as unactivated difluoroalkyl bromides were also suitable substrates (3i–3l), with 3.5 equiv. of alkene and 1.8 equiv. of 6 needed. Replacing silylalkene 5a with siylalkene 5b led to comparable yields (3d, 3g, and 3j), suggesting that 5a and 5b possess similar reactivity. Notably, the steric difluoroalkyl iodide substituted with a cyclohexyl group was also applicable to the reaction (3m). In addition to XCF2–alkyl (X = I, Br), the reaction of 5a with functionalized difluoroalkyl bromides proceeded smoothly (3n–3r). Bromodifluoroacetate and bromodifluoroacetamide showed higher reactivity than those of XCF2–alkyl, while gem-difluoroproparyl bromides and heteroaryl substituted difluoromethyl bromide provided comparable yields. The reaction is readily scalable, as demonstrated by the gram-scale synthesis of 3d and 3g without the erosion of the reaction efficiency. Chlorodifluoroacetate was also examined, but poor yield (19%) was obtained by reaction with 5a and 2a.
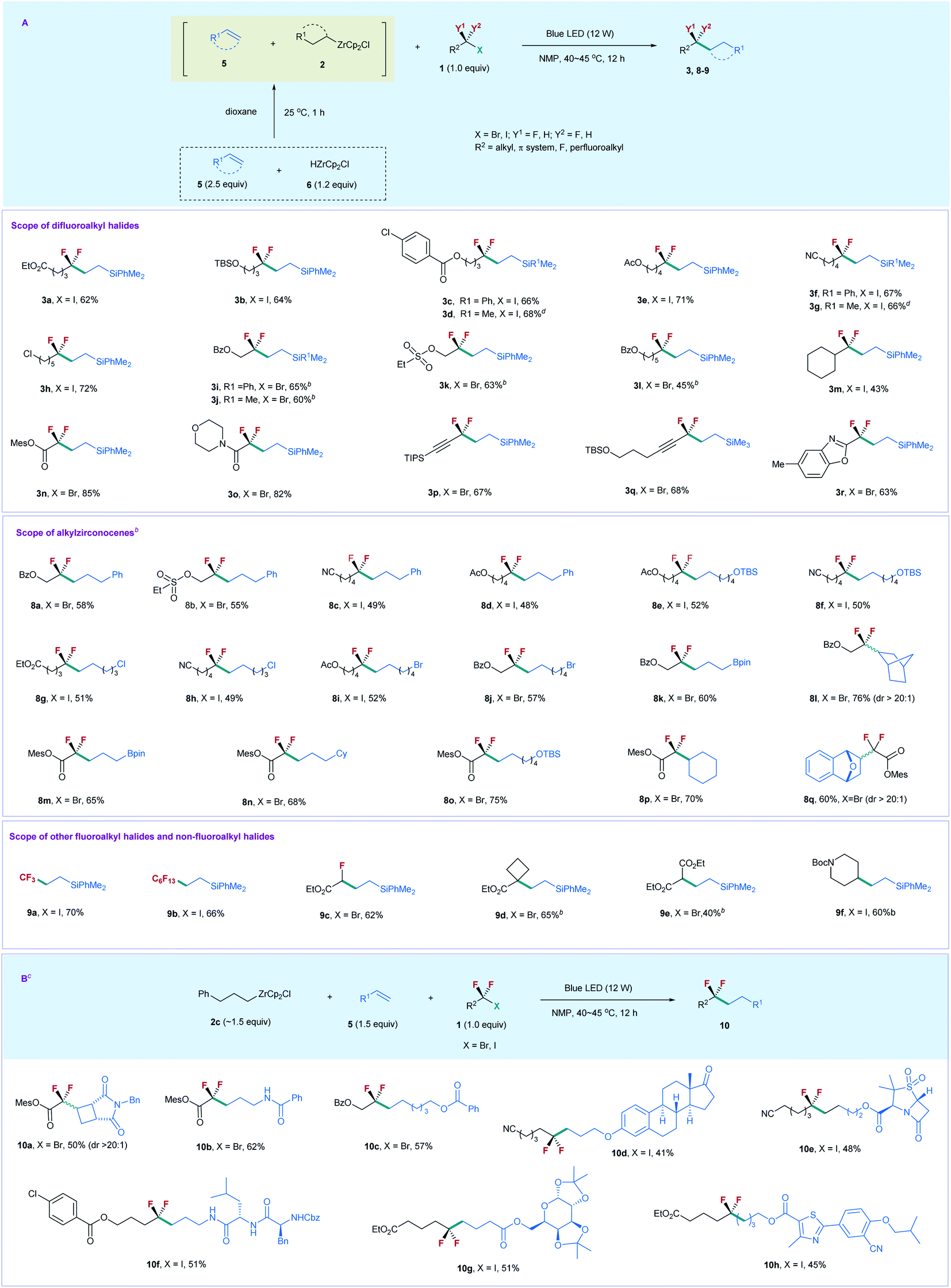 | ||
| Scheme 2 (A) lkylzirconocene promoted fluoroalkylation of alkenes with (fluoro)alkyl halides under the irradiation of blue light. (B) Alkylzirconocene 2c promoted fluoroalkylation of alkenes with difluoroalkyl halides under irradiation of blue light. a Reaction conditions: 1 (0.3 mmol, 1.0 equiv.), 5 (2.5 equiv.), 6 (1.2 equiv.), and NMP (3 mL). b5 (3.5 equiv.) and 6 (1.8 equiv.) were used. c2c (∼1.5 equiv.), 5 (1.5 equiv.), and 6 (2.0 equiv.), and NMP (3 mL) were used. d Gram scale synthesis. | ||
We next examined the substrate scope of alkenes 5. The reaction of XCF2–alkyl (X = I, Br) 1 with compounds 5 bearing a different chain length furnished the corresponding products smoothly (8a–8k). However, styrene was not applicable to the reaction. The successful production of 8i and 8j with alkyl bromide intact demonstrates that the alkyl bromides are less reactive than the difluoroalkyl iodides and bromides, due to the strong electron-withdrawing effect of the CF2 group that enables the difluoroalkyl iodides and bromides to relatively more easily accept an electron than their nonfluorinated counterparts via a SET pathway. Most importantly, a boronate-containing substrate was also applicable to the reaction (8k and 8m). Since boronate is a useful synthetic handle in organic synthesis, the resulting boronate-containing products should serve as a versatile building block for organic synthesis. Notably, the cyclic alkene did not affect the reaction efficiency, and norbornene provided compound 8l in 76% yield. In addition, the reaction of difluoroacetate bromide with alkenes, including linear and cyclic alkenes, proceeded smoothly, and afforded the corresponding products 8m–8q with high efficiency. The reaction can also be extended to trifluoromethyl, perfluoroalkyl, and monofluoroacetyl iodides and bromides, with moderate to good yields obtained (9a–9c). Remarkably, nonfluorinated alkyl halides were also applicable to the reaction. Tertiary alkyl bromide bearing an ester group (9d) and 2-bromomalonate (9e) underwent the coupling efficiently; even Boc-protected 4-iodopiperidine afforded 9f in good yield, thus demonstrating the generality and advantage of this catalyst free, visible light induced process.
Given that the alkylzirconocene is essential in promoting the reaction, but is not the substrate for generating the desired product, we envisioned that alkylzirconocene may functionalize an initiator in the reaction. In this way, we can use a simple alkylzirconocene in combination with different alkenes and fluoroalkyl halides to construct alkylCF2–alkyl bonds. Compared with the above synthetic procedure, this alternative process does not need the preparation of different alkylzirconocenes and enables fluoroalkylation of a variety of alkenes bearing different functional groups that are sensitive to HZrCp2Cl, thus expanding the substrate scope of the current protocol. Accordingly, we prepared alkylzirconocene 2c by reaction of allylbenzene with HZrCp2Cl. As shown in Scheme 2B, the treatment of 2c with bromodifluoroacetate and the cyclobutene derivative under the irradiation of blue light afforded product 10a in 50% yield. Since a four-membered ring and difluoroacetyl are important structural motifs in medicinal chemistry,21 compound 10a would have applications in the synthesis of biologically active molecules. The terminal alkene bearing an amide proton was also applicable to the reaction, providing 10b in good yield. In addition to bromodifluoroacetate, unactivated difluoroalkyl bromides and iodides were also competent coupling partners to react with a variety of complex molecule derived alkenes (10c–10h). Estone- and sulbactam-containing alkenes underwent the difluoroalkylation reaction smoothly (10d and 10e). Peptide and carbohydrate derivatives were also suitable substrates (10f and 10g); even febuxostat bearing a heteroarene was also amenable to the reaction (10h). Thus, the current protocol represents a facile route for applications in medicinal chemistry.
To demonstrate the synthetic utility of this method, transformations of the resulting difluoroalkylated compounds were conducted (Scheme 3). The silyl group on compounds 3 can be readily converted into a hydroxyl. As shown in Scheme 3A, the treatment of compounds 3e, 3i, and 3m with TBAF, followed by KF and H2O2, afforded alcohols 11d, 11h, and 11l with high efficiency.22 Given the important applications of alcohols in organic synthesis and the unique properties of the CF2 moiety, these CF2-containing alcohols offer good opportunities in the synthesis of bioactive molecules. The resulting difluoroalkylated products can also serve as a useful building block for organic synthesis. As shown in Scheme 3B, the gram-scale synthesis of compound 3p under standard reaction conditions proceeded smoothly. The selective deprotection of 3p provided terminal alkyne 12 in 85% yield. The transformation of its carbon–carbon triple bond afforded α,β-unsaturated ketone 13 efficiently.23 The reaction of 13 with N–Me indole in the presence of BF3·Et2O afforded 14 in 70% yield, which was subsequently converted into alcohol 15 in 56% yield.
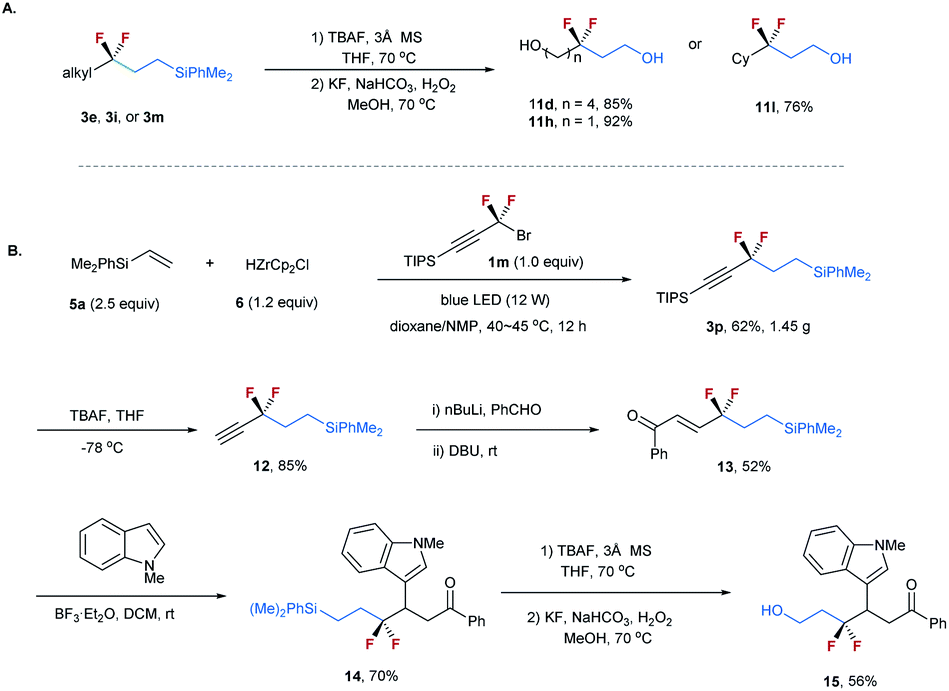 | ||
| Scheme 3 (A) Transformations of difluoroalkylated compounds 3e, 3i and 3m. (B) Gram-scale synthesis of 3p and its transformations. | ||
To gain mechanistic insight into the reaction, we conducted several radical clock experiments with α-cyclopropylstyrene 16 as a probe (Fig. 2A).24 When compound 16 was added to the reaction mixture of difluoroalkyl bromide 1g and alkylzirconocene 2b, two ring expanded products 17 (8% yield) and 18 (13% yield) were obtained under standard reaction conditions (Fig. 2A(a)). The treatment of 2b with 16 under the irradiation of blue light also provided 17 in 15% yield, but no 17 was observed in the absence of blue light (Fig. 2A(b)). In addition, no 18 was provided when 1a was treated with 16 under the irradiation of blue light (Fig. 2A(c)). These results suggest that visible light can induce alkylzirconocene 2b to generate an alkyl radical; in the meanwhile, 2b can also induce difluoroalkyl bromide to generate a difluoroalkyl radical under the irradiation of blue light. Because an alkyl radical (I) can be generated by photolysis of alkylzirconocene under the irradiation of blue light, we proposed that a Zr(III) species [ZrCp2Cl] was formed simultaneously during this process, as illustrated in Fig. 2B.25 The Zr(III) subsequently reacted with fluoroalkyl halide to produce a fluoroalkyl radical via a SET pathway. We also conducted light–dark experiments under standard reaction conditions (see the ESI†), and found that no reaction occurred in the dark, suggesting the essential role of blue light in promoting the reaction. This result demonstrates that a radical chain mechanism is unlikely involved in the reaction. After the fluoroalkyl radical was generated, we proposed that it directly reacted with the alkene to generate a new alkyl radical (II), which subsequently abstracted a proton from the solvent to produce the final product (Fig. 2B).26 As for the radical (I) generated from alkylzirconocene, it can also be quenched by the solvent or undergo a homo-coupling reaction. This deduction was supported by the GC-MS analysis of the reaction, in which homo-coupling product III and a large amount of alkane IV were observed (for details, see Fig. S2†).
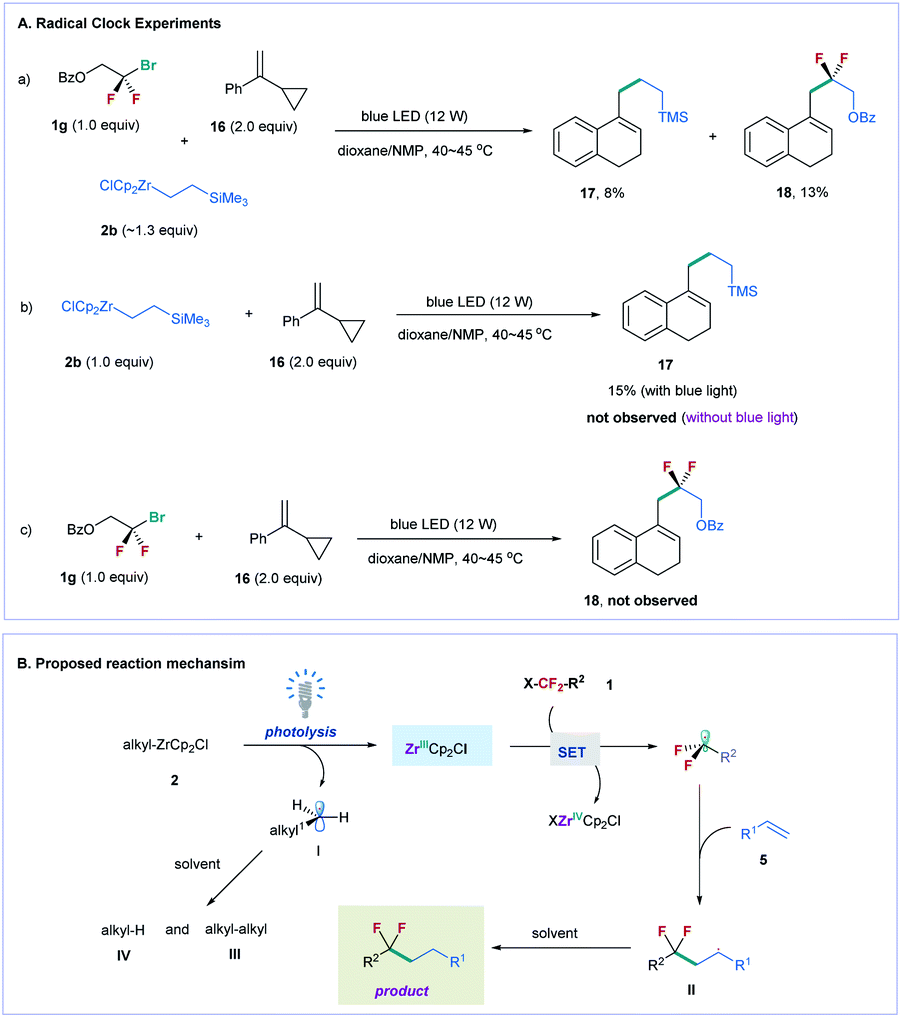 | ||
| Fig. 2 Mechanistic studies and proposed reaction mechanism. (A) Radical clock experiments. (B) Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a general and catalyst-free method for the fluoroalkylation of alkenes promoted by photolysis of alkylzirconocenes. The reaction exhibits high functional group tolerance and broad substrate scope. A wide range of silyl- and alkyl-alkenes as well as fluoroalkyl halides, including difluoroalkyl, trifluoromethyl, perfluoroalkyl, and monofluoroalkyl bromides and iodides, were suitable substrates. In particular, the adaptability of the readily available unactivated difluoroalkyl halides and aliphatic alkenes to the reaction paves a new way for the construction of alkylCF2–alkyl bonds. The reaction can also be applied to nonfluorinated alkyl halides, thus demonstrating the generality of this protocol further. Furthermore, the use of simple alkylzirconocene, instead of generating different alkylzirconocenes between HZrCp2Cl and a series of alkenes, significantly expands the substrate scope, including a variety of complex molecules that are sensitive to HZrCp2Cl. The synthetic utility of this protocol has also been demonstrated by the transformations of difluoroalkylated compounds, providing good opportunities in medicinal chemistry. Preliminary mechanistic studies reveal that a SET pathway induced by a Zr(III) species is involved in the reaction, in which the Zr(III) species is generated by the photolysis of alkylzirconocenes with blue light. This intriguing pathway may prompt great interest in using the photolysis of organozirconium for organic synthesis.Data availability
The authors declare that all the data supporting the findings of this study are available within the paper and its ESI. For characterization data and all NMR spectra for all new compounds see ESI.†Author contributions
X. Z. and X. R. conceived and designed the experiments. X. Z. directed the project. X. R. performed the experiments and mechanistic studies. X. G. and Q.-Q. M. prepared some starting materials and performed some reactions for the preparation of compounds 3 and 8–9. X. Z. wrote the paper. S. Z. revised the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (21931013 and 21790362), the Science and Technology Committee of Shanghai Municipality (21XD1404400 and 21ZR1476600), and Zhengzhou University.Notes and references
- (a) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (c) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed; (d) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (e) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (b) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed; (c) Y. Wang, R. Callejo, A. M. Z. Slawin and D. O'Hagan, Beilstein J. Org. Chem., 2014, 10, 18–25 CrossRef PubMed.
- N. M. F. S. A. Cerqueira, P. A. Fernandes and M. J. Ramos, Chem.–Eur. J., 2007, 13, 8507–8515 CrossRef CAS PubMed.
- J.-P. Bégué and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992–1012 CrossRef.
- S. J. Teague, Drug Discovery Today, 2011, 16, 398–411 CrossRef PubMed.
- (a) F. Zhang, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef PubMed; (b) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2015, 21, 12836–12865 CrossRef CAS PubMed; (c) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS PubMed; (d) B. Chen and D. Vicic, Top. Organomet. Chem., 2014, 52, 113 CrossRef; (e) H. Uno, K. Kawai, M. Shiro and N. Shibata, ACS Catal., 2020, 10, 14117–14126 CrossRef CAS; (f) Y. Sumii, T. Nagasaka, J. Wang, H. Uno and N. Shibata, J. Org. Chem., 2020, 85, 15699–15707 CrossRef CAS PubMed.
- (a) T. Taguchi, O. Kitagawa, T. Morikawa, T. Nishiwaki, H. Uehara, H. Endo and Y. Kobayashi, Tetrahedron Lett., 1986, 27, 6103–6106 CrossRef CAS; (b) K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, Org. Lett., 2011, 13, 5560–5563 CrossRef CAS PubMed; (c) Z. Feng, F. Chen and X. Zhang, Org. Lett., 2012, 14, 1938–1941 CrossRef CAS PubMed; (d) Q.-Q. Min, Z. Yin, Z. Feng, W.-H. Guo and X. Zhang, J. Am. Chem. Soc., 2014, 136, 1230–1233 CrossRef CAS PubMed; (e) Z. Feng, Q.-Q. Min, Y.-L. Xiao, B. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1669–1673 CrossRef CAS PubMed; (f) S. Ge, S. I. Arlow, M. G. Mormino and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 14401–14404 CrossRef CAS PubMed.
- W. J. Middleton, J. Org. Chem., 1975, 40, 574–578 CrossRef CAS.
- (a) D. J. Burton and Z.-Y. Yang, in Chemistry of Organic Fluorine Compounds II: A Critical Review, ed. M. Hudlický and A. E. Pavlath, American Chemical Society, ACS Monograph 187, Washington, DC, 1995, p. 684 Search PubMed; (b) Y. Shen and M. Qi, J. Fluorine Chem., 1994, 67, 229–232 CrossRef CAS; (c) T. Taguchi, O. Kitagawa, Y. Suda, S. Ohkawa, A. Hashimoto, Y. Iitaka and Y. Kobayashi, Tetrahedron Lett., 1988, 29, 5291–5294 CrossRef CAS; (d) K. Iseki, Y. Kuroki, D. Asada and Y. Kobayashi, Tetrahedron Lett., 1997, 38, 1447–1448 CrossRef CAS; (e) W. Kashikura, K. Mori and T. Akiyama, Org. Lett., 2011, 13, 1860–1863 CrossRef CAS PubMed.
- C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- (a) D. V. Sevenard, P. Kirsch, G.-V. Roschenthaler, V. N. Movchun and A. A. Kolomeitsev, Synlett, 2001, 3, 379–381 CrossRef; (b) W. Tyrra, D. Naumann, S. Quadt, S. Buslei, Y. L. Yagupolskii and M. M. Kremlev, J. Fluorine Chem., 2007, 128, 813–817 CrossRef CAS; (c) G. K. S. Prakash, J. Hu, Y. Wang and G. A. Olah, Angew. Chem., Int. Ed., 2004, 43, 5203–5206 CrossRef CAS PubMed; (d) V. Petrik and D. Cahard, Tetrahedron Lett., 2007, 48, 3327–3330 CrossRef CAS.
- (a) A. A. Zemtsov, N. S. Kondratyev, V. V. Levin, M. I. Struchkova and A. D. Dilman, J. Org. Chem., 2014, 79, 818–822 CrossRef CAS PubMed; (b) L. An, C. Xu and X. Zhang, Nat. Commun., 2017, 8, 1460–1468 CrossRef PubMed; (c) L. An, F.-F. Tong, S. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 11884–11892 CrossRef CAS PubMed.
- (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed; (b) C. Yu, N. Iqbal, S. Park and E. J. Cho, Chem. Commun., 2014, 50, 12884–12887 RSC; (c) V. I. Supranovich, V. V. Levin, M. I. Struchkova, J. Hu and A. D. Dilman, Beilstein J. Org. Chem., 2018, 14, 1637–1641 CrossRef CAS PubMed.
- (a) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909–9913 CrossRef CAS PubMed; (b) Y.-L. Xiao, Q.-Q. Min, C. Xu, R.-W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5837–5841 CrossRef CAS PubMed.
- For the preparation of unactivated difluoroalkyl iodides, see: V. V. Levin, A. A. Zemtsov, M. I. Struchkova and A. D. Dilman, Org. Lett., 2013, 15, 917–919 CrossRef CAS PubMed.
- (a) B. Kautzner, P. C. Wailes and H. Weigold, J. Chem. Soc. D, 1969, 1105a RSC; (b) D. W. Hart and J. Schwartz, J. Am. Chem. Soc., 1974, 96, 8115–8116 CrossRef CAS; (c) E. I. Negishi and T. Takahashi, Acc. Chem. Res., 1994, 27, 124–130 CrossRef CAS; (d) J. Barluenga, F. Rodríguez, L. Álvarez-Rodrigo and F. J. Fañanás, Chem. Soc. Rev., 2005, 34, 762–768 RSC; (e) X. Yan and C. Xi, Coord. Chem. Rev., 2017, 350, 275–284 CrossRef CAS.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- J. I. Bardagi, V. A. Vaillard and R. A. Rossi, The SRN1 Reaction, Encycloedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, 2012 Search PubMed.
- M. Moss, X. Han and J. M. Ready, Angew. Chem., Int. Ed., 2016, 55, 10017–10021 CrossRef CAS PubMed.
- P. J. Chirik, M. W. Day, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 1999, 121, 10308–10317 CrossRef CAS.
- (a) V. M. Dembitsky, Phytomedicine, 2014, 21, 1559–1581 CrossRef CAS PubMed; (b) V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CAS.
- B. Nelson, W. Hiller, A. Pollex and M. Hiersemann, Org. Lett., 2011, 13, 4438–4441 CrossRef CAS PubMed.
- A. N. E. Dine, A. Khalaf, D. Grée, O. Tasseau, F. Fares, N. Jaber, P. Lesot, A. Hachem and R. Grée, Beilstein J. Org. Chem., 2013, 9, 1943–1948 CrossRef PubMed.
- J. E. Baldwin, Chem. Rev., 2003, 103, 1197–1212 CrossRef CAS PubMed.
- (a) A. Hudson, M. F. Lappert and R. Pichon, J. Chem. Soc., Chem. Commun., 1983, 7, 374–376 RSC; (b) C. Yang, C. Jiang and X. Qi, Synthesis, 2021, 53, 1061–1076 CrossRef CAS.
- GC-MS analysis of the reaction showed that trace amount of side product was formed from the reaction of II with alkene 5..
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and analytical data for all new compounds. See DOI: 10.1039/d1sc07061d |
| This journal is © The Royal Society of Chemistry 2022 |