
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel/Brønsted acid dual-catalyzed regio- and enantioselective hydrophosphinylation of 1,3-dienes: access to chiral allylic phosphine oxides†
Jiao
Long
a,
Yuqiang
Li
a,
Weining
Zhao
*b and
Guoyin
Yin
 *a
*a
aThe Institute for Advanced Studies, Wuhan University, Wuhan 430072, China. E-mail: yinguoyin@whu.edu.cn
bCollege of Pharmacy, Shenzhen Technology University, Shenzhen 518118, China. E-mail: zhaoweining@sztu.edu.cn
First published on 28th December 2021
Abstract
While chiral allylic organophosphorus compounds are widely utilized in asymmetric catalysis and for accessing bioactive molecules, their synthetic methods are still very limited. We report the development of asymmetric nickel/Brønsted acid dual-catalyzed hydrophosphinylation of 1,3-dienes with phosphine oxides. This reaction is characterized by an inexpensive chiral catalyst, broad substrate scope, and high regio- and enantioselectivity. This study allows the construction of chiral allylic phosphine oxides in a highly economic and efficient manner. Preliminary mechanistic investigations suggest that the 1,3-diene insertion into the chiral Ni–H species is a highly regioselective process and the formation of the chiral C–P bond is an irreversible step.
Introduction
Chiral organophosphorus compounds are widely utilized as synthetic building blocks for accessing bioactive molecules and functional materials.1 Indeed, as chiral ligands, they also play significant roles in transition metal-catalyzed2 and organocatalytic2h,3 asymmetric transformations (Fig. 1a). Chiral allylic organophosphorus compounds contain pendant unsaturation that might be leveraged for downstream synthesis applications. For instance, they could undergo hydrogenation,4 hydrofunctionalization5 or difunctionalization6 to deliver a series of structurally diverse chiral phosphines (Fig. 1b). Despite the versatility of chiral allylic organophosphorus compounds, efficient methods for their preparation are rather limited.7 Asymmetric allylic substitution of P–H with electrophiles containing a good leaving group was an efficient strategy to deliver chiral allylic organophosphorus compounds (Fig. 1c, left),7b,8 wherein P-stereogenic organophosphorus compounds are usually obtained.8 Recently, the enantioselective hydrofunctionalization of conjugated 1,3-dienes, allenes and alkynes with C-,9 N-,10 or S-nucleophiles11 has attracted broad attention and has been developed as a powerful tool to synthesize chiral allylic compounds.12 Asymmetric hydrophosphination (including hydrophosphonylation and hydrophosphinylation)13 of 1,3-dienes with P–H nucleophiles is undoubtedly a straightforward and atom-efficient approach to construct chiral allylic organophosphorus compounds. A seminal study of enantioselective coupling of terminal 1,3-dienes and phosphine oxides affording a series of chiral α-methyl allylic phosphine oxides by palladium catalysis was demonstrated by Dong and coworkers in 2018 (Fig. 1c, right).14 When we were preparing this manuscript, an elegant example of Pd-catalyzed hydrophosphinylation of allenes with phosphine oxides producing chiral allylic phosphine oxides was disclosed by Wang.15 Given their multifunctional utility, the development of new catalytic systems based on inexpensive metal catalysts for the efficient synthesis of chiral allylic organophosphorus compounds not only greatly benefits synthetic chemists and pharmacologists, but also represents a significant advance in metal catalysis. The past few decades have witnessed tremendous advances in the nickel catalyst.16 Apart from low cost and ready availability, the nickel catalyst holds a unique reactivity profile.17 As a continuation of our previous studies on nickel-catalyzed functionalization of alkenes,18 particularly the asymmetric hydroamination of 1,3-dienes19 and inspired by Dong's work,14 we speculated that the application of a smaller nickel catalyst would allow the hydrophosphinylation reaction to occur under milder conditions and has better stereocontrol, leading to the development of a highly enantioselective nickel-catalyzed reaction. Herein we describe our success in the above speculations, resulting in the development of asymmetric nickel/Brønsted acid-catalyzed hydrophosphinylation of 1,3-dienes with secondary phosphine oxides (Fig. 1d). This methodology tolerates both 1,4-disubstituted acyclic dienes and terminal 1,3-dienes, and is characterized by an inexpensive catalyst and high regio- and enantioselectivity. This study provides an efficient access to a diversity of chiral allylic organophosphorus compounds from readily available starting materials.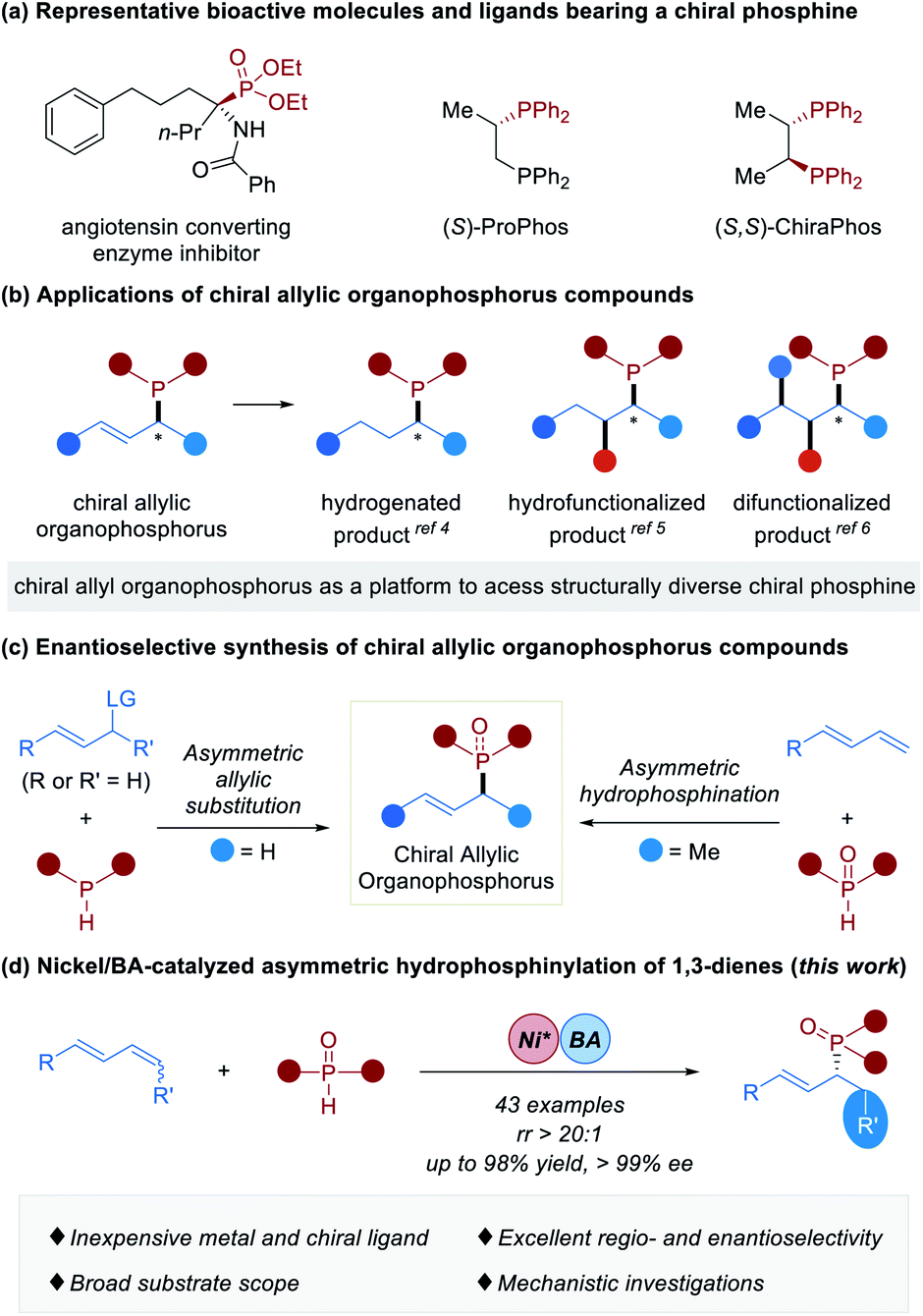 | ||
| Fig. 1 (a) Representative bioactive molecules and ligands bearing a chiral phosphine; (b) applications of chiral allylic organophosphorus compounds; (c) enantioselective synthesis of chiral allylic organophosphorus compounds; (d) nickel/BA-catalyzed asymmetric hydrophosphinylation of 1,3-dienes (this work). | ||
Results and discussion
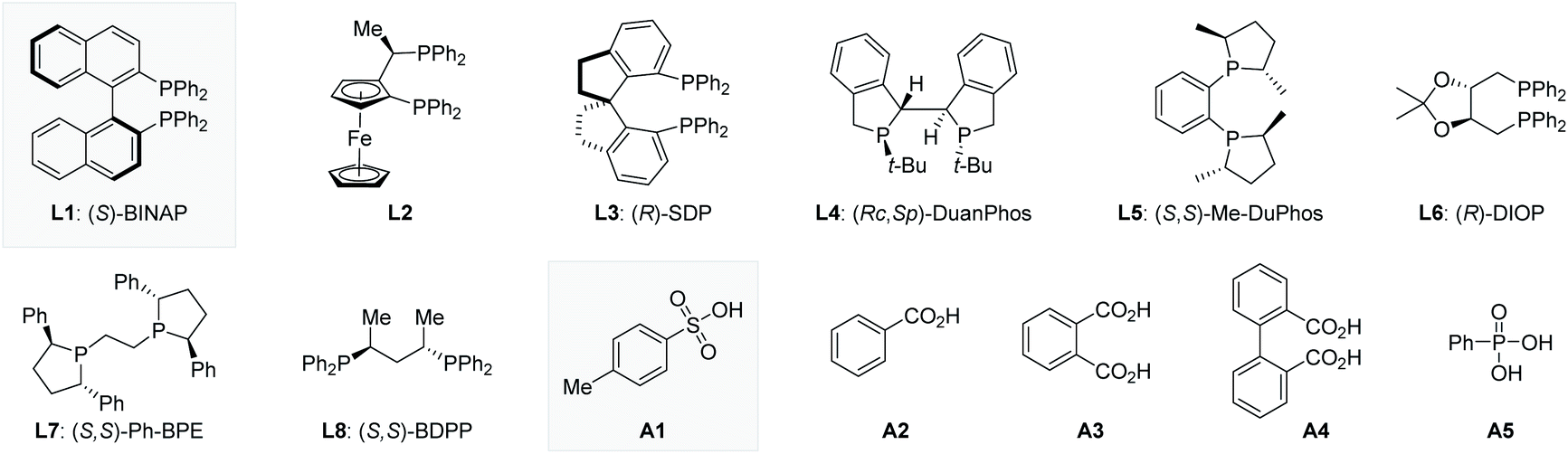
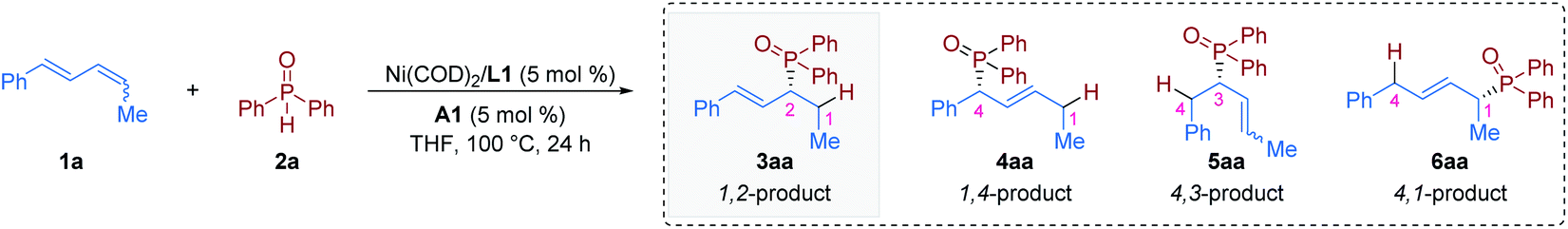
We began our studies by carrying out reactions of the E,Z/E,E mixture of 1,4-disubstituted 1,3-diene 1a20 with commercially available diphenylphosphine oxide 2a (Table 1). After careful evaluation of all reaction parameters, we found that a combination of 5 mol% Ni(COD)2, 5 mol% (S)-BINAP (L1), and 5 mol% TsOH·H2O (A1) in tetrahydrofuran (THF) at 100 °C provided the 1,2-hydrophosphinylation product 3aa in high yield with excellent enantioselectivity (98% yield and >99% ee, entry 1). The use of plane chiral ligand L2, spiro ligand (R)-SDP (L3), P chiral ligand (RC,SP)-DuanPhos (L4), rigid ligand (S,S)-Me-DuPhos (L5), flexible ligand (R)-DIOP (L6), (S,S)-Ph-BPE (L7), or (S,S)-BDPP (L8) instead of axially chiral biaryl bisphosphorus ligands (S)-BINAP (L1) provided the 1,2-hydrophosphinylation product 3aa with either low yield or poor enantioselectivity (entries 2–8). Control experiments indicated that the acid cocatalyst TsOH·H2O (A1) was essential for the success of this hydrophosphinylation reaction; when switching from TsOH·H2O (A1) to other Brønsted acids, the reaction was almost inert (entries 9–13). In addition, the choice of the solvent had an obvious effect on the yield, but little effect on the enantioselectivity (entries 14–19). The reaction activity decreased obviously when the temperature was lowered (entries 20–22). Specifically, no other regioisomers (4aa, 5aa or 6aa) were detected in these reactions (entries 1–22).|
|
||||
|---|---|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) | rr (3aa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4aa + 5aa + 6aa) :4aa + 5aa + 6aa) |
ee (%) |
|
a Unless otherwise noted, all reactions were carried out with 0.24 mmol 1a (E,Z:E,E = 3.0:1), 0.20 mmol 2a, 5.0 mol% Ni(COD)2, 5.0 mol% ligand, and 5.0 mol% acid in 1 mL solvent at 100 °C for 24 h. Yields were determined by gas chromatography analysis, using naphthalene as the internal standard. The rr (regioisomer ratio) was determined by 31P NMR analysis of the reaction mixture. The ee (enantiomeric excess) values were determined by HPLC analysis using a chiral stationary phase. ND, not determined. NP, no product.
b Isolated yield.
|
||||
| 1 | None | 98b | >20:1 |
>99 |
| 2 | L2 instead of L1 | 35 | >20:1 |
−47 |
| 3 | L3 instead of L1 | 6 | ND | ND |
| 4 | L4 instead of L1 | 67 | >20:1 |
61 |
| 5 | L5 instead of L1 | 24 | >20:1 |
−68 |
| 6 | L6 instead of L1 | 16 | >20:1 |
15 |
| 7 | L7 instead of L1 | 62 | >20:1 |
93 |
| 8 | L8 instead of L1 | 31 | >20:1 |
36 |
| 9 | A2 instead of A1 | Trace | ND | ND |
| 10 | A3 instead of A1 | Trace | ND | ND |
| 11 | A4 instead of A1 | Trace | ND | ND |
| 12 | A5 instead of A1 | Trace | ND | ND |
| 13 | Without A1 | NP | — | — |
| 14 | Toluene instead of THF | 68 | >20:1 |
>99 |
| 15 | EtOAc instead of THF | 71 | >20:1 |
95 |
| 16 | DCE instead of THF | 6 | ND | ND |
| 17 | DMF instead of THF | 9 | ND | ND |
| 18 | 1,4-Dioxane instead of THF | 59 | >20:1 |
98 |
| 19 | MTBE instead of THF | 38 | >20:1 |
94 |
| 20 | 80 °C | 92 | >20:1 |
>99 |
| 21 | 50 °C | 33 | >20:1 |
>99 |
| 22 | 25 °C | Trace | ND | ND |
|
||||
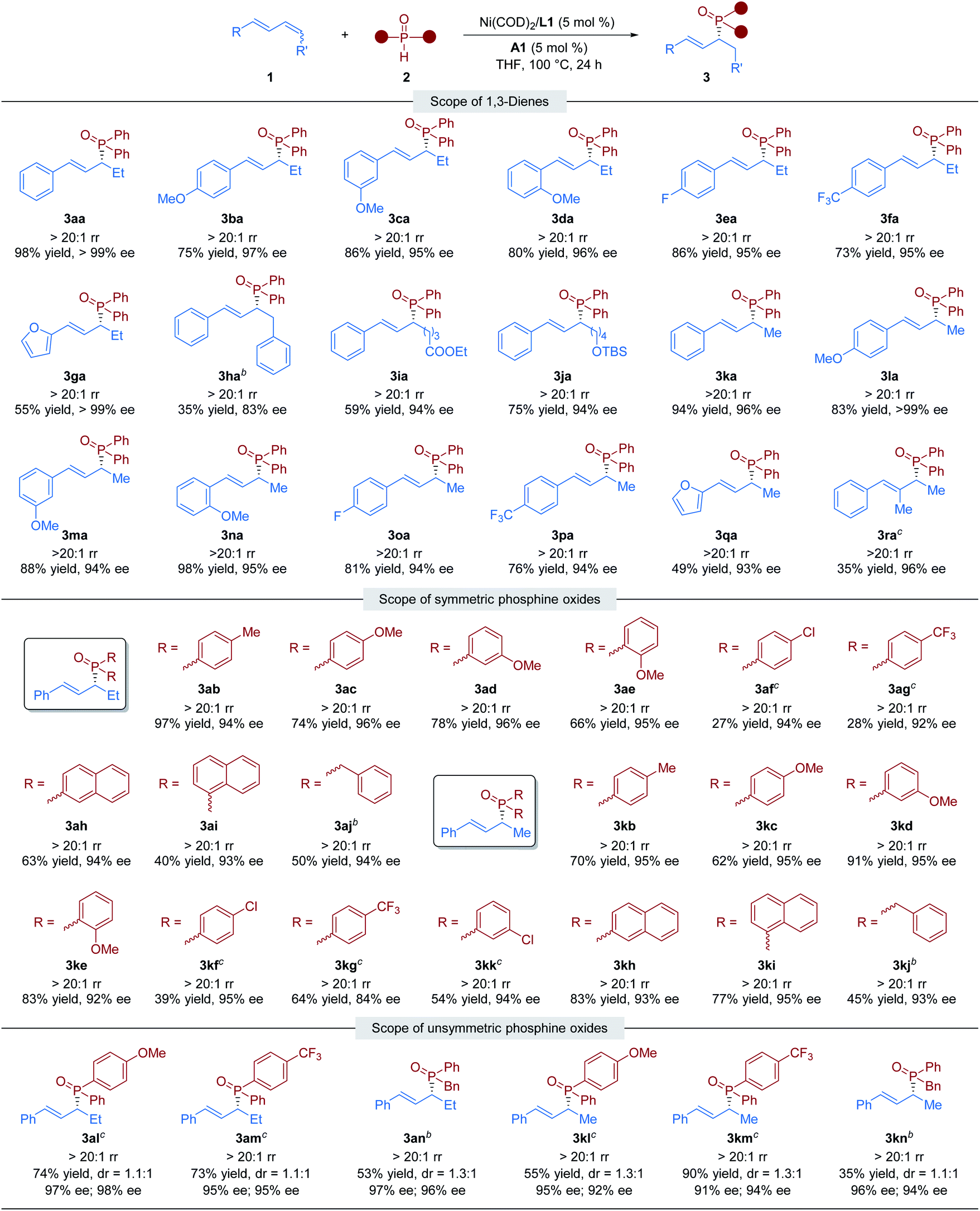
With the optimal conditions in hand, we shifted our attention to investigate the generality of this nickel/Brønsted acid-catalyzed asymmetric hydrophosphinylation reaction. Utilizing 2a, we initially examined the scope of the internal 1,3-dienes. As shown in Table 2, we began with a series of 1-methyl-4-substituted phenyl-1,3-butadiene substrates. Regardless of the electronic properties or the position of the substituent on the benzene ring, all the tested aryl–alkyl-substituted 1,3-dienes could be transformed into the desired chiral allylic phosphine oxides in high yields with excellent regio- and enantioselectivity (3aa–3fa). 1,3-Dienes with a furan ring substituent also gave the corresponding hydrophosphinylation product (3ga) in moderate yield with perfect regio- and enantioselectivity (55% yield, >20:1 rr and >99% ee). In particular, symmetric aryl–aryl-substituted 1,3-diene, which is a challenging substrate in transition metal-catalyzed hydrofunctionalizations,12a reacted smoothly with diphenylphosphine oxides to give the desired product 3ha with >20:1 rr and 83% ee, although in low yield. In addition, the substrate bearing an alkyl chain with a terminal ethyl ester (COOEt) or silyl ether (OTBS) group afforded the desired products 3ia and 3ja in 59% and 75% yields respectively, both with >20:1 rr and 94% ee. We also investigated the hydrophosphinylation of various terminal 1,3-dienes with phosphine oxide 2a. We found that a variety of aryl-substituted dienes could be transformed into chiral allylic phosphine oxide products 3ka–3ra with moderate to high reactivity (35%–98% yield) and excellent selectivity (>20:1 rr, 93–99% ee). Unfortunately, alkyl–alkyl-substituted 1,3-dienes gave the corresponding hydrophosphinylation products with poor regioselectivity (see the ESI† for details).
| a See the ESI for details. Data are presented as mean values from two experiments. Isolated yields are reported. The rr was determined by 31P NMR analysis of the reaction mixture. The dr (diastereomer ratio) was determined by 1H NMR or 31P NMR analysis. ee values were determined by HPLC analysis using a chiral stationary phase. b 48 h. c 80 h. |
|---|
|
Subsequently, the scope of secondary phosphine oxides was studied. A set of symmetric secondary phosphine oxides were examined with both internal and terminal dienes under the optimal conditions. As shown in Table 2, this hydrophosphinylation tolerates both aryl and benzyl phosphine oxides, thus produced a series of structurally and electronically different chiral allylic phosphine oxides (3ab–3aj and 3kb–3kk). Overall, electron-deficient phosphine oxides provided lower yields than electron-rich ones (3af, 3ag, 3kf, 3kg and 3kkvs.3ac–3ae and 3kc–3ke), probably due to their weaker nucleophilicity. Notably, fused ring motifs, which are the basis of a large class of ligand scaffolds,21 can also be incorporated in the phosphine oxide partner to generate 1,2-hydrophosphinylation products (3ah, 3ai, 3kh, and 3ki). Regrettably, dialkyl phosphine oxides were scarcely tolerated in the transformation (see the ESI† for details). We also studied the dynamic kinetic asymmetric transformation of unsymmetric secondary phosphine oxides under the standard conditions, which could construct vicinal C-stereogenic and P-stereogenic synthons simultaneously. The hydrophosphinylation of 1,4-disubstituted 1,3-diene 1a with racemic unsymmetric phosphine oxides (±)-2l, (±)-2m, and (±)-2n gave the expected products in moderate to good yield (53–74% yield) with excellent regio- and enantioselectivity (>20:1 rr, 95–98% ee), but with no diastereocontrolling (dr = 1.1:1–1.3:1) (3al–3an). The similar results were obtained in the transformation of terminal 1,3-diene 1k with unsymmetric phosphine oxides (3kl–3kn). To further determine whether the reaction undergoes kinetic resolution or dynamic kinetic resolution,22 the hydrophosphinylation of model 1,3-diene substrates 1a and 1k with an excess of unsymmetric secondary phosphine oxide (±)-2n was conducted under the standard reaction conditions respectively. The desired chiral allylic phosphine oxide products 3an and 3kn were obtained with a diastereomeric ratio close to 1:1, and the recovered 2n was totally racemic in both cases (Fig. 2). Thus, neither the kinetic resolution nor dynamic kinetic resolution phenomenon exists in this nickel/Brønsted acid dual-catalyzed hydrophosphinylation.
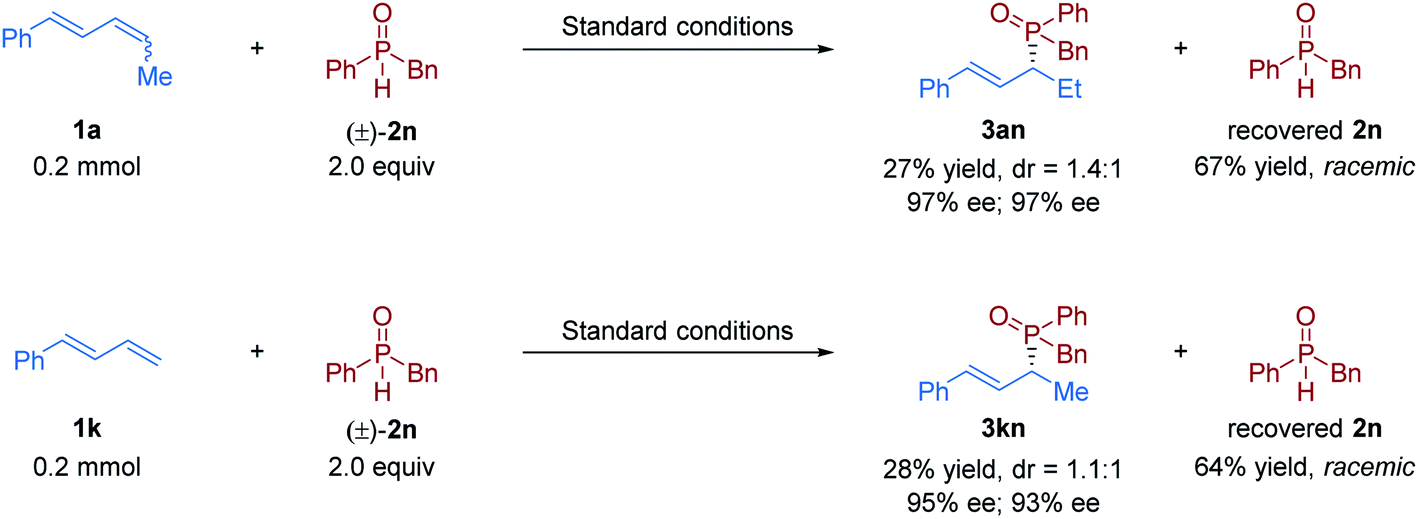 | ||
| Fig. 2 Kinetic resolution/dynamic kinetic resolution investigations. | ||
Furthermore, to demonstrate the practicability of this protocol, a scale-up experiment was performed next. The product 3aa was obtained in the same level of yield and enantioselectivity (90% yield with >99% ee) from a gram-scale experiment of the model reaction (Fig. 3). The chiral allylic phosphine oxide product could be further manipulated to access various useful functionalities. Then derivatization of the product was conducted. Upon exposure of 3aa to a H2 atmosphere with the Pd/C catalyst, the hydrogenation product 3aaa was obtained in quantitative yield without loss of enantioselectivity.
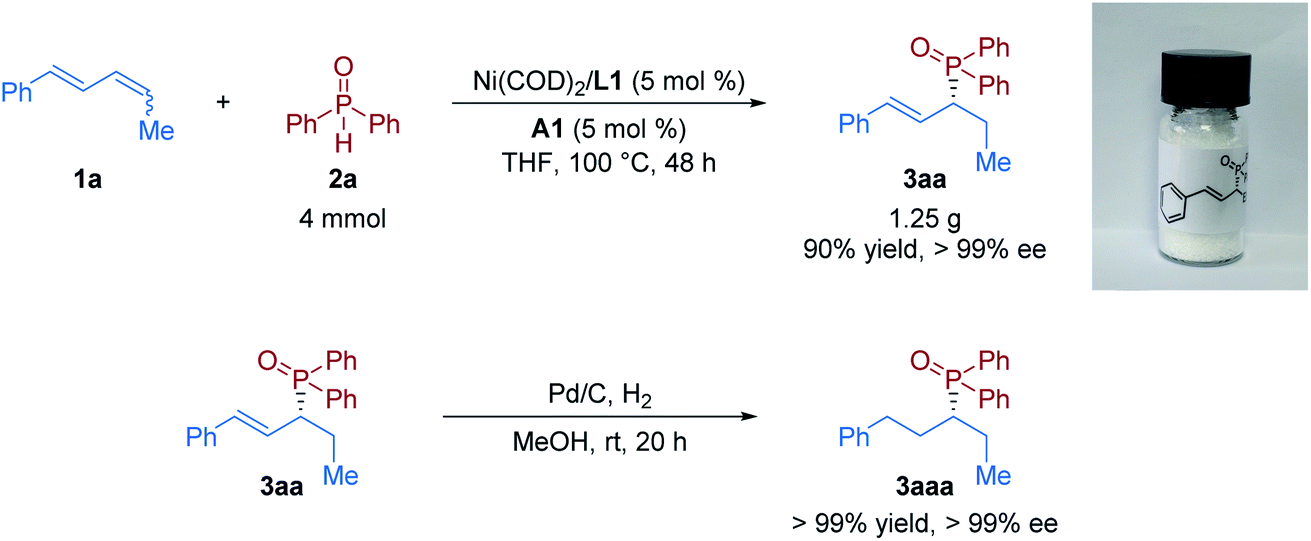 | ||
| Fig. 3 Scale-up reaction and derivatization of the product. | ||
To obtain further information on this transformation, mechanistic investigations were conducted. First, we performed hydrophosphinylation of internal diene 1b with deuterium-labeled phosphine oxide d-2a under standard reaction conditions, which afforded d-3ba with deuterium only at the C1 position; similar results were obtained when the terminal diene 1l was used (Fig. 4a). These results disclosed that the hydrogen source is from the secondary phosphine oxide, and no reversible 1,4-addition occurs in this nickel-catalyzed hydrophosphinylation, which is inconsistent with the Pd-catalyzed system.14 To determine if C–P bond formation is a reversible step in this reaction, we conducted the cross-over experiments by subjecting a mixture of 3aa and 2b or 3ab and 2a to the standard conditions. However, possible crossover products 3ab and 3aa were not observed (Fig. 4b). These experiments suggest that this nickel/Brønsted acid-catalyzed hydrophosphinylation reaction is not a reversible transformation. Furthermore, the results of time-course experiments (Fig. 4c) showed that there was no alteration of enantioselectivity during the reaction. This result further indicates that the formation of C–P bonds is irreversible, which is inconsistent with a previous report on the nickel-catalyzed hydroamination reaction.19
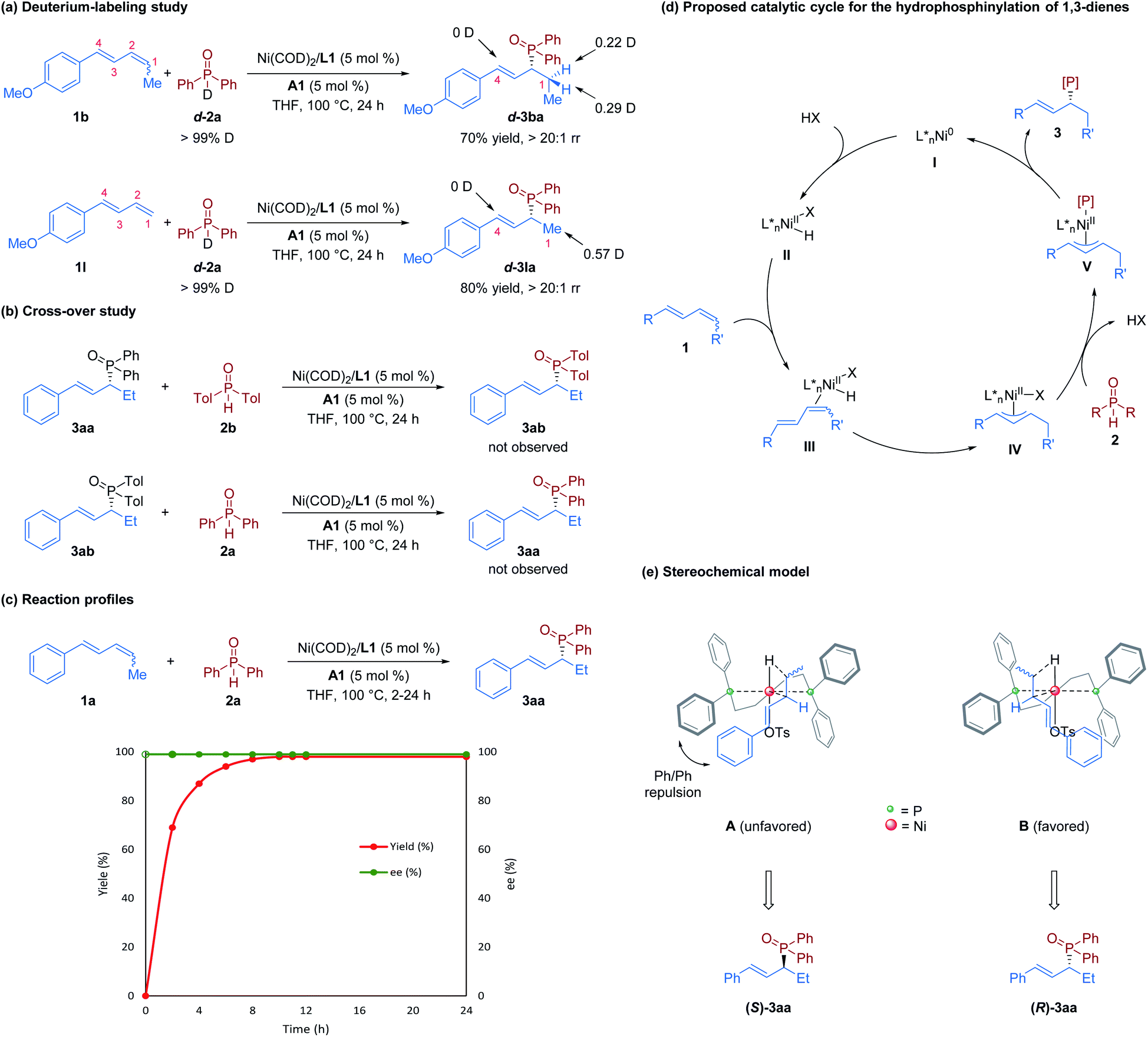 | ||
| Fig. 4 Mechanistic investigations: (a) deuterium-labeling study; (b) cross-over study; (c) reaction profiles; (d) proposed catalytic cycle for the hydrophosphinylation of 1,3-dienes; (e) stereochemical model. | ||
On the basis of our observations and previous reports,14,19 we proposed a catalytic cycle for this nickel-catalyzed hydrophosphinaylation reaction, as depicted in Fig. 4d. The Ni(0) precatalyst undergoes ligand substitution with (S)-BINAP to form chiral monomeric species I, and subsequent oxidative addition to TsOH·H2O (HX) forms Ni–H species II. Then, selective insertion of 1,3-diene 1 provides the key π-allyl nickel intermediate IVvia species III. Species IV then undergoes a ligand exchange or nucleophilic attack with secondary phosphine oxide 2 to form species V. Finally, species V undergos reductive elimination to furnish allylic phosphine oxide 3 and regenerates the Ni-catalyst I. To explore the stereochemical control of the reactions, we proposed a stereochemical model of the transition states of the diene insertion step, as shown in Fig. 4e. Because of Ph/Ph repulsion, model B is favored to afford the R configuration product.
In this hydrophosphinylation reaction, high enantioselectivity was observed regardless of the E,Z/E,E ratio of the starting 1,3-dienes. To explore the origin of the enantioselectivity, we also calculated the energies of the transition states of the diene insertion step for the reaction of (E,Z)-1a and (E,E)-1a to afford (R)-3aa and (S)-3aa, respectively (see the ESI† for details). The results suggested that both (E,Z)-1a and (E,E)-1a underwent an enantiodetermining step to give (R)-3aa as the predominant product.
Conclusions
In summary, we have developed an asymmetric nickel/Brønsted acid-catalyzed hydrophosphinylation reaction of 1,3-dienes with secondary phosphine oxides, featuring highly regio- and enantioselective control with an inexpensive chiral catalyst. This protocol allows the synthesis of a broad range of chiral allylic phosphine oxides in a step and atom-economical fashion from easily accessible materials. Good functional group tolerance and scalability demonstrate the potential of this method in synthesis. Preliminary mechanistic studies indicate that C–P bond formation is an irreversible step. We believe that this chemistry will greatly benefit the biomedical chemistry and chiral ligand development.Data availability
The data that support the findings of this study are available within the article and its ESI files.†Author contributions
J. Long and Y. Li performed the experiments, collected and analyzed the data. W. Zhao and G. Yin conceived and directed the project. J. Long and G. Yin wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21871211 and 22122107), the Fundamental Research Funds for Central Universities (2042019kf0208), and China Postdoctoral Science Foundation (2021M702519). We thank Prof. Qingquan Lu at Wuhan University for sharing the basic instruments. We thank Dr Austin Schultz, from Liwen Bianji (www.liwenbianji.cn/), for editing the language of the draft of this manuscript. The numerical calculations in this paper have been done on the supercomputing system in the Supercomputing Centre of Wuhan University.Notes and references
- (a) Phosphorus chemistry I asymmetric synthesis and bioactive compounds, Topics in current chemistry 360, ed. J.-L. Montchamp, Springer, Switzerland, 2015 Search PubMed; (b) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC.
- (a) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2769 CrossRef CAS PubMed; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3069 CrossRef CAS PubMed; (c) X. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS PubMed; (d) T. Imamoto, J. Synth. Org. Chem., Jpn., 2007, 65, 1060–1069 CrossRef CAS; (e) P. W. N. M. van Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Chem. Rev., 2011, 111, 2077–2118 CrossRef CAS PubMed; (f) Y.-M. He, Y. Feng and Q.-H. Fan, Acc. Chem. Res., 2014, 47, 2894–2906 CrossRef CAS PubMed; (g) W. Zhao, D. Yang and Y. Zhang, Chin. J. Org. Chem., 2016, 36, 2301–2316 CrossRef CAS; (h) G. Xu, C. H. Senanayake and W. Tang, Acc. Chem. Res., 2019, 52, 1101–1112 CrossRef CAS PubMed; (i) F. Wan and W. Tang, Chin. J. Chem., 2021, 39, 954–968 CrossRef CAS.
- (a) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS; (b) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719–724 RSC; (c) S. J. Connon, Angew. Chem., Int. Ed., 2006, 45, 3909–3912 CrossRef CAS PubMed; (d) M. Benaglia and S. Rossi, Org. Biomol. Chem., 2010, 8, 3824–3830 RSC; (e) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005–1018 CrossRef CAS PubMed; (f) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed; (g) R. Xu, H. Yang and W. Tang, Chin. J. Org. Chem., 2020, 40, 1409–1422 CrossRef CAS.
- E. E. Coyle, B. J. Doonan, A. J. Holohan, K. A. Walsh, F. Lavigne, E. H. Krenske and C. J. O'Brien, Angew. Chem., Int. Ed., 2014, 53, 12907–12911 CrossRef CAS PubMed.
- (a) J. M. Gil, J. H. Hah, K. Y. Park and D. Y. Oh, Synth. Commun., 2000, 30, 789–794 CrossRef CAS; (b) S. Demay, M. Lotz, K. Polborn and P. Knochel, Tetrahedron: Asymmetry, 2001, 12, 909–914 CrossRef CAS; (c) M. Stankevic, M. Jaklinska and K. M. Pietrusiewicz, J. Org. Chem., 2012, 77, 1991–2000 CrossRef CAS PubMed; (d) M. Jaklinska, M. Cordier and M. Stankevic, J. Org. Chem., 2016, 81, 1378–1390 CrossRef CAS PubMed.
- (a) N. J. S. Harmat and S. Warren, Tetrahedron Lett., 1990, 31, 2743–2746 CrossRef CAS; (b) P. Camps, G. Colet, M. Font-Bardia, V. Muñoz-Torrero, X. Solansb and S. Vázqueza, Tetrahedron, 2002, 58, 3473–3484 CrossRef CAS; (c) P. Camps, G. Colet, M. Font-Bardia, V. Muñoz-Torrero, X. Solansb and S. Vázqueza, Tetrahedron: Asymmetry, 2002, 13, 759–778 CrossRef CAS; (d) J. H. Hah, B. S. Lee, S. Y. Lee and H.-Y. Lee, Tetrahedron Lett., 2003, 44, 5811–5814 CrossRef CAS.
- (a) T. Nishimura, S. Hirabayashi, Y. Yasuhara and T. Hayashi, J. Am. Chem. Soc., 2006, 128, 2556–2557 CrossRef CAS PubMed; (b) P. Butti, R. Rochat, A. D. Sadow and A. Togni, Angew. Chem., Int. Ed., 2008, 47, 4878–4881 CrossRef CAS PubMed; (c) T. Nishimura, X.-X. Guo and T. Hayashi, Chem.–Asian J., 2008, 3, 1505–1510 CrossRef CAS PubMed; (d) T. Kawamoto, S. Hirabayashi, X.-X. Guo, T. Nishimura and T. Hayashi, Chem. Commun., 2009, 3528–3530 RSC; (e) L. Hong, W. Sun, C. Liu, D. Zhao and R. Wang, Chem. Commun., 2010, 46, 2856–2858 RSC; (f) Y. Huang, R. J. Chew, S. A. Pullarkat, Y. Li and P. H. Leung, J. Org. Chem., 2012, 77, 6849–6854 CrossRef CAS PubMed; (g) M. Hatano, T. Horibe and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 4549–4553 CrossRef CAS PubMed; (h) X.-Y. Yang, W. S. Tay, Y. Li, S. A. Pullarkat and P.-H. Leung, Organometallics, 2015, 34, 5196–5201 CrossRef CAS; (i) W. Hu, E.-Q. Li, Z. Duan and F. Mathey, J. Org. Chem., 2020, 85, 14772–14778 CrossRef CAS PubMed.
- (a) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, J. Am. Chem. Soc., 2019, 141, 16584–16589 CrossRef CAS PubMed; (b) H. Qiu, Q. Dai, J. He, W. Li and J. Zhang, Chem. Sci., 2020, 11, 9983–9988 RSC; (c) S. Zhang, J.-Z. Xiao, Y.-B. Li, C.-Y. Shi and L. Yin, J. Am. Chem. Soc., 2021, 143, 9912–9921 CrossRef CAS PubMed.
- (a) N. J. Adamson, K. C. E. Wilbur and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 2761–2764 CrossRef CAS PubMed; (b) L. Cheng, M.-M. Li, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2018, 140, 11627–11630 CrossRef CAS PubMed; (c) L. Cheng, M.-M. Li, B. Wang, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, Chem. Sci., 2019, 10, 10417–10421 RSC; (d) L. Lv, D. Zhu, Z. Qiu, J. Li and C.-J. Li, ACS Catal., 2019, 9, 9199–9205 CrossRef CAS; (e) S. Park, N. J. Adamson and S. J. Malcolmson, Chem. Sci., 2019, 10, 5176–5182 RSC.
- (a) O. Löber, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4366–4367 CrossRef PubMed; (b) N. J. Adamson, E. Hull and S. J. Malcolmson, J. Am. Chem. Soc., 2017, 139, 7180–7183 CrossRef CAS PubMed; (c) X.-H. Yang and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1774–1777 CrossRef CAS PubMed; (d) S. Park and S. J. Malcolmson, ACS Catal., 2018, 8, 8468–8476 CrossRef CAS; (e) G. Tran, W. Shao and C. Mazet, J. Am. Chem. Soc., 2019, 141, 14814–14822 CrossRef CAS PubMed.
- (a) X.-H. Yang, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 10443–10446 CrossRef CAS PubMed; (b) X.-H. Yang, R. T. Davison, S.-Z. Nie, F. A. Cruz, T. M. McGinnis and V. M. Dong, J. Am. Chem. Soc., 2019, 141, 3006–3013 CrossRef CAS PubMed; (c) Q. Zhang, D. Dong and W. Zi, J. Am. Chem. Soc., 2020, 142, 15860–15869 CrossRef CAS PubMed; (d) M.-M. Li, L. Cheng, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2021, 60, 2948–2951 CrossRef CAS PubMed.
- (a) N. J. Adamson and S. J. Malcolmson, ACS Catal., 2019, 10, 1060–1076 CrossRef; (b) R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545–13598 CrossRef CAS PubMed; (c) G. Li, X. Huo, X. Jiang and W. Zhang, Chem. Soc. Rev., 2020, 49, 2060–2118 RSC.
- (a) L.-B. Han, N. Choi and M. Tanaka, Organometallics, 1996, 15, 3259–3261 CrossRef CAS; (b) F. Mirzaei, L.-B. Han and M. Tanaka, Tetrahedron Lett., 2001, 42, 297–299 CrossRef CAS; (c) L.-B. Han and C.-Q. Zhao, J. Org. Chem., 2005, 70, 10121–10123 CrossRef CAS PubMed; (d) D. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095–2108 RSC; (e) S. Pullarkat, Synthesis, 2015, 48, 493–503 CrossRef; (f) Z.-W. Lu, H. Zhang, Z. Yang, N. Ding, L. Meng and J. Wang, ACS Catal., 2019, 9, 1457–1463 CrossRef CAS; (g) Q. Dai, L. Liu, Y. Qian, W. Li and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20645–20650 CrossRef CAS PubMed; (h) Z. Yang, X. Gu, L.-B. Han and J. Wang, Chem. Sci., 2020, 11, 7451–7455 RSC.
- S.-Z. Nie, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 16450–16454 CrossRef CAS PubMed.
- Z. Yang and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 27288–27292 CrossRef CAS PubMed.
- (a) H. Zhang, Q. Gu and S. You, Chin. J. Org. Chem., 2019, 39, 15–27 CrossRef CAS; (b) A. L. Clevenger, R. M. Stolley, J. Aderibigbe and J. Louie, Chem. Rev., 2020, 120, 6124–6196 CrossRef CAS PubMed; (c) H. Dai, F. Wu and D. Bai, Chin. J. Org. Chem., 2020, 40, 1423–1436 CrossRef CAS; (d) Y. Liu, X.-Q. Dong and X. Zhang, Chin. J. Org. Chem., 2020, 40, 1096–1104 CrossRef CAS; (e) A. D. Marchese, T. Adrianov and M. Lautens, Angew. Chem., Int. Ed., 2021, 60, 16750–16762 CrossRef CAS PubMed; (f) C. Zhu, H. Yue, J. Jia and M. Rueping, Angew. Chem., Int. Ed., 2021, 60, 17810–17831 CrossRef CAS PubMed.
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (b) V. P. Ananikov, ACS Catal., 2015, 5, 1964–1971 CrossRef CAS; (c) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed; (d) S. Manna, K. K. Das, S. Nandy, D. Aich, S. Paul and S. Panda, Coord. Chem. Rev., 2021, 448, 214165 CrossRef CAS.
- (a) Y. Li, H. Pang, D. Wu, Z. Li, W. Wang, H. Wei, Y. Fu and G. Yin, Angew. Chem., Int. Ed., 2019, 58, 8872–8876 CrossRef CAS PubMed; (b) W. Wang, C. Ding, Y. Li, Z. Li, Y. Li, L. Peng and G. Yin, Angew. Chem., Int. Ed., 2019, 58, 4612–4616 CrossRef CAS PubMed; (c) Y. Li, H. Wei, D. Wu, Z. Li, W. Wang and G. Yin, ACS Catal., 2020, 10, 4888–4894 CrossRef CAS; (d) W. Wang, C. Ding and G. Yin, Nat. Catal., 2020, 3, 951–958 CrossRef CAS; (e) Z. Li, D. Wu, C. Ding and G. Yin, CCS Chem., 2021, 3, 576–582 CrossRef CAS; (f) B. Zhao, Y. Li, H. Li, M. Belal, L. Zhu and G. Yin, Sci. Bull., 2021, 66, 570–577 CrossRef CAS; (g) C. Ding, Y. Ren, C. Sun, J. Long and G. Yin, J. Am. Chem. Soc., 2021, 143, 20027–20034 CrossRef CAS PubMed; (h) M. Belal, Z. Li, L. Zhu and G. Yin, Sci. China: Chem., 2021 DOI:10.1007/s11426-021-1172-x; (i) H. Li, J. Long, Y. Li, W. Wang, H. Pang and G. Yin, Eur. J. Org. Chem., 2021, 2021, 1424–1428 CrossRef CAS.
- J. Long, P. Wang, W. Wang, Y. Li and G. Yin, iScience, 2019, 22, 369–379 CrossRef CAS PubMed.
- The substrate was inert in our previous nickel-catalyzed asymmetric hydroamination: ref. 19.
- (a) Q. Wang, Q. Gu and S.-L. You, Acta Chim. Sin., 2019, 77, 690–704 CrossRef CAS; (b) C. Claver, E. Fernandez, A. Gillon, K. Heslop, D. J. Hyett, A. Martorell, A. G. Orpen and P. G. Pringle, Chem. Commun., 2000, 961–962 Search PubMed; (c) C. Li and S. Yang, Chin. J. Org. Chem., 2020, 40, 2159–2160 CrossRef CAS.
- (a) Q. Dai, W. Li, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 20556–20564 CrossRef CAS PubMed; (b) Q. Dai, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 27247–27252 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental details and characterization data, see DOI: 10.1039/d1sc05651d |
| This journal is © The Royal Society of Chemistry 2022 |