
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring a new class of singlet fission fluorene derivatives with high-energy triplets†
Letizia
Mencaroni
 ,
Benedetta
Carlotti
*,
Fausto
Elisei
,
Assunta
Marrocchi
and
Anna
Spalletti
,
Benedetta
Carlotti
*,
Fausto
Elisei
,
Assunta
Marrocchi
and
Anna
Spalletti
Department of Chemistry Biology and Biotechnology and CEMIN, University of Perugia, via elce di sotto n. 8, 06123, Perugia, Italy. E-mail: benedetta.carlotti@unipg.it
First published on 2nd February 2022
Abstract
In this study, we report strong experimental evidence for singlet fission (SF) in a new class of fluorene-based molecules, exhibiting two-branched donor–acceptor structures. The time-resolved spectroscopic results disclose ultrafast formation of a double triplet state (occurring in few picoseconds) and efficient triplet exciton separation (up to 145% triplet yield). The solvent polarity effect and the role of intramolecular charge transfer (ICT) on the SF mechanism have been thoroughly investigated with several advanced spectroscopies. We found that a stronger push–pull character favors SF, as long as the ICT does not act as a trap by opening a competitive pathway. Within the context of other widely-known SF chromophores, the unconventional property of generating high-energy triplet excitons (ca. 2 eV) via SF makes these materials outstanding candidates as photosensitizers for photovoltaic devices.
Introduction
Singlet fission (SF) is an intriguing process in which a high-energy singlet exciton down-converts into two independent triplet excitons, initially coupled into an overall zero spin state.1–3 SF thus enables multiple exciton generation by absorption of just a single photon. By integrating SF materials into photovoltaic devices, the Shockley–Queisser limit can be pushed from 32% to approximately 45% improving the solar cell efficiency.4–6 For practical implementation in high-performance devices, SF systems should show high triplet energies for use as sensitizers to lower band gap semiconductors or for charge extraction to acceptors.7,8 Although this phenomenon has become a hot topic in chemical research, the families of SF capable materials remain relatively limited9 and several mechanistic details of the SF process remain unclear.10–12 Notably, the influence of intramolecular charge transfer (ICT) on the SF has been the object of recent investigations carried out on dimers of tetracene,13 pentacene,14–18 terrylenediimide,19–23 diketopyrrolopyrrole,24–28 on tetracene and perylenediimide oligomers or dendrimers29–31 and on push–pull polymers.32–34 These studies generally consider either the effect of solvent polarity or the effect of tuning the electron donor/acceptor group strength on the SF. In some cases SF was found to occur efficiently only in non-polar solvents19 whereas in other cases SF was strongly favored in polar media.16 It is thus apparent that the solvent effect cannot be easily generalized, as it highly depends on the nature of the SF active chromophore. For instance, the role played by CT states on the SF was found to be subtly tuned by the side-group engineering, either directly participating or merely assisting the excited state deactivation depending on the particular pentacene dimer structure.15 In another research concerning push–pull polymers,32 strong intramolecular donor–acceptor interactions were found to positively impact the SF and were thus highlighted as key design features for organic SF candidates. Within the framework of these literature works, our present investigation constitutes a further attempt to get to a clear-cut overview of the effect of the ICT on the SF.Here, we aim at investigating at the same time the effect of the solvent polarity and the donor/acceptor group strength in a series of push–pull compounds. The experimental study of both the SF efficiency and rate has been carried out by employing several time-resolved spectroscopies, with nanosecond (ns) and femtosecond (fs) time resolution, in a joint effort with TD-DFT simulations. The accurate data analysis of the results has allowed to gain a deep insight into the excited state deactivation mechanism of a new class of SF materials. Three push–pull fluorene derivatives have been here taken into consideration. The two-branched molecules under study (Chart 1) show a central fluorene unit symmetrically functionalized with acetylene-bridged phenyl rings. Compound F shows a highly symmetrical D–π–D structure where both lateral phenyls are substituted with electron donating alkoxy groups. AF and NF, instead, are dipolar A–π–D systems with the lateral acceptor unit being a formyl or a nitro group, respectively. While AF and NF had been prepared for previous studies about their ICT and NLO properties,35–37 compound F has been designed and newly-synthetized for this work. Recently, computational design research,38 magnetic field effect studies on photovoltaic devices39 and spectroscopic investigations of polymer-based thin films40 have highlighted fluorene as a suitable novel SF-motif. To the best of our knowledge, except for recent reports of pentacene dimers with fluorene as the linker,41,42 this is the first experimental study addressing the photophysics of SF-active fluorene-based small molecules in solution.
 | ||
| Chart 1 Investigated molecules. | ||
Results and discussion
All the investigated compounds exhibit large absorption coefficients of ca. 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 (see Table S1†). Fig. 1 shows the effect of solvent polarity on the absorption and emission spectra of the investigated molecules, as well as on their fluorescence quantum yields (ΦF). The solvent effect on the absorption spectra is quite small for all the samples. However, a highly structured absorption profile is observed for F while a non-structured and red-shifted band is observed for AF and NF (Fig. 1 and Table S2†). The emission spectrum loses vibrational structure and clearly red-shifts upon increasing the solvent polarity.43,44 The positive fluorosolvatochromism is the largest for NF, still remarkable but smaller for AF and only hinted for F. These results, in agreement with previous studies,35–37 suggest the strongest push–pull character for NF, an intermediate but still significant push–pull character for AF and a negligible push–pull character for F. The ΦF values were found to be quite low in cyclohexane (CH): 3% for F, 37% for AF and 0.3% for NF. Efficient non-radiative decay pathways for the excited states of these compounds are thus operative in non-polar solvents. A further decrease of ΦF for AF and NF in polar media (0.2% and <0.01%, respectively) may be due to the opening of ICT processes for these dipolar systems.36,45,46 In contrast, the slight increase of the fluorescence efficiency observed for F with the solvent polarity (from 3% in CH to 8% in dimethylformamide, DMF) is in line with the negligible push–pull character inferred from its small solvatochromism.
000 M−1 cm−1 (see Table S1†). Fig. 1 shows the effect of solvent polarity on the absorption and emission spectra of the investigated molecules, as well as on their fluorescence quantum yields (ΦF). The solvent effect on the absorption spectra is quite small for all the samples. However, a highly structured absorption profile is observed for F while a non-structured and red-shifted band is observed for AF and NF (Fig. 1 and Table S2†). The emission spectrum loses vibrational structure and clearly red-shifts upon increasing the solvent polarity.43,44 The positive fluorosolvatochromism is the largest for NF, still remarkable but smaller for AF and only hinted for F. These results, in agreement with previous studies,35–37 suggest the strongest push–pull character for NF, an intermediate but still significant push–pull character for AF and a negligible push–pull character for F. The ΦF values were found to be quite low in cyclohexane (CH): 3% for F, 37% for AF and 0.3% for NF. Efficient non-radiative decay pathways for the excited states of these compounds are thus operative in non-polar solvents. A further decrease of ΦF for AF and NF in polar media (0.2% and <0.01%, respectively) may be due to the opening of ICT processes for these dipolar systems.36,45,46 In contrast, the slight increase of the fluorescence efficiency observed for F with the solvent polarity (from 3% in CH to 8% in dimethylformamide, DMF) is in line with the negligible push–pull character inferred from its small solvatochromism.
 | ||
| Fig. 1 Normalized absorption (upper panel, concentration ca. 2 × 10−5 M) and emission (bottom panel, concentration ca. 2 × 10−6 M) spectra of the investigated compounds in solvents of different polarity: cyclohexane (CH, black), toluene (Tol, red), ethyl acetate (EtAc, green) and dimethylformamide (DMF, magenta). Emission spectra obtained by exciting at the relative absorption maximum. Fluorescence quantum yields (ΦF) are also reported in CH and DMF (AF and NF values are taken from ref. 36). | ||
Possible competitive non-radiative decay pathways in non-polar media, such as triplet production, have been investigated by ns laser flash photolysis. Fig. 2 and S5† show the time-resolved absorption spectra obtained for the three molecules in CH. The detected transient absorption (TA) signals have been assigned to the lowest excited triplet states, given the significant oxygen effect on the lifetime of these species36 (Table S6†) and the resemblance with the spectra obtained through sensitization experiments from a high-energy triplet donor as well as their ability to sensitize a low-energy triplet acceptor (Fig. S8 and S10†). In the case of AF and NF, the triplet spectra show two absorption bands: a narrow peak at 530 nm and a broad band centered around 730/780 nm. For F, just a single narrow triplet absorption is observed, peaked at 490 nm. TD-DFT calculations have been employed to predict the triplet absorption spectra (ESI-Section 4†). The computationally predicted transitions together with their molecular orbital configurations are also reported in Fig. 2 and S5.† The agreement between the computational and experimental spectra is very good for the A–π–D systems NF and AF, while a poorer theoretical prediction of the triplet absorption is obtained by employing the same CAM-B3LYP functional in the case of the symmetrical F. For NF, the high-energy triplet absorption band is described by transitions localized on just one arm of the molecular structure while the broad bathochromic band by a transition with a clear CT character. Indeed, the second broad CT band is revealed only for the case of the highly dipolar AF and NF and not for F, in agreement with the other findings discussed so far.
 | ||
| Fig. 2 Transient absorption spectra of NF in de-aerated CH (ca. 1 × 10−5 M) obtained by ns flash photolysis experiments with λpump = 355 nm and theoretical triplet absorption spectra (violet bars) obtained by quantum mechanical calculations. The molecular orbitals associated to the most intense T1 → Tn electronic transitions are also shown. | ||
Sensitization experiments with several sensitizers were performed through ns-TA to access the triplet energy (ET) of the fluorenes (see the ESI,† Section 3.3.1). From the recorded kinetics the quenching rate constants were obtained for each sensitizer/fluorene pair. The rate constant values suggest a diffusional quenching of the sensitizers with ET >2.4 eV due to efficient triplet energy transfer to the fluorene acceptors. On the other hand, the fluorene derivatives act as triplet energy donors, with a diffusional rate, when considering an acceptor with ET <1.9 eV. By analyzing the results obtained with the sensitizers characterized by 1.9 eV < ET < 2.4 eV in detail, the triplet energy of the fluorene derivatives was found to follow the trend: F (ca. 2.2 eV) > NF (ca. 2.0 eV) > AF (<2.0 eV). For the case of compound F, this estimation of the triplet energy was also confirmed by measuring the phosphorescence spectrum in rigid matrix at 77 K, showing a maximum around 2.2 eV (Fig. S4†).
The triplet sensitization and relative actinometry measurements have also allowed the triplet absorption coefficients (εT – see the ESI, Section 3.3.2, and Table S4†)47 and the triplet quantum yields (ΦT – see the ESI,† Section 3.3.3, and Table 1) for the investigated chromophores to be measured. The ΦT values are remarkable in non-polar solvents, such as CH and toluene (Tol), while being significantly reduced in the more polar solvents ethyl acetate (EtAc) and DMF, especially for AF and NF. Moreover, ΦT exhibits a clear trend to increase with the sample concentration in CH and Tol (Table 1). This finding suggests that bimolecular processes may be involved in the lowest excited triplet state (T1) production. This can also be inferred by the observed concentration effect on the triplet lifetime (Table S9†), which shows values of ca. 100 μs in dilute purged CH and Tol solutions and is largely reduced for the case of more concentrated samples.48 Interestingly, the ΦT values were found to exceed 100% in the most concentrated solutions (2–9 × 10−5 M) when non-polar media are considered (Table 1). In particular, in concentrated CH solutions, ΦT reaches values of 136%, 117% and 145% for F, AF and NF, respectively, pointing to the possibility that multiexciton generation processes, such as SF, may be involved in the production of the T1 state.
| C/10−5 M | Φ T in CH | Φ T in Tol | Φ T in EtAc | Φ T in DMF | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | AF | NF | F | AF | NF | F | AF | NF | F | AF | NF | |
| 0.25 | 0.46 ± 0.07 | 0.57 ± 0.09 | 0.73 ± 0.11 | 0.53 ± 0.08 | 0.52 ± 0.08 | |||||||
| 0.75 | 0.66 ± 0.10 | 0.86 ± 0.13 | 0.95 ± 0.14 | 0.86 ± 0.13 | 0.58 ± 0.09 | 0.72 ± 0.11 | 0.76 ± 0.11 | 0.22 ± 0.03 | 0.32 ± 0.05 | 0.72 ± 0.11 | 0.05 ± 0.01 | 0.23 ± 0.03 |
| 1.0 | 0.80 ± 0.12 | 1.06 ± 0.16 | 1.05 ± 0.16 | 1.03 ± 0.15 | 0.80 ± 0.12 | 0.90 ± 0.13 | ||||||
| 2.5 | 0.94 ± 0.14 | 1.17 ± 0.18 | 1.45 ± 0.22 | 1.14 ± 0.17 | 0.87 ± 0.13 | 1.10 ± 0.17 | 0.83 ± 0.12 | 0.44 ± 0.07 | 0.70 ± 0.11 | 0.85 ± 0.13 | 0.04 ± 0.01 | 0.29 ± 0.04 |
| 4.5 | 1.26 ± 0.19 | |||||||||||
| 9.0 | 1.36 ± 0.20 | |||||||||||
In order to examine this hypothesis in depth, the triplet formation mechanism has been investigated by means of fs-TA experiments. The results obtained for the case of NF in CH and Tol are shown in Fig. 3. The time-resolved absorption spectra (in panel B) initially show an excited state absorption (ESA) signal at ca. 650 nm associated to the lowest excited singlet state (S1) populated by light absorption. This S1 ESA exhibits an ultrafast decay while a simultaneous ultrafast rise of an ESA peaked at 530 and 750 nm is observed, resembling the spectrum assigned to T1 through the ns-TA measurements. The kinetics (inset of panel B) recorded at ca. 650 nm display the fast S1 decay, while those recorded at ca. 530 or above 700 nm exhibit a peculiar profile: a fast rise followed by a slower decay to reach a residual ΔA, which persists at the longest investigated delays (3.2 ns). The results of the global fitting of these data are reported in panel C of the graphs. The shortest components are associated to solvent relaxation or vibrational cooling processes (in black). The component characterized by a lifetime of 5.6 ps in CH and 45 ps in Tol is assigned to the relaxed S1 state populated by light absorption (in green). The component showing an infinite lifetime in the investigated time-window corresponds to the T1 state (rest, in wine), given the similarity with the spectra revealed for this component by ns-TA. Surprisingly, an additional species (in blue) is revealed by the fitting characterized by a lifetime of ca. 400 ps, typical of a singlet excited state, and by a spectral profile analogous to the triplet spectrum. For the most fluorescent AF compound, fluorescence up conversion (FUC) measurements have also been carried out (Fig. 4). Interestingly, the transient showing a lifetime of ca. 500 ps and a triplet-like ESA detected during the TA has been revealed even by the FUC. This finding points out that this transient is an emissive state, characterized by a peculiar fluorescence spectrum distinct from that of S1 and by a significantly lower intensity (Fig. S18†).33,49–51 Given its double nature, the blue transient has been associated to the SF intermediate1(TT): a double triplet state with an overall singlet spin multiplicity and therefore able to emit.49,52 This assignment thus justifies the ultrafast formation in the picosecond time-range as the S1 → 1(TT) transition is spin allowed.53,54 For NF in CH, the S1 → 1(TT) transition occurs in 5.6 ps, as resulting from the optimal fitting of the data by Global Analysis and by assuming a consecutive kinetic model. On the other hand, for NF in Tol the best fitting has been achieved by Target Analysis and considering a branching in the excited state deactivation (Fig. S14†). Therefore, the double triplet state is actually formed in 7.9 ps from the non-relaxed S1 state in Tol. A similar dynamic, remarkably reproduced by Global Analysis with a consecutive kinetic model, has been revealed by fs-TA for AF and F in CH (ESI-Section 3.4†), resulting in a slower formation of the 1(TT) (77 ps for AF and 127 ps for F) relative to NF.
 | ||
| Fig. 3 Fs-TA measurements of NF in cyclohexane (CH, left) and toluene (Tol, right) solutions (ca. 1 × 10−4 M) with λpump = 400 nm. Panel A: experimental 3D matrix; panel B: representative spectra at different delay times and representative kinetics (inset) at different wavelengths; panel C: Evolution Associated Spectra (EAS) obtained by Global Analysis or Species Associated Spectra (SAS) obtained by Target Analysis. | ||
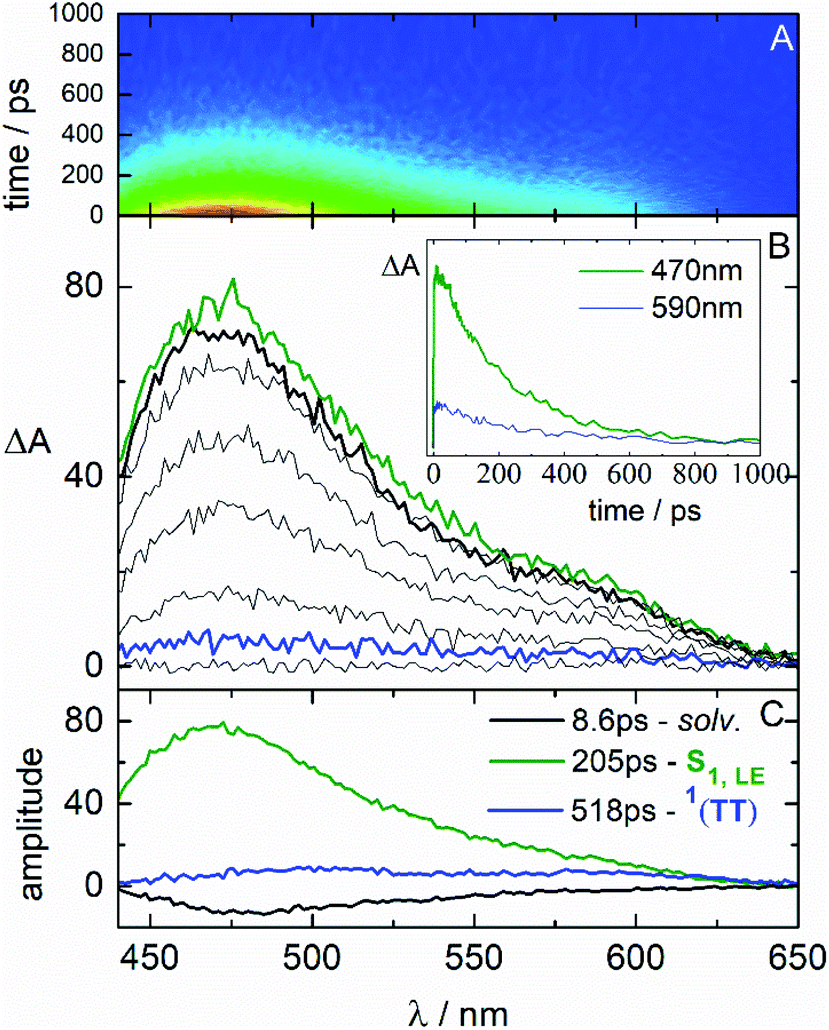 | ||
| Fig. 4 Femtosecond fluorescence up conversion measurement of AF in toluene (Tol) solution (ca. 1 × 10−4 M) with λpump = 400 nm. | ||
The ultrafast spectroscopic results in non-polar solvents agree with the excited singlet and triplet state energies as obtained by the TD-DFT calculations (Table 2), which predict the Franck-Condon singlet state (S1,FC) at ca. 3.5 eV, the relaxed singlet state (S1,REL) at ca. 3.2 eV and the relaxed T1 state (T1,REL) at ca. 1.8 eV. These computational results are consistent with the experimental assessment of the triplet energy through sensitization (ESI, Section 3.3.1†) and phosphorescence (ESI, Section 3.2†) measurements. In particular, not only the calculations reproduce the trend experimentally observed for the triplet energy (F > NF > AF), but also the absolute ET values within the intrinsic error associated to the experiments and the computations. Very interestingly, our experimental and theoretical results suggest higher energetic triplet excitons relative to literature SF-materials (1.2 eV at the most).7,8 According to the estimated ΔETT-S,ad = 2E(T1,REL)–E(S1,REL), an endothermic SF may take place in these fluorene derivatives.13,55 In particular, for the case of NF, the fs-TA experiments suggest that SF likely occurs from a non-relaxed singlet state, in agreement with a reduced computational endothermicity when considering the ΔETT-S = 2E(T1,REL)–E(S1,FC) (Table 2). The smallest ΔETT-S for NF (0.23 eV) would be in line with its fastest and most efficient SF among the investigated systems.
The SF rate evaluated through the ultrafast spectroscopic data (kSF = 1–79 × 109 s−1 for the three molecules in CH, see the details in Table 3) is found to be higher than the diffusional rate (kdiff × c ≃ 6.7 × 109 M−1 s−1 × 2.5 × 10−5 M = 1.7 × 105 s−1) governing intermolecular interactions between distinct molecules in solution. Hence, the peculiar double triplet formation kinetics of these molecules may be related to intramolecular processes occurring at short time delays after excitation, rather than slower diffusion-controlled bimolecular encounters. The ultrafast 1(TT) formation, in competition with the S1 decay by ordinary intersystem crossing (ISC) and fluorescence, may reflect intramolecular SF events occurring in the investigated two-branched molecules, showing approximately a dimer-like structure. An intramolecular 1(TT) production is also consistent with the absence of significant concentration effects on the steady-state absorption and emission spectra (Fig. S1 and S2,† respectively), implying the formation of both aggregates and excimers to be excluded. However, important concentration effects were observed on ΦT measured via ns-TA. To rationalize this effect, the triplets generated by ISC have been quantitatively distinguished from those generated by SF (see Table S10†). The triplet yield found in dilute solution and at cryogenic temperature56 may be associated to the ISC yield of the isolated monomer. In dilute solution, the double triplet state decays back to the ground state mainly by internal conversion. When the solution concentration increases, the presence of a second nearby chromophore may favor the independent triplet separation, accomplishing the second step of SF more efficiently. This leads to the observed increase of the triplet yield with the concentration. To better verify this model, femtosecond transient absorption measurements were performed in solutions of different concentrations for the representative case of NF in CH (Table S12†). The investigated concentration range parallels that in which the triplet yields have been determined (Table 1). As a result, the S1 lifetime is practically unaffected by the concentration, while the 1(TT) decay is slowed down in the most dilute solutions. These results may be a further indication of an ultrafast INTRAmolecular double triplet formation followed by an INTERmolecular independent triplet separation.50,57–60 Preliminary ultrafast spectroscopic results obtained for the investigated compounds in thin film seem to confirm the occurrence of SF also in the solid state.
| τ SF/ps | τ TT/ps | Φ SF | k SF /109 s−1 | |
|---|---|---|---|---|
a
Φ
SF obtained as the difference between ΦT in the most concentrated and ΦT in the most diluted solution, with this latter corresponding to ΦISC.
b
k
SF = ΦSF × kS/2 where 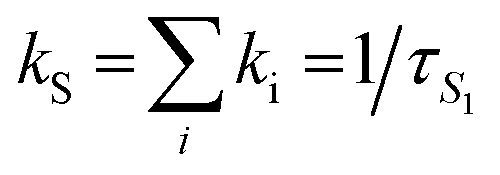 that is the sum of the rates of all the processes deactivating S1.63 that is the sum of the rates of all the processes deactivating S1.63
|
||||
| Structure effect (in CH) | ||||
| F | 127 | 640 | 0.28 | 1.1 |
| AF | 77 | 620 | 0.71 | 4.6 |
| NF | 5.6 | 400 | 0.88 | 79 |
|
||||
| Solvent effect (NF) | ||||
| CH | 5.6 | 400 | 0.88 | 79 |
| Tol | 7.9 | 390 | 0.58 | 37 |
| EtAc | 7.6 | 100 | 0.38 | 25 |
| DMF | 0.59 | 260 | 0.06 | 51 |
The fs-TA measurements, when carried out for NF in polar solvents (Fig. 5), have revealed a significant blue shift of the ESA at early delays after photoexcitation. The global fitting of the data has uncovered the presence of a further transient species in addition to those already detected also in non-polar solvents (solvation, S1,LE, 1(TT) and T1). This additional component, characterized by an ESA peaked at ca. 560 nm (in red in panel C), is assigned to the S1,ICT state stabilized in polar solvents.61 An optimal fitting of the data could be achieved by Target Analysis and by considering a branching in the excited state deactivation after the S1,LE to populate either the S1,ICT or the 1(TT) (Fig. S15†). The opening of the ICT channel, competitive to SF (Chart 2), justifies the significant decrease of ΦT measured for NF in polar solvents.62 Analogously, an ICT state has been evidenced for AF in polar media (Fig. S17†), leading in this case to the complete suppression of SF in the most polar DMF solvent. Conversely, no ICT has been observed for F in a polar solvent (Fig. S16†) in agreement with its lower push–pull character and almost solvent-independent SF behavior.
 | ||
| Fig. 5 Fs-TA measurements of NF in ethyl acetate (EtAc, left) and dimethylformamide (DMF, right) solutions (ca. 1 × 10−4 M) with λpump = 400 nm. | ||
 | ||
| Chart 2 Competition between SF and ICT for NF as evidenced by the solvent effect on the excited states dynamics. | ||
In summary, the time-resolved spectroscopic results allow to get a comprehensive picture of the SF efficiency and rate (Table 3) for three fluorene derivatives characterized by a different push–pull character: F ≪ AF < NF. The lifetime associated to the first step of SF (S1 → 1(TT), τSF) decreases upon increasing the push–pull character, while the lifetime associated to the second step of SF (1(TT) → T1, τTT) is less affected by the molecular structure (Table 3). Moreover, a significant increase of the ΦSF is observed upon increasing the push–pull character (28% for F, 71% for AF and 88% for NF) when considering a non-polar solvent where the ICT likely acts as a high-energy virtual state. The molecular structure effect on both τSF and ΦSF leads to a SF rate constant (kSF) strongly enhanced with the push–pull character: 1.1 × 109 s−1 for F, 4.6 × 109 s−1 for AF and 79 × 109 s−1 for NF. With this study, we could also uncover the solvent polarity effect on the SF yield and rate. For the representative case of NF (Table 3), τSF was found to be 6–8 ps and slightly affected by the solvent when low polarity solvents are considered (ε ∼ 2–6). However, the triplet yield undergoes a large drop below 100% as soon as the ICT becomes a real intermediate state: ΦSF is 88% in CH, 58% in Tol, 38% in EtAc and 6% in DMF. In a highly polar solvent such as DMF (ε = 37), even though a significant decrease of the τSF and consequent increase of the kSF is observed, the SF yield is lowered due to the strong competition of the ICT to the SF.
Conclusions
In conclusion, we report strong experimental evidence for ultrafast double triplet formation followed by efficient triplet separation observed for the first time in small push–pull fluorene derivatives. Our time-resolved spectroscopic results show that a stronger push–pull character favors SF as long as the ICT does not become a real competitive path. The investigated compounds are highly photostable systems and show remarkable light absorption ability, thus exhibiting outstanding features for photovoltaic applications. Also, in comparison to the well-known SF literature materials, these molecules manifest the unique property of SF-generated high-energy triplet excitons (ca. 2 eV) and are thus extremely promising as new SF photosensitizers in solar cell devices.Data availability
All the data associated with this article are already reported in the ms and in the ESI.†Author contributions
L. M.: data curation, investigation, methodology, writing-original draft, writing-review & editing; B. C.: conceptualization, investigation, supervision, writing-original draft, writing-review & editing; F. E.: investigation, funding acquisition, writing-review & editing; A. M.: investigation, resources, writing-review & editing; A. S.: conceptualization, data curation, supervision, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Italian “Ministero per l’Università e la Ricerca Scientifica e Tecnologica”, MIUR (Rome, Italy) under the “Dipartimenti di Eccellenza 2018–2022” (grant AMIS) program and from the University of Perugia under the FRB2019 program.References
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- K. C. Krishnapriya, A. J. Musser and S. Patil, ACS Energy Lett., 2019, 4, 192–202 CrossRef CAS.
- R. Casillas, I. Papadopoulos, T. Ullrich, D. Thiel, A. Kunzmann and D. M. Guldi, Energy Environ. Sci., 2020, 13, 2741–2804 RSC.
- J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Thompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300–1311 CrossRef CAS PubMed.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, D. N. Congreve and L. M. Campos, Adv. Mater., 2017, 29, 1701416 CrossRef PubMed.
- J. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. Liu, L. M. Campos and T. Sun, Adv. Mater., 2017, 29, 1601652 CrossRef PubMed.
- T. Ullrich, D. Munz and D. M. Guldi, Chem. Soc. Rev., 2021, 50, 3485–3518 RSC.
- N. Monahan and X.-Y. Zhu, Annu. Rev. Phys. Chem., 2015, 66, 601–618 CrossRef CAS PubMed.
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS PubMed.
- E. J. Taffet, D. Beljonne and G. D. Scholes, J. Am. Chem. Soc., 2020, 142, 20040–20047 CrossRef CAS PubMed.
- A. M. Alvertis, S. Lukman, T. J. H. Hele, E. G. Fuemmeler, J. Feng, J. Wu, N. C. Greenham, A. W. Chin and A. J. Musser, J. Am. Chem. Soc., 2019, 141, 17558–17570 CrossRef CAS PubMed.
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss, J. Wu, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS.
- S. Lukman, K. Chen, J. M. Hodgkiss, D. H. P. Turban, N. D. M. Hine, S. Dong, J. Wu, N. C. Greenham and A. J. Musser, Nat. Commun., 2016, 7, 13622 CrossRef CAS PubMed.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, S. R. Reddy, B. T. Phelan, M. D. Krzyaniak, M. K. Volland, P. B. Coto, R. M. Young, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Chem, 2018, 4, 1092–1111 CAS.
- A. Aster, F. Zinna, C. Rumble, J. Lacour and E. Vauthey, J. Am. Chem. Soc., 2021, 143, 2361–2371 CrossRef CAS PubMed.
- I. Papadopoulos, J. Zirzlmeier, C. Hetzer, Y. J. Bae, M. D. Krzyaniak, M. R. Wasielewski, T. Clark, R. R. Tykwinski and D. M. Guldi, J. Am. Chem. Soc., 2019, 141, 6191–6203 CrossRef CAS PubMed.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef CAS PubMed.
- M. Chen, Y. J. Bae, C. M. Mauck, A. Mandal, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 9184–9192 CrossRef CAS PubMed.
- M. Chen, J. Y. Shin, R. M. Young and M. R. Wasielewski, J. Chem. Phys., 2020, 153, 094302 CrossRef CAS PubMed.
- J. D. Schultz, J. Y. Shin, M. Chen, J. P. O'Connor, R. M. Young, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2021, 143, 2049–2058 CrossRef CAS PubMed.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845–7857 CrossRef CAS PubMed.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed.
- C. E. Miller, M. R. Wasielewski and G. C. Schatz, J. Phys. Chem. C, 2017, 121, 10345–10350 CrossRef CAS.
- C. M. Mauck, Y. J. Bae, M. Chen, N. Powers-Riggs, Y.-L. Wu and M. R. Wasielewski, ChemPhotoChem, 2018, 2, 223–233 CrossRef CAS.
- I. Papadopoulos, M. J. Álvaro-Martins, D. Molina, P. M. McCosker, P. A. Keller, T. Clark, Á. Sastre-Santos and D. M. Guldi, Adv. Energy Mater., 2020, 10, 2001496 CrossRef CAS.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed.
- B. Carlotti, I. K. Madu, H. Kim, Z. Cai, H. Jiang, A. K. Muthike, L. Yu, P. M. Zimmerman and T. Goodson, Chem. Sci., 2020, 11, 8757–8770 RSC.
- A. K. Muthike, B. Carlotti, I. K. Madu, H. Jiang, H. Kim, Q. Wu, L. Yu, P. M. Zimmerman and T. Goodson, J. Phys. Chem. B, 2021, 125, 5114–5131 CrossRef CAS PubMed.
- J. Kim, H. T. Teo, Y. Hong, J. Oh, H. Kim, C. Chi and D. Kim, Angew. Chem., Int. Ed., 2020, 59, 20956–20964 CrossRef CAS PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X.-Y. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426–433 CrossRef CAS PubMed.
- G. He, E. Busby, K. Appavoo, Q. Wu, J. Xia, L. M. Campos and M. Y. Sfeir, J. Chem. Phys., 2020, 153, 244902 CrossRef CAS PubMed.
- L. Wang, X. Liu, X. Shi, C. L. Anderson, L. M. Klivansky, Y. Liu, Y. Wu, J. Chen, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 17892–17896 CrossRef CAS PubMed.
- R. Flamini, I. Tomasi, A. Marrocchi, B. Carlotti and A. Spalletti, J. Photochem. Photobiol., A, 2011, 223, 140–148 CrossRef CAS.
- B. Carlotti, R. Flamini, A. Spalletti, A. Marrocchi and F. Elisei, ChemPhysChem, 2012, 13, 724–735 CrossRef CAS PubMed.
- B. Carlotti, R. Flamini, I. Kikaš, U. Mazzucato and A. Spalletti, Chem. Phys., 2012, 407, 9–19 CrossRef CAS.
- S. Naskar and M. Das, Chem. Phys. Lett., 2020, 749, 137368 CrossRef CAS.
- S. Kawata, Y.-J. Pu, A. Saito, Y. Kurashige, T. Beppu, H. Katagiri, M. Hada and J. Kido, Adv. Mater., 2016, 28, 1585–1590 CrossRef CAS PubMed.
- Y. Tamai, H. Ohkita, H. Benten and S. Ito, J. Phys. Chem. C, 2013, 117, 10277–10284 CrossRef CAS.
- J. K. G. Karlsson, A. Atahan, A. Harriman, S. Tojo, M. Fujitsuka and T. Majima, J. Phys. Chem. Lett., 2018, 9, 3934–3938 CrossRef CAS.
- J. K. G. Karlsson, A. Atahan, A. Harriman, N. V. Tkachenko, A. D. Ward, F. A. Schaberle, C. Serpa and L. G. Arnaut, J. Phys. Chem. A, 2021, 125, 1184–1197 CrossRef CAS PubMed.
- C. Bonaccorso, A. Cesaretti, F. Elisei, L. Mencaroni, A. Spalletti and C. G. Fortuna, ChemPhysChem, 2018, 19, 1917–1929 CrossRef CAS.
- O. Mongin, L. Porrès, C. Katan, T. Pons, J. Mertz and M. Blanchard-Desce, Tetrahedron Lett., 2003, 44, 8121–8125 CrossRef CAS.
- B. Carlotti, A. Cesaretti, P. L. Gentili, A. Marrocchi, F. Elisei and A. Spalletti, Phys. Chem. Chem. Phys., 2016, 18, 23389–23399 RSC.
- A. Cesaretti, B. Carlotti, F. Elisei, C. G. Fortuna and A. Spalletti, Phys. Chem. Chem. Phys., 2018, 20, 2851–2864 RSC.
- L. Mencaroni, B. Carlotti, A. Cesaretti, F. Elisei, A. Grgičević, I. Škorić and A. Spalletti, Photochem. Photobiol. Sci., 2020, 19, 1665–1676 CrossRef CAS PubMed.
- T. Sakuma, H. Sakai, Y. Araki, T. Mori, T. Wada, N. V. Tkachenko and T. Hasobe, J. Phys. Chem. A, 2016, 120, 1867–1875 CrossRef CAS PubMed.
- A. J. Musser and J. Clark, Annu. Rev. Phys. Chem., 2019, 70, 323–351 CrossRef CAS PubMed.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019–1024 CrossRef CAS PubMed.
- H. L. Stern, A. J. Musser, S. Gelinas, P. Parkinson, L. M. Herz, M. J. Bruzek, J. Anthony, R. H. Friend and B. J. Walker, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7656–7661 CrossRef CAS PubMed.
- C. K. Yong, A. J. Musser, S. L. Bayliss, S. Lukman, H. Tamura, O. Bubnova, R. K. Hallani, A. Meneau, R. Resel, M. Maruyama, S. Hotta, L. M. Herz, D. Beljonne, J. E. Anthony, J. Clark and H. Sirringhaus, Nat. Commun., 2017, 8, 15953 CrossRef CAS PubMed.
- H. Kim and P. M. Zimmerman, Phys. Chem. Chem. Phys., 2018, 20, 30083–30094 RSC.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X.-Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- M. H. Farag and A. I. Krylov, J. Phys. Chem. C, 2018, 122, 25753–25763 CrossRef CAS.
- G. Cacioppa, B. Carlotti, F. Elisei, P. L. Gentili, A. Marrocchi and A. Spalletti, Phys. Chem. Chem. Phys., 2016, 18, 285–294 RSC.
- M. T. Trinh, Y. Zhong, Q. Chen, T. Schiros, S. Jockusch, M. Y. Sfeir, M. Steigerwald, C. Nuckolls and X. Zhu, J. Phys. Chem. C, 2015, 119, 1312–1319 CrossRef CAS.
- N. V. Korovina, N. F. Pompetti and J. C. Johnson, J. Chem. Phys., 2020, 152, 040904 CrossRef CAS PubMed.
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617–627 CrossRef CAS PubMed.
- N. V. Korovina, J. Joy, X. Feng, C. Feltenberger, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2018, 140, 10179–10190 CrossRef CAS PubMed.
- W. Akemann, D. Laage, P. Plaza, M. M. Martin and M. Blanchard-Desce, J. Phys. Chem. B, 2008, 112, 358–368 CrossRef CAS PubMed.
- E. Sundin, R. Ringström, F. Johansson, B. Küçüköz, A. Ekebergh, V. Gray, B. Albinsson, J. Mårtensson and M. Abrahamsson, J. Phys. Chem. C, 2020, 124, 20794–20805 CrossRef CAS.
- D. Rais, P. Toman, J. Pfleger, U. Acharya, Y. R. Panthi, M. Menšík, A. Zhigunov, M. A. Thottappali, M. Vala, A. Marková, S. Stříteský, M. Weiter, M. Cigánek, J. Krajčovič, K. Pauk, A. Imramovský, A. Zaykov and J. Michl, ChemPlusChem, 2020, 85, 2689–2703 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc07175k |
| This journal is © The Royal Society of Chemistry 2022 |