DOI:
10.1039/D1SC06803B
(Edge Article)
Chem. Sci., 2022,
13, 1706-1714
Why intermolecular nitric oxide (NO) transfer? Exploring the factors and mechanistic aspects of NO transfer reaction†
Received
6th December 2021
, Accepted 17th December 2021
First published on 11th January 2022
Abstract
Small molecule activation and their transfer reactions in biological or catalytic reactions are greatly influenced by the metal-centers and the ligand frameworks. Here, we report the metal-directed nitric oxide (NO) transfer chemistry in low-spin mononuclear {Co(NO)}8, [(12-TMC)CoIII(NO−)]2+ (1-CoNO, S = 0), and {Cr(NO)}5, ([(BPMEN)Cr(NO)(Cl)]+) (4-CrNO, S = 1/2) complexes. 1-CoNO transfers its bound NO moiety to a high-spin [(BPMEN)CrII(Cl2)] (2-Cr, S = 2) and generates 4-CrNOvia an associative pathway; however, we did not observe the reverse reaction, i.e., NO transfer from 4-CrNO to low-spin [(12-TMC)CoII]2+ (3-Co, S = 1/2). Spectral titration for NO transfer reaction between 1-CoNO and 2-Cr confirmed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction stoichiometry. The NO transfer rate was found to be independent of 2-Cr, suggesting the presence of an intermediate species, which was further supported experimentally and theoretically. The experimental and theoretical observations support the formation of μ-NO bridged intermediate species ({Cr–NO–Co}4+). Mechanistic investigations using 15N-labeled-15NO and tracking the 15N-atom established that the NO moiety in 4-CrNO is derived from 1-CoNO. Further, to investigate the factors deciding the NO transfer reactivity, we explored the NO transfer reaction between another high-spin CrII-complex, [(12-TMC)CrII(Cl)]+ (5-Cr, S = 2), and 1-CoNO, showing the generation of the low-spin [(12-TMC)Cr(NO)(Cl)]+ (6-CrNO, S = 1/2); however, again there was no opposite reaction, i.e., from Cr-center to Co-center. The above results advocate clearly that the NO transfer from Co-center generates thermally stable and low-spin and inert {Cr(NO)}5 complexes (4-CrNO & 6-CrNO) from high-spin and labile Cr-complexes (2-Cr & 5-Cr), suggesting a metal-directed NO transfer (cobalt to chromium, not chromium to cobalt). These results explicitly highlight that the NO transfer is strongly influenced by the labile/inert behavior of the metal-centers and/or thermal stability rather than the ligand architecture.
:1 reaction stoichiometry. The NO transfer rate was found to be independent of 2-Cr, suggesting the presence of an intermediate species, which was further supported experimentally and theoretically. The experimental and theoretical observations support the formation of μ-NO bridged intermediate species ({Cr–NO–Co}4+). Mechanistic investigations using 15N-labeled-15NO and tracking the 15N-atom established that the NO moiety in 4-CrNO is derived from 1-CoNO. Further, to investigate the factors deciding the NO transfer reactivity, we explored the NO transfer reaction between another high-spin CrII-complex, [(12-TMC)CrII(Cl)]+ (5-Cr, S = 2), and 1-CoNO, showing the generation of the low-spin [(12-TMC)Cr(NO)(Cl)]+ (6-CrNO, S = 1/2); however, again there was no opposite reaction, i.e., from Cr-center to Co-center. The above results advocate clearly that the NO transfer from Co-center generates thermally stable and low-spin and inert {Cr(NO)}5 complexes (4-CrNO & 6-CrNO) from high-spin and labile Cr-complexes (2-Cr & 5-Cr), suggesting a metal-directed NO transfer (cobalt to chromium, not chromium to cobalt). These results explicitly highlight that the NO transfer is strongly influenced by the labile/inert behavior of the metal-centers and/or thermal stability rather than the ligand architecture.
Introduction
In the biological system, the choice of metal-centers and the ligand frameworks significantly influence coordination chemistry, redox property, and various metalloenzymes' reactivity.1 When it comes to small molecule activation, enzymatic and catalytic reactions, these properties become more prominent for selective biological reactions. For example, heme systems of particular proteins (NiRs) bind with nitric oxide (NO);2 however, other proteins with similar heme groups do not show NO-activation,3 and it is rare to have NO activation on different metal centers having the same coordination sphere.4 Together, these parameters provide an excellent opportunity or challenge to the scientific community to study/explore the small molecules' interactions with metals in different ligand frameworks. Consequently, the transfer of coordinated ligands to another metal center has become the area of interest for the past few decades. Halide transfer reactions are one of the most explored reactions.5 Some examples of intermolecular transfer of bound carbon monoxide (CO),6 superoxide (O2˙−),7 and peroxide (O22−)8 were also reported. Nowadays, atom transfer reactions are an area of fundamental importance, particularly apparent in biological systems.9 A number of metalloenzymes were reported to mediate the oxygen atom transfer (OAT) reactions,10 specially molybdoenzymes such as nitrate reductase (NRs)11 and C–H activation reactions.12,13
NO activation became an area of interest for chemists and biochemists due to the vast presence of metal-nitrosyls (M-NOs) in the biological system. Being a radical species, NO is widely known as a signal transduction molecule for its involvement in major biological processes, such as neurotransmission, vascular regulation, platelet disaggregation, and immune response to bacterial infections.13 The deficiency of NO in the natural system hamper the processes described above and leads to several biological disorders, including atherothrombosis,14 diabetic hypertension,15 and chronic kidney disease.16,17 In this regard, nitric oxide synthases (NOSs)18 and nitrite reductases (NiRs)2,19 accomplish the constant biosynthesis of NO. NOSs are a family of enzymes that catalyze the guanidine nitrogen oxidation in L-arginine to produce NO.20 Whereas, in some mammalian and bacterial systems, NiRs catalyze the production of NO by NO2− reduction in the presence of protons, i.e., NO2− + e− + 2H+ → NO + H2O.21 In the biological system, NO acts as a double-edged sword, and overproduction can lead to the generation of several cytotoxic molecules.22
The coordination chemistry of M-NOs has a long history of understanding the fundamental aspects of binding, electronic arrangements, and reactivity.23 Until now, several M-NOs have been synthesized and explored for different reactivities,4b,24 including even a few reports on intermolecular NO transfer;25 however, the mechanistic aspects of these reactions have rarely been explored. An extensive study of the NO transfer process may enlighten the NO transport process in the biological system. Earlier, Armor reported acid-promoted metal-dependent NO transfer from CoIII to CrII involving a labile aqua complex.25e Further studies suggest the transfer of the NO ligand at high acidic conditions due to axial NO ligand protonation.26 Caulton and co-workers, in a study of NO transfer reactivity from Co to various metal complexes of Fe, Ru, Rh, and Co complexes, proposed that the NO transfers readily to the coordinatively unsaturated metal complexes, either via a simple NO transfer or via halide exchange pathway.25c Mu and Kadish speculated the effect of metal-center on a NO transfer reaction.27 An intermediate species formation was also proposed before NO transfer, suggesting an association mechanism.25a–e Cook and co-workers, for the first time, reported a dissociative pathway in a NO transfer reaction from Co(NO)(dmgH)2 to Hemoglobin (Hb).28 Lippard and co-workers also testified a dissociated NO transfer pathway from Mn to Fe bearing a tropocarnado ligand.25d A μ-NO bridge intermediate was discussed in a NO transfer reaction of Ru to Fe-center of Hb and myoglobin (Mb); however, this has not been well identified or confirmed with experimental evidence.25a Recently, Nam and co-workers reported NO transfer reactivity influenced by ligand size and the spin-state of the acceptor/donor metal complexes.29 These results suggest that the reaction mechanism and the intermediate involved in the intermolecular NO transfer reaction are inconsistent and depend on various factors; however, these reports lack a detailed mechanistic investigation. Here, the spectroscopic signatures for the proposed μ-NO bridge intermediate species need to be characterized in detail, which will surely help the scientific community understand the mechanistic insights of NO transfer reactions.
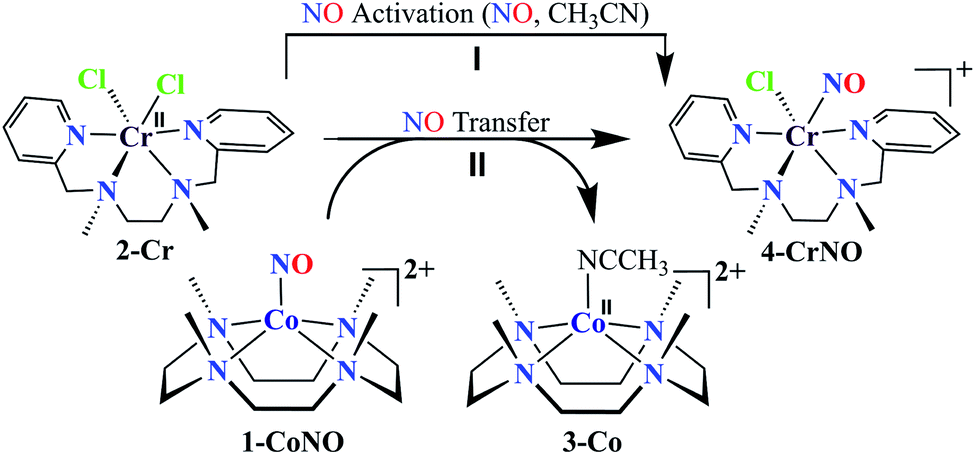
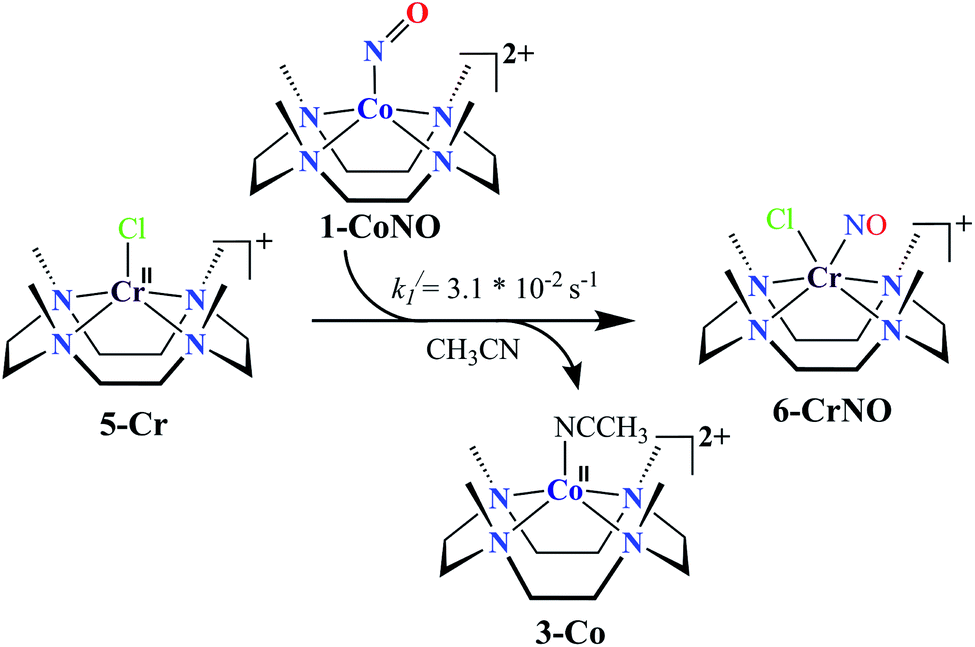
We herein report the NO transfer reactivity of a CoIII-nitrosyl complex, [(12-TMC)CoIII(NO−)]2+ (1-CoNO),4b,30 bearing a 12-TMC ligand (12-TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane) with a CrII-complex, [(BPMEN)CrII(Cl)2] (2-Cr), bearing BPMEN ligand (BPMEN = N,N′-bis(2-pyridylmethyl)-1,2-diaminoethane), and this reaction generates [(12-TMC)CoII]2+ (3-Co) and [(BPMEN)Cr(Cl)(NO)]+ (4-CrNO) (Scheme 1, reaction II). Mechanistic insight into the reaction suggests that the reaction goes through an associative pathway and is also supported by determining various physical parameters (Scheme 2, reaction I & II). We have observed the generation of an intermediate, proposed to be μ-NO bridge intermediate species ({Cr–NO–Co}4+), formation prior to the intermolecular NO transfer and well-characterized spectroscopically for the first time. The ligand framework plays a critical role in the NO transfer chemistry;25f however, we can not ignore the metal-centers' properties in M-NOs, i.e., spin-states and labile/inert behavior, and also the M-NOs thermal stability. To understand the factors deciding the NO transfer chemistry, we performed NO transfer reactivity from 1-CoNO to [(12-TMC)CrII(Cl)]+ (5-Cr), which generates ([(12-TMC)Cr(NO)(Cl)]+) (6-CrNO) (Scheme 3), and vice versa (6-CrNO to 3-Co; no NO transfer). This chemistry established the labile-inert behavior of metal-centers through a significant impact in the NO transfer reactions rather than the influence of the ligand framework.
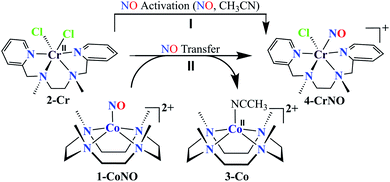 |
| | Scheme 1 | |
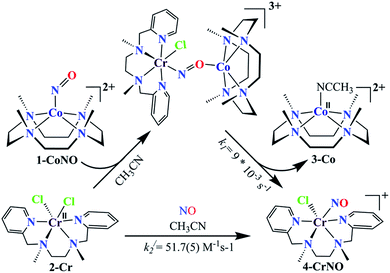 |
| | Scheme 2 | |
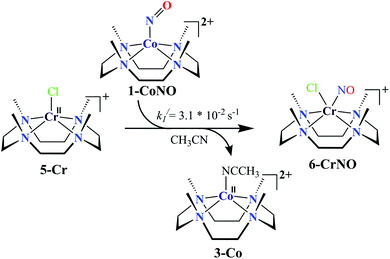 |
| | Scheme 3 | |
Results
Synthesis of Cr-nitrosyl complex [(BPMEN)Cr(NO)(Cl)]Cl (4-CrNO)
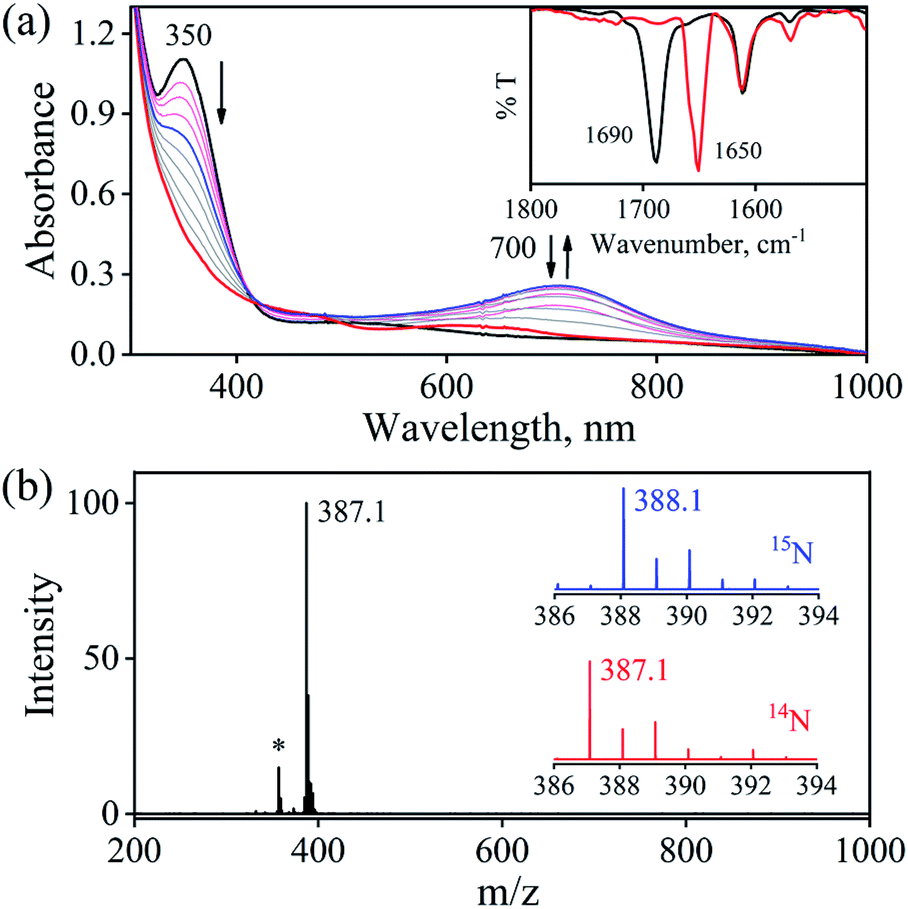
The initial CrII-complex [(BPMEN)CrII(Cl)2] was synthesized by adding BPMEN ligand to a stirring solution of CrCl2 (ESI,† Experimental Section (ES)). The addition of excess NO(g) (Fig. S1 in the ESI† shows a schematic diagram of the NO(g) purification and handling process) to the CH3CN solution of 2-Cr at RT under an Ar atmosphere resulted in the generation of 4-CrNO (λmax = 600, ε = 110 M−1 cm−1, red line) within 5 min and isolated as a solid product (Fig. 1a and S2a, ESI,† ES) (Scheme 1, reaction I). Electrospray ionization mass spectrometry (ESI-MS) of 4-CrNO showed a prominent ion peak at m/z 387.1, whose mass and isotope distribution patterns correspond to [(BPMEN)Cr(NO)(Cl)]+ (calcd m/z 387.1) (Fig. 1b). When the reaction was carried out with 15N-labeled NO (15NO), the mass peak corresponding to [Cr(BPMEN)(15NO)(Cl)]+ appears at m/z 388.1 (calcd m/z 388.1) (inset, Fig. 1b and S2b; ESI†). This one unit shift in mass value upon substitution of NO by 15NO indicates that 4-CrNO contains an NO moiety attached to the Cr-center. The FT-IR spectrum is also consistent with the exact formulation of 4-CrNO, revealing a linear NO with a typical M-NO stretching at 1690 cm−1, which shifted to 1650 cm−1 when 4-CrNO was prepared using 15NO (inset, Fig. 1a, S2c and S2d; ESI†).31 The 1H NMR of 4-CrNO does not exhibit any signal for aromatic protons, suggesting a paramagnetic Cr-center (ESI,† Fig. S3a). Further, the magnetic moment calculated by Evans' method revealed a magnetic moment of 1.68, which corresponds to a low-spin Cr-centre (d5, S = 1/2) in complex 4-CrNO (ESI,† Fig. S3b), which can be assigned as {Cr(NO)}5.32 Further, 4-CrNO was structurally characterized via single-crystal X-ray crystallography. The NO ligand is coordinated to a Cr-center in an end-on fashion with a distorted octahedral geometry. The Cr(1)–N(5)–O(1) bond angle of 4-CrNO was found to be 174.0(11)° suggesting sp hybridization of N-atom, as explained in other reports.31a Hence, NO coordination to the low-spin Cr-center was confirmed, which was further consistent with our assignment of this complex as {Cr(NO)}5 species (Fig. 2, S4, Tables S1 and S2; ESI,† ES).33
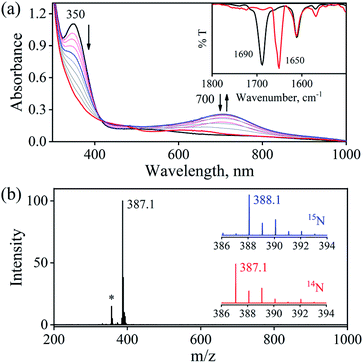 |
| | Fig. 1 (a) UV-Visible spectral changes of 2-Cr (1 mM, black line) upon addition of NO(g) in CH3CN under Ar at 298 K. Inset: FT-IR spectra of 4-CrNO (black line) and 4-Cr15NO (red line) in KBr. (b) ESI-MS spectra of 4-CrNO The peak at m/z 387.1 is assigned to [(BPMEN)Cr(NO)(Cl)]+ (calcd m/z 387.1). Inset: isotopic distribution pattern for 4-CrNO (red line) and 4-Cr15NO (blue line). The peak at m/z 357.1 marked with asterisk is assigned to [(BPMEN)Cr(Cl)]+, which is the starting CrII-complex. | |
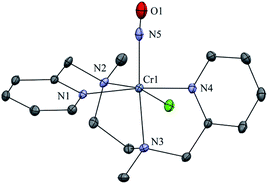 |
| | Fig. 2 Displacement ellipsoid plot (20% probability) of 4-CrNO at 296 K. Disorder in N-atom of NO ligand is refined, anion and H-atoms have been removed for clarity. | |
NO transfer reaction from 1-CoNO to 2-Cr
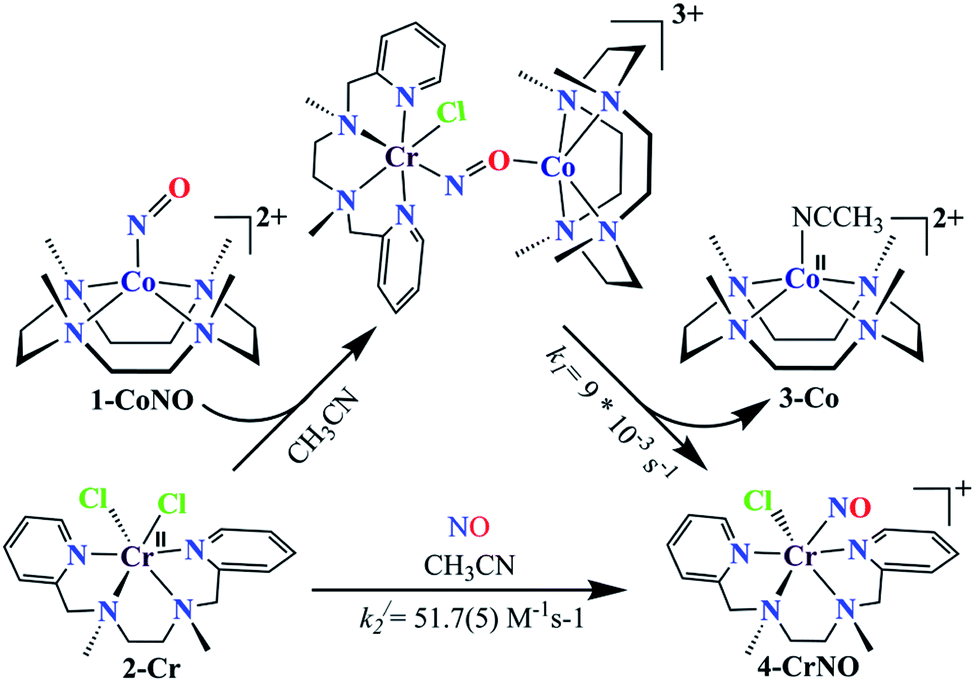
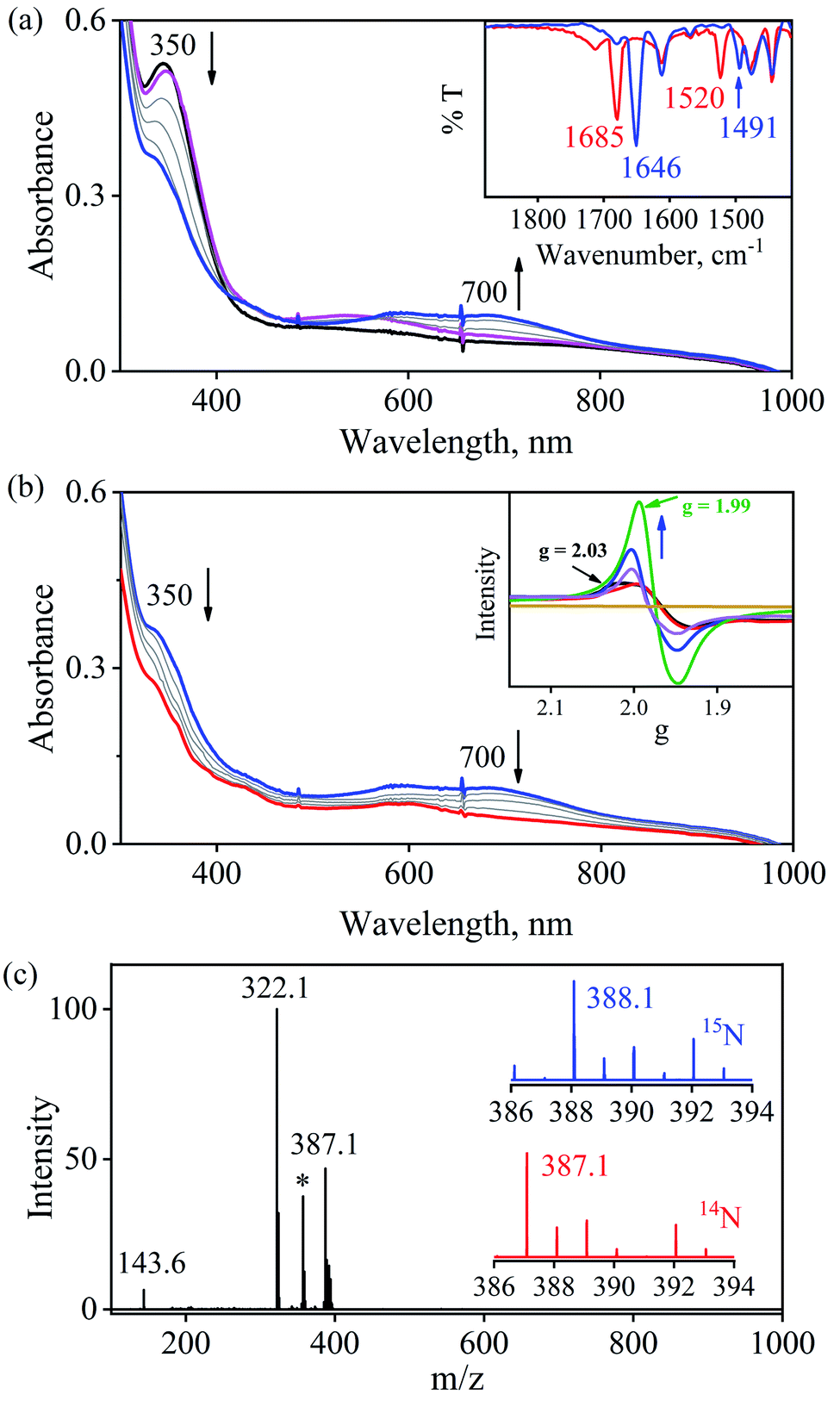
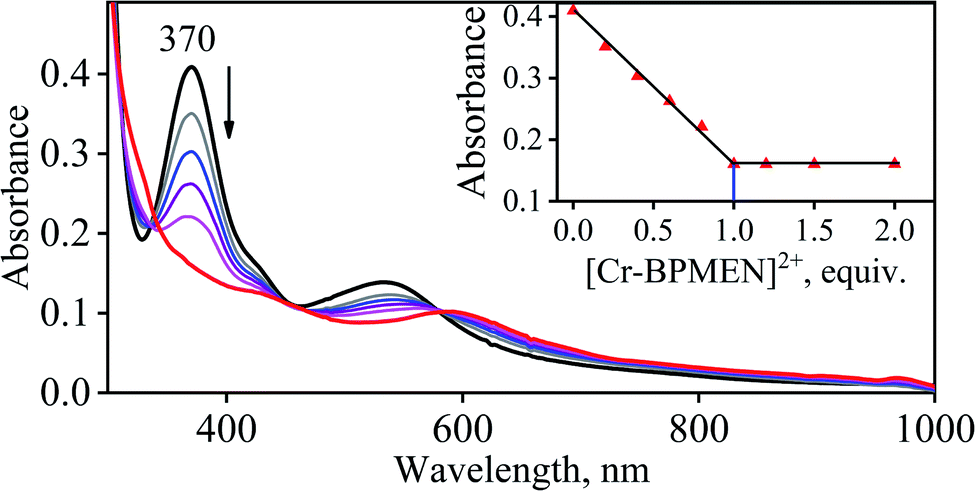
To further understand the NO-coordination chemistry, we explored NO transfer reactions between cobalt (1-CoNO) and chromium complexes (2-Cr & 5-Cr), having different or alike ligand frameworks to determine the fundamental parameters behind NO transfer chemistry. When CH3CN solution of 4-CrNO reacted with an equivalent amount of 3-Co under Ar atmosphere, we did not observe any significant change in the UV-Visible spectra except a new band at 485 nm, corresponding to 3-Co (ESI,† Fig. S5). In contrast to the NO transfer from complex 4-CrNO to 3-Co, the reverse reaction, i.e., NO transfer from 1-CoNO to 2-Cr, showed a visible color change from pink to bluish-green. The addition of one equivalent of 2-Cr to 1-CoNO immediately showed the generation of a new band (λmax = 700 nm, blue line) (Fig. 3a), proposed to be an intermediate, which then slowly changed to a new absorption band (λmax = 600 nm, red line), which corresponds to the absorption spectrum of 4-CrNO (Fig. 3b and S6a; ESI†) (Scheme 2, reaction I & II). The final UV-Visible spectrum of the above NO transfer reaction was confirmed as the sum of complexes 3-Co and 4-CrNO, further supporting the NO transfer from Co to Cr-center (ESI,† Fig. S6b). The NO transfer reaction was established by the spectroscopic titration at 370 nm for the decomposition of 1-CoNO, demonstrating that the ratio-metric equivalent of complex 2-Cr to 1-CoNO was 1:1 (Fig. 4). We further characterized the generation of this intermediate by FT-IR spectroscopy to follow the reaction mechanism. The addition of 2-Cr to the solution of 1-CoNO showed a new peak at 1520 cm−1, which shifted to 1491 cm−1 when the reaction was performed with 1-Co15NO (Inset, Fig. 3a), suggesting the generation of μ-NO bridged species34 {BPMEN-Cr–NO–Co-12-TMC}4+ ({Cr–NO–Co}4+), in the NO transfer reaction from Co to Cr-center. Additionally, EPR measurements were performed to trap the intermediate. In the low-temperature EPR measurements, we observed the formation of an intermediate species (g = 2.03) that shifted to a signal (g = 1.99) (inset, Fig. 3b) characteristic to the EPR signal of 4-CrNO. This EPR spectrum suggests that the NO moiety is more on the Cr-center than Co-center in the {Cr–NO–Co}4+ intermediate species, as both 1-CoNO and 2-Cr are EPR silent and only 4-CrNO showed a peak in this range of g-values. Then, we further tried to characterize the intermediate via ESI-MS. However, being a thermally unstable moiety, the intermediate species decomposes under the mass spectrometry instrumental environment even at low temperatures and gives a peak for 4-CrNO and 3-Co, and efforts to characterize the intermediate become futile.
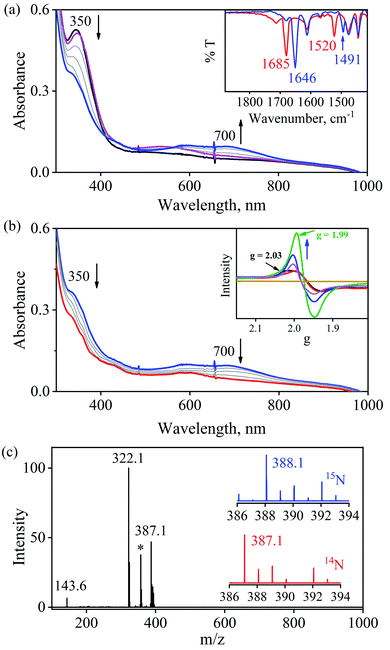 |
| | Fig. 3 (a) UV-Visible spectral changes showing the formation of an intermediate species (blue line) in the reaction of 2-Cr (0.50 mM, black line) with 1-CoNO (0.50 mM) under an Ar atmosphere in CH3CN at 273 K. Inset: FT-IR spectra of the reaction solution of 1-CoNO with 2-Cr (red line) and 1-Co15NO with 2-Cr (blue line). (b) Decomposition of the intermediate species to 4-CrNO (red line). Inset: time-dependent EPR spectra of intermediate formation (black line) in the reaction of 2-Cr and 1-CoNO and finally conversion to end product (4-CrNO, green line) in CH3CN at 77 K (c) ESI-MS spectra of the reaction mixture obtained in the reaction of 1-CoNO with 2-Cr. The peaks at m/z 387.1, 322.1, 143.6 are assigned to [(BPMEN)Cr(NO)(Cl)]+ (calcd m/z 387.1), [(12-TMC)Co(Cl)]+ (calcd m/z 322.1) and [(12-TMC)Co]2+ (calcd m/z 143.6). The peak at m/z 357.1 marked with an asterisk is assigned to [(BPMEN)Cr(Cl)]+. Isotopic distribution pattern for 4-CrNO (red line) and 4-Cr15NO (blue line). | |
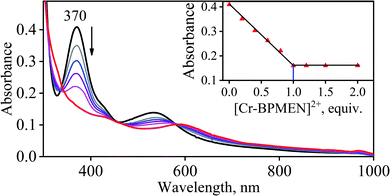 |
| | Fig. 4 UV-Vis spectral changes observed in the reaction 1-CoNO with 2-Cr (in the increments of 0, 0.20, 0.40, 0.60, 0.80, 1.0, 1.2, 1.5, 2.0 equivalent) in CH3CN under Ar at 298 K. | |
Furthermore, intermolecular NO transfer was confirmed by various spectroscopic techniques. The FT-IR spectrum of the reaction mixture obtained after the completion of the reaction of 2-Cr with 1-CoNO, in the KBr palette, exhibited a peak at 1690 cm−1, which shifted to 1650 cm−1 when the reaction was carried out using 15N labelled-NO complex 1-CoNO (1-Co15NO) (ESI,† Fig. S7a). The aforesaid FT-IR spectrum superimposes the FT-IR spectrum of an independently prepared equimolar mixture of 3-Co and 4-CrNO, again confirming the generation of {Cr(NO)}5 (ESI,† Fig. S7b). The ESI-MS of the above reaction mixture showed prominent ion peaks at m/z 143.6, 322.1, and 387.1, respectively, and their mass and isotope distribution patterns correspond to [(12-TMC)Co]2+ (calcd m/z 143.6), [(12-TMC)Co(Cl)]+ (calcd m/z 322.1), and [(BPMEN)Cr(Cl)(NO)]+ (calcd m/z 387.1) (Fig. 3c). When the above reaction was performed by using 1-Co15NO, the mass peak shifted by one unit and exhibited a prominent peak at m/z 388.1, which corresponds to [(BPMEN)Cr(Cl)(15NO)]+ and confirms the transfer of Co-bound NO to Cr-center (Fig. 3c and S7c, ESI†). The 1H-NMR of the reaction mixture does not show any signals for the protons of the 12-TMC ring, which was initially present for 1-CoNO (d6, S = 0), suggesting the generation of 3-Co (low spin d7, S = 1/2) (ESI,† Fig. S8).4b,25f Further, 1H-NMR titration also confirmed the 1:1 stoichiometry of 2-Cr to 1-CoNO (ESI,† Fig. S9). Based on the above evidence, we can conclude that complexes 3-Co and 4-CrNO were generated in the reaction of 1-CoNO with 2-Cr.
DFT calculations and experimental details for mechanistic insight into the NO transfer reaction of 1-CoNO with 2-Cr
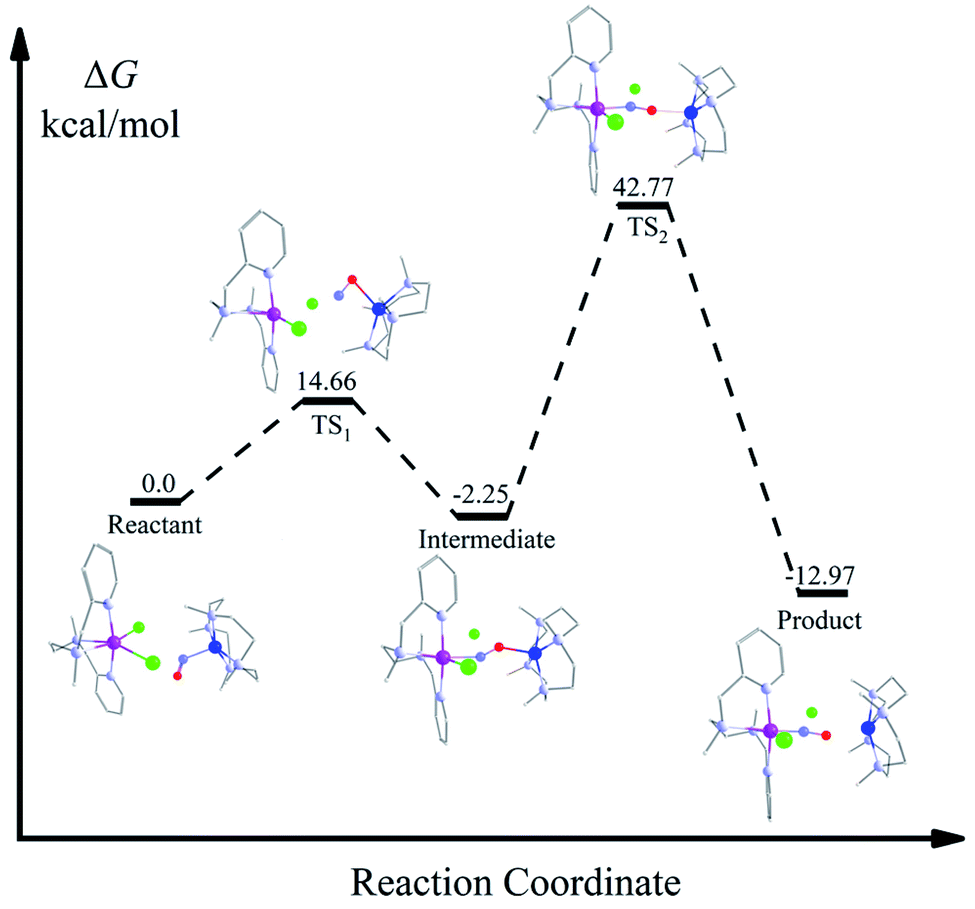
From the spectroscopic and kinetic results, it is evident that the reaction is going through the formation of a bridged NO intermediate species. However, due to the instability of this intermediate, we were unable to characterize it structurally. Therefore, we investigated the reaction pathway of the intermediate formation by carrying out a DFT calculation. DFT calculation suggests the formation of an isonitrosyl-Co complex in the first transition state (TS1) with an energy barrier of 14.66 kcal mol−1 (Fig. 5). In the next step, NO binds to the Cr via the N-donor site, which is −2.25 kcal mol−1 lower in energy than the reactant and therefore formed a thermodynamically favored side-on bound {Cr–NO–Co}4+ intermediate species.35 The experimental results well justified this; the influence of both the metal-centers leads to the lower stretching frequency from 1703 cm−1 for 1-CoNO to 1520 cm−1 for the side on bonded {Cr–NO–Co}4+ species. In addition to IR spectroscopy, a parallel trend was observed in low-temperature EPR measurements. When we reacted 1-CoNO with 2-Cr, a new peak formation for the intermediate species was observed with g = 2.03, corresponding to a low spin Cr-center having a strong interaction of the N-atom of NO moiety to the Cr-center than Co-center (distance of N-atom of NO in intermediate, Cr–N = 228 pm and Co–N = 303 pm). This peak shifted to g = 1.99 (inset: Fig. 3b), corresponding to 4-CrNO when the NO transfer completes. Further, the coordinated NO moiety undergoes dissociation leading to the final NO transfer product. The highest lying transition state TS2 along the reaction profile is 42.77 kcal mol−1, more elevated than the reactant, significantly higher than the 14.66 kcal mol−1 barrier of TS1, indicating that the dissociation of the intermediate to the product is the rate-determining step. This high-energy TS2 leads the reaction to a first-order reaction independent of the concentration of both 1-CoNO and 2-Cr.
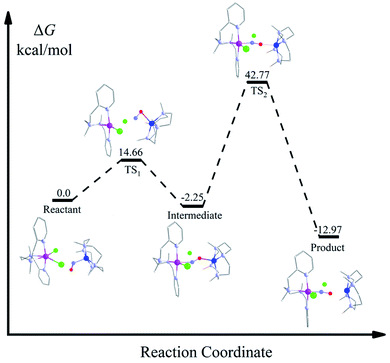 |
| | Fig. 5 : DFT-calculated associative reaction profile of NO-transfer from 1-CoNO to 2-Cr initiated from a μ-NO-bridged intermediate {Cr–NO–Co}4+. | |
NO transfer reaction from 1-CoNO to 5-Cr
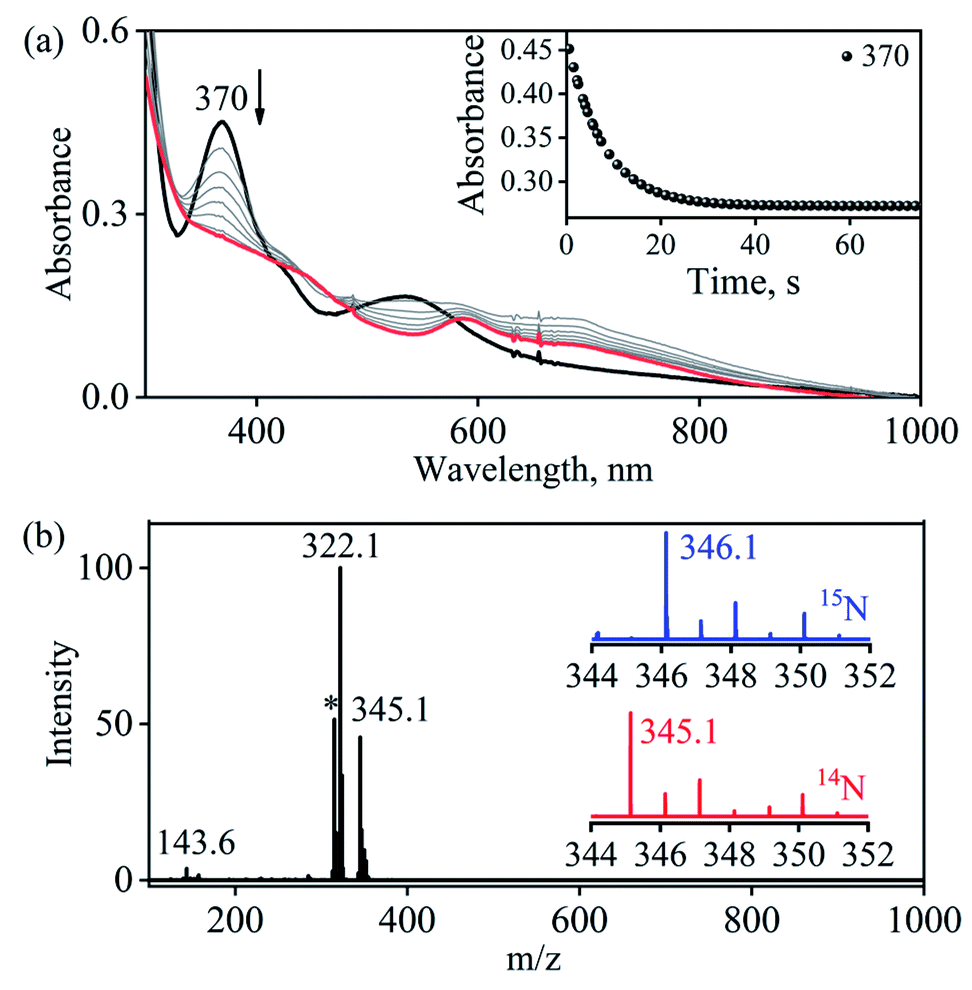
Complex 1-CoNO eventually showed the NO transfer reactivity with [(12-TMC)Cr(Cl)]+ (5-Cr) (Scheme 3). The addition of one equivalent of 5-Cr to the solution of 1-CoNO leads to the disappearance of the band at 370 nm (Fig. 6a). Further analysis of the reaction mixture by the ESI-MS measurements showed prominent ion peaks at m/z 143.6, 345.1 and 322.1, corresponding to [(12-TMC)Co]2+ (calcd m/z 143.6), [(12-TMC)Cr(NO)(Cl)]+ (6-CrNO) (calcd m/z 345.1), and [(12-TMC)Co(Cl)]+ (calcd m/z 322.1), respectively (Fig. 6b). The mass peaks shifted by one unit to m/z 346.1 corresponds to [(12-TMC)Cr(15NO)(Cl)]+ (calcd m/z 346.1) when the reaction is performed with 1-Co15NO confirms the NO transfer from complex 1-CoNO to 5-Cr (ESI,† Fig. S12). Further, stoichiometric titration for NO transfer reaction was followed at 370 nm for the decomposition of 1-CoNO, which showed that the ratio-metric equivalent of complex 5-Cr to 1-CoNO was 1:1 (ESI,† Fig. S13).
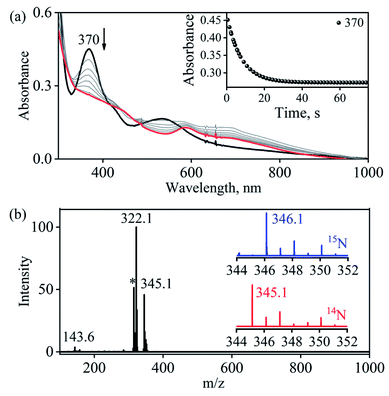 |
| | Fig. 6 (a) UV-Visible spectral changes obtained in the NO transfer from 1-CoNO (0.50 mM; black line) to 5-Cr (0.50 mM) to form 6-CrNO (brick red line) under an Ar atmosphere in CH3CN at 273 K. Inset: time course of the decomposition of 1-CoNO monitored at 370 nm (black circles) (b) ESI-MS spectra of the reaction mixture obtained in the reaction of 5-Cr with 1-CoNO. The peaks at m/z 345.1, 322.1, 143.6 are assigned to [(12-TMC)Cr(NO)(Cl)]+ (calcd m/z 345.1) [(12-TMC)Co(Cl)]+ (calcd m/z 322.1) and [(12-TMC)Co]2+ (calcd m/z 143.6). The peak at m/z 314.1 marked with asterisk is assigned to [(12-TMC)CrII(Cl)]+ (calcd m/z 314.1). Inset: isotopic distribution pattern for 6-CrNO (red line) and 6-Cr15NO (blue line). | |
Discussion
The mechanistic insights of the NO transfer reaction
To elaborate on the chemistry involved in the NO transfer reaction of 1-CoNO with 2-Cr, why is it happening? We then explored the NO transfer reaction's mechanistic insight. There are two possible mechanistic approaches for the NO transfer reactions in M-NOs, i.e., associative or dissociative pathways (vide infra).25a,25d,25f As shown in Fig. 3, the reaction of 1-CoNO with 2-Cr is proceeding via the formation of an intermediate species (Fig. 3a, blue line) before generating the final product (Fig. 3b, red line), suggesting an associative mechanistic pathway. The UV-Visible spectrum of this intermediate species is different from the sum of the spectra of complexes 1-CoNO + 2-Cr, indicating that it is a new species rather than the mixture of 1-CoNO & 2-Cr (ESI,† Fig. S10). Suppose NO transfer is going through an associative pathway, then the dissociation of the intermediate species should be the rate-determining step. In that case, the reaction rate will be independent of 2-Cr.25f Kinetics study revealed that the reaction rate was independent of the concentration of 2-Cr, suggesting an associative intermediate species before NO transfer. In addition, the DFT calculation suggests an energy barrier of 42.77 kcal mol−1 in TS2, a 28 kcal mole−1 higher-lying transition state than TS1. Due to this higher energy barrier, the formation of the product from intermediate via TS2 becomes slow and makes the reaction first order. Further, the formation of a peak at 1520 cm−1 in IR spectroscopy also supports the generation of a μ-NO bridged34 transient species (Scheme 2) in the above reaction.25b,25d,36
The NO transfer reaction between 1-CoNO and 2-Cr was found to be following the first-order kinetic with a first-order rate constant (k1) of 9 × 10−3 s−1. Hence, the dissociation of the proposed {Cr–NO–Co}4+ intermediate is supposed to be a rate-determining step, which is ∼103 times slower than the earlier reported second-order formation rate-constant of 1-CoNO (k2 = 12(1) M−1 s−1).4b To find the driving forces of the NO transfer reaction, the second-order rate constant (k2/) and equilibrium constant (Keq/) of the formation of 4-CrNO were determined and found to be 51.7(5) M−1 s−1 and 2.27 × 103 M−1 (ESI,† Fig. S11), respectively, which are ∼4 times and ∼2.5 times larger than the k2 (12(1) M−1 s−1) and Keq (8.90 × 102 M−1) of 1-CoNO, respectively. This comparison of the rate-constants and equilibrium constants suggests clearly that the dissociation of the {Cr–NO–Co}4+ intermediate should generate thermodynamically more stable molecules 4-CrNO & 3-Co, as proposed by the second-order rate constant and equilibrium constant for 1-CoNO & 4-CrNO.
Further, various thermodynamic properties were also determined for the generation of 1-CoNO & 4-CrNO (Table 1), which suggests that the formation of 4-CrNO (ΔG‡ = 15.71 kcal mol−1, ΔG0 = −4.57 kcal mol−1) is thermodynamically more favorable than the formation of 1-CoNO (ΔG‡ = 16.43 kcal mol−1, ΔG0 = −4.02 kcal mol−1). The thermodynamic stability of these molecules can also be explained based on the labile and inert behavior of the metal-centers.37 The generation of an inert species 4-CrNO (low spin d5, S = 1/2) from a highly labile 2-Cr (high spin d4, S = 2) is one of the major driving forces of NO transfer, as reported in many other reports.25f,37 In addition to labile and inert parameters and second-order formation rate constants for 1-CoNO & 4-CrNO, thermodynamic parameters (Table 1) further support our hypothesis as the generation of 3-Co & 4-CrNO from 1-CoNO & 2-Cr is due to the lower free energy for 4-CrNO than 1-CoNO, making the NO transfer reaction more spontaneous. Thus, apart from the various parameters (vide supra), other factors may affect the NO transfer reactivity. Logically thinking about the NO transfer chemistry clarifies that this reaction depends not only on various metal-center parameters,25d but also on the ligand frameworks, which may influence the transfer of NO from Co to Cr metal-center.25f
Table 1 Activation Parameters for 1-CoNO, 4-CrNO and 6-CrNO
| Complex |
ΔH‡ (kcal mol−1) |
ΔS‡ (cal mol−1 K) |
ΔG‡298 K (kcal mol−1) |
|
1-CoNO
|
3.93 ± 0.2 |
−41.94 ± 0.6 |
16.43 ± 0.2 |
|
4-CrNO
|
7.32 ± 0.3 |
−28.15 ± 1.4 |
15.71 ± 0.3 |
|
6-CrNO
|
5.21 ± 0.06 |
−36.77 ± 0.3 |
16.17 ± 0.12 |
With an aim to determine the effect of the ligand frameworks, the NO transfer reaction was performed between Co and Cr bearing the same 12-TMC ligand framework. Complex 1-CoNO also showed NO transfer to 5-Cr and generated 6-CrNO. While further exploring the reaction mechanism, we have investigated the reaction of 5-Cr with 1-CoNO in various ratios. The characteristic UV-Visible peaks of 1-CoNO overlap with 5-Cr made it difficult to perform the kinetic comparison in the presence of the higher amounts of 5-Cr. Complex 1-CoNO is thermodynamically stable and does not show any natural decay (ESI,† Fig. S14); hence, the NO transfer's plausible mechanism from 1-CoNO to 5-Cr suggests that the reaction may go through a transient intermediate species. We did not observe any intermediate species formation in the UV-Visible or FT-IR spectral measurements (Fig. 6a), which suggests that the strain between the 12-TMC ligand frameworks of 1-CoNO & 5-Cr causes a fast transfer of NO from Co to Cr-center. Since the NO transfer is first-order in nature, the formation of 6-CrNO is believed to be the rate-determining step, which is equal to the overall reaction rate with a rate constant (k1/) of 3.1 × 10−2 s−1. The second-order rate constant (k2//) and equilibrium constant (Keq//) of the formation of 6-CrNO were also determined and found to be 29.3(2) M−1 s−1 and 1.48 × 103 M−1, respectively, which are ∼2.5 times and ∼1.6 times larger than the second-order rate constant and equilibrium constant of 1-CoNO,25f hence explaining why NO transfers from Co to Cr-center. In addition, other thermodynamic parameters for 1-CoNO (ΔG‡ = 16.43 kcal mol−1, ΔG0 = −4.02 kcal mol−1) and 6-CrNO (ΔG‡ = 16.17 kcal mol−1, ΔG0 = −4.32 kcal mol−1) further supports why NO transfer reaction between 1-CoNO & 5-Cr generates 6-CrNO (Table 1). In addition to the above experimental results and their interpretations, labile and inert nature of more stable 6-CrNO (low spin d5, S = 1/2, inert) (Fig. S15a†) and less stable 5-Cr (high spin d4, S = 2, labile) (Fig. S15b†) suggests a huge change in the spin state, which promotes the reaction to move in the forward direction.37,38 The above discussion suggests that the ligand frameworks may influence the intermediate's stability and the rate of NO transfer; however, when it comes to the net transfer of coordinated NO moiety from one metal to another, it largely depends upon the stability of the metal-center and various other physical parameters.
Conclusion
In this report, for the very first time, we have demonstrated the NO transfer reactivity of CoIII-nitrosyl, which can be regulated by (a) the thermal stability of the end products, (b) by varying the ligand frameworks (open chain/cyclic) of the supporting ligands, and (c) the change in the labile/inert nature of Cr metal-center before and after NO coordination. Here, we explored the NO transfer reaction of [(12-TMC)Co(NO)]2+ (1-CoNO) with a CrII-complex, [(BPMEN)CrII(Cl)2] (2-Cr). For the very first time, we spectroscopically characterized (by UV-Visible, FT-IR, and EPR) the formation of a μ-NO bridged intermediate {BPMEN-Cr–NO–Co-12-TMC}4+ ({Cr–NO–Co}4+) before the NO transfer from 1-CoNO to 2-Cr, generating [(BPMEN)Cr(NO)(Cl)]+ (4-CrNO) (Scheme 2), with no NO transfer of Cr-bound NO. These results were interpreted based on the significant thermal stability of {CrNO}5, explained based on various physical parameters, i.e., rate constant (k), Gibbs free energy of activation (ΔG), and labile-inert nature of metal-centers. The second-order rate constant (k2/) and equilibrium constant (Keq/) of the formation of 4-CrNO was ∼4 times and ∼2.5 times that of 1-CoNO; subsequently, it pushes the NO transfer from Co to Cr-center to generate 4-CrNO. The free energy of activation of 4-CrNO is lower than that of 1-CoNO; hence, driving the NO transfer reaction towards the forward direction to generate thermodynamically more inert/stable 4-CrNO. Also, the formation of an inert 4-CrNO (low spin d5, S = 1/2) from a labile 2-Cr (high spin d4, S = 2) is another major driving force of NO transfer to achieve more stability.25f,37 Additionally, the structure of the intermediate was predicted from a DFT calculation. The results from DFT calculations suggest a μ-NO bridged intermediate that binds in a side-on fashion to both Co and Cr-center, having a strong interaction of NO with Cr-center than Co-center, and is in qualitative agreement with other experimental results. NO transfer reaction was also analyzed by taking [(12-TMC)CrII(Cl)]+ (5-Cr) to evaluate the effect of the ligand framework. Complex 1-CoNO showed the transfer of its NO moiety to 5-Cr without observing any intermediate species generating thermodynamically stable 6-CrNO (vide supra), which may be due to the high strain caused by the methyl groups in the 12-TMC ligand and its cyclic nature.25f The BPMEN is an open-chain ligand with only two methyl groups, probably making it adapt/change its geometry to form the proposed intermediate. This is quite interesting because two CrII-complexes show NO transfer reactivity due to the higher stability of the final {CrNO}5 adduct; however, the ligand frameworks significantly influence the stability of the intermediate and hence control the reaction's fate. The present results describe original findings, where the generation of μ-NO bridged intermediate was observed before NO transfer and characterized spectroscopically and theoretically.
Data availability
All the required data already provided in the ESI† and manuscript.
Author contributions
PK & SD discovered/conceptualized the initial project. SD, Kulbir, & SG carried out the different experiments and gathered the data. SR & PM carried out the DFT calculation, and PM analyzed the data. PK & TD interpreted the experimental results. PK & SD wrote the first draft of the article. PK, TD & PM corrected the manuscript, finalized the final draft, and guided during the revision. PK followed and guided the whole project work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grants-in-Aid (Grant No. EEQ/2016/000466) from SERB-DST and DAAD (ID: 57552334). S. D., Kulbir, S. G., S. R. thank IISER Tirupati for their fellowship. PM thanks DST, SERB, GoI (SRG/2020/001354) for financial support. We thank Mr Vamsi Katta (IISER Tirupati) and Mr S. Subramanian (SAIF, IIT Madras) for doing some specific experiments. Thanks to SAIF (IIT Madras) for the EPR facility.
References
-
(a)
L. R. MacGillivray and C. M. Lukehart, Metal–organic framework materials, John Wiley & Sons, 2014 Search PubMed;
(b) H. J. Kruger, G. Peng and R. H. Holm, Inorg. Chem., 2002, 30, 734–742 CrossRef;
(c) X. J. Yao, G. Velez Ruiz, M. R. Whorton, S. G. Rasmussen, B. T. DeVree, X. Deupi, R. K. Sunahara and B. Kobilka, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9501–9506 CrossRef CAS PubMed;
(d) K. Oettl and R. E. Stauber, Br. J. Pharmacol., 2007, 151, 580–590 CrossRef CAS PubMed;
(e) J. Anastassopoulou, J. Mol. Struct., 2003, 651–653, 19–26 CrossRef CAS.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. Murphy, Science, 2004, 304, 867–870 CrossRef CAS PubMed.
-
(a) J. B. Weinberg, Y. Chen, N. Jiang, B. E. Beasley, J. C. Salerno and D. K. Ghosh, Free Radicals Biol. Med., 2009, 46, 1626–1632 CrossRef CAS PubMed;
(b) R. A. Firth, H. A. O. Hill, J. M. Pratt, R. G. Thorp and R. J. P. Williams, J. Chem. Soc. A, 1969, 381–386, 10.1039/j19690000381.
-
(a) A. Yokoyama, K. B. Cho, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2013, 135, 14900–14903 CrossRef CAS PubMed;
(b) P. Kumar, Y. M. Lee, Y. J. Park, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2015, 137, 4284–4287 CrossRef CAS PubMed.
-
(a)
T. J. Meyer and H. Taube, in Comprehensive Coordination Chemistry, Pergamon Press, Oxford, ed Wilkinson G., 1987, p. 331 Search PubMed;
(b) T. M. Shea, S. P. Deraniyagala, D. B. Studebaker and T. D. Westmoreland, Inorg. Chem., 1996, 35, 7699–7703 CrossRef CAS.
- T. Morimoto, K. Fuji, K. Tsutsumi and K. Kakiuchi, J. Am. Chem. Soc., 2002, 124, 3806–3807 CrossRef CAS PubMed.
- S. Hong, K. D. Sutherlin, J. Park, E. Kwon, M. A. Siegler, E. I. Solomon and W. Nam, Nat. Commun., 2014, 5, 5440 CrossRef CAS PubMed.
-
(a) J. Cho, R. Sarangi, H. Y. Kang, J. Y. Lee, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2010, 132, 16977–16986 CrossRef CAS PubMed;
(b) J. Cho, R. Sarangi, J. Annaraj, S. Y. Kim, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, Nat. Chem., 2009, 1, 568–572 CrossRef CAS PubMed.
- L. K. Woo, Chem. Rev., 2002, 93, 1125–1136 CrossRef.
-
(a)
P. R. O. De Montellano, Cytochrome P450: structure, mechanism, and biochemistry, Springer Science & Business Media, 2005 CrossRef;
(b)
I. Schuster, Cytochrome P-450: biochemistry and biophysics, Hemisphere Pub, 1989 Search PubMed;
(c) R. H. Holm, Chem. Rev., 1987, 87, 1401–1449 CrossRef CAS;
(d) R. H. Holm, Coord. Chem. Rev., 1990, 100, 183–221 CrossRef CAS.
-
(a) L. I. Hochstein and G. A. Tomlinson, Annu. Rev. Microbiol., 1988, 42, 231–261 CrossRef CAS PubMed;
(b) W. H. Campbell, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1999, 50, 277–303 CrossRef CAS PubMed.
-
(a) K. D. Karlin, Science, 1993, 261, 701–708 CrossRef CAS PubMed;
(b) C. Perez-Rizquez, A. Rodriguez-Otero and J. M. Palomo, Org. Biomol. Chem., 2019, 17, 7114–7123 RSC.
-
(a) G. B. Richter-Addo, P. Legzdins and J. Burstyn, Chem. Rev., 2002, 102, 857–860 CrossRef CAS PubMed;
(b) C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed;
(c) L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221–226 CrossRef CAS PubMed;
(d) R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS;
(e) L. J. Ignarro, Biosci. Rep., 1999, 19, 51–71 CrossRef CAS PubMed;
(f)
L. J. Ignarro, Nitric Oxide: Biology and Pathobiology, Academic press, 2000 Search PubMed.
- B. Voetsch, R. C. Jin and J. Loscalzo, Histochem. Cell Biol., 2004, 122, 353–367 CrossRef CAS PubMed.
- C. S. Wilcox, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2005, 289, R913–R935 CrossRef CAS PubMed.
- C. Baylis, Am. J. Physiol. Ren. Physiol., 2008, 294, F1–F9 CrossRef CAS PubMed.
-
(a) F. Vargas, J. M. Moreno, R. Wangensteen, I. Rodriguez-Gomez and J. Garcia-Estan, Eur. J. Endocrinol., 2007, 156, 1–12 CAS;
(b) S. Cook, O. Hugli, M. Egli, P. Vollenweider, R. Burcelin, P. Nicod, B. Thorens and U. Scherrer, Swiss Med. Wkly., 2003, 133, 360–363 Search PubMed.
-
(a) L. Castillo, T. C. deRojas, T. E. Chapman, J. Vogt, J. F. Burke, S. R. Tannenbaum and V. R. Young, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 193–197 CrossRef CAS PubMed;
(b) R. M. Palmer, D. S. Ashton and S. Moncada, Nature, 1988, 333, 664–666 CrossRef CAS PubMed.
- B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed.
-
(a) Q. Liu and S. S. Gross, Methods Enzymol., 1996, 268, 311–324 CAS;
(b) C. Nathan and Q. W. Xie, J. Biol. Chem., 1994, 269, 13725–13728 CrossRef CAS PubMed.
-
(a) B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed;
(b) J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Nat. Rev. Drug Discovery, 2008, 7, 156–167 CrossRef CAS PubMed.
-
(a) R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef CAS PubMed;
(b) C. H. Lim, P. C. Dedon and W. M. Deen, Chem. Res. Toxicol., 2008, 21, 2134–2147 Search PubMed.
-
(a)
G. B. Richter-Addo and P. Legzdins, Metal nitrosyls, Oxford University Press, 1992 Search PubMed;
(b) P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993–1018 CrossRef CAS PubMed;
(c) G. R. Wyllie and W. R. Scheidt, Chem. Rev., 2002, 102, 1067–1090 CrossRef CAS PubMed;
(d) N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121(24), 14682–14905 CrossRef CAS PubMed.
-
(a) S. Das, Kulbir, S. Ghosh, S. Chandra Sahoo and P. Kumar, Chem. Sci., 2020, 11, 5037–5042 RSC;
(b) B. Mondal, D. Borah, R. Mazumdar and B. Mondal, Inorg. Chem., 2019, 58, 14701–14707 CrossRef CAS PubMed;
(c) S. Ghosh, H. Deka, Y. B. Dangat, S. Saha, K. Gogoi, K. Vanka and B. Mondal, Dalton Trans., 2016, 45, 10200–10208 RSC.
-
(a) G. Metzker, P. P. Lopes, A. C. da Silva, S. C. da Silva and D. W. Franco, Inorg. Chem., 2014, 53, 4475–4481 CrossRef CAS PubMed;
(b) A. Sacco, G. Vasapollo and P. Giannoccaro, Inorg. Chim. Acta, 1979, 32, 171–174 CrossRef CAS;
(c) C. B. Ungermann and K. G. Caulton, J. Am. Chem. Soc., 1976, 98, 3862–3868 CrossRef CAS;
(d) K. J. Franz and S. J. Lippard, Inorg. Chem., 2000, 39, 3722–3723 CrossRef CAS PubMed;
(e) J. Armor, Inorg. Chem., 1973, 12, 1959–1961 CrossRef CAS;
(f) P. Kumar, Y. M. Lee, L. Hu, J. Chen, Y. J. Park, J. Yao, H. Chen, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2016, 138, 7753–7762 CrossRef CAS PubMed.
- R. L. Roberts, D. W. Carlyle and G. L. Blackmer, Inorg. Chem., 2002, 14, 2739–2744 CrossRef.
- X. H. Mu and K. M. Kadish, Inorg. Chem., 1990, 29, 1031–1036 CrossRef CAS.
- M. P. Doyle, R. A. Pickering, R. L. Dykstra and B. R. Cook, J. Am. Chem. Soc., 1982, 104, 3392–3397 CrossRef CAS.
- P. Kumar, Y. M. Lee, L. Hu, J. Chen, Y. J. Park, J. Yao, H. Chen, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2016, 138, 7753–7762 CrossRef CAS PubMed.
- Kulbir, S. Das, T. Devi, M. Goswami, M. Yenuganti, P. Bhardwaj, S. Ghosh, S. C. Sahoo and P. Kumar, Chem. Sci., 2021, 12, 10605–10612 RSC.
-
(a)
H. Lewandowska, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine I, ed. D. M. P. Mingos, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, DOI:10.1007/430_2013_109, pp. 115–165;
(b) A. Yokoyama, J. E. Han, J. Cho, M. Kubo, T. Ogura, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2012, 134, 15269–15272 CrossRef CAS PubMed.
-
(a) J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS;
(b) A. P. Hunt and N. Lehnert, Acc. Chem. Res., 2015, 48, 2117–2125 CrossRef CAS PubMed.
-
D. J. Thomas and N. Lehnert, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2017, DOI:10.1016/b978-0-12-409547-2.11678-6.
-
(a) J. R. Norton, J. P. Collman, G. Dolcetti and W. T. Robinson, Inorg. Chem., 1972, 11, 382–388 CrossRef CAS;
(b) J. L. Brumaghim, J. G. Priepot and G. S. Girolami, Organometallics, 1999, 18, 2139–2144 CrossRef CAS;
(c) K. A. Kubat-Martin, M. E. Barr, B. Spencer and L. F. Dahl, Organometallics, 1987, 6, 2570–2579 CrossRef CAS;
(d) R. M. Kirchner, T. J. Marks, J. S. Kristoff and J. A. Ibers, J. Am. Chem. Soc., 1973, 95, 6602–6613 CrossRef CAS.
-
(a) R. Mas-Balleste, A. R. McDonald, D. Reed, D. Usharani, P. Schyman, P. Milko, S. Shaik and L. Que Jr, Chem.–Eur. J., 2012, 18, 11747–11760 CrossRef CAS PubMed;
(b) S. K. Sharma, A. W. Schaefer, H. Lim, H. Matsumura, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2017, 139, 17421–17430 CrossRef CAS PubMed.
- J. A. McCleverty, Chem. Rev., 1979, 79, 53–76 CrossRef CAS.
-
G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, Pearson, fifth edn, 2014, pp. 439–441 Search PubMed.
- M. Yenuganti, S. Das, Kulbir, S. Ghosh, P. Bhardwaj, S. S. Pawar, S. C. Sahoo and P. Kumar, Inorg. Chem. Front., 2020, 7, 4872–4882 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2076723. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06803b |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Kulbir
a,
Kulbir