
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the C55-P targeting lipopeptide antibiotics revealed by structure–activity studies and high-resolution crystal structures†
Thomas M.
Wood‡
 ab,
Matthieu R.
Zeronian‡
c,
Ned
Buijs
a,
Kristine
Bertheussen
a,
Hanieh K.
Abedian
a,
Aidan V.
Johnson
a,
Nicholas M.
Pearce
c,
Martin
Lutz
c,
Johan
Kemmink
d,
Tjalling
Seirsma
e,
Leendert W.
Hamoen
e,
Bert J. C.
Janssen
*c and
Nathaniel I.
Martin
*a
ab,
Matthieu R.
Zeronian‡
c,
Ned
Buijs
a,
Kristine
Bertheussen
a,
Hanieh K.
Abedian
a,
Aidan V.
Johnson
a,
Nicholas M.
Pearce
c,
Martin
Lutz
c,
Johan
Kemmink
d,
Tjalling
Seirsma
e,
Leendert W.
Hamoen
e,
Bert J. C.
Janssen
*c and
Nathaniel I.
Martin
*a
aBiological Chemistry Group, Institute of Biology Leiden, Leiden University, Sylviusweg 72, 2333 BE Leiden, The Netherlands. E-mail: n.i.martin@biology.leidenuniv.nl
bDepartment of Chemical Biology & Drug Discovery, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
cStructural Biochemistry, Bijvoet Centre for Biomolecular Research, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands. E-mail: b.j.c.janssen@uu.nl
dFaculty of Science and Engineering, University of Groningen, 9747 AG Groningen, The Netherlands
eBacterial Cell Biology and Physiology Group, Swammerdam Institute for Life Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
First published on 21st February 2022
Abstract
The continued rise of antibiotic resistance is a global concern that threatens to undermine many aspects of modern medical practice. Key to addressing this threat is the discovery and development of new antibiotics that operate by unexploited modes of action. The so-called calcium-dependent lipopeptide antibiotics (CDAs) are an important emerging class of natural products that provides a source of new antibiotic agents rich in structural and mechanistic diversity. Notable in this regard is the subset of CDAs comprising the laspartomycins and amphomycins/friulimicins that specifically target the bacterial cell wall precursor undecaprenyl phosphate (C55-P). In this study we describe the design and synthesis of new C55-P-targeting CDAs with structural features drawn from both the laspartomycin and amphomycin/friulimicin classes. Assessment of these lipopeptides revealed previously unknown and surprisingly subtle structural features that are required for antibacterial activity. High-resolution crystal structures further indicate that the amphomycin/friulimicin-like lipopeptides adopt a unique crystal packing that governs their interaction with C55-P and provides an explanation for their antibacterial effect. In addition, live-cell microscopy studies provide further insights into the biological activity of the C55-P targeting CDAs highlighting their unique mechanism of action relative to the clinically used CDA daptomycin.
Introduction
The rapid emergence of multidrug resistant bacteria presents a growing threat to human health and is considered a top priority of the World Health Organization.1 The most effective way to address this threat is to identify antibiotics that operate by unique, unexploited mechanisms.2 While the so-called “golden age” of antibiotic discovery spanning the 1940s–1960s delivered a plethora of life-saving drugs, in the subsequent 60 years only two new antibiotic classes operating with truly novel modes of action have been introduced.3 Among these is the macrocyclic lipopeptide daptomycin, the preeminent calcium-dependent antibiotic (CDA), which entered the clinic as a first-in-class agent in 2004.4,5 Despite its clinical success, daptomycin's precise mechanism of action remains a topic of continued investigation.6–9 By comparison, the mode of action of other structurally similar CDAs like laspartomycin C, friulimicin B, and amphomycin (Fig. 1) are more fully understood.10–13 These CDAs specifically target the unique bacterial phospholipid undecaprenyl phosphate (C55-P). In bacteria, C55-P plays an essential role as a lipid carrier in cell wall biosynthesis.14 Specifically, on the inner surface of the bacterial membrane, the enzyme MraY couples C55-P with UDP-MurNAc-pentapeptide to form lipid I. The membrane anchored lipid I is next converted to lipid II by action of MurG. Lipid II is then flipped to the periplasm where the disaccharide-pentapeptide motif is incorporated into the growing peptidoglycan layer and the phospholipid carrier is released, first as the pyrophosphate (C55-PP) which is subsequently converted to C55-P by action of the phosphatase UppP/BacA.15,16 For another cycle to begin, C55-P must first be flipped back to the cytoplasm where it can again be used as a membrane anchor for peptidoglycan synthesis. Compounds capable of binding to and sequestering C55-P on the outer surface of the bacterial membrane therefore have the capacity to function as antibacterial agents. Notably, while C55-P plays a central role in peptidoglycan synthesis, there are no clinically approved antibiotics that operate by directly binding C55-P.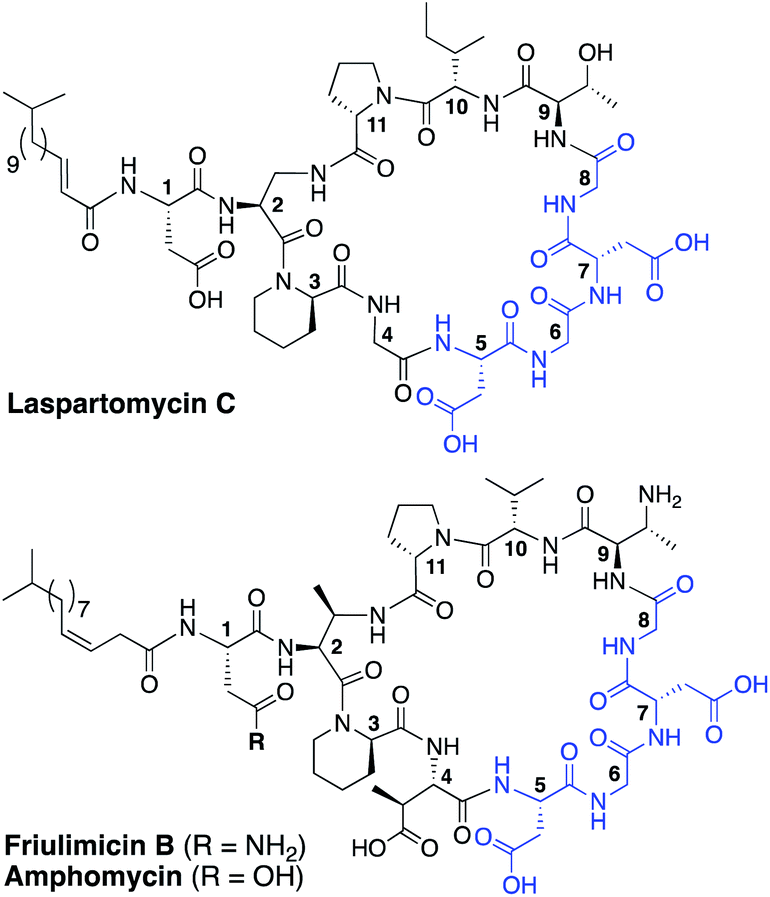 | ||
| Fig. 1 Structures of laspartomycin C, friulimicin B, and amphomycin. Highlighted in blue is the Asp–X–Asp–Gly calcium binding motif conserved throughout the CDAs. Laspartomycin C differs from friulimicin B and amphomycin at positions 2, 4, 9, and 10. | ||
To date, more than forty structurally distinct CDAs have been reported with varying antibacterial activities and mechanisms of action.17 A number of structural features are common among the CDAs, including specifically positioned D-amino acids and the highly conserved Asp–X–Asp–Gly motif, essential for binding of calcium (Fig. 1).18 Apart from the recently reported malacidins,19,20 all CDAs contain 10 amino acids in their macrocycle. An interesting sub-class of CDAs are those wherein the peptide macrocycle is closed by a lactam linkage, a group comprised of laspartomycin C, friulimicin B, and amphomycin (Fig. 1). Considering their structural similarities, it is perhaps not surprising that all three share the same target (C55-P) as part of their antibacterial mechanisms. There are, however, a number of subtle structural features that distinguish the friulimicins/amphomycins from laspartomycin C. For example, the length and geometry of the N-terminal lipid in friulimicin B and amphomycin differs slightly from that found in laspartomycin C. In addition, while both laspartomycin C and amphomycin contain an Asp residue at position 1, in friulimicin B this is Asn. A more notable difference is observed within the peptide macrocycles of these CDAs. Laspartomycin C contains diamino-propionic acid (Dap), Gly, D-allo-Thr, and Ile at positions 2, 4, 9, and 10 respectively whereas in the friulimicin/amphomycin class the same position are filled by (2S,3R)-diamino-butyric acid (Dab), (2S,3S)-3-methyl-Asp, (2R,3R)-diamino-butyric acid (D-Dab), and Val respectively (Fig. 1).
Previous findings from our group revealed that for laspartomycin C the side chains of residues 4, 9, and 10 are not directly involved in coordinating the C55-P phosphate head group or the bridging calcium ions.12 This knowledge, coupled with the structural differences between the laspartomycin and friulimicin/amphomycin class at these positions, prompted us to investigate the impact of introducing structural features present in friulimicin/amphomycin into laspartomycin C. Specifically, we here describe the impact of systematically introducing amino acids containing carboxylate and amino side chains into the laspartomycin macrocycle at positions 4 and 9 respectively, as found in the friulimicin/amphomycin class. Notably, the calcium-dependent activity of these new compounds was found to be highly influenced by a previously unknown steric factor associated with the side chain of the residue at position 10. An explanation for these observations was subsequently provided by high-resolution X-ray crystal structures of the new lipopeptide variants in complexation with C10-P and Ca2+. These findings provide new insights into the mechanism of action of the friulimicin/amphomycin class of CDAs and the subtle differences with that of the laspartomycin family.
Results and discussion
To evaluate the impact of introducing amino acids specific to the friulimicins/amphomycin class into laspartomycin C we applied a flexible synthetic route wherein the linear peptide precursor, including the N-terminal lipid, was first assembled on solid support using the acid sensitive 2-chlorotrityl resin (Scheme 1).11,21,22 Notably, Gly residues at positions 6 and 8 were incorporated using the corresponding Fmoc-DMB-Gly building block to avoid aspartamide formation. On resin removal of the Alloc group on the Dap2 side chain was followed by mild acid cleavage to yield the protected linear peptide intermediate. Formation of the macrocycle was achieved by treatment with BOP/DIPEA under high-dilution conditions, after which global deprotection and RP-HPLC purification provided lipopeptides 1–9. The first structural variation explored involved the swapping of the exocyclic Asp1 found in laspartomycin C for Asn1 as in friulimicin B. This analogue (2) showed no appreciable difference in minimum inhibitory concentration (MIC) when compared to laspartomycin C (Table 1). This is not surprising as this exocyclic amino acid is also the only difference between the amphomycin and friulimicin class of CDAs which are reported to have similar activities.11,13 We next focused our attention to the differing amino acids contained within the peptide macrocycles of the laspartomycin and friulimicins/amphomycin classes. To this end lipopeptides 3–5 were prepared to assess the contribution of the acidic and basic residues unique to positions 4 and 9 in the amphomycin/friulimicin class. Interestingly, these new variants bearing either Asp4 or D-Dap9, or both substitutions, demonstrated severely reduced antibacterial activities relative to laspartomycin C and friulimicin B. Lipopeptide 6 was next synthesized to probe the role of Val10 present in the amphomycin/friulimicin class compared to the slightly bulkier Ile reside found at the same position in laspartomycin C. Somewhat surprisingly, the subtle substitution of Val for Ile at position 10 led to a significant enhancement in the antibacterial activity of lipopeptide 6 relative to compound 5. In the presence of 10 mM Ca2+, 6 was found to exhibit an MIC of 1 μg mL−1 against MRSA, an activity on par/slightly better than that measured for both laspartomycin C and friulimicin B. Given the increased activity observed for 6, analogue 7, bearing Asn at position 1, was also prepared and found to also demonstrate a similarly enhanced antibacterial activity.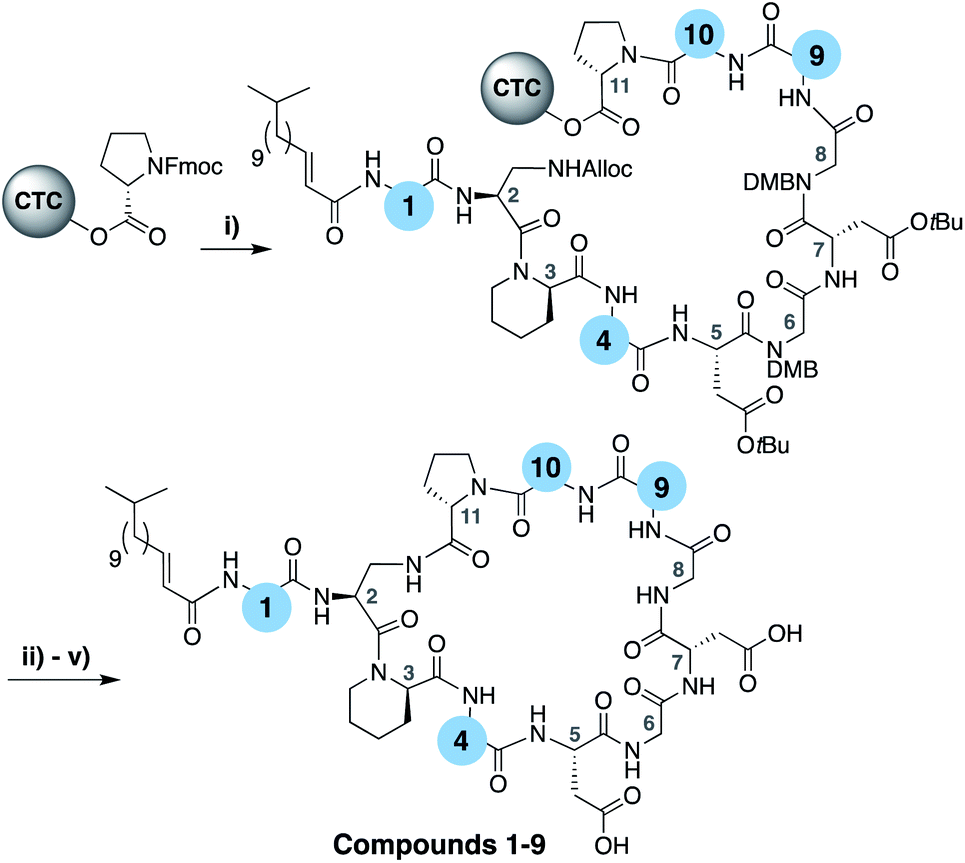 | ||
| Scheme 1 (i) Fmoc SPPS; (ii) Pd(PPh3)4, PhSiH3, CH2Cl2, 1 h; (iii) HFIP, CH2Cl2, 1 h; (v) BOP, DIPEA, CH2Cl2, 16 h; (v) TFA, TIS, H2O, 1 h. | ||
| Compound | AA1 | AA4 | AA9 | AA10 | MICa |
|---|---|---|---|---|---|
| a Minimum inhibitory concentration reported in μg mL−1 against MRSA USA 300 at a Ca2+ concentration of 10 mM. All compounds tested in triplicate. b Natural product. | |||||
| 1 (LaspC) | L-Asp | Gly | D-allo-Thr | L-Ile | 2 |
| 2 | L-Asn | Gly | D-allo-Thr | L-Ile | 4 |
| 3 | L-Asp | L-Asp | D-allo-Thr | L-Ile | 16 |
| 4 | L-Asp | Gly | D-Dap | L-Ile | 4 |
| 5 | L-Asp | L-Asp | D-Dap | L-Ile | 8 |
| 6 | L-Asp | L-Asp | D-Dap | L-Val | 1 |
| 7 | L-Asn | L-Asp | D-Dap | L-Val | 2 |
| 8 | L-Asp | L-Asp | D-allo-Thr | L-Val | 4 |
| 9 | L-Asp | Gly | D-Dap | L-Val | 8 |
| Friulimicin Bb | L-Asn | MeAsp | D-Dab | L-Val | 1–2 |
Our findings with lipopeptides 6 and 7 indicate that the increased antibacterial activity exhibited by these more friulimicin/amphomycin-like analogues is the product of a combined effect dependent on an acidic side chain at AA4, a basic side chain at AA9, and a slightly less bulky side chain in AA10. This reasoning was further probed by the preparation of analogues 8 and 9 wherein the acidic and basic residues at positions 4 and 9 were independently exchanged for the uncharged Gly and D-allo-Thr residues found in laspartomycin C. The reduced activity measured for these compounds further confirms a role for both the acidic and basic residues at positions 4 and 9, in combination with the optimized sterics of Val10, in achieving full antibacterial effect. The activities of lipopeptides 6 and 7 were further assessed across a range of Ca2+ concentrations and against an expanded panel of Gram-positive pathogens including vancomycin-resistant and daptomycin-resistant isolates further demonstrating their antibacterial effects (ESI Tables S1–S3†).
Mechanistic and crystallographic studies
The activity observed for lipopeptides 6 and 7 led us to investigate the underlying mechanism responsible. To do so we first examined their ability to interfere with bacterial cell wall synthesis. Specifically, an assay was used that detects accumulation of the cell wall precursor UDP-MurNAc pentapeptide in response to cell-wall targeting antibiotics. As the last soluble precursor in the lipid II cycle, UDP-MurNAc pentapeptide serves as a convenient diagnostic for compounds that disrupt cell wall synthesis. When S. aureus cells were treated with laspartomycin C and lipopeptides 6 and 7, a clear accumulation of this precursor was observed (ESI Fig. S1†). Interestingly, no such accumulation of the UDP-MurNAc pentapeptide species is observed for cells treated with daptomycin.To gain molecular level insights into the differences in activity observed for lipopeptide 6 and 7 relative to analogue 5, all three were taken forward for crystal structure determination. In doing so the lipopeptides were incubated with C10-P, a more soluble analogue of C55-P, in buffers containing Ca2+. Under these conditions, 5 and 7 gave crystals of sufficient quality for structural analysis, diffracting to a resolution of 1.04 Å and 1.14 Å, respectively. The structures of the complexes obtained for both 5 and 7 with C10-P and Ca2+ share many similarities with the structure previously reported for the laspartomycin C complex.12 As illustrated in Fig. 2A, the complex itself consists of one lipopeptide molecule, one geranyl phosphate ligand, and two calcium ions which play key roles both in establishing the conformation of the peptide as well as mediating binding of the phosphate head group. Notable interactions observed in the complex include hydrogen bonds formed between the C10-phosphate group and the backbone and side chain amides of Dap2 as well as the backbone amide of Gly8. Each calcium ion also provides an interaction with the phosphate moiety. Of the two calcium ions in the complex, one is more centrally coordinated via multiple interactions with the lipopeptide including four backbone carbonyls (Dap2, Gly6, Gly8, Ile10/Val10) and one aspartic acid side chain (Asp5). The peripheral Ca2+ is bound via interactions with the side chains of Asp1/Asn1 and Asp7 and the N-terminal fatty acid carbonyl group along with one water molecule.
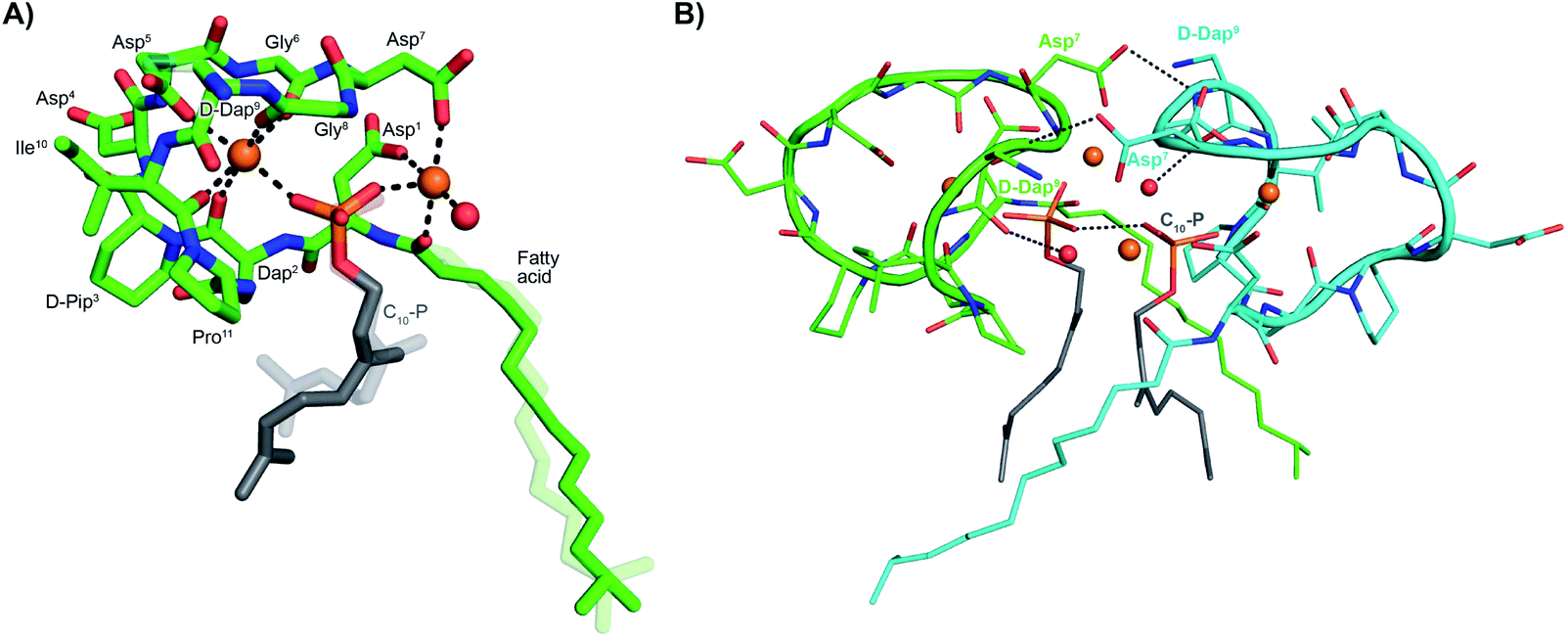 | ||
| Fig. 2 (A) Structure of the ternary complex with lipopeptide 5 (green stick representation), two bound Ca2+ ions (orange spheres), a bound water molecule (red sphere), and the C10-P ligand (lipid in grey). Major and minor conformations of the D-Dap9 side chain, the C10-P lipid and the lipopeptide fatty acid tail are shown in the structure (indicated with dark and light colouring respectively). (B) Lipopeptide 5 adopts a saddle-shaped conformation when complexed with two Ca2+ ions and C10-P that forms a dimer in the crystal. For clarity only major conformations are shown. ESI Fig. S2† presents the same views for lipopeptide 7. | ||
Collectively, these interactions cause the lipopeptides to adopt a saddle-shaped fold wherein the cavity created envelops the C10-P phosphate head group and the two calcium ions. As also observed for laspartomycin C, the complexes formed by both 5 and 7 with C10-P and Ca2+ organize as dimers stabilized by a number of intermolecular interactions. As shown in Fig. 2B, dimerization is largely driven by hydrogen bonding interactions between the D-Dap9 backbone amide of one lipopeptide molecule and the Asp7 side chain carboxylate of the other. Additional indirect hydrogen bonding interactions are mediated by interaction of the D-Dap9 backbone carbonyl, and the water molecules coordinated by the peripheral Ca2+ of the other ternary unit. In this dimer complex, the two C10-P phosphate head groups are fully coordinated and completely sequestered from the solvent. A comparison of the conformation of the peptide backbones and location of the C10-P and Ca2+ in the dimers formed by 5 and 7 with that of laspartomycin C reveals a high degree of similarity (ESI Fig. S3†). Notable, however, was the finding that the differing side chains at positions 4, 9, and 10 in lipopeptides 5 and 7 induce and stabilize a unique, higher-ordered assembly not observed for laspartomycin C.
As noted above, the amphomycin/friulimicin class of lipopeptide antibiotics differs from the laspartomycin class at positions 4, 9, and 10. Lipopeptides 5, 6, and 7 were generated to specifically probe the roles played by the side chains of these different amino acids. The crystal structures obtained with 5 and 7 indeed reveal that the presence of Asp4 and D-Dap9 result in additional inter-dimer interactions not possible for laspartomycin C. Particularly striking was the finding that when coordinated with C10-P and Ca2+, lipopeptides 5 and 7 both formed higher-ordered complexes that are not observed for laspartomycin under similar conditions (Fig. 3A). Specifically, interactions between Asp4 and D-Dap9 in 5 and 7 serve to stabilize this higher-ordered arrangement wherein the side chain carboxylate and backbone carbonyl of one Asp4 residue in one dimer interacts with the amino side chain of a D-Dap9 residue in an adjacent dimer (Fig. 3B). Furthermore, the same Asp4 also interacts with the proximal calcium coordinated by the second dimer further stabilizing this arrangement. Interestingly, the dimer of dimers thus formed is precisely oriented so as to make the same interactions with the Asp4 and D-Dap9 side chains of a third dimer to generate a trimer of dimers. This repeating “trimer of dimers” motif is not observed in the crystal packing formed by laspartomycin C in complex with C10-P and Ca2+ as it lacks the Asp4 and D-Dap9 required to do so. Also different from laspartomycin C is the finding that lipopeptides 5 and 7 form alternating layers in the crystal, with a peptide macrocycle layer inducing a strong packing in cis (within the same layer), sandwiched by a hydrophobic layer constituted of lipids (geranyl phosphate and the peptide N-terminal lipid) and by a hydrophilic layer composed of water molecules, both inducing a weak packing in trans (between adjacent layers) (ESI Fig. S4†).
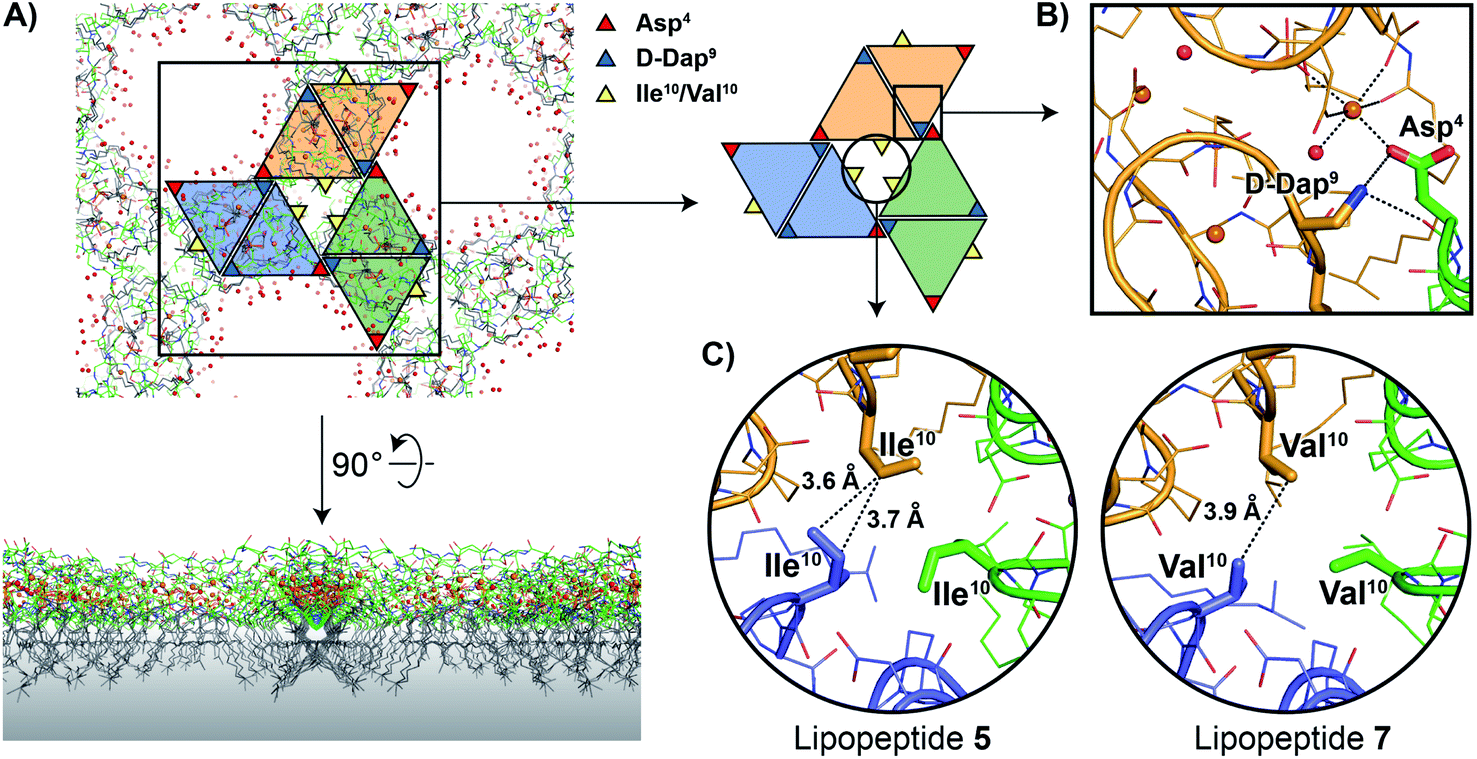 | ||
| Fig. 3 (A) In the crystal packing lipopeptides 5 and 7 adopt higher-ordered structures not observed with laspartomycin C. In this arrangement the lipids of both the lipopeptides and C10-P are oriented in the same direction while the peptide macrocycles interact to form a repeating trimer of dimers motif as indicated by the colored triangles. A proposed orientation of the multimeric assembly in the bacterial membrane (indicated with a grey gradient) is shown. (B) Interactions between the D-Dap9 and Asp4 residues present in lipopeptides 5 and 7, but absent in laspartomycin C, stabilize the trimer of dimers. (C) The presence of a hydrophobic core formed at the center of the trimer of dimers motif suggests that the side chain of Val10 present in the biologically more active lipopeptide 7 more optimally suits the steric requirements of this motif vs. the slightly bulkier Ile10 in lipopeptide 5. For clarity only major conformations are shown. | ||
The higher-ordered trimer of dimers motif formed by both 5 and 7 in complex with C10-P and Ca2+ also points to an explanation for the notable enhancement in the biological activity of lipopeptides 6 and 7 relative to 5. As described above, the peptide macrocycles of 6 and 7 contain a Val residue at position 10 while in lipopeptide 5 the same position is filled by a slightly bulkier Ile residue. This subtle structural difference results in an 8-fold increase in the activity for 6 and 7 relative to 5. Careful inspection of the trimer of dimers formed by both peptides 5 and 7 reveals a hydrophobic pocket at center of the trimer where the side chains of Val10/Ile10 meet (Fig. 3C). This finding suggests that the Val10 side chain in 6 and 7 (and as found naturally in the amphomycin/friulimicin class) allows for optimal packing of the trimer, enhancing the interaction with the C55-P bacterial target, and drives the antibiotic activity observed. By comparison, the slightly bulkier Ile10 side chain in 5 may impinge upon the precise steric requirements of the hydrophobic pocket formed at the trimer interface and in doing so destabilize the interaction with C55-P resulting in reduced antibacterial activity.
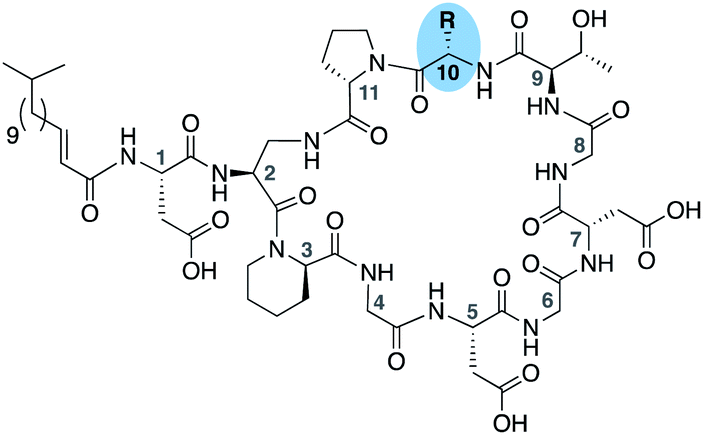
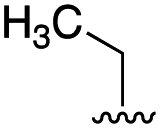
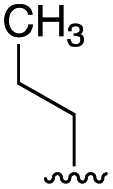
To further probe the role of side chain sterics at position 10, laspartomycin C analogues 10–15 were next prepared wherein Ile10 was systematically replaced by amino acids with smaller or larger side chains (Table 2). Replacement of Ile10 with Gly completely abolished calcium-dependent activity. Replacement by Ala also led to a severe loss of activity but did yield a species that was calcium dependent. The incremental addition of methylene groups achieved by incorporating Abu, Nval, and Val led in turn to a stepwise enhancement of antibacterial activity. In contrast, introduction of Phe at position 10 was found to have a detrimental effect on activity indicating that the side chain sterics at position 10 are indeed finely balanced. Taken together, these findings provide new insight into the mechanism of action of amphomycin/friulimicin class of calcium dependent antibiotics and how they compare to the laspartomycin family.

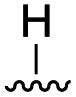



Live cell imaging
To gain additional insights into the impact of these lipopeptide antibiotics on live bacteria, laspartomycin C, lipopeptide 6, and daptomycin were evaluated in a set of comprehensive mode-of-action studies conducted using the model organism Bacillus subtilis and imaged using fluorescence light microscopy.8,23 These studies reveal that laspartomycin C and lipopeptide 6 both interfere with bacterial membrane integrity by delocalizing key membrane proteins and/or interfering with lipid organization in a manner that is distinct from that observed for daptomycin. An extensive bacterial cytological profiling study previously showed that daptomycin causes the clustering of ‘fluid lipids’, i.e. lipids with short, branched or unsaturated fatty acyl chains, into large aggregates, lowering the membrane fluidity outside these aggregates.8 This has a severe effect on the binding of peripheral membrane proteins with essential functions, including the N-acetylglucosamine transferase MurG responsible for the last synthesis step of the peptidoglycan precursor lipid II. To further compare the effect of laspartomycin C with that of daptomycin, we performed bacterial cytological profiling using a broad set of B. subtilis reporter strains expressing GFP-tagged proteins involved in DNA replication (DNA polymerase subunit PolC), transcription (RNA polymerase subunit RpoC), translation (ribosome subunit RpsB), ATP synthesis (F1F0-ATPase subunit AtpA), cell division (FtsZ), cell wall synthesis coordination (MreB), cell division regulation (MinD) and peptidoglycan synthesis (MurG). The reporters MreB, MinD, and MurG are all peripheral membrane proteins. Cells were incubated with the lipopeptide antibiotics at 2× MIC and observed by fluorescent light microscopy after 10 min and 30 min incubation. The reporter strains indicated that neither DNA, RNA and protein synthesis, nor cell division and the localization of F1F0-ATPase AtpA were affected by laspartomycin C (Fig. 4) or lipopeptide 6 (ESI Fig. S5†), findings that are in keeping with those previously observed for daptomycin (ESI Fig. S6†).8 Likewise, the delocalization of MreB by laspartomycin C and lipopeptide 6 is similar to the effect seen with daptomycin (ESI Fig. S7†).8 A notable difference, however, was the finding that the localization of MinD was unaffected by laspartomycin C and lipopeptide 6, whereas this protein rapidly detaches from the membrane when treated with daptomycin (see ESI Fig. S8†). Another clear difference is the delocalization of MurG, which detaches from the cell membrane in the presence of daptomycin, whereas laspartomycin C and lipopeptide 6 appear to dissolve the large MurG clusters so that the protein diffuses throughout the cell membrane (Fig. 4, ESI Fig. S5 and S6†). | ||
| Fig. 4 Bacterial cytological profiling analysis of laspartomycin C. The GFP-tagged marker proteins represents the following cellular activities: DNA polymerization (PolC), RNA polymerization (RpoC), protein synthesis (RpsB), F0F1 ATPase (AtpA), lateral cell wall synthesis regulation (MreB), cell division (FtsZ), cell division regulation (MinD) and peptidoglycan precursor synthesis (MurG). Left panels schematically show the normal localization patterns of the different GFP fusions. Strains were grown in LB medium supplemented with 2 mM CaCl2 at 30 °C. 2× MIC concentration was added (0 min) and samples for microscopy were taken after 10 and 30 min incubation, respectively. Scale bars indicate 2 μm. Lipopeptide 6 also showed a similar bacterial cytological profile (ESI Fig. S5†). | ||
The differences observed for laspartomycin C and lipopeptide 6vs. daptomycin may be explained by the multifaceted mechanism of action attributed to daptomycin. Recent investigations have revealed that in the presence of phosphatidylglycerol, daptomycin can interact with C55-P, C55-PP, and the peptidoglycan precursor lipid II.9 As a result, insertion of daptomycin into the cell membrane not only affects lipid II synthesis, but also causes a dramatic rearrangement of lipids in the cell membrane resulting in the detachment of peripheral membrane proteins, including MinD and MurG.8 Conversely, the binding of laspartomycin C to C55-P is not facilitated by phosphatidylglycerol or any other phospholipids11 and is therefore more likely to specifically interfere with lipid II synthesis and not with the distribution of other phospholipids in the membrane. However, the activity of proteins that rely on access to C55-P including MurG and MreB will still be affected, explaining the dissolution of MurG clusters and the delocalization of MreB observed in our studies. This rationale is also in line with a recent report revealing MreB membrane association to be dependent on the presence of lipid-linked peptidoglycan precursors and that when such precursors are depleted, MreB filaments disassemble, and peptidoglycan synthesis is disrupted.24
Conclusions
In summary, a series of lipopeptides inspired by the laspartomycin and friulimicin/amphomycin class of C55-P targeting antibiotics were synthesized to probe the effects associated with the structural differences found in this unique class of CDAs. The antibacterial activities measured, and the high-resolution crystal structures obtained for these lipopeptides, reveal a previously unknown interplay between the side chains of residues at positions 4, 9, and 10 in the peptide macrocycle. Interestingly, the amino acid side chains present at these positions in the friulimicin/amphomycin class contribute to the formation of higher-order assemblies when in complex with Ca2+ and the bacterial target, an effect not seen for the other well-characterized C55-P binding CDA laspartomycin C. In addition, live cell imaging studies reveal subtle differences in the activities of laspartomycin C and daptomycin. Compared to daptomycin, laspartomycin C and the other C55-P targeting lipopeptides here studied appear to have a more narrowly defined range of cellular targets. Particularly notable is the ability of laspartomycin C to dissolve large clusters of MurG in the cell membrane, an effect not seen in daptomycin. Taken together, our results provide new insights into the mechanisms of action associated with the C55-P-targeting subfamily of CDAs and expand our current understanding of this promising class of lipopeptide antibiotics.Data availability
Coordinates and structure factors of lipopeptides 5 and 7 in complex with Ca2+ and C10-P have been deposited to the Protein Data Bank (PDB) under the accession codes 7AG5 and 7ANY, respectively.Author contributions
TMW, NB, KB, HKA, and AVJ synthesized peptides. TMW, NB, and KB, performed the biological assays. MRZ, NMP, and ML conducted the crystallographic studies. TMW and JK performed the NMR data acquisition and analyses. TS and LWH performed the live cell microscopy studies. BJCJ and NIM designed and led the study. TMW, MRZ, BJCJ and NIM prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support provided by the Netherlands Organization for Scientific Research (NWO PhD grant to T. M. W.) and the European Research Council (ERC consolidator grant to N. I. M., grant agreement no. 725523, and ERC starting grant to B. J. C. J., grant agreement no. 677500). We thank Paolo Innocenti for performing HRMS analyses and Fons Lefeber for assistance in acquiring NMR spectra. Professor Tanja Schneider is kindly thanked for providing an authentic sample of friulimicin B. We thank the staff at beamlines I04-1 and I04 of the Diamond Light Source (Harwell, United Kingdom) for diffraction data collection.Notes and references
- P. Beyer and S. Paulin, Bull. W. H. O., 2020, 98, 151 CrossRef PubMed.
- M. F. Chellat, L. Raguz and R. Riedl, Angew. Chem., Int. Ed., 2016, 55, 6600–6626 CrossRef CAS PubMed.
- M. N. Gwynn, A. Portnoy, S. F. Rittenhouse and D. J. Payne, Ann. N. Y. Acad. Sci., 2010, 1213, 5–19 CrossRef PubMed.
- R. Sauermann, M. Rothenburger, W. Graninger and C. Joukhadar, Pharmacology, 2008, 81, 79–91 CrossRef CAS PubMed.
- R. M. Humphries, S. Pollett and G. Sakoulas, Clin. Microbiol. Rev., 2013, 26, 759–780 CrossRef CAS PubMed.
- R. Moreira, J. Wolfe and S. D. Taylor, Org. Biomol. Chem., 2021, 19, 3144–3153 RSC.
- S. D. Taylor and M. Palmer, Bioorg. Med. Chem., 2016, 24, 6253–6268 CrossRef CAS PubMed.
- A. Muller, M. Wenzel, H. Strahl, F. Grein, T. N. V. Saaki, B. Kohl, T. Siersma, J. E. Bandow, H. G. Sahl, T. Schneider and L. W. Hamoen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7077–E7086 CrossRef PubMed.
- F. Grein, A. Muller, K. M. Scherer, X. Liu, K. C. Ludwig, A. Klockner, M. Strach, H. G. Sahl, U. Kubitscheck and T. Schneider, Nat. Commun., 2020, 11, 1455 CrossRef CAS PubMed.
- T. Schneider, K. Gries, M. Josten, I. Wiedemann, S. Pelzer, H. Labischinski and H. G. Sahl, Antimicrob. Agents Chemother., 2009, 53, 1610–1618 CrossRef CAS PubMed.
- L. H. Kleijn, S. F. Oppedijk, P. t. Hart, R. M. van Harten, L. A. Martin-Visscher, J. Kemmink, E. Breukink and N. I. Martin, J. Med. Chem., 2016, 59, 3569–3574 CrossRef CAS PubMed.
- L. H. J. Kleijn, H. C. Vlieg, T. M. Wood, J. Sastre Torano, B. J. C. Janssen and N. I. Martin, Angew. Chem., Int. Ed., 2017, 56, 16546–16549 CrossRef CAS PubMed.
- A. Diehl, T. M. Wood, S. Gebhard, N. I. Martin and G. Fritz, Antibiotics, 2020, 9, 729 CrossRef CAS PubMed.
- G. Manat, S. Roure, R. Auger, A. Bouhss, H. Barreteau, D. Mengin-Lecreulx and T. Touze, Microb. Drug Resist., 2014, 20, 199–214 CrossRef CAS PubMed.
- M. El Ghachi, N. Howe, C. Y. Huang, V. Olieric, R. Warshamanage, T. Touze, D. Weichert, P. J. Stansfeld, M. Wang, F. Kerff and M. Caffrey, Nat. Commun., 2018, 9, 1078 CrossRef PubMed.
- S. D. Workman, L. J. Worrall and N. C. J. Strynadka, Nat. Commun., 2018, 9, 1159 CrossRef PubMed.
- T. M. Wood and N. I. Martin, MedChemComm, 2019, 10, 634–646 RSC.
- M. Strieker and M. A. Marahiel, ChemBioChem, 2009, 10, 607–616 CrossRef CAS PubMed.
- B. M. Hover, S. H. Kim, M. Katz, Z. Charlop-Powers, J. G. Owen, M. A. Ternei, J. Maniko, A. B. Estrela, H. Molina, S. Park, D. S. Perlin and S. F. Brady, Nat. Microbiol., 2018, 3, 415–422 CrossRef CAS PubMed.
- Z. Sun, Z. Shang, N. Forelli, K. H. L. Po, S. Chen, S. F. Brady and X. Li, Angew. Chem., Int. Ed., 2020, 59, 19868–19872 CrossRef CAS PubMed.
- L. Corcilius, N. T. Elias, J. L. Ochoa, R. G. Linington and R. J. Payne, J. Org. Chem., 2017, 82, 12778–12785 CrossRef CAS PubMed.
- T. M. Wood, K. Bertheussen and N. I. Martin, Org. Biomol. Chem., 2020, 18, 514–517 RSC.
- M. Wenzel, M. P. Dekker, B. Wang, M. J. Burggraaf, W. Bitter, J. R. T. van Weering and L. W. Hamoen, Commun. Biol., 2021, 4, 306 CrossRef CAS PubMed.
- K. Schirner, Y. J. Eun, M. Dion, Y. Luo, J. D. Helmann, E. C. Garner and S. Walker, Nat. Chem. Biol., 2015, 11, 38–45 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and analytical data for all new compounds. Figures and tables for: 2D-NMR spectra, analytical RP-HPLC traces, HRMS data, antibacterial assays, UDP-MurNAc-pentapeptide accumulation assays, crystallographic studies, and bacterial cytological profiling. See DOI: 10.1039/d1sc07190d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |