
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-independent activation of photocycloadditions using two colours of light†
Philipp W.
Kamm
abc,
Leona L.
Rodrigues
ab,
Sarah L.
Walden
ab,
James P.
Blinco
 *ab,
Andreas-Neil
Unterreiner
*c and
Christopher
Barner-Kowollik
*ab
*ab,
Andreas-Neil
Unterreiner
*c and
Christopher
Barner-Kowollik
*ab
aCentre for Materials Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: j.blinco@qut.edu.au; christopher.barnerkowollik@qut.edu.au
bSchool of Chemistry and Physics, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia
cMolecular Physical Chemistry Group, Institute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, Geb. 30.44, Karlsruhe 76131, Germany. E-mail: andreas.unterreiner@kit.edu
First published on 14th December 2021
Abstract
We exploit two reactive chromophores to establish sequence-independent photochemical activation, employing ortho-methyl benzaldehyde (oMBA) and N,N-(dimethylamino)pyrene aryl tetrazole (APAT) with N-(2-hydroxy)ethyl maleimide (NHEM), without any additives. Critically, the order of the irradiation sequence is irrelevant, as the shorter wavelength does not activate the higher wavelength activated species. Therefore, full sequence-independent λ-orthogonality is achieved through differences in both the reaction quantum yields (Φr,oMBA and Φr,APAT) and wavelength-dependent reactivity profiles of the employed chromophores.
Introduction
Spatio-temporal resolution, energy efficiency and innocuousness are hallmarks of photochemistry.1–5 By manipulating the wavelength and intensity of light, chemists are moving towards precise control over multi-component reaction systems.6,7 Independently addressing specific photoreactive moieties alongside each other is called λ-orthogonality,8 wavelength selectivity,9,10 or chromatic orthogonality.11 In recent years, λ-orthogonal systems have gained interest because their one-pot nature offers simplicity over conventional multi-step protection-group chemistry, while maintaining modularity.12 This is demonstrated by a multitude of applications such as advanced synthetic protocols,13–15 post-functionalisation of polymers,16 or tuning of material properties.17 Attaining full λ-orthogonality, however, is a challenging endeavour. It requires exclusively addressing one photo-active site and reaching high conversions, while the other component remains unaffected, and vice versa. This is mostly achieved through designing red-shifted chromophores with distinct absorption spectra.17,18 Yet even red-shifted chromophores usually display significant absorption in the short-wavelength region. Thus, spectral overlap in multi-component arrays is often unavoidable, and irradiation at shorter wavelengths usually triggers both photoactive units.8 The lack of spectral disparity therefore necessitates irradiation with different colours of light in a predefined sequence. Thus, low-energy light is applied first until complete conversion of the long-wavelength selective moiety is achieved, followed by high-energy light to convert the short-wavelength selective moiety.19–23 Exceptions to this constraint mostly involve photocleavable protecting groups or uncaging units,24–26 however examples of fully sequence-independent λ-orthogonal systems for covalent bond formation remain scarce.Two common types of visible or UV light-activated covalent bond forming reactions are the nitrile imine-mediated tetrazole-ene cycloaddition (NITEC) and the o-quinodimethane-mediated Diels–Alder reaction of o-methyl-benzaldehydes (oMBA). Upon irradiation with visible or UV light, tetrazoles eject molecular nitrogen, forming a highly reactive nitrile imine intermediate that can react with a range of substitutes.27 Upon UV irradiation, ortho-methylbenzaldehydes undergo photoisomerization, generating hydroxy-o-quinodimethanes that are able to perform Diels–Alder cycloadditions with electron-deficient enes. Our group recently introduced a combination of 2,5-diphenyltetrazole and oMBA thioether for sequence-independent ligation with 285 nm and 382 nm light.28 In the presence of water, the reaction quantum yield of the latter is significantly lowered and irradiation with 285 nm light therefore exclusively leads to conversion of the tetrazole, meaning that sequence-independence is achieved through switching solvents prior to changing the colour of light. Reducing this preparative complexity is a matter of priority.
When designing a λ-orthogonal reaction system, not only absorption spectra, but also reaction quantum yields must be taken into account.29 This is especially relevant since the wavelength-dependent reactivity often deviates from the absorption profiles of the respective chromophores.30,31 To study wavelength-dependent reactivity, we employed a wavelength-tuneable ns-pulsed laser system as a monochromatic light source to induce photoreactions at specific wavelengths, offering fine control over parameters such as wavelength, energy, and photon count.30
Results and discussion
Herein, we introduce a fully sequence-independent λ-orthogonal reaction system that solely relies on differences in absorption profiles and reaction quantum yields, without the need of altering reaction conditions, or using any kind of additive. We carefully probed the degree of λ-orthogonality at different wavelengths, using a combination of two UV and visible light activated photoactive molecules. Having identified the most suitable wavelengths, we employ a set of two appropriate LEDs to demonstrate the sequence-independence of the reaction system. As the UV-active component we chose a methoxy-substituted oMBA, which is well known to react cleanly and with high reaction quantum yield, forming a cycloadduct with N-(2-hydroxy)ethyl maleimide (Fig. 1, bottom left arrow).32 As the visible light responsive component, we employed a recently developed red-shifted pyrene based tetrazole ((N,N-dimethyl)-aminopyrene aryl tetrazole, APAT) that is able to undergo NITEC reaction upon blue or green light irradiation (Fig. 1, top left arrow).33 We hypothesized that APAT could be a suitable candidate for a sequence-independent λ-orthogonal system when combined with a shorter-wavelength photoreactive compound of higher quantum yield.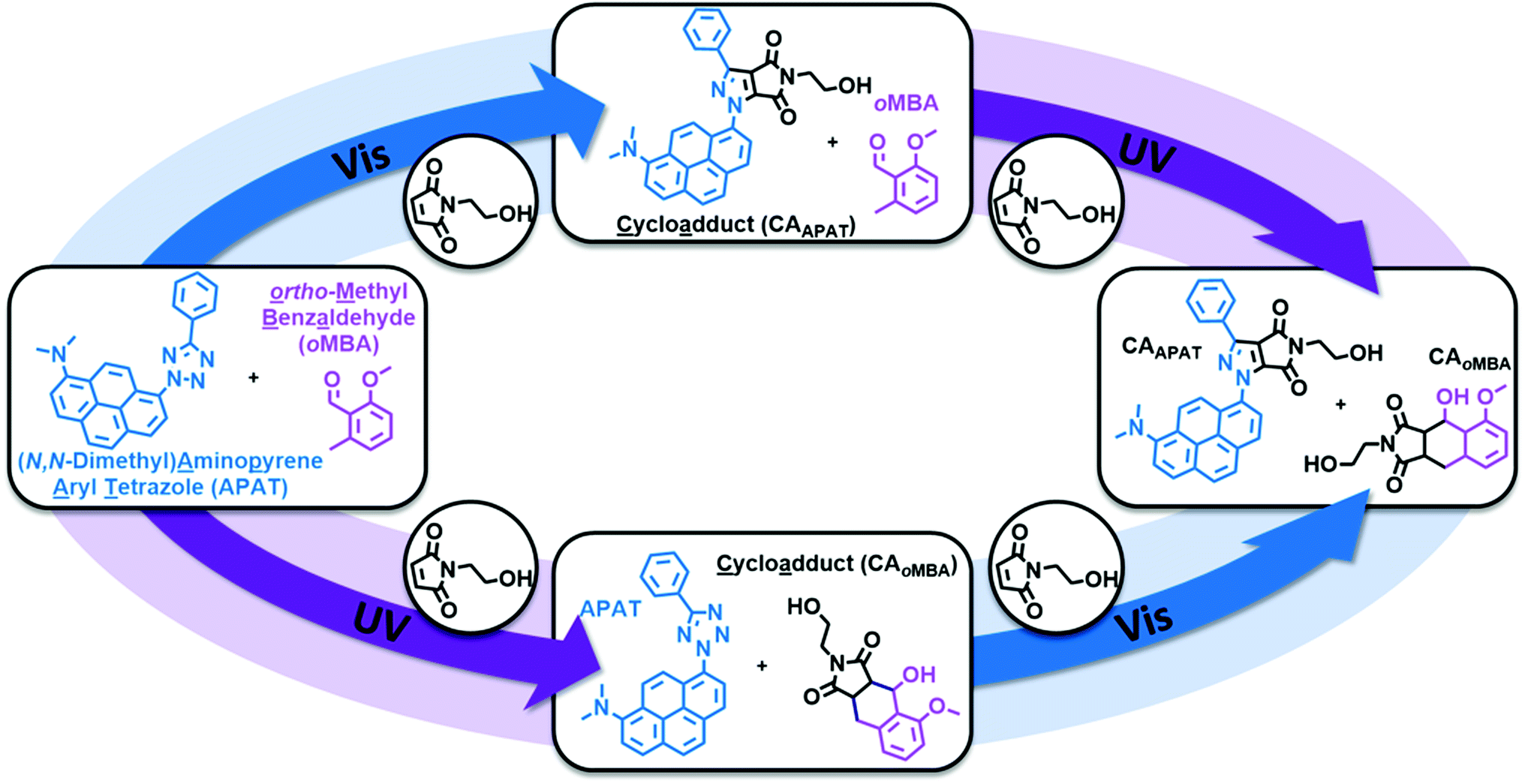 | ||
| Fig. 1 Orthogonal photo-induced reactions of (N,N-dimethyl) aminopyrene aryl tetrazole (APAT) and o-methyl benzaldehyde (oMBA) with N-(2-hydroxy)ethyl maleimide (NHEM) in MeCN. Top left arrow: visible light activated nitrile imine-mediated tetrazole-ene cycloaddition (NITEC). Bottom left arrow: UV light activated o-quinodimethane-mediated Diels–Alder reaction of oMBA. | ||
To determine reaction quantum yields for oMBA and APAT, small molecule ligation reactions were conducted in acetonitrile (MeCN), using N-(2-hydroxy)ethyl maleimide (NHEM) as a ligation partner for both reactions. To obtain a complete picture, we chose a range of wavelengths (Fig. 2a). From previous studies, 320 nm and 450 nm were determined to be among the most efficient wavelengths for triggering the respective photoreactions.32,33 360 nm and 390 nm were chosen as intermediate wavelengths. The reaction products were analysed via liquid chromatography coupled with high-resolution mass spectrometry (LC-MS). oMBA, APAT, as well as their respective cycloadducts CAoMBA and CAAPAT were quantified by correlating the integrals of the chromatogram peaks to the absorption coefficient ε214 nm of the respective species, where 214 nm is the scanning wavelength of the UV detector (refer to Fig. S1†). NHEM was used as a reaction partner because is it a common dipolarophile, and the hydroxy group provides sufficient separation of all involved species on the column, minimising peak overlap (Fig. 2b and c). Photo-induced NITEC yields pyrazoline-derivatives, which can undergo oxidation, forming aromatic pyrazoles. The degree of re-aromatization varies and is difficult to predict. While in most cases the irradiation experiments exclusively formed the pyrazole–cycloadduct, non-aromatic pyrazoline-species were observed as well (Fig. S2†). Attempts to isolate the cycloadduct led to the fully rearomatized pyrazole species. For analysis of the chromatograms, we therefore treated both species as one.
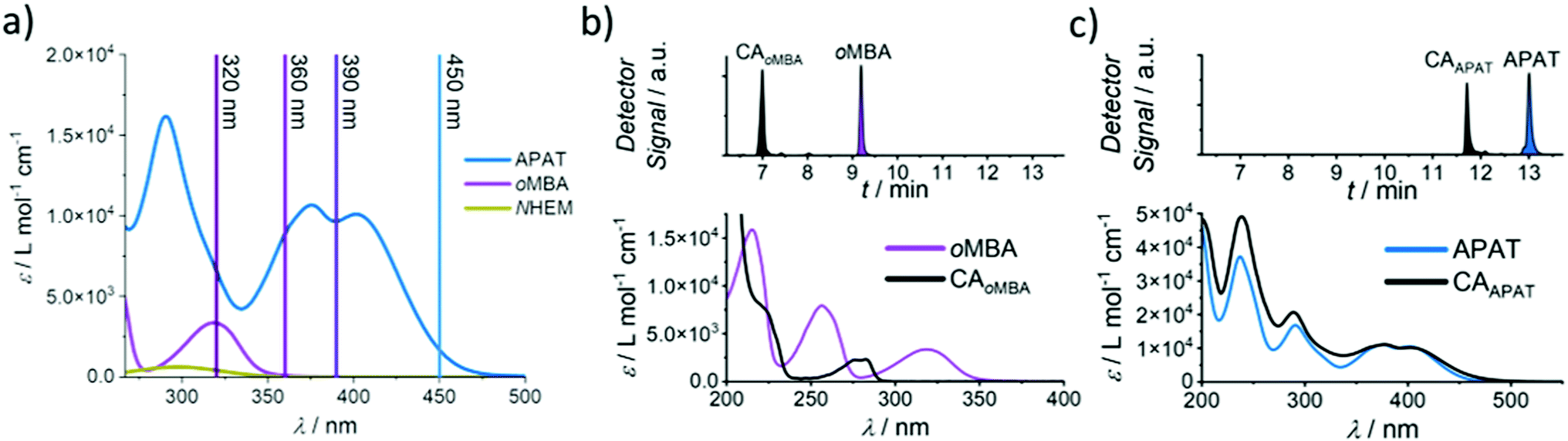 | ||
Fig. 2 (a) Absorption spectra of APAT, oMBA and NHEM in MeCN, as well as employed laser wavelengths. (b) (top) Chromatogram of oMBA and CAoMBA after 25 s at 320 nm irradiation. (bottom) Absorption spectra of oMBA (89 μM, in MeCN) and CAoMBA (49 μM, in MeCN/H2O 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8). (c) (top) Chromatogram of APAT and CAAPAT after 30 min of 450 nm irradiation. (bottom) Absorption spectra of APAT (55 μM) and CAAPAT (36 μM) in MeCN. :8). (c) (top) Chromatogram of APAT and CAAPAT after 30 min of 450 nm irradiation. (bottom) Absorption spectra of APAT (55 μM) and CAAPAT (36 μM) in MeCN. | ||
In Fig. 3, the photoreaction yield is plotted as a function of time. Since output energies were employed at different wavelengths, irradiation time is not the appropriate measure to compare the reaction kinetics. Instead, the top x-axis was adjusted to give the incident photon equivalents in relation to the number of chromophores in the solution. From Fig. 3a, it becomes clear that oMBA and NHEM cleanly form the cycloadduct, yielding 89% after 50 s (ΦoMBA = 0.18 ± 0.02, for a detailed description of the determination of reaction quantum yields, refer to the ESI†). During this time, no detectable amounts of CAAPAT are formed. We therefore assign a wavelength selectivity of at least 89% at 320 nm. The absorption coefficients of both compounds differ by a factor of two (εoMBA320 nm = 3340 ± 20 L mol−1 cm−1, εAPAT320 nm = 6570 ± 30 L mol−1 cm−1), but due to the strongly disparate reaction quantum yields, oMBA reaches high conversion before APAT commences conversion. Similarly, NHEM in 8-fold excess acts as a competing chromophore (εNHEM320 nm = 418 ± 2 L mol−1 cm−1). At 360 nm irradiation (Fig. 3b), 91% yield of CAoMBA are reached after 19 min (ΦoMBA = 0.10 ± 0.02). We attribute the lower reactivity to two factors:
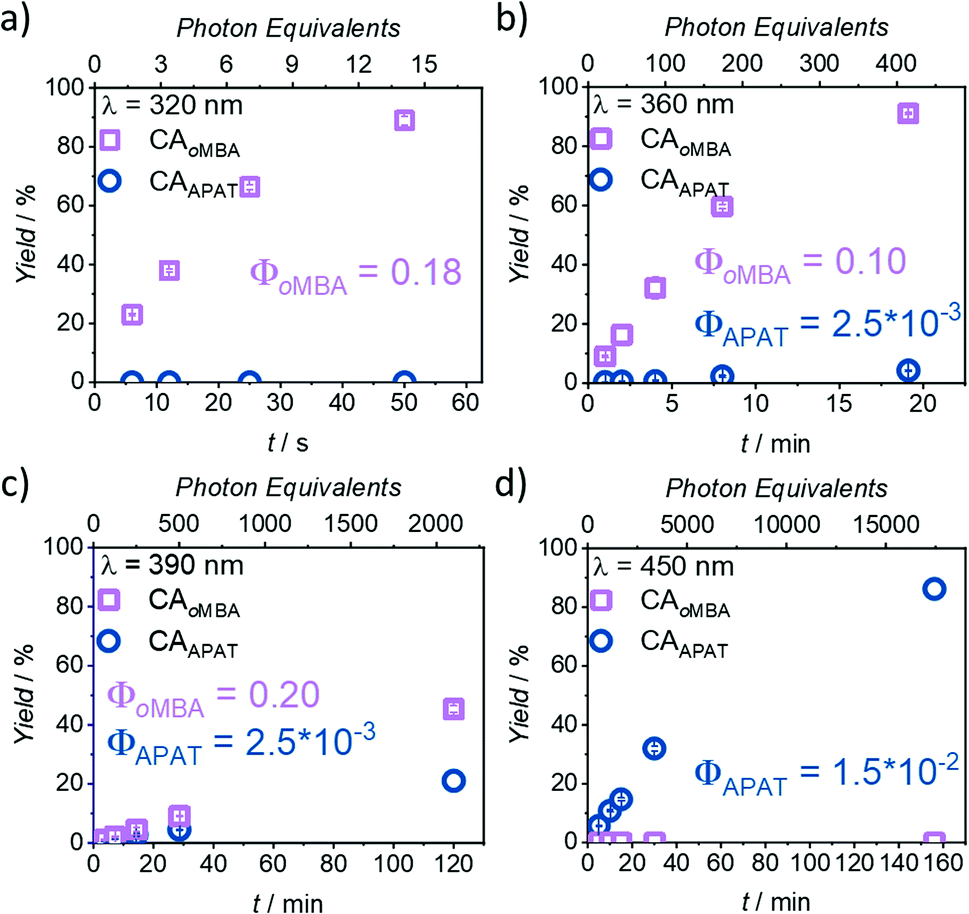 | ||
| Fig. 3 Kinetic plots after irradiation with (a) 320 nm, (b) 360 nm, (c) 390 nm, and (d) 450 nm laser light. Photoreactions of oMBA (0.23 mM) and APAT (0.23 mM) with NHEM (1.82 mM) were carried out in degassed MeCN, and irradiation times were chosen to cover a wide range of conversions. Each experiment was repeated 3× to obtain error bars. Differences in the top and bottom x-axes stem from different laser energies and irradiation times. For detailed information, refer to ESI.† | ||
First, the absorptivity of oMBA at 360 nm is considerably lower (εoMBA360 nm = 87 ± 3 L mol−1 cm−1) compared to 320 nm. Secondly, the absorptivity of APAT (εAPAT360 nm = 8920 ± 50 L mol−1 cm−1) is ∼90× higher. While oMBA alone at 360 nm retained approximately 60% of its reactivity at 320 nm in a previous study,32 competing absorption of APAT further reduces the reactivity. Moreover, the reaction time is sufficiently long for APAT to reach 4% conversion (ΦAPAT = (2.5 ± 0.5) × 10−3), lowering the overall degree of λ-orthogonality at this wavelength. Interestingly, in our initial study, APAT was close to six times more reactive at 320 nm (∼13% conversion) compared to 360 nm (∼2%).33
The same trend is observed at 390 nm (Fig. 3c). An even higher discrepancy between absorption coefficients and longer irradiation times led to rather similar conversions of both compounds after 120 min (45% CAoMBA, ΦoMBA = 0.20 ± 0.18; 21% CAAPAT, ΦAPAT = (2.5 ± 0.5) × 10−3). The large error in ΦoMBA stems from the uncertainty of εoMBA390 nm (refer to ESI†). Finally, at 450 nm (Fig. 3d), no CAoMBA was detected even after 156 min of irradiation, while exclusively APAT was converted, yielding 86% of CAAPAT (ΦAPAT = (1.5 ± 0.5) × 10−2). We therefore assign a wavelength-selectivity of at least 86% at 450 nm. Clearly, of the investigated wavelengths, 390 nm is the least suitable for λ-orthogonality. This finding is especially interesting, because 390 nm is close to the absorption maxima of APAT (376 nm and 402 nm, respectively). Traditionally, wavelengths in λ-orthogonal reaction systems are determined by the absorption maxima of the involved chromophores. However, we herein show that 450 nm is the most appropriate wavelength, consistent with the reported findings of our preceding study on the reactivity of APAT. Yet, while our previous study reported increased reactivity at 320 nm,33 herein, no conversion is observed within the reaction time of 50 s.
Having established the wavelengths with the highest selectivity (320 nm and 450 nm), we explored the λ-orthogonal reaction system in a batch reaction, administering different colours of light in a defined sequence (refer to Fig. 4). The sequence includes both possible pathways (UV → vis, vis → UV) as well as a dark period. A 0.67 W LED, centred around 325 nm, was used to activate oMBA, while a 10 W LED, centred around 445 nm, was used to activate APAT (for emission spectra of the LEDs, refer to the ESI†).
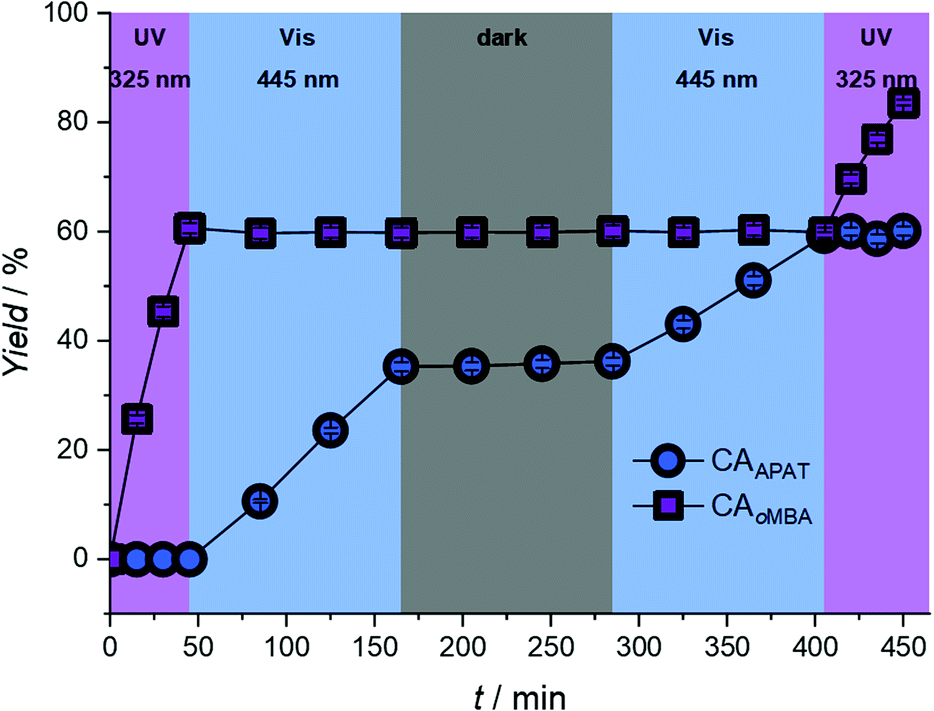 | ||
| Fig. 4 Photoproduct yield of CAoMBA and CAAPAT after sequential irradiation with two LEDs (325 nm, 0.67 W and 445 nm, 10 W), as well as a dark period. oMBA reacts exclusively upon 325 nm irradiation, while APAT only reacts to 445 nm light. | ||
We initially tested the UV → vis sequence, starting with the 325 nm LED. Although receiving significantly less photons per time unit due to the lower output power of the 325 nm LED, oMBA reacts close to five times faster than APAT due to the higher reaction quantum yield. Thus, samples were taken in 15 minute intervals. After a combined 45 min we found 60% yield for CAoMBA, while no APAT cycloadduct was detected. After switching to a 445 nm 10 W LED, samples were taken every 40 min due to the longer reaction times required for APAT. To ensure that no thermal reaction was induced by the high-powered LED, we heated a solution of APAT and a dipolarophile in MeCN to 80 °C, yet found no change of the starting materials even after hours of heating (refer to ESI, Fig. S3†). After 120 min of irradiation (3 × 40 min) at 445 nm, a CAAPAT yield of 35% was recorded, whereas the amount of CAoMBA remained constant. However, we found small amounts of side product (refer to ESI, Fig. S4†). We were not able to isolate the side product, however mass spectrometry indicates that it stems from the reaction between the nitrile imine and water (refer to Fig. S5†).34 Repeated penetration of the rubber septum while withdrawing samples from the reaction mixture probably allowed small amounts of moisture to enter the reaction vessel. In the following 120 min dark period no increase in either of the species was observed. Subsequently, the vis → UV sequence was tested, starting with 445 nm irradiation. During visible light irradiation APAT continued to react and finally yielded 59% of CAAPAT after another 120 min, while oMBA remained unaffected. Switching to 325 nm irradiation yielded a final 84% of CAoMBA, with the amount of CAAPAT remaining constant.
Conclusions
In summary, we introduce a fully sequence-independent λ-orthogonal reaction system, based on oMBA as a UV-activated photoactive moiety with high quantum yield, and APAT as a visible light activated unit with a low quantum yield. Initially, we probed the system at a range of specific wavelengths, using a tuneable ns-laser system, recording kinetic reaction plots, and determining the most suitable wavelengths for λ-orthogonality. Counterintuitively, 390 nm is the least suitable wavelength, despite being close to the absorption maxima of APAT. Instead, 450 nm led to exclusive conversion of APAT, whereas 320 nm addressed exclusively oMBA. Finally, we employed two spectrally broad LEDs (centred 325 nm and 445 nm) for sequential irradiation of both compounds within the reaction mixture, clearly demonstrating the individually addressable nature of the system. Our findings open an avenue for the construction of λ-orthogonal photoresists for 3D laser lithography.35Data availability
Experimental data is available in the ESI.† Additional data is available upon request by contact with the corresponding author.Author contributions
P. W. K. conducted the majority of all syntheses and the photochemical experiments. L. L. R. assisted with the syntheses as well as optimized and conducted the liquid chromatography experiments. S. L. W. conducted the quantum yield determination. A. N. U., J. P. B. and C. B.-K. conceptualized the study and were responsible for writing the grants on which the study is based (ARC Laureate C. B.-K., ARC Discovery C. B.-K. and J. P. B., KIT funding A. N. U.). All authors discussed the data and co-edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Philipp Neidinger (Karlsruhe Institute of Technology) for the measurement of absorption spectra. C. B.-K. acknowledges funding by the Australian Research Council (ARC), in the context of a Laureate Fellowship underpinning his photochemical research program. C. B.-K. and J. P. B. acknowledge support via an ARC Discovery project targeted at red-shifting photoligation chemistry. Additional support by the Queensland University of Technology (QUT) and the Karlsruhe Institute of Technology (KIT) is gratefully acknowledged.Notes and references
- G. S. Kumar and Q. Lin, Chem. Rev., 2020, 121, 6991–7031 CrossRef PubMed.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Adv. Mater., 2020, 32, 1903850 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- J. J. Schwartz and A. J. Boydston, Nat. Commun., 2019, 10, 791 CrossRef CAS PubMed.
- N. D. Dolinski, Z. A. Page, E. B. Callaway, F. Eisenreich, R. V. Garcia, R. Chavez, D. P. Bothman, S. Hecht, F. W. Zok and C. J. Hawker, Adv. Mater., 2018, 30, 1800364 CrossRef PubMed.
- K. Hiltebrandt, T. Pauloehrl, J. P. Blinco, K. Linkert, H. G. Börner and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2015, 54, 2838–2843 CrossRef CAS PubMed.
- V. X. Truong, F. Li, F. Ercole and J. S. Forsythe, ACS Macro Lett., 2018, 7, 464–469 CrossRef CAS.
- P. Lu, D. Ahn, R. Yunis, L. Delafresnaye, N. Corrigan, C. Boyer, C. Barner-Kowollik and Z. A. Page, Matter, 2021, 4, 2172–2229 CrossRef CAS.
- N. Corrigan, M. Ciftci, K. Jung and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 1748–1781 CrossRef CAS PubMed.
- C. G. Bochet, Isr. J. Chem., 2021, 61, 486–495 CrossRef CAS.
- A. Eibel, D. E. Fast, J. Sattelkow, M. Zalibera, J. Wang, A. Huber, G. Müller, D. Neshchadin, K. Dietliker, H. Plank, H. Grützmacher and G. Gescheidt, Angew. Chem., Int. Ed., 2017, 56, 14306–14309 CrossRef CAS PubMed.
- S. Bialas, L. Michalek, D. E. Marschner, T. Krappitz, M. Wegener, J. Blinco, E. Blasco, H. Frisch and C. Barner-Kowollik, Adv. Mater., 2019, 31, 1807288 CrossRef PubMed.
- A. Ohtsuki, L. Lei, M. Tanishima, A. Goto and H. Kaji, J. Am. Chem. Soc., 2015, 137, 5610–5617 CrossRef CAS PubMed.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS.
- J. L. Pelloth, P. A. Tran, A. Walther, A. S. Goldmann, H. Frisch, V. X. Truong and C. Barner-Kowollik, Adv. Mater., 2021, 33, 2102184 CrossRef CAS PubMed.
- X. Zhang, L. Cox, Z. Wen, W. Xi, Y. Ding and C. N. Bowman, Polymer, 2018, 156, 162–168 CrossRef CAS PubMed.
- M. Wu, X. Lin, X. Tan, J. Li, Z. Wei, D. Zhang, Y. Zheng, A.-X. Zheng, B. Zhao, Y. Zeng, X. Liu and J. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 19416–19427 CrossRef CAS PubMed.
- X. Zhang, W. Xi, S. Huang, K. Long and C. N. Bowman, Macromolecules, 2017, 50, 5652–5660 CrossRef CAS PubMed.
- R. R. Batchelor, E. Blasco, K. N. R. Wuest, H. Lu, M. Wegener, C. Barner-Kowollik and M. H. Stenzel, Chem. Commun., 2018, 54, 2436–2439 RSC.
- I. Elamri, C. Abdellaoui, J. K. Bains, K. F. Hohmann, S. L. Gande, E. Stirnal, J. Wachtveitl and H. Schwalbe, J. Am. Chem. Soc., 2021, 143, 10596–10603 CrossRef CAS PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- J. A. Peterson, D. Yuan and A. H. Winter, J. Org. Chem., 2021, 86, 9781–9787 CrossRef CAS PubMed.
- W. A. Velema, J. P. van der Berg, W. Szymanski, A. J. M. Driessen and B. L. Feringa, ACS Chem. Biol., 2014, 9, 1969–1974 CrossRef CAS PubMed.
- C. G. Bochet, Angew. Chem., Int. Ed., 2001, 40, 2071–2073 CrossRef CAS PubMed.
- J. S. Clovis, A. Eckell, R. Huisgen and R. Sustmann, Chem. Ber., 1967, 100, 60–70 CrossRef CAS.
- J. P. Menzel, F. Feist, B. Tuten, T. Weil, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 7470–7474 CrossRef CAS PubMed.
- M. J. Hansen, W. A. Velema, M. M. Lerch, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2015, 44, 3358–3377 RSC.
- D. E. Fast, A. Lauer, J. P. Menzel, A.-M. Kelterer, G. Gescheidt and C. Barner-Kowollik, Macromolecules, 2017, 50, 1815–1823 CrossRef CAS.
- D. E. Marschner, P. W. Kamm, H. Frisch, A.-N. Unterreiner and C. Barner-Kowollik, Chem. Commun., 2020, 56, 14043–14046 RSC.
- J. P. Menzel, B. B. Noble, A. Lauer, M. L. Coote, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2017, 139, 15812–15820 CrossRef CAS PubMed.
- P. W. Kamm, J. P. Blinco, A.-N. Unterreiner and C. Barner-Kowollik, Chem. Commun., 2021, 57, 3991–3994 RSC.
- S.-L. Zheng, Y. Wang, Z. Yu, Q. Lin and P. Coppens, J. Am. Chem. Soc., 2009, 131, 18036–18037 CrossRef CAS PubMed.
- L. Yang, F. Mayer, U. H. F. Bunz, E. Blasco and M. Wegener, Light: Advanced Manufacturing, 2021, 2, 1–17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, instrumentation, synthetic procedures, supporting spectroscopic data. See DOI: 10.1039/d1sc06154b |
| This journal is © The Royal Society of Chemistry 2022 |