
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolable fluorinated triphenylmethyl cation salts of [HCB11Cl11]−: demonstration of remarkable hydride affinity†
S. Olivia
Gunther
,
Chun-I
Lee
,
Ellen
Song
,
Nattamai
Bhuvanesh
and
Oleg V.
Ozerov
 *
*
Department of Chemistry, Texas A&M University, 3255 TAMU, College Station, Texas 77842, USA. E-mail: ozerov@chem.tamu.edu
First published on 4th April 2022
Abstract
Significantly fluorinated triarylmethyl cations have long attracted attention as potentially accessible highly reactive carbocations, but their isolation in a convenient form has proved elusive. We show that abstraction of chloride with a cationic silylium reagent leads to the facile formation of di-, tetra-, and hexafluorinated trityl cations, which could be isolated as analytically pure salts with the [HCB11Cl11]− counterion and are compatible with (halo)arene solvents. The F6Tr+ cation carrying six meta-F substituents was computationally predicted to possess up to 20% higher hydride affinity than the parent triphenylmethyl cation Tr+. We report that indeed F6Tr+ displays reactivity unmatched by Tr+. F6Tr+ at ambient temperature abstracts hydrides from the C–H bonds in tetraethylsilane, mesitylene, methylcyclohexane, and catalyzes Friedel–Crafts alkylation of arenes with ethylene, while Tr+ does none of these.
Introduction
The triphenylmethyl or trityl cation (Ph3C+ or Tr+) is a textbook example of a carbocation that is isolable owing to the high degree of benzylic conjugation and the steric protection afforded to the central carbon by the three phenyl substituents.1 In organometallic chemistry and catalysis, salts of Tr+ are frequently used to study the thermodynamics and kinetics of hydride transfer,2,3,4 or to generate reactive main-group and transition-metal cations through hydride or alkyl anion abstraction.5,6,7Tr+ can also serve as a convenient one-electron oxidant.8 Trityl cation derivatives bearing stabilizing electron-donating groups can even exist in aqueous solutions, with a rich history of use as indicators and dyes.9 The trityl cation versions bearing electron-withdrawing substituents have proven more challenging to obtain. Fluorinated trityl cations, up to (C6F5)3C+ (A, Fig. 1), have been of particular fundamental interest,10–12 including as isoelectronic analogs of the widely used borane (C6F5)3B,13,14 and more recently have been studied by Horn and Mayr15 and Dutton et al.16 The more reactive A or other ortho- and/or meta-fluorinated trityl cations were not isolated in those studies, but were generated in situ, or their intermediacy was indicated by kinetic studies. While generation of fluorinated trityl cations in oleum and other superacidic media,10–12,16 or by in situ abstraction of halides with element halide Lewis acids15 is possible, these media and counteranions are not fully compatible with either the more electron-deficient trityl cations themselves or with their potential use in the synthesis of other reactive main-group or transition metal cations. Thus, the full extent of the reactivity of the fluorinated trityl cations can only be accessed when paired with more robust weakly coordinating anions in weakly coordinating solvents.17 The only example of an isolated trityl-type cation fluorinated in the ortho-/meta-positions is B (Fig. 1), obtained by Douvris and Reed in an undefined yield, and not studied further.18 The perchlorotrityl cation has also been isolated.19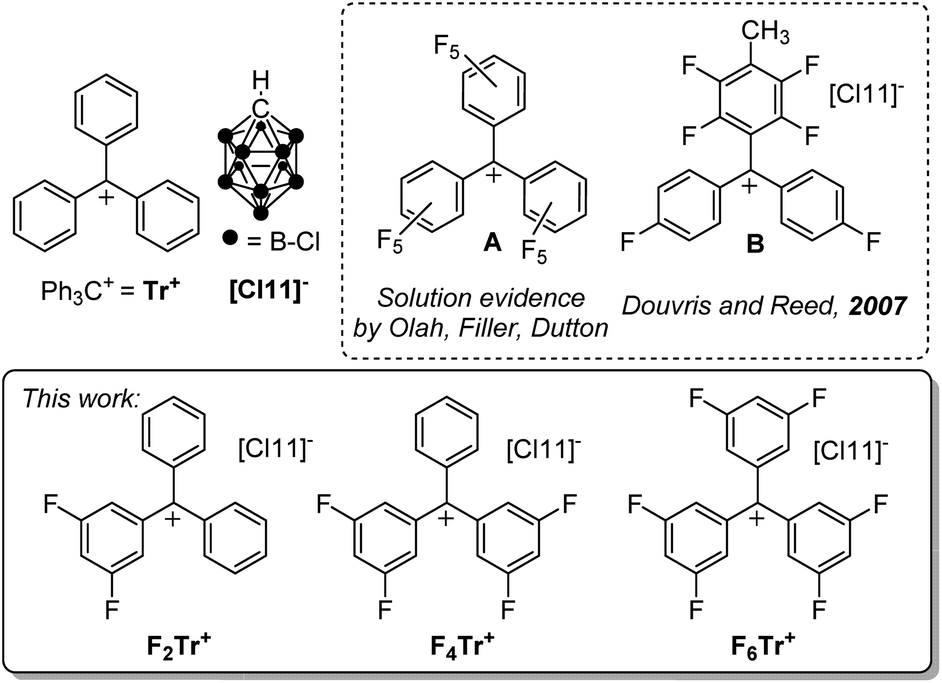 | ||
| Fig. 1 The parent trityl cation Tr+, selected literature examples of fluorinated trityls, and the fluorinated trityl salts prepared and studied in this work. | ||
Our group has been attracted to the highly reactive carbo- and main-group cations in the context of our work on silylium and alumenium cation-catalyzed activation of aliphatic C–F bonds,20–23 which permitted exhaustive defluorination of perfluoroalkyl groups under mild conditions. The chemistry of abstraction of fluoride from certain fluoroarenes with silylium cations has led to innovative reactivity, as well.24,25 Trialkylsilylium cations are typically generated by hydride abstraction from trialkylsilanes (R3SiH) using Tr+,26 but our theoretical analysis suggested that the parent Tr+ only barely has the thermodynamic hydride affinity (HA) to abstract hydrides from even the relatively electron-rich SiH bonds in trialkylsilanes. Given the perceived challenge10,16 in the isolation of the fully fluorinated (C6F5)3C+, we decided to first explore the partially fluorinated derivatives. Here, we report the isolation of analytically pure di-, tetra-, and hexafluorosubstituted trityl cation salts, and the remarkable contrast in the hydride abstraction reactivity with the parent Tr+.
Results and discussion
Theoretical HA analysis
Wilson and Dutton calculated gas-phase and CH2Cl2 solvent continuum HA values for a series of symmetric polychloro- and polyfluorosubstituted trityl cations.27 They discussed the fit to the known experimental values provided by the various computational methods and settled on the use of B3LYP/aug-cc-pVTZ//B3LYP/def2-TZVPP.28,29The Wilson–Dutton calculations showed that replacement of H with F in the para-position has an essentially zero effect on HA, whereas introduction of each ortho- or a meta-fluorine increases HA by ca. 2.4–2.7 kcal mol−1 (CH2Cl2 continuum) or ca. 3.5 kcal mol−1 (gas phase). This is in line with the more negative pKR+ values for the various ortho- and meta-fluorinated trityls compared to Tr+ or the para-F substituted trityls, determined by Filler et al.10 The ortho- and para-CF positions are conjugated to the central carbon by resonance and the para-CF has been identified as a site of alternative nucleophilic attack on (C6F5)3C+ related to its decomposition pathways.10,16 We decided to avoid fluorination in the ortho- or para-positions and focus on meta-fluorination. The Wilson–Dutton HA values for F6Tr+ (213.0 and 108.3 kcal mol−1) were 11% and 17% higher than for Tr+ (191.4 and 92.5 kcal mol−1) in the gas phase and CH2Cl2 continuum, respectively.
In 2011,30 we analyzed the HA and FA values for a series of cations relevant to the silylium-catalyzed HDF using the M05-2X functional with the basis sets 6-311+G(d) for F, and 6-31++G(d,p) for C and H.31 Utilizing the DFT approach from our 2011 paper, we calculated the gas-phase and the chlorobenzene solvent continuum HA values for F6Tr+ to be 229.4 and 135.0 kcal mol−1, representing a 13% and a 20% increase vs.Tr+. These relative increases are similar to those in the Wilson–Dutton work.27 The substantial increase suggests that the HA of F6Tr+ is thermodynamically sufficient to abstract a hydride from a range of Si–H containing molecules, and rivals the HA values calculated (also in PhCl) for Me3C+ (126.6 kcal mol−1), PhCH2+ (137.8 kcal mol−1), and Me2CH+ (138.9 kcal mol−1).30 Without assessing quantitative accuracy, we nonetheless surmised that F6Tr+ might be able to abstract hydrides from tertiary and possibly secondary and benzylic C(sp3)–H bonds.
Synthesis and characterization of FxTr+ salts
We envisioned the synthesis of fluorinated trityl cations partnered with the exceptionally robust and weakly coordinating [HCB11Cl11]− anion ([Cl11], Fig. 1)18,32–35via abstraction of a chloride anion from the corresponding F2TrCl, F4TrCl, and F6TrCl.36,37 Na[Cl11] can abstract a chloride from TrCl38 and from F2TrCl in o-C6H4Cl2 at ambient temperature, giving a 97% isolated yield of F2Tr[Cl11] after workup. Attempts to use Na[Cl11] to generate F4Tr[Cl11] and F6Tr[Cl11] were unsuccessful and we moved to a more powerful39–41 chloride abstractor [(Me3Si)2OTf][Cl11] (Si[Cl11]).38,42Indeed, treatment of F2TrCl with Si[Cl11] in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 C6D6/o-C6H4Cl2 solvent mixture at ambient temperature (Fig. 2) resulted in rapid color change. Analysis of the resultant solution by NMR spectroscopy after 10 min revealed the expected formation of equimolar amounts of Me3SiCl and Me3SiOTf and 96% yield of F2Tr+ (19F NMR evidence, δ −104.6 ppm). The analogous reactions with F4TrCl and F6TrCl also proceeded smoothly. The resultant F4Tr[Cl11] and especially F6Tr[Cl11] are less soluble than F2Tr[Cl11] or Tr[Cl11], and precipitate readily out of fluorobenzene, allowing isolation of analytically pure solids in 96% and 70% yields.
:1 C6D6/o-C6H4Cl2 solvent mixture at ambient temperature (Fig. 2) resulted in rapid color change. Analysis of the resultant solution by NMR spectroscopy after 10 min revealed the expected formation of equimolar amounts of Me3SiCl and Me3SiOTf and 96% yield of F2Tr+ (19F NMR evidence, δ −104.6 ppm). The analogous reactions with F4TrCl and F6TrCl also proceeded smoothly. The resultant F4Tr[Cl11] and especially F6Tr[Cl11] are less soluble than F2Tr[Cl11] or Tr[Cl11], and precipitate readily out of fluorobenzene, allowing isolation of analytically pure solids in 96% and 70% yields.
 | ||
| Fig. 2 Synthesis of fluorinated trityl cation salts and their appearance. | ||
The 13C NMR chemical shifts of the central carbons of F2Tr[Cl11], F4Tr[Cl11], and F6Tr[Cl11] in the 208–210 ppm range,43 as well as the 1H and 19F NMR spectral data did not suggest any significant interaction of the cations with the [Cl11]− anion, the arene or CD2Cl2 solvents, or the Me3SiCl/Me3SiOTf by-products. Single-crystal X-ray diffractometry (Fig. 3) revealed that the central carbons in F2Tr[Cl11] and F6Tr[Cl11] possessed a planar environment in all the crystallographically independent cations (the sums of C–C–C angles are ca. 360°), and the aryl groups splay out in a pinwheel pattern about the central carbon. The closest approach of any chlorine to the central carbon in F2Tr[Cl11] is at least 3.7 Å, and 3.25 Å in F6Tr[Cl11], consistent with the well-separated, ionic nature of the FxTr[Cl11] salts.
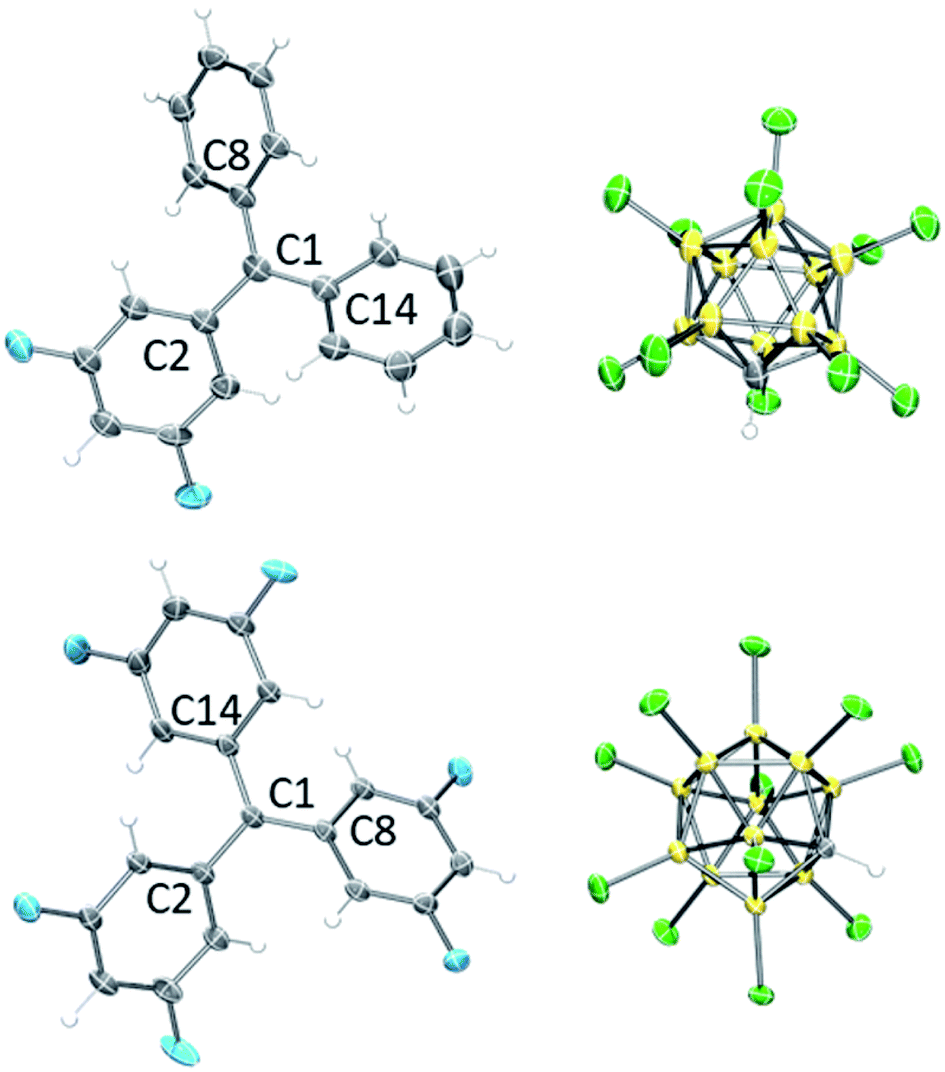 | ||
| Fig. 3 POV-Ray rendition of the ORTEP (50% probability ellipsoids) drawing of F2Tr[Cl11] (top) and F6Tr[Cl11] (bottom). Only one cation and one anion from each asymmetric unit is shown. Solvent and disorder are omitted for clarity. | ||
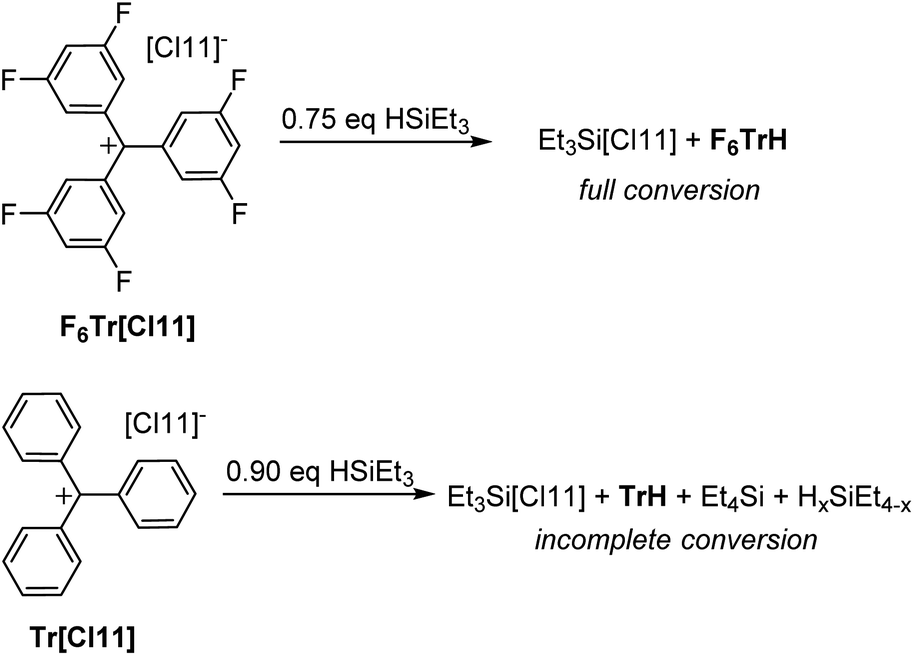 | ||
| Fig. 4 Reactions of Tr+ and F6Tr+ with a substoichiometric amount of HSiEt3. | ||
Reactivity of F6Tr+vs. Tr+ with Et3Si–H
It was previously shown that Tr+ is not capable of abstracting the full hydride equivalent from Et3SiH in non-coordinating solvents and that two equivalents of R3SiH are needed for complete formation of TrH.44 Our observations are similar: treatment of either F6Tr[Cl11] or Tr[Cl11] with two equivalents of Et3SiH in a C6D6/o-C6H4Cl2 solvent mixture led to the quantitative formation of F6TrH or TrH, respectively. The fate of the “Et3Si+” species in arene solvents is not straightforward, as has been studied in detail45 by Heinekey and coworkers: the presence of varying amounts of Et4Si betrays complexity arising from the H/Et redistribution in the Si species and/or reactions with the arenes.The reaction of Tr[Cl11] with a substoichiometric (0.9 equiv.) amount of Et3SiH did not lead to the complete disappearance of the Si–H moiety (16% of the original Si–H intensity remained) and only 82% of the possible TrH was observed (Fig. 4). In contrast, the reaction of F6Tr[Cl11] with substoichiometric (0.75 equiv.) amount of Et3SiH led to the production of the expected quantity of F6TrH, the complete disappearance of the Si–H signals, and without the concomitant observation of Et4Si.
H–D exchange
In the reactions of F6Tr[Cl11] with Et3SiH, significant H/D scrambling was observed among the neutral aromatic compounds present in solution: C6D6, o-C6H4Cl2, and F6TrH (but the C(sp3)–H bond in F6TrH was never deuterated). The extent of H–D exchange was analyzed via1H, 13C, or 19F46 NMR spectroscopy (see ESI† for details). The mechanism of the H/D exchange likely involves the generation of superacidic protonated arenes in situ,47 which should enable rapid H/D exchange via H+/D+ shuttling (Fig. S4†).47,48 The product of addition of either Et3Si+ or F6Tr+ to a neutral arene can be alternatively viewed as a protonated arene.47 It is also possible that analogous cations are accessed via reactions involving the minor components of the mixture. The Oestreich group recently examined this type of H/D exchange catalysis in greater detail.49Abstraction of hydride from C–H bonds
Given the computational prediction of the enhanced hydride affinity of F6Tr+vs.Tr+, we wished to examine their reactivity towards benzylic and aliphatic C–H bonds. As expected, no reaction was observed between Tr[Cl11] and (1) 1 equiv. of mesitylene or (2) 1 equiv. of methylcyclohexane in o-C6H4Cl2 after 1 week at ambient temperature. In contrast, the reaction of F6Tr[Cl11] with mesitylene (as solvent) resulted in 66% yield (NMR evidence) or F6TrH after 48 h. We propose that hydride abstraction from mesitylene by F6Tr[Cl11] generates a 3,5-dimethylbenzyl cation, which rapidly undergoes Friedel–Crafts20,21 addition to mesitylene. GC-MS analysis of the mixture after quenching with water showed the presence of a m/z signal at 238, consistent with compound 4 (Fig. 5). Treatment of F6Tr[Cl11] in o-C6H4Cl2 with 1 equiv. of methylcyclohexane resulted in the >95% yield (NMR evidence) of F6TrH after 96 h. The aliphatic region of the 1H NMR spectrum presented a large number of overlapping aliphatic signals, indicating a complex mixture (Fig. 5b).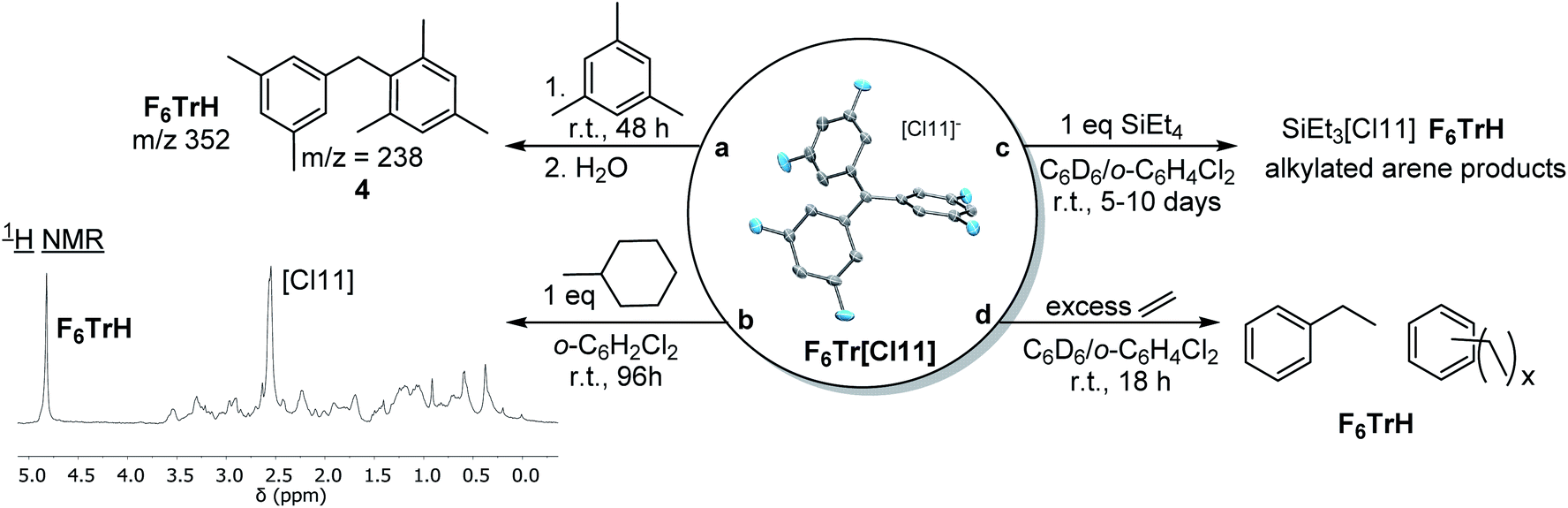 | ||
| Fig. 5 Reactions of F6Tr[Cl11] resulting in the abstraction of a hydride from C(sp3)–H bonds. | ||
The methylcyclohexyl cation presumed to be formed initially may undergo isomerization50 and Friedel–Crafts addition to o-C6H4Cl2, with many potential products. Abstraction of a hydride from alkanes, with generation of rearranged tertiary carbocations, was previously reported by the Reed group using Me[HCB11Me5Br6].51,52 The key difference between Reed's “Me+” reagents and the F6Tr+ reported here is that the latter can be prepared in bulk analytical purity and is stable in haloarene solutions.
Abstraction of a hydride from the β-position in trialkylaluminums with Tr+ has been used to generate reactive alumenium (R2Al+) cations.6,22,53 The analogous abstraction of β-hydride from alkylsilanes by Tr+ is not known, and we have confirmed that no reaction takes place between Tr[Cl11] and Et4Si in C6D6/o-C6H4Cl2. However, an analogous reaction of Et4Si with F6Tr[Cl11] resulted in the formation of 82% F6TrH after 96 h (and complete disappearance of Et4Si after 10 d). The major Si product appeared to be “Et3Si”, but instead of the stoichiometric complement of free ethylene, we observed ethane and other aliphatic resonances. Ethane may result from the protonolysis of Et4Si by the highly Brønsted acidic cations generated in the reaction (extensive H/D exchange was concomitantly observed), a process reported on by Oestrich and co-workers.54 As a control experiment, we examined the reaction of F6Tr[Cl11] with 6.3 equiv. of ethylene in C6D6/o-C6H4Cl2. Within 18 h at ambient temperature, all ethylene had been consumed, with the concomitant generation of ethylbenzene (1.8 equiv.) and other alkylarenes, and quantitative production of F6TrH. It is reasonable to propose that F6Tr[Cl11] abstracts a hydride from the benzylic positions of ethylbenzene or other alkylarenes generated through Friedel–Crafts alkylation. In complete contrast, no reaction occurred between Tr[Cl11] and ethylene under analogous conditions.
Conclusion
Introduction of six meta-F substituents in F6Tr+ brought about remarkable contrast with the reactivity of the parent triphenylmethyl (Tr+) cation, understood primarily through the greatly enhanced hydride affinity of especially the hexafluorinated F6Tr+. Interestingly, while F6Tr+ catalyzes the Friedel–Crafts alkylation of arenes with ethylene, and generates alkyl cations via hydride abstraction which then readily engage in Friedel–Crafts addition, F6Tr+ itself is stable in combination with (halo)arene solvents and dichloromethane. This shows that fluorinated trityl cations represent a promising class of reagents for achieving the extremes of hydride affinity while minimizing reactivity with other potential substrates.Data availability
Data for this manuscript are available in the ESI.†Author contributions
S. O. G. and C. I. L. performed the syntheses and obtained the characterization data. E. S. performed the DFT calculations. N. B. carried out the X-ray diffraction studies on the crystals grown by S. O. G. S. O. G. and O. V. O. wrote the manuscript with assistance of the other co-authors. O. V. O. directed the overall effort.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Naval Research Laboratory (grant N00173-20-1-G010 to O. V. O.). We thank Dr Manoj Kolel-veetil for insightful discussions. The authors acknowledge the High-Performance Research Computing Center at Texas A&M University for providing access to supercomputing clusters, software, and computing time. We thank R. A. Gholson for assistance with manuscript formatting.Notes and references
- G. A. Olah and G. K. S. Prakash, Carbocation Chemistry; John Wiley & Sons, Inc., 2004 Search PubMed.
- F. A. Carey and H. S. Tremper, J. Am. Chem. Soc., 1968, 90, 2578–2583 CrossRef CAS.
- T.-Y. Cheng and M. R. Bullock, Organometallics, 1995, 14, 4031–4033 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1432 CrossRef CAS PubMed.
- K.-C. Kim, C. A. Reed, G. S. Long and A. Sen, J. Am. Chem. Soc., 2002, 124, 7662–7663 CrossRef CAS PubMed.
- K.-C. Kim, C. A. Reed, D. W. Elliott, L. J. Mueller, F. Tham, L. Lin and J. B. Lambert, Science, 2002, 297, 825–827 CrossRef CAS PubMed.
- P. M. C. MacInnis, J. C. DeMott, E. M. Zolnhofer, J. Zhou, K. Meyer, R. P. Hughes and O. V. Ozerov, Chem, 2016, 1, 902–920 Search PubMed.
- T. Gessner and U. Mayer, Triarylmethane and Diarylmethane Dyes, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2002 Search PubMed.
- R. Filler, C. Wang, M. A. McKinney and F. N. Miller, J. Am. Chem. Soc., 1967, 89, 1026 CrossRef CAS.
- G. A. Olah and M. B. Comisarow, J. Am. Chem. Soc., 1967, 89, 1027–1028 CrossRef CAS.
- V. Shrikant, R. Schure and R. Filler, J. Am. Chem. Soc., 1973, 95, 1859–1864 CrossRef.
- J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed.
- G. Erker, Dalton Trans., 2005, 11, 1883–1890 RSC.
- M. Horn and H. Mayr, J. Phys. Org. Chem., 2012, 25, 979–988 CrossRef CAS.
- E. G. Delany, S. Kaur, S. Cummings, K. Basse, J. D. Wilson and J. L. Dutton, Chem.–Eur. J., 2019, 25, 5298–5302 CrossRef CAS PubMed.
- I. M. Riddlestone, A. Kraft, J. Schaefer and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 13982–14024 CrossRef CAS PubMed.
- C. Douvris, E. S. Stoyanov, F. S. Tham and C. A. Reed, Chem. Commun., 2007, 40, 145–1147 Search PubMed.
- E. Molins, M. Mas, W. Manjukiewicz, M. Ballester and J. Castaner, Acta Crystallogr. C, 1996, 52, 2412–2414 CrossRef.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946–4953 CrossRef CAS PubMed.
- W. Gu, M. R. Haneline, C. Douvris and O. V. Ozerov, J. Am. Chem. Soc., 2009, 131, 11203–11212 CrossRef CAS PubMed.
- J. C. L. Walker, H. F. T. Klare and M. Oestreich, Nat. Rev. Chem., 2020, 4, 54–62 CrossRef CAS.
- J. S. Siegel, O. Allemann, S. Duttwyler, P. Romanato and K. K. Baldridge, Science, 2011, 332, 574–577 CrossRef PubMed.
- B. Shao, A. L. Bagdasarian, S. Popov and H. M. Nelson, Science, 2017, 355, 1403–1407 CrossRef CAS PubMed.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestrich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed.
- S. A. Couchman, D. J. D. Wilson and J. L. Dutton, Eur. J. Org. Chem., 2014, 3902–3908 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- D. G. Gusev and O. V. Ozerov, Chem.–Eur. J., 2011, 17, 634–640 CrossRef CAS PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- E. S. Stoyanov, K.-C. Kim and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 8500–8508 CrossRef CAS PubMed.
- S. P. Fisher, A. W. Tomich, S. O. Lovera, J. F. Kleinasser, J. Guo, H. M. Asay and V. Lavallo, Chem. Rev., 2019, 119, 8262–8290 CrossRef CAS PubMed.
- C. Douvris and J. Michl, Chem. Rev., 2013, 113, PR179–PR233 CrossRef PubMed.
- W. Gu, B. J. McCulloch, J. H. Reibenspies and O. V. Ozerov, Chem. Commun., 2010, 46, 282–2822 RSC.
- J. R. Bour, J. C. Green, V. J. Winton and J. B. Johnson, J. Org. Chem., 2013, 78, 1665–1669 CrossRef CAS PubMed.
- M. Horn, C. Metz and H. Mayr, Eur. J. Org. Chem., 2011, 6476–6485 CrossRef CAS.
- L. P. Press, B. J. McCulloch, W. Gu, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2015, 51, 14034–14037 RSC.
- D. J. Liston, Y. J. Lee, W. R. Scheidt and C. A. Reed, J. Am. Chem. Soc., 1989, 111, 6643–6648 CrossRef CAS.
- A. Martens, P. Weis, M. C. Krummer, M. Kreuzer, A. Meierhöfer, S. C. Meier, J. Bohnenberger, H. Scherer, I. Riddlestone and I. Krossing, Chem. Sci., 2018, 9, 7058–7068 RSC.
- C. A. Reed, Acc. Chem. Res., 1998, 31, 133–139 CrossRef CAS.
- A. Schulz, J. Thomas and A. Villinger, Chem. Commun., 2010, 46, 3696–3698 RSC.
- G. A. Olah, A. L. Berrier and G. K. S. Prakash, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 1998–2002 CrossRef CAS PubMed.
- M. Nava and C. A. Reed, Organometallics, 2011, 30, 4798–4800 CrossRef CAS PubMed.
- S. J. Connelly, W. Kaminsky and D. M. Heinekey, Organometallics, 2013, 32, 7478–7748 CrossRef CAS.
- V. Salamanca and A. C. Albeniz, Eur. J. Org. Chem., 2020, 3206–3212 CrossRef CAS.
- H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2021, 143, 15490–15507 CrossRef CAS PubMed.
- D. Munz, M. Webster-Gardiner, R. Fu, T. Strassner, W. A. Goddard III and T. B. Gunnoe, ACS Catal., 2015, 5, 769–775 CrossRef CAS.
- T. He, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2022, 144(11), 4734–4738 CrossRef CAS PubMed.
- I. D. Mackie and J. Govindhakannan, J. Mol. Struct., 2010, 939, 53–58 CrossRef CAS.
- T. Kato and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 2908–2911 CrossRef CAS PubMed.
- C. A. Reed, Acc. Chem. Res., 2010, 43, 121–128 CrossRef CAS PubMed.
- M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2021, 51, 7323–7326 CrossRef PubMed.
- Q. Wu, Z. W. Qu, L. Omann, E. Irran, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 9176–9179 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and pictorial NMR spectra, details of the computational studies and the coordinate files. CCDC 2079761 and 2079762. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d1sc05936j |
| This journal is © The Royal Society of Chemistry 2022 |