
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical C–H arylation of heteroarenes for DNA-encoded library synthesis†
Matthias
Krumb‡
 a,
Lisa Marie
Kammer‡
a,
Shorouk O.
Badir
a,
María Jesús
Cabrera-Afonso
a,
Victoria E.
Wu
b,
Minxue
Huang
b,
Adam
Csakai
b,
Lisa A.
Marcaurelle
b and
Gary A.
Molander
*a
a,
Lisa Marie
Kammer‡
a,
Shorouk O.
Badir
a,
María Jesús
Cabrera-Afonso
a,
Victoria E.
Wu
b,
Minxue
Huang
b,
Adam
Csakai
b,
Lisa A.
Marcaurelle
b and
Gary A.
Molander
*a
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
bEncoded Library Technologies/NCE Molecular Discovery, R&D Medicinal Science and Technology, GlaxoSmithKline, 200 Cambridge Park Drive, Cambridge, MA 02140, USA
First published on 21st December 2021
Abstract
DNA-encoded library (DEL) technology has emerged as a time- and cost-efficient technique for the identification of therapeutic candidates in the pharmaceutical industry. Although several reaction classes have been successfully validated in DEL environments, there remains a paucity of DNA-compatible reactions that harness building blocks (BBs) from readily available substructures bearing multifunctional handles for further library diversification under mild, dilute, and aqueous conditions. In this study, the direct C–H carbofunctionalization of medicinally-relevant heteroarenes can be accomplished via the photoreduction of DNA-conjugated (hetero)aryl halides to deliver reactive aryl radical intermediates in a regulated fashion within minutes of blue light illumination. A broad array of electron-rich and electron-poor heteroarene scaffolds undergo transformation in the presence of sensitive functional groups.
Introduction
The identification of potent drug candidates against novel biological targets is a critical, resource-intensive, and time-consuming challenge in the pharmaceutical industry. Although high-throughput screening (HTS),1–4 is routinely used to identify specific binding molecules within drug discovery efforts, a drawback of this approach is the sheer cost and extensive labor required per library member. Originally envisioned by Brenner and Lerner in 1992,5 DNA-encoded library (DEL) technology has been recently enlisted as an innovative interrogation format in pharmaceutical settings to overcome this limitation.6,7 A DEL library describes a collection of small molecule ligands covalently bound to a DNA barcode bearing the information with respect to the identity and structural composition of discrete library members. These libraries can be prepared by an initial reaction of diverse building blocks (BBs) with a short DNA sequence conjugated to an organic functional group (the DNA headpiece). This is followed by several split and pool cycles of chemistry to synthesize and simultaneously encode additional units (Scheme 1).8,9 Upon library completion, the generated structures are incubated against an immobilized target protein, after which the low-affinity or non-binding ligands are washed away.10,11 The DNA barcode of the remaining high-affinity ligands are then amplified using polymerase chain reaction (PCR), and the corresponding chemical identities can be decoded through next generation sequencing of the DNA barcode. Using this interrogation technique, combinatorial libraries of remarkable size (>106 to 1012 molecules) are screened against a protein target of interest,12 while only vanishingly small quantities of the library are used per experiment.13–16 Given that DEL technology is currently in its early stages of development relative to traditional HTS, it is remarkable that DEL success stories within medicinal chemistry are already known. These include the discovery of GlaxoSmithKline's inhibitor of receptor interacting protein 1 (RIP1) kinase GSK2982772 against inflammatory diseases in 2016,14,17 as well as AstraZeneca, Heptares Therapeutics, and X-Chem's inhibitor for protease-activated receptor 2 (PAR2) AZ3451, all fruitful outcomes of DEL screening.18,19 These examples emphasize the potential of DELs as readily accessible and effective screening modalities in drug discovery research. | ||
| Scheme 1 Schematic representation of DNA-encoded library construction and hit identification. | ||
Although various synthetic hurdles have been addressed, the intrinsic presence of the DNA molecule poses restrictions regarding the nature of chemical transformations amenable in DELs.20 In particular, strong (Lewis)-acids, oxidizing reagents, elevated temperatures, and chemical transformations requiring anhydrous and inert conditions enforce a high barrier to practical implementation in high-throughput settings. Recently, our groups and others have developed DNA-compatible reactions that harness diverse chemical feedstocks including carboxylic acids,21–23 (α-silyl)amines,24–26 1,4-dihydropyridines (DHPs),27 and alkyl bromides.26,28 Despite the vast chemical space available for drug discovery today,29,30 transformations for the assembly of C(sp2)–C(sp2) bonds remain an integral part of the toolbox of medicinal chemists.31,32 The development of such transformations from commodity and medicinally relevant BBs would further advance DEL technology (Scheme 2).
 | ||
| Scheme 2 Medicinally relevant heteroarene scaffolds containing (hetero)arene C(sp2)–C(sp2) bonds (top). On-DNA heteroarylation protocols using palladium two-electron cross-coupling processes (middle). This strategy: direct photochemical C–H heteroarylation through the intermediacy of aryl radicals (bottom). | ||
Notably, progress to achieve C(sp2)–C(sp2) cross-coupled products on DNA using (hetero)aryl halides or aryl fluorosulfonates with organoboron reagents in homogeneous or micellar systems has been reported (Scheme 2, middle).33–35 These procedures, however, typically require elevated temperatures or highly activated electrophiles for the reaction to proceed under traditional palladium-catalyzed, two-electron cross-coupling conditions. To address these inherent mechanistic limitations, the rise of photoredox catalysis has provided access to alternative single-electron-transfer (SET) reaction pathways facilitating C–C and C–X bond formation via open shell intermediates under exceptionally mild conditions at room temperature, platforms that continue to render a significant impact in DEL settings.36–40
As part of a research program centered on the development of novel synthetic methods to expand chemical space in DELs,12,26,27 herein we report a photochemical strategy for the assembly of C(sp2)–C(sp2) linkages using DNA-conjugated (hetero)aryl halides41 as carbon-based radical precursors (Scheme 2, bottom). This Minisci-type C–H arylation harnesses medicinally-relevant heteroarenes as acceptors, bypassing the need for prefunctionalization of library BBs.
Traditionally, activation of carbon–halogen bonds relies on harsh reaction conditions such as metal–halogen exchange,42 reaction with metal hydrides,43 or transition metal-catalyzed dehalogenation reactions.44,45 By contrast, photoexcited transition metal complexes, such as fac-tris[2-phenylpyridinato-C2,N]iridium(III) [Ir(ppy)3], can reach powerful reduction potentials ( = −1.88 V, Ered = −2.23 V vs. SCE46), facilitating a direct reductive dehalogenation event of aryl electrophiles under mild conditions.44 In this context, the photoreduction of DNA-conjugated aryl halides presents a powerful scenario to access multifunctional subunits on DNA from readily available building blocks. Although Minisci-type alkylation processes generally require protonation of the nitrogen to facilitate lowering of the LUMO energy for radical attack,47 aryl radicals are known to undergo functionalization under neutral conditions.48 We reasoned this reactivity profile would be ideal for on-DNA chemistries that benefit from user-friendly processes amenable to high-throughput platforms.
= −1.88 V, Ered = −2.23 V vs. SCE46), facilitating a direct reductive dehalogenation event of aryl electrophiles under mild conditions.44 In this context, the photoreduction of DNA-conjugated aryl halides presents a powerful scenario to access multifunctional subunits on DNA from readily available building blocks. Although Minisci-type alkylation processes generally require protonation of the nitrogen to facilitate lowering of the LUMO energy for radical attack,47 aryl radicals are known to undergo functionalization under neutral conditions.48 We reasoned this reactivity profile would be ideal for on-DNA chemistries that benefit from user-friendly processes amenable to high-throughput platforms.
Discussion
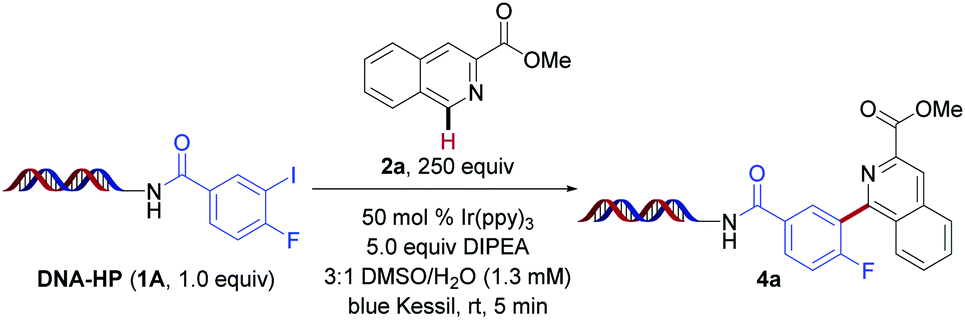
Inspired by the pioneering work of Beckwith,49 followed by seminal publications from Stephenson,44 König,50 and others,51–55 we investigated the feasibility of this photochemical proposal in DEL environments under photoredox conditions. Initial experiments were performed using on-DNA conjugated 4-fluoro-3-iodobenzoic acid (1A) with isoquinoline derivative 2a as the radical acceptor in the presence of Ir(ppy)3 under blue light irradiation (Table 1). Good conversion to the desired arylated product was observed within five minutes of blue light irradiation under open-to-air conditions (entry 1). Next, the influence of the amine reductant was investigated (entries 2–5). Although product formation was detected in the presence of triethylamine, Hantzsch ester (HE, diethyl-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate), diisopropylamine (DIPA), and tetramethylethylenediamine (TMEDA), optimal reactivity was accomplished using 5.0 equivalents of N,N-diisopropylethylamine (DIPEA). As expected, the exclusion of photocatalyst as well as light led to no conversion (entries 14 and 15). Notably, the major side product observed under the developed conditions stems from protodehalogenation of the aryl halide formed through rapid hydrogen atom transfer (HAT). To supress this competitive process,44 higher loadings of the heteroarene were utilized (250 equivalents). This strategy can be quite powerful in the context of DEL reactions, which are conducted on minute scale (e.g., 10–25 nmol), rendering the chemical processes highly selective with side products that can be readily identified.|
|
||
|---|---|---|
| Entry | Deviation from std conditions | % Conversionb |
a Reaction conditions: DNA-conjugated aryl halide 1A (1.0 equiv., 25 nmol), methyl isoquinoline-3-carboxylate 2a (250 equiv., 6.25 μmol), Ir(ppy)3 (0.5 equiv., 12.5 nmol), DIPEA (5.0 equiv., 125 nmol), 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO/H2O (1.25 mM), 5 min irradiation with blue Kessil lamps (λmax = 456 nm, 40 W).
b Conversion to 4a was determined by LC/MS analysis (see ESI). :1 DMSO/H2O (1.25 mM), 5 min irradiation with blue Kessil lamps (λmax = 456 nm, 40 W).
b Conversion to 4a was determined by LC/MS analysis (see ESI).
|
||
| 1 | None | 60 |
| 2 | Et3N | 56 |
| 3 | Hantzsch ester | 48 |
| 4 | DIPA | 41 |
| 5 | TMEDA | 43 |
| 6 | 1 equiv. DIPEA | 57 |
| 7 | 10 equiv. DIPEA | 51 |
| 8 | 20 equiv. DIPEA | 41 |
| 9 | 0.25 equiv. Ir(ppy)3 | 60 |
| 10 | 1.0 equiv. Ir(ppy)3 | 56 |
| 11 | 0.25 equiv. Ir(ppy)3 + 10 equiv. DIPEA | 53 |
| 12 | 0.10 equiv. Ir(ppy)3 + 15 equiv. DIPEA | 43 |
| 13 | No amine | 34 |
| 14 | No Ir(ppy)3 | 0 |
| 15 | No light | 0 |
The scope of the photochemical reaction was evaluated on a broad cross-section of pharmaceutically-relevant heterocyclic space employing DNA-conjugated aryl iodide 1A (Scheme 3). In general, successful (hetero)arylation of both electron-deficient and electron-rich heteroaromatics was accomplished in the presence of structural scaffolds displaying a high density of pendant functional groups. First, the C–H carbofunctionalization of electron-deficient isoquinolines and quinolines was examined. Substitution at the 3- and 4-positions of the isoquinoline moiety was successful (4a–4c). Aryl radical addition to 2-aminoquinoline (4d) occurs with retention of the free amine handle, delivering linchpins that drive derivatization in DEL-library settings.29,56 Of note, non-substituted heteroarenes, such as quinoline, were found to be reactive under the developed reaction conditions, but were excluded from our study for practical reasons because of the formation of regioisomers upon radical addition. Next, a palette of electron-rich pyrroles (4e, 4f, 4g, 4i, 5h, 6h) as well as indoles (4j–4q) was investigated. Successful arylation was observed in the absence of nitrogen protecting groups. The method further facilitates the incorporation of multifunctional handles including esters (4a, 4e, 4z), nitriles (4f, 4i, 4k, 4l, 4u), ketones (4g, 4o), aldehydes (4m, 4n), and an amide (4q).
 | ||
| Scheme 3 Evaluation of heteroarene scope. Conversion was determined by LC/MS analysis (see ESI†). Reaction conditions: DNA-conjugated aryl halide (1.0 equiv., 25 nmol), heteroaryl subunit (250 equiv., 6.25 μmol), Ir(ppy)3 (0.5 equiv., 12.5 nmol), DIPEA (5.0 equiv., 125 nmol), 3:1 DMSO/H2O (1.3 mM), 5 min irradiation with blue Kessil lamps (λmax = 456 nm, 40 W). a DNA-conjugated aryl halide derived from 4-chloro-3-iodo-benzoic acid (1B) was used. b DNA-conjugated aryl halide derived from 2-bromoisonicotinic acid (1C) was used. c Reaction was irradiated for 10 min. | ||
Medicinally-relevant scaffolds57 including pyridazine (4r), pyrimidines (4s, 6s), as well as pyrazine (6t) were efficiently incorporated, underscoring the versatility of this photochemical heteroarylation. Furthermore, five-membered ring analogues such as thiophene (4u), benzothiazoles (4v, 4w), indazole (4x), benzimidazoles (4y, 4z), and imidazole (4aa) served as competent substrates under the developed conditions. In contrast, pyridine derivatives were found to be non-productive radical acceptors under the developed reaction conditions. Given the importance of peptides and their corresponding macrocycles in drug discovery efforts,58,59 the carbofunctionalization of tryptophan derivatives was examined. Under the developed conditions, effective C–H arylation of the electron-rich indole heterocycles in tryptophan–proline dipeptide 4ab and monomeric tryptophan 6ad was observed. In addition to free amine handles, N-Boc-proline moiety 4ab was incorporated. These initial findings may present underexplored opportunities for the conjugation of peptide derivatives in DEL-settings using tryptophan as an efficient radical trap. Finally, the modification of biomolecules displaying higher degrees of molecular complexity, including caffeine (4ac) and dihydroquinine (6ae), proved successful. Contrary to these findings, derivatives of nucleobases, such as adenine or adenosine, showed no conversion in our hands. Of pertinent note, the ease of identification of reaction components in DEL synthesis is valuable for efficacious library composition. Under the developed photochemical heteroarylation, the corresponding DNA-bound, dehalogenated aryl subunit, stemming from C–X bond reduction followed by a rapid hydrogen atom transfer event, is typically observed as the major by-product in cases where the yield is compromised.60 Finally, good regioselectivity with respect to aryl radical additions can be expected based on precedented Minisci-type radical functionalizations.61–65
To examine the influence of the halogenated aryl moiety toward the developed transformation, various DNA-bound headpieces (HPs) using pyrrole 2g as an acceptor (Scheme 4, top) were examined. Aryl iodides (1B, 1D, 1G, and 1H) all served as competent substrates. Despite having an intrinsically higher reduction potential,50 (hetero)aryl bromides (1C, 1E, and 1F) readily undergo reduction in the presence of the photocatalyst Ir(ppy)3. Given the commercial availability of these building blocks, they serve as crucial feedstocks in drug discovery efforts and therefore are abundant in DEL libraries.66
 | ||
| Scheme 4 Evaluation of DNA-conjugated (hetero)aryl halides and heteroarenes. Conversion was determined by LC/MS analysis (see ESI†). Reaction conditions: DNA-conjugated aryl halide (1.0 equiv., 25 nmol), heteroaryl subunit (250 equiv., 6.25 μmol), Ir(ppy)3 (0.5 equiv., 12.5 nmol), DIPEA (5.0 equiv., 125 nmol), 3:1 DMSO/H2O (1.3 mM), 5 min irradiation with blue Kessil lamps (λmax = 456 nm, 40 W). | ||
To validate the robustness of the method further, the reactivity of various HPs with selected heteroaryl BBs was examined (Scheme 4, bottom). Isoquinoline 5a displayed good reactivity with a total of six different HPs (1B, 1C, 1D, 1I, 1J, and 1K). Notably, complete retention of the chloride handle in 1B and 1K, and similarly selective reduction of the C–I bond over the C–Br handle in 1J, showcase the direct availability of electrophilic groups for subsequent cross-couplings. The C–H arylation of indole-5-carboxaldehyde was further evaluated with a total of five different HPs (1E, 1I, 1J, 1K, and 1L) in comparable conversions. For the series employing iodo-chloro-substituted HP (1B), diverse functional groups were compatible, including ester 5a, free aniline 5d, nitriles 5i–l, ketone 5o, and phenol 5af. A different selection of BBs exhibited comparable reactivity using 2-bromoisonicotinic acid HP (1C, examples 6a–6ag). Remarkably, effective heteroaryl radical addition to electron-deficient isoquinoline 2a was observed. This is likely a result of the pronounced nucleophilicity of pyridyl radicals in DMSO as the solvent.54 Electron-rich pyrrolyl-, pyrazolyl-, or imidazolyl-halide derivatives were found to be non-productive substrates. Furthermore, the investigation of DNA-bound styrene system 1I demonstrates that the aryl radical additions occur exclusively to the heteroarene in preference to the activated olefin. Overall, these findings demonstrate that myriad aryl halides on-DNA can be successfully reduced to achieve the direct C–H functionalization of electron-rich as well as electron-deficient heterocycles.
As the aryl radical intermediates generated in these transformations are highly reactive and potentially damaging to DNA, it was necessary to demonstrate that the integrity of the DNA remained intact during the indicated photochemical processes to ensure fidelity in the barcoding that relates the DNA sequence to the building blocks installed. To this end, a model DNA conjugate composed of an exemplary headpiece ligated to a 4-cycle tag and equipped with a 2-base 5′ overhang was subjected to the standard reaction conditions under blue light irradiation. This model DNA conjugate was also reacted in the absence of either DIPEA, photocatalyst, or light. Upon completion of reactions under these conditions, these DNA conjugates were elongated by ligation to introduce the essential PCR primers and then quantified by qPCR. No substantial difference in qPCR amplification was observed across the various reaction samples, indicating DNA compatibility under the developed photochemical heteroarylation conditions (see ESI†).
Conclusion
In summary, a robust protocol for the direct C–H carbofunctionalization of heterocycles was achieved via the photoreduction of DNA-bound halogenated aryl subunits. The reaction proceeds at room temperature and is completed within minutes of blue light illumination under open-to-air conditions. The developed method is amenable to both electron-rich as well as electron-deficient medicinally-relevant heterocyclic building blocks, thereby expanding the chemical space available for DEL synthesis. This heteroarylation proceeds without the need for pre-functionalization of the heteroarene moiety or the presence of acidic additives, rendering this process suitable in DEL settings. Of note, electrophilic handles, including aryl chlorides, remain untouched during the course of the reaction, providing avenues for subsequent cross-couplings on-DNA. Furthermore, free alcohol and amine handles are tolerated, presenting opportunities for rapid library diversification. These findings demonstrate a general blueprint toward the synthesis of heteroaryl subunits, a complementary radical approach to traditional two-electron cross-coupling chemistry for C(sp2)–C(sp2) bond construction on-DNA.Conflicts of interest
There are no conflicts to declare.Data availability
Preparation of on-DNA substrates, synthesis of heteroarene derivatives, reaction workflow, procedures for photoinduced transformations, qPCR, PCR and sequencing data, NMR spectra, and UPLC/MS data have been uploaded as the ESI† material.Author contributions
Dr Shorouk O. Badir conceived the topic. Dr Matthias Krumb, Dr Lisa Marie Kammer, Dr Shorouk O. Badir, and Dr María Jesús Cabrera-Alonso completed experiments with input from Professor Gary A. Molander. Dr Matthias Krumb, Dr Lisa Marie Kammer, and Dr Shorouk O. Badir prepared the manuscript with input from Professor Gary A. Molander and Dr Lisa A. Marcaurelle. All authors contributed to ideation and discussion of results.Acknowledgements
The authors are grateful for financial support provided by NIGMS (R35 GM 131680 to G. M.) and GlaxoSmithKline. Dr Shorouk O. Badir is supported by the Bristol-Meyer-Squibb Graduate Fellowship for Synthetic Organic Chemistry. Dr María Jesús Cabrera-Afonso acknowledges the Fundación Ramón Areces for a postdoctoral fellowship. The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of NMRs used in this study. We thank Dr Melissa C. Grenier-Davies (GSK), Dr Chris Dimitri (GSK), Dr Pradeep Bandaru (GSK), and Haleh Kazemi (GSK) for stimulating discussions. We thank Dr Charles W. Ross, III (UPenn) for mass spectral data. Johnson Matthey is acknowledged for donation of iridium(III) chloride used in this study, and Kessil is thanked for the donation of lamps.Notes and references
- A. A. Shelat and R. K. Guy, Nat. Chem. Biol., 2007, 3, 442–446 CrossRef CAS PubMed.
- J. R. Broach and J. Thorner, Nature, 1996, 384, 14–16 CrossRef CAS PubMed.
- A. Carnero, Clin. Transl. Oncol., 2006, 8, 482–490 CrossRef CAS PubMed.
- K. P. Mishra, L. Ganju, M. Sairam, P. K. Banerjee and R. C. Sawhney, Biomed. Pharmacother., 2008, 62, 94–98 CrossRef CAS PubMed.
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CrossRef CAS PubMed.
- P. R. Fitzgerald and B. M. Paegel, Chem. Rev., 2021, 121(12), 7155–7177 CrossRef CAS PubMed.
- D. Madsen, C. Azevedo, I. Micco, L. K. Petersen and N. J. V. Hansen, in Prog. Med. Chem., ed. D. R. Witty and B. Cox, Elsevier, 2020, vol. 59, pp. 181–249 Search PubMed.
- D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- Y. Shi, Y.-r. Wu, J.-q. Yu, W.-n. Zhang and C.-l. Zhuang, RSC Adv., 2021, 11, 2359–2376 RSC.
- M. Song and G. T. Hwang, J. Med. Chem., 2020, 63, 6578–6599 CrossRef CAS PubMed.
- A. L. Satz, J. Cai, Y. Chen, R. Goodnow, F. Gruber, A. Kowalczyk, A. Petersen, G. Naderi-Oboodi, L. Orzechowski and Q. Strebel, Bioconjugate Chem., 2015, 26, 1623–1632 CrossRef CAS PubMed.
- S. Patel, S. O. Badir and G. A. Molander, Trends Chem., 2021, 3, 161–175 CrossRef CAS PubMed.
- A. Mullard, Nature, 2016, 530, 367–369 CrossRef CAS PubMed.
- P. A. Harris, B. W. King, D. Bandyopadhyay, S. B. Berger, N. Campobasso, C. A. Capriotti, J. A. Cox, L. Dare, X. Dong and J. N. Finger, J. Med. Chem., 2016, 59, 2163–2178 CrossRef CAS PubMed.
- A. Litovchick, C. E. Dumelin, S. Habeshian, D. Gikunju, M.-A. Guié, P. Centrella, Y. Zhang, E. A. Sigel, J. W. Cuozzo and A. D. Keefe, Sci. Rep., 2015, 5, 1–8 Search PubMed.
- M. A. Clark, R. A. Acharya, C. C. Arico-Muendel, S. L. Belyanskaya, D. R. Benjamin, N. R. Carlson, P. A. Centrella, C. H. Chiu, S. P. Creaser and J. W. Cuozzo, Nat. Chem. Biol., 2009, 5, 647–654 CrossRef CAS PubMed.
- P. A. Harris, S. B. Berger, J. U. Jeong, R. Nagilla, D. Bandyopadhyay, N. Campobasso, C. A. Capriotti, J. A. Cox, L. Dare, X. Dong, P. M. Eidam, J. N. Finger, S. J. Hoffman, J. Kang, V. Kasparcova, B. W. King, R. Lehr, Y. Lan, L. K. Leister, J. D. Lich, T. T. MacDonald, N. A. Miller, M. T. Ouellette, C. S. Pao, A. Rahman, M. A. Reilly, A. R. Rendina, E. J. Rivera, M. C. Schaeffer, C. A. Sehon, R. R. Singhaus, H. H. Sun, B. A. Swift, R. D. Totoritis, A. Vossenkämper, P. Ward, D. D. Wisnoski, D. Zhang, R. W. Marquis, P. J. Gough and J. Bertin, J. Med. Chem., 2017, 60, 1247–1261 CrossRef CAS PubMed.
- R. K. Y. Cheng, C. Fiez-Vandal, O. Schlenker, K. Edman, B. Aggeler, D. G. Brown, G. A. Brown, R. M. Cooke, C. E. Dumelin, A. S. Doré, S. Geschwindner, C. Grebner, N.-O. Hermansson, A. Jazayeri, P. Johansson, L. Leong, R. Prihandoko, M. Rappas, H. Soutter, A. Snijder, L. Sundström, B. Tehan, P. Thornton, D. Troast, G. Wiggin, A. Zhukov, F. H. Marshall and N. Dekker, Nature, 2017, 545, 112–115 CrossRef CAS PubMed.
- X. Huang, B. Ni, Y. Xi, X. Chu, R. Zhang and H. You, Aging, 2019, 11, 12532–12545 CrossRef CAS PubMed.
- M. L. Malone and B. M. Paegel, ACS Comb. Sci., 2016, 18, 182–187 CrossRef CAS PubMed.
- D. K. Kölmel, R. P. Loach, T. Knauber and M. E. Flanagan, ChemMedChem, 2018, 13, 2159–2165 CrossRef PubMed.
- D. K. Kölmel, J. Meng, M.-H. Tsai, J. Que, R. P. Loach, T. Knauber, J. Wan and M. E. Flanagan, ACS Comb. Sci., 2019, 21, 588–597 CrossRef PubMed.
- Z. Fan, S. Zhao, T. Liu, P.-X. Shen, Z.-N. Cui, Z. Zhuang, Q. Shao, J. S. Chen, A. S. Ratnayake, M. E. Flanagan, D. K. Kölmel, D. W. Piotrowski, P. Richardson and J.-Q. Yu, Chem. Sci., 2020, 11, 12282–12288 RSC.
- A. Gironda-Martínez, D. Neri, F. Samain and E. J. Donckele, Org. Lett., 2019, 21, 9555–9558 CrossRef PubMed.
- Y. Li, E. Gabriele, F. Samain, N. Favalli, F. Sladojevich, J. r. Scheuermann and D. Neri, ACS Comb. Sci., 2016, 18, 438–443 CrossRef CAS PubMed.
- S. O. Badir, J. Sim, K. Billings, A. Csakai, X. Zhang, W. Dong and G. A. Molander, Org. Lett., 2020, 22, 1046–1051 CrossRef CAS PubMed.
- J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed.
- D. K. Kölmel, A. S. Ratnayake and M. E. Flanagan, Biochem. Biophys. Res. Commun., 2020, 533, 201–208 CrossRef PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- J.-L. Reymond and M. Awale, ACS Chem. Neurosci., 2012, 3, 649–657 CrossRef CAS PubMed.
- G. I. Stevenson, A. L. Smith, S. Lewis, S. G. Michie, J. G. Neduvelil, S. Patel, R. Marwood, S. Patel and J. L. Castro, Bioorg. Med. Chem. Lett., 2000, 10, 2697–2699 CrossRef CAS PubMed.
- D. Chianelli, P. V. Rucker, J. Roland, D. C. Tully, J. Nelson, X. Liu, B. Bursulaya, E. D. Hernandez, J. Wu, M. Prashad, T. Schlama, Y. Liu, A. Chu, J. Schmeits, D. J. Huang, R. Hill, D. Bao, J. Zoll, Y. Kim, T. Groessl, P. McNamara, B. Liu, W. Richmond, I. Sancho-Martinez, A. Phimister, H. M. Seidel, M. K. Badman, S. B. Joseph, B. Laffitte and V. Molteni, J. Med. Chem., 2020, 63, 3868–3880 CrossRef CAS PubMed.
- Y. Ding, G. J. Franklin, J. L. DeLorey, P. A. Centrella, S. Mataruse, M. A. Clark, S. R. Skinner and S. Belyanskaya, ACS Comb. Sci., 2016, 18, 625–629 CrossRef CAS PubMed.
- H. Xu, F. Ma, N. Wang, W. Hou, H. Xiong, F. Lu, J. Li, S. Wang, P. Ma and G. Yang, Adv. Sci., 2019, 6, 1901551 CrossRef CAS PubMed.
- J. H. Hunter, L. Prendergast, L. F. Valente, A. Madin, G. Pairaudeau and M. J. Waring, Bioconjugate Chem., 2020, 31, 149–155 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef.
- J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- F. Nicholas, G. Bassi, T. Zanetti, J. Scheuermann and D. Neri, Helv. Chim. Acta, 2019, 102, e1900033 CrossRef PubMed.
- W. F. Bailey and J. J. Patricia, J. Organomet. Chem., 1988, 352, 1–46 CrossRef CAS.
- N. M. Yoon, Pure Appl. Chem., 1996, 68, 843–848 CAS.
- J. D. Nguyen, E. M. D'amato, J. M. Narayanam and C. R. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed.
- J. Chen, Y. Zhang, L. Yang, X. Zhang, J. Liu, L. Li and H. Zhang, Tetrahedron, 2007, 63, 4266–4270 CrossRef CAS.
- Y. Wu, D. Kim and T. S. Teets, Synlett, 2021 DOI:10.1055/a-1390-9065.
- J. Tauber, D. Imbri and T. Opatz, Molecules, 2014, 19, 16190–16222 CrossRef PubMed.
- J. Zhang, J. Chen, X. Zhang and X. Lei, J. Org. Chem., 2014, 79, 10682–10688 CrossRef CAS PubMed.
- A. L. Beckwith and S. H. Goh, J. Chem. Soc., Chem. Commun., 1983, 907 RSC.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- R. Aycock, D. Vogt and N. T. Jui, Chem. Sci., 2017, 8, 7998–8003 RSC.
- R. Aycock, H. Wang and N. Jui, Chem. Sci., 2017, 8, 3121–3125 RSC.
- J. I. Bardagi, I. Ghosh, M. Schmalzbauer, T. Ghosh and B. König, Eur. J. Org. Chem., 2018, 2018, 34–40 CrossRef CAS.
- A. J. Boyington, M.-L. Y. Riu and N. T. Jui, J. Am. Chem. Soc., 2017, 139, 6582–6585 CrossRef CAS PubMed.
- C. P. Seath, D. B. Vogt, Z. Xu, A. J. Boyington and N. T. Jui, J. Am. Chem. Soc., 2018, 140, 15525–15534 CrossRef CAS PubMed.
- P. Slobbe, E. Ruijter and R. V. Orru, MedChemComm, 2012, 3, 1189–1218 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- A. M. White and D. J. Craik, Expert Opin. Drug Discovery, 2016, 11, 1151–1163 CrossRef CAS PubMed.
- Z. Zhu, A. Shaginian, L. C. Grady, T. O'Keeffe, X. E. Shi, C. P. Davie, G. L. Simpson, J. A. Messer, G. Evindar, R. N. Bream, P. P. Thansandote, N. R. Prentice, A. M. Mason and S. Pal, ACS Chem. Biol., 2018, 13, 53–59 CrossRef CAS PubMed.
- A. L. Satz, ACS Comb. Sci., 2016, 18, 415–424 CrossRef CAS PubMed.
- A. Arora and J. D. Weaver, Org. Lett., 2016, 18, 3996–3999 CrossRef CAS PubMed.
- A. S. Kyei, K. Tchabanenko, J. E. Baldwin and R. M. Adlington, Tetrahedron Lett., 2004, 45, 8931–8934 CrossRef CAS.
- M. M. Nebe, D. Loeper, F. Fürmeyer and T. Opatz, Eur. J. Org. Chem., 2018, 2018, 2471–2476 CrossRef CAS.
- J.-H. Ye, L. Zhu, S.-S. Yan, M. Miao, X.-C. Zhang, W.-J. Zhou, J. Li, Y. Lan and D.-G. Yu, ACS Catal., 2017, 7, 8324–8330 CrossRef CAS.
- E. C. Swift, T. M. Williams and C. R. J. Stephenson, Synlett, 2016, 27, 754–758 CrossRef CAS.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, as well as spectral data. See DOI: 10.1039/d1sc05683b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |