DOI:
10.1039/D1SC04595D
(Edge Article)
Chem. Sci., 2022,
13, 1398-1407
Synthesis of bioactive fluoropyrrolidines via copper(I)-catalysed asymmetric 1,3-dipolar cycloaddition of azomethine ylides†
Received
19th August 2021
, Accepted 6th December 2021
First published on 7th December 2021
Abstract
Chiral pyrrolidinyl units are important building blocks in biologically active natural products and drugs, and the development of efficient methods for the synthesis of diverse structured pyrrolidine derivatives is of great importance. Meanwhile, incorporating fluorine containing groups into small molecules often changes their activities to a great extent due to the special physicochemical properties of fluorine atoms. Herein, we report an efficient route to obtain enantioenriched 3,3-difluoro- and 3,3,4-trifluoropyrrolidinyl derivatives by Cu(I)-catalysed enantioselective 1,3-dipolar cycloaddition of azomethine ylides with less active 1,1-difluoro- and 1,1,2-trifluorostyrenes. A series of new fluorinated pyrrolidines have been prepared in high yields (up to 96%) and with excellent stereoselectivities (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 97% ee), and these unique structural blocks could be readily introduced into some natural compounds and pharmaceuticals. Additionally, antifungal activity investigation against four common plant fungi showed that some products possess general and high biological activities; comparison with the low antifungal activities of corresponding nonfluorinated compounds revealed that the fluorine atoms at the pyrrolidinyl rings play a crucial role in the antifungal activity.
:1 dr and 97% ee), and these unique structural blocks could be readily introduced into some natural compounds and pharmaceuticals. Additionally, antifungal activity investigation against four common plant fungi showed that some products possess general and high biological activities; comparison with the low antifungal activities of corresponding nonfluorinated compounds revealed that the fluorine atoms at the pyrrolidinyl rings play a crucial role in the antifungal activity.
Introduction
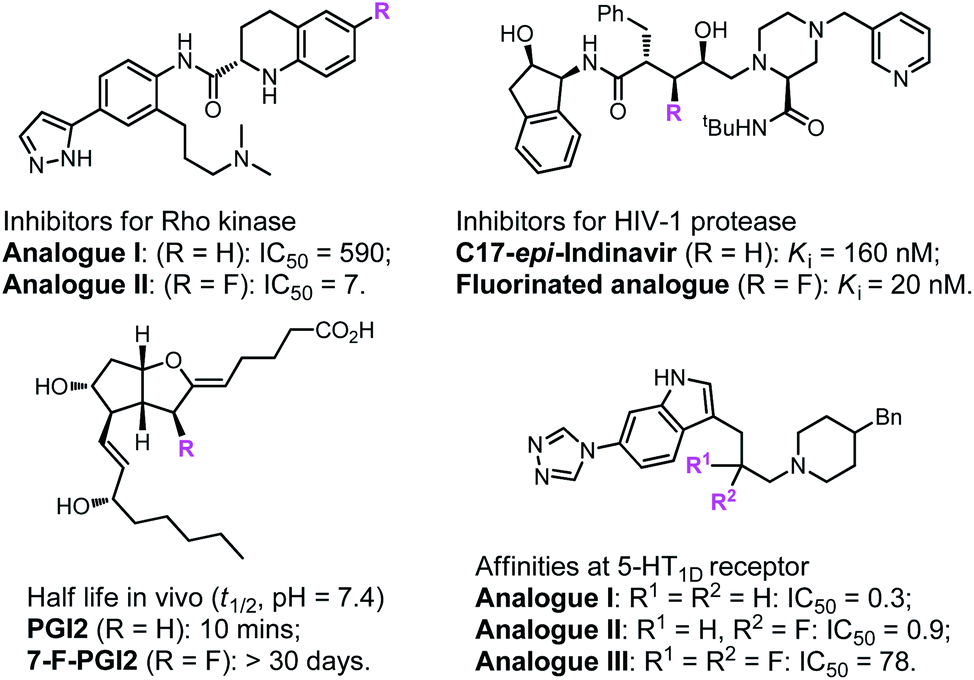
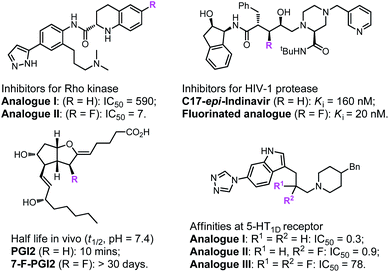
Fluoro-organic chemistry is one of the most fiery-hot areas in modern chemistry, and has currently become crucial in the evolution of many transdisciplinary research fields.1–4 The determinant impact of fluorine is undoubtedly associated with the pharmaceutical and agrochemical industries. Due to their special properties, the introduction of fluorine-containing groups into compounds often greatly affects their biological activity (Fig. 1) and thus has always been regarded as a powerful tactic in agrochemicals and creation of pharmaceuticals.5–14 According to a recent survey, approximately half of newly developed pesticides and 35% of new drug candidates in phase II–III clinical trials contain distinct kinds of fluorine-substituted moieties.15–17 Over the past few decades, the remarkable success of and great demand for fluorine compounds in various fields have provided infinite vitality for organic chemists to explore methods for the construction of fluorinated molecules.18–22
 |
| | Fig. 1 Fluorine effects on biological activity. | |
On the other hand, enantioenriched pyrrolidinyl groups, which are important building blocks in many biologically active natural alkaloids and pharmaceuticals, have attracted much attention in the last few decades.23–28 The transition-metal-catalysed asymmetric 1,3-dipolar cycloaddition of azomethine ylides with electron deficient alkenes represents one of the most powerful methods for the stereocontrolled synthesis of these molecules.29–34 Considering the crucial properties of fluorine atoms, incorporating fluorine atoms into heterocycle rings in chiral pyrrolidine derivatives would change their bioactivities to a great extent. In principle, the asymmetric 1,3-dipolar cycloaddition of azomethine ylides with fluorostyrenes could lead to multiple fluorinated pyrrolidines, however, few studies on the construction and bioactivity evaluation of such molecules have been reported to date.
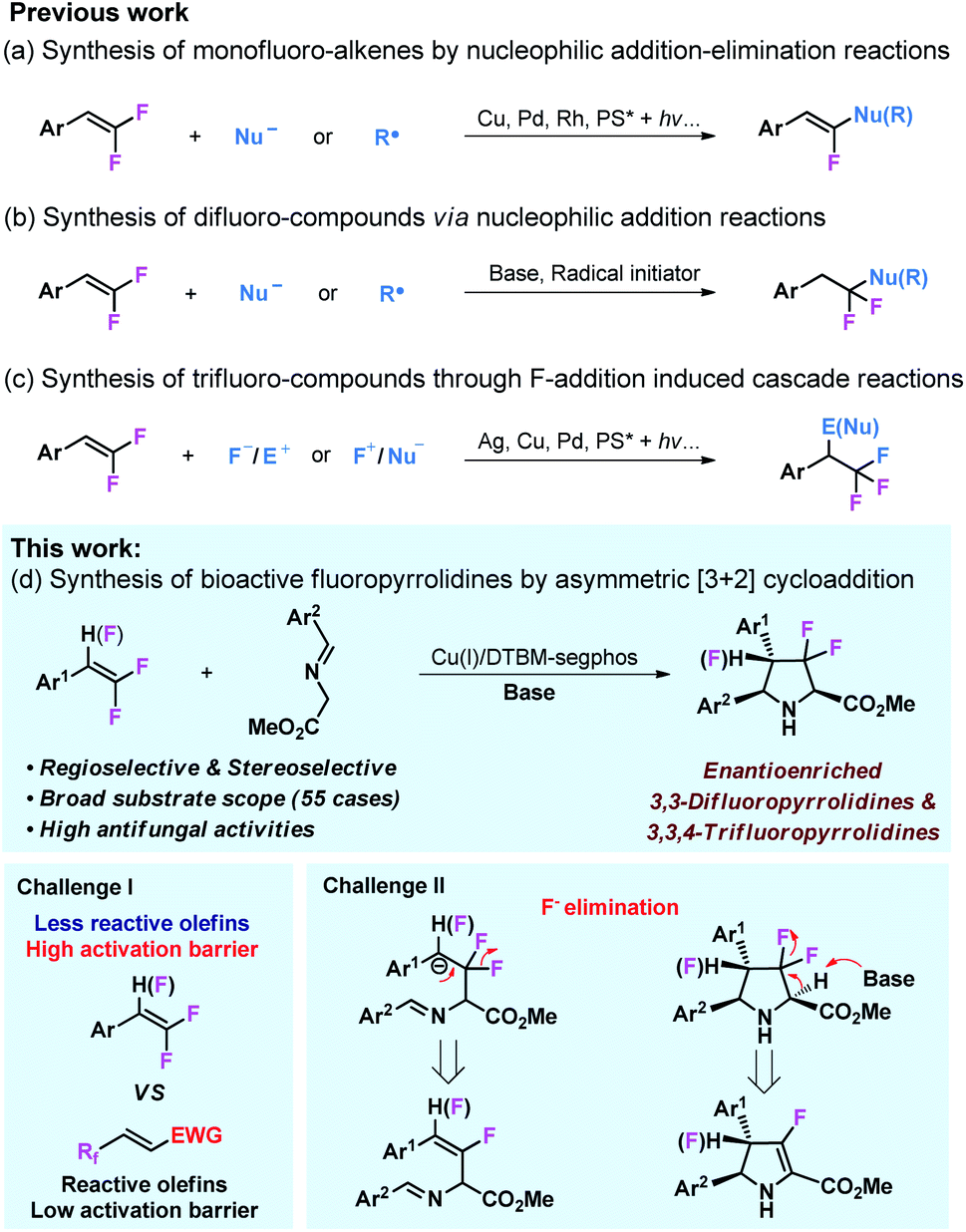
Owing to their multiple reactivities, readily available gem-difluorostyrenes35 can participate in various organic transformations to access structurally diversified fluorocompounds.36–43 Addition or addition–elimination of nucleophiles or free radicals to gem-difluoro-styrenes could lead to difluorocompounds44–46 and monofluoro-alkenes47–55 (Schemes 1a and b). Taking advantage of gem-difluorostyrenes as reliable trifluoromethyl (CF3) precursors through F+/F− addition, several cascade reactions have been achieved to access trifluoromethyl compounds (Scheme 1c).56–60 Despite the comprehensive application of gem-difluorostyrenes in the synthesis of fluoro-compounds, the only example of the asymmetric transformation of gem-difluorostyrenes via fluoroarylation was reported by Zhang and coworkers,61 and asymmetric cycloaddition with these substrates still remains blank.
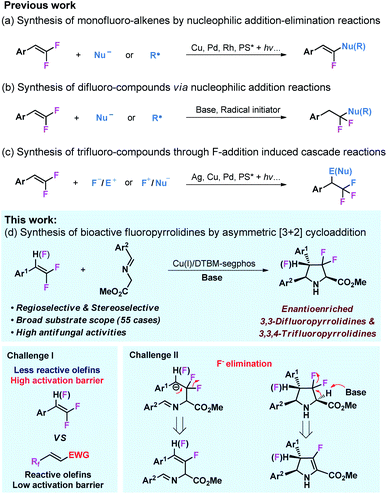 |
| | Scheme 1 Synthesis of fluorochemicals from fluorostyrenes. | |
In view of the importance of fluoro-compounds and chiral N-heterocycles, we envisioned that enantioselective 1,3-dipolar cycloaddition of azomethine ylides with gem-1,1-difluorostyrenes and 1,1,2-trifluorostyrenes would lead to a broad scope of biologically active fluoropyrrolidines.62–68 To achieve this transformation, two challenges need to be considered (Scheme 1d). It is well known that the dipolarophiles of the 1,3-dipolar cycloaddition of azomethine ylide are generally limited to highly activated olefins bearing strong electron withdrawing substituents,29–34 and only a few cases are associated with less reactive olefins, however, the utilization of inactive alkenes as dipolarophiles remains a great challenge.69–72 In addition, the potential elimination of fluorine atoms in intermediates or products is another challenge for this reaction design.73
Results and discussion
Reaction condition optimization
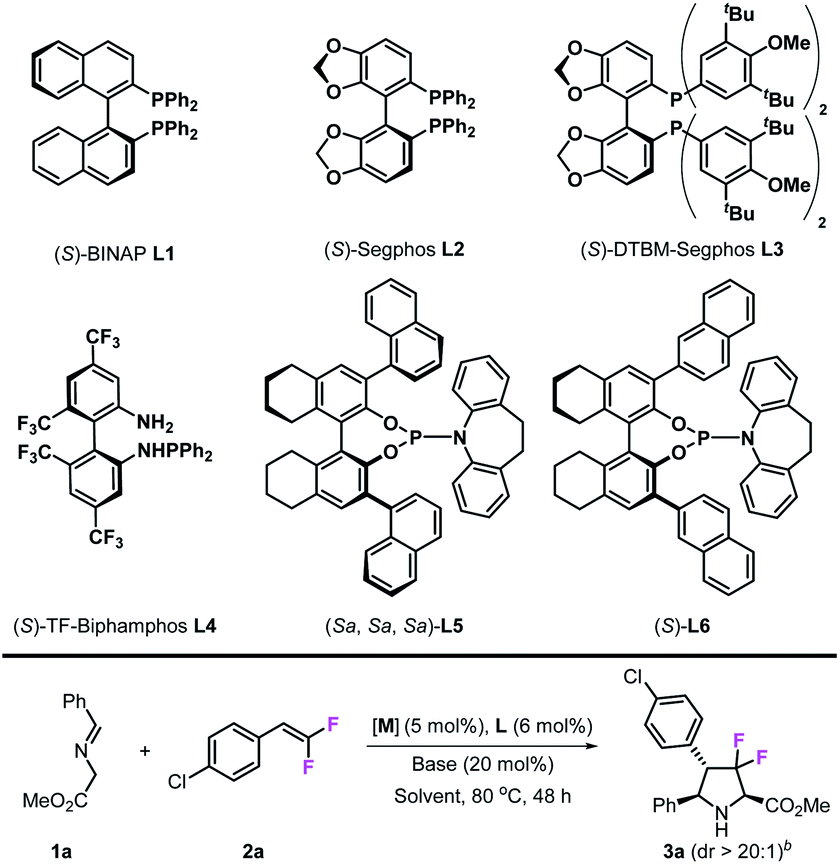
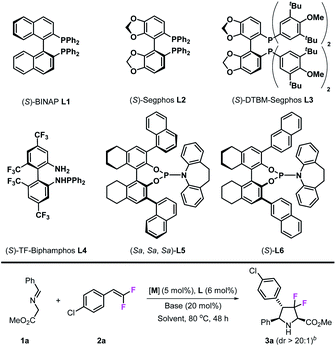
In the initial studies, imino ester 1a was chosen as the azomethine ylide precursor and 4-Cl-gem-difluorostyrene 2a was chosen as the dipolarophile. In the presence of the CuPF6/(S)-BINAP complex (5 mol%) and KOtBu (20 mol%) in toluene, only trace amounts of cycloadduct 3a could be detected at room temperature (Table 1, entry 1); when the reaction temperature was increased to 80 °C, the desired product 3a was obtained in 60% yield with >20:1 dr and 60% ee after 48 h (Table 1, entry 2). Encouraged by these results, we next tested other chiral diphosphine ligands, (S)-segphos L2 gave similar catalytic reactivity with (S)-BINAP (61% yield) but with poor enantioselectivity (28% ee) (Table 1, entry 3). To our delight, (S)-DTBM-segphos L3 exhibited high reactivity in this reaction, which made this reaction happen readily at room temperature and gave 3a in 48% yield and 96% ee (Table 1, entry 4). When the reaction was carried out at 80 °C, the desired product 3a was obtained in 91% yield with >20:1 dr and 95% ee (Table 1, entry 5). Chiral N–P ligands are not effective in this transformation; (S)-TF-biphamphos L4, which served as an efficient ligand in azomethine ylide-involved 1,3-dipolar cycloaddition, gave very poor results in this transformation (Table 1, entry 6), while chiral N–P ligands L5 and L6 also showed low reactivities (Table 1, entries 7 and 8). Besides ligands, metal salts also play an important role in this reaction; when CuBF4 was used, the yield decreased from 91% to 57%, although stereoselectivities were maintained at the same level (>20:1 dr and 92% ee) (Table 1, entry 9), while neither CuOAc nor Cu(OAc)2 could cause this reaction to occur (Table 1, entries 10 and 11). Other metal salts, such as AgOAc, also worked in this transformation, however, they provided the corresponding product 3a with moderate enantioselectivity (Table 1, entry 12). Next, we examined various common solvents such as tetrahydrofuran, ethyl acetate, and 1,4-dioxane, in which, catalytic reactivities decreased to various degrees while stereoselectivities remained at the same level. It seems that bases did not affect stereo-control at all; however, in the presence of K2CO3, 3a was obtained only in 42% yield, which may be due to the poor solubility of K2CO3 in toluene (Table 1, entry 16). Through the experiments above, we finally confirmed the optimal reaction conditions: 5 mol% Cu(CH3CN)4PF6, 6 mol% (S)-DTBM-segphos, 20 mol% KOtBu, and 2 mL of toluene, at 80 °C.
Table 1 Investigation of optimal reaction conditionsa
|
|
| Entry |
[M]c |
L
|
Solvent |
Base |
T (°C) |
Yieldd (%) |
Eee (%) |
|
All reactions were carried out with 0.40 mmol of 1a and 0.20 mmol of 2a in 2 mL solvent.
Dr was determined by crude 1H NMR.
CuPF6 = Cu(CH3CN)4PF6, CuBF4 = Cu(CH3CN)4BF4.
Isolated yield.
Ee was determined by chiral HPLC analysis.
|
| 1 |
CuPF6 |
L1
|
Toluene |
KOtBu |
25 |
Trace |
— |
| 2 |
CuPF6 |
L1
|
Toluene |
KOtBu |
80 |
60 |
60 |
| 3 |
CuPF6 |
L2
|
Toluene |
KOtBu |
80 |
61 |
28 |
| 4 |
CuPF6 |
L3
|
Toluene |
KOtBu |
25 |
48 |
96 |
| 5 |
CuPF
6
|
L3
|
Toluene
|
KO
t
Bu
|
80
|
91
|
95
|
| 6 |
CuPF6 |
L4
|
Toluene |
KOtBu |
80 |
28 |
28 |
| 7 |
CuPF6 |
L5
|
Toluene |
KOtBu |
80 |
28 |
65 |
| 8 |
CuPF6 |
L6
|
Toluene |
KOtBu |
80 |
34 |
94 |
| 9 |
CuBF4 |
L3
|
Toluene |
KOtBu |
80 |
57 |
92 |
| 10 |
CuOAc |
L3
|
Toluene |
KOtBu |
80 |
Trace |
— |
| 11 |
Cu(OAc)2 |
L3
|
Toluene |
KOtBu |
80 |
Trace |
— |
| 12 |
AgOAc |
L3
|
Toluene |
KOtBu |
80 |
85 |
77 |
| 13 |
CuPF6 |
L3
|
THF |
KOtBu |
80 |
81 |
95 |
| 14 |
CuPF6 |
L3
|
AcOEt |
KOtBu |
80 |
61 |
94 |
| 15 |
CuPF6 |
L3
|
1,4-Dioxane |
KOtBu |
80 |
48 |
91 |
| 16 |
CuPF6 |
L3
|
Toluene |
K2CO3 |
80 |
42 |
95 |
| 17 |
CuPF6 |
L3
|
Toluene |
DIPEA |
80 |
90 |
95 |
Substrate scope of azomethine ylides and gem-difluorostyrenes
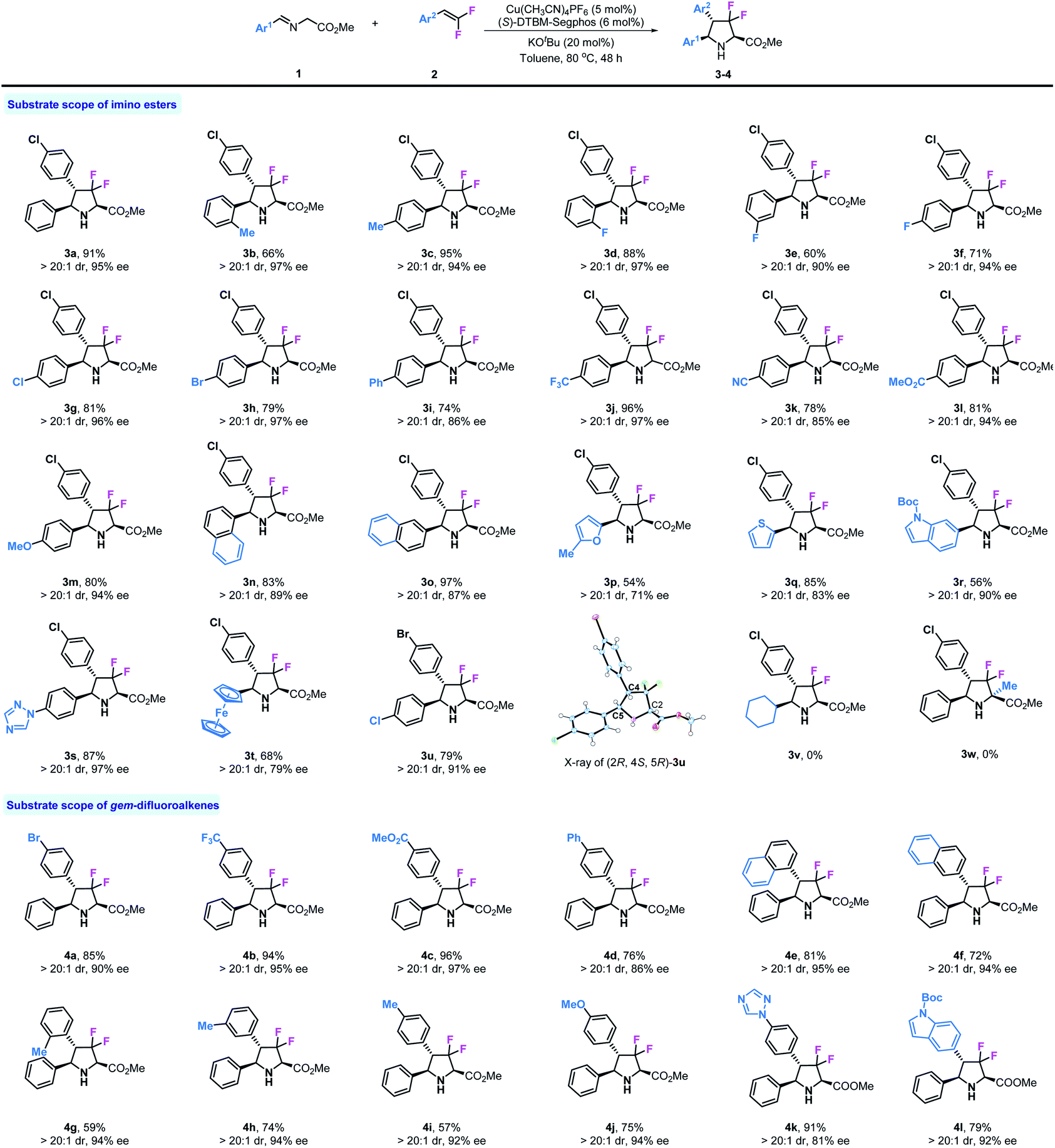
Then, we investigated the reaction of various substituted imino esters 1 with 2a under optimal conditions, and the results are summarized in Table 2. In addition to product 3a, various substituted chiral difluoropyrrolidines can be prepared as a single diastereomer (>20:1 dr) by this method. The steric effect of substituents did not influence the stereoselectivities, and ortho- and para-Me substituted products 3b and 3c were obtained with excellent enantioselectivities (97% and 94% ee respectively). Fluoro-substituted products 3d–3f were obtained with moderate to good yields and with high stereoselectivities. Other halogen substituents, such as chloride or bromide, also tolerated these reaction conditions, providing corresponding products 3g and 3h in good yields (81%, 79%) and with high enantioselectivities (96%, 97% ee). Biphenyl derivative 3i was produced in 74% yield and with high enantioselectivity (86% ee). As shown, substrates bearing strong electron withdrawing substituents such as –CF3, –CN, and –CO2Me performed very well in this reaction, leading to desired products 3j–3l with good reactivities (78–96% yields) and stereoselectivities (>20:1 dr, 85–97% ee). The strong electron donating group –OMe also showed no influence on this reaction (3m). Both 1-naphthyl and 2-naphthyl substituted difluoropyrrolidines 3n and 3o were readily prepared with satisfactory results. Considering the important role of heterocycle units in biologically active compounds, we investigated four substrates containing furyl, thienyl, indolyl and triazolyl in this transformation; to our delight, all corresponding products 3p, 3q, 3r and 3s could be obtained in moderate to good yields (54–87%) and with good enantioselectivities (71–97% ee). We also tried an imino ester bearing a ferrocene group (3t), which worked but yielded the product in moderate yield (68%) and with only 79% ee. To confirm the absolute configuration of 3u, X-ray analysis was carried out and the stereo-configuration of 3u was determined to be (2R, 4S, 5R). Finally, more challenging substrates derived from cyclohexanal 1v and alanine 1w were tested, unfortunately, neither of them could participate in this reaction. Encouraged by the results of imino esters, we next examined a series of substituted gem-difluorostyrenes. As mentioned in the beginning, normal aromatic alkenes could not be used in the 1,3-dipolar cycloaddition of azomethine ylide, except those activated by strong electron withdrawing substituents on phenyl rings; however, due to the electron withdrawing inductive effect of fluorine atoms, all substrates with electron-rich, neutral and deficient substituents can be used in this reaction. Product 4a with a bromide substituent was prepared with similar result to 3a (85% yield, >20:1 dr, 90% ee). As expected, substrates with electron withdrawing groups such as –CF3 and –CO2Me performed very well in this reaction and provided corresponding products 4b and 4c in high yields (94%, 96%) and with excellent enantioselectivities (95%, 97% ee). Diphenyl alkene can be used in this transformation although the enantioselectivity is somewhat lower (86% ee). It seems that steric effect does not affect stereoselective control in this reaction; for example, 1-naphthyl and 2-napthyl substituted gem-difluorostyrenes gave similar results (81%, 95% ee and 72%, 94% ee). Meanwhile, ortho-, meta- and para-methyl substituted products 4g–4i could be created by this method with the same enantioselectivities (94%, 94%, 92% ee) but in different yields (59%, 74%, 57%). The substrate with a strong electron donating group –OMe proceeded smoothly and gave product 4j with >20:1 dr and 94% ee despite a moderate yield (75%). In addition, two gem-difluorostyrenes bearing 1-triazolyl and 6-indolyl substituents were synthesized and tested in this transformation, and both of the desired products 4k and 4l were prepared with satisfactory results.
Table 2 Substrate scope of imino esters 1 and gem-difluorostyrene 2a
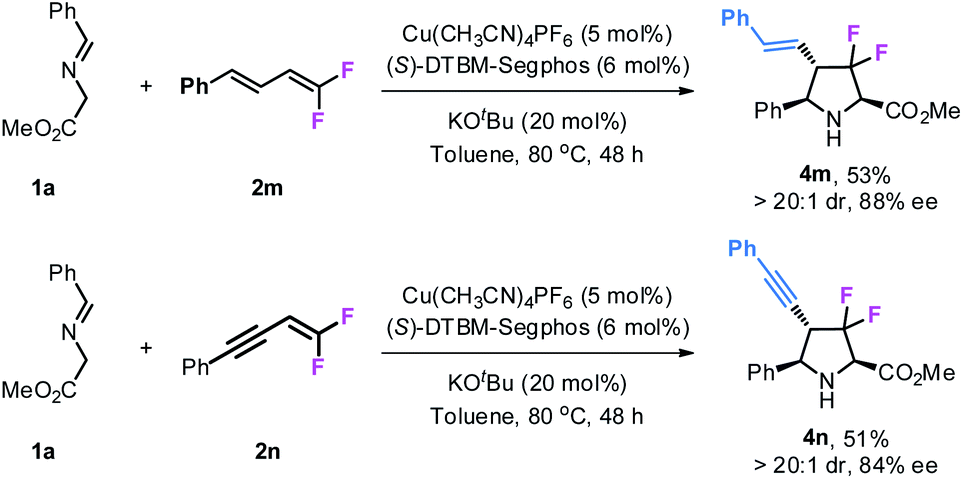
Gem-difluorodiene 2m derived from cinnamaldehyde also participated in this reaction, giving product 4m bearing a styryl group at the 4-position with >20:1 dr and 88% ee although in moderate yield (53%). Similar results were also obtained with gem-difluoroenyne 2n as the fluorinating precursor to access the corresponding product 4n with an alkynyl group (Scheme 2).
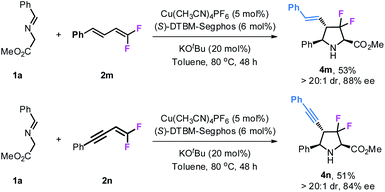 |
| | Scheme 2 Reactions of gem-difluorodiene 2m and difluoroenyne 2n. | |
Modification of natural compounds and drugs
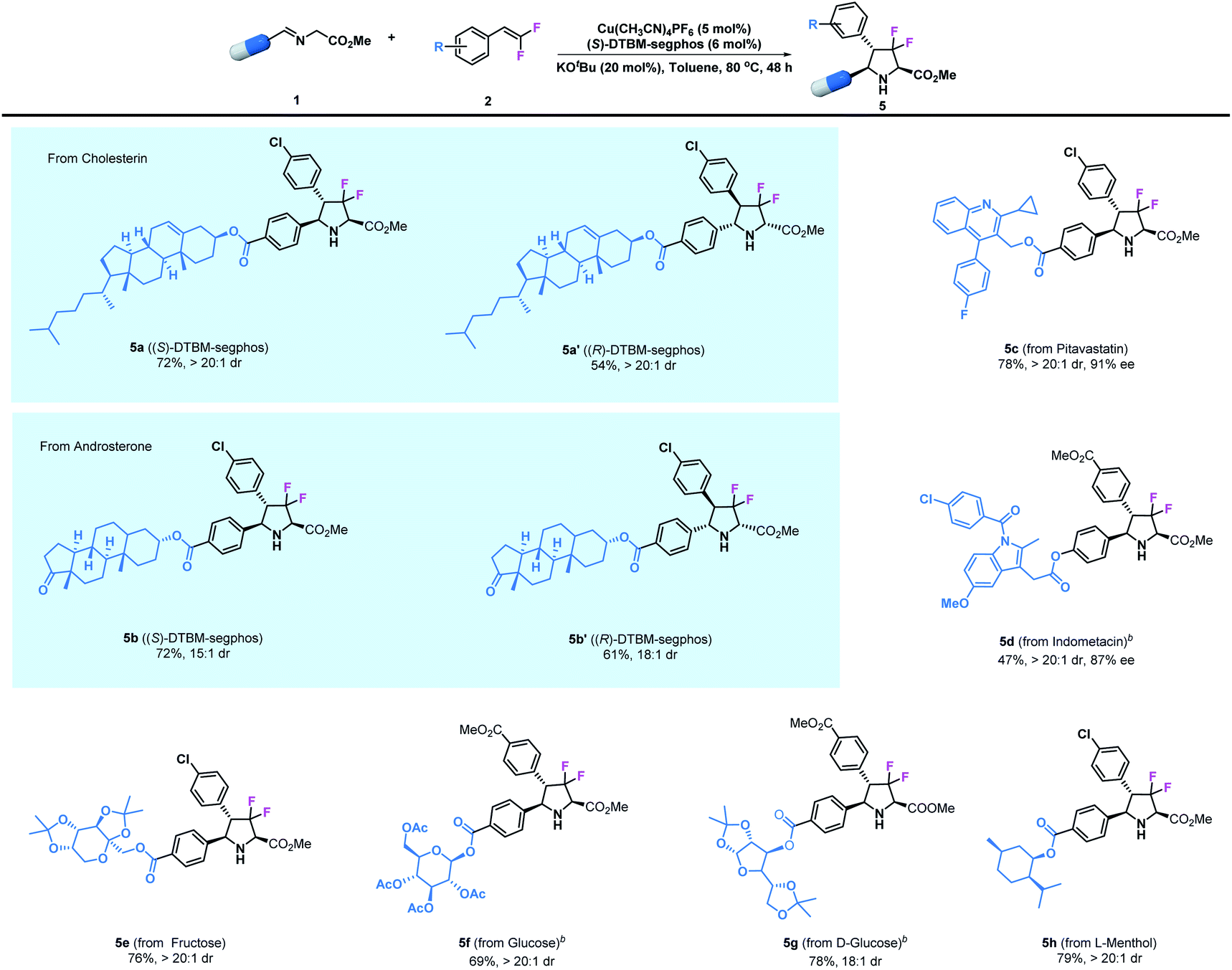
Additionally, since the modification of biologically active compounds is an important method for the development of agrochemicals and pharmaceuticals, the introduction of certain units into biologically active molecules is of great significance. Therefore, we further demonstrate the broad applicability of the asymmetric transformation protocol by examining imino esters derived from natural compounds and currently existing synthetic drugs. As shown in Table 3, the desired product 5a bearing a cholesterin unit could be readily prepared in 72% yield and with >20:1 dr under the optimized reaction conditions; similarly, an androsterone derived imino ester could participate in this reaction to access adduct 5b. Notably, the diastereomers 5a′ and 5b′ could also be prepared in high yield and stereoselectivity by using Cu(I)/(R)-DTBM-segphos as the catalyst. Pitavastatin, which is an important building skeleton in drugs, could also be employed in this reaction to give 5c in good yield (78%) and with excellent stereoselectivity (>20:1 dr and 91% ee). Meanwhile, the well-known commercial analgesic indomethacin has been modified by this method, leading to the corresponding derivative 5d in moderate yield (47%) together with excellent stereoselectivity (>20:1 dr and 87% ee). Encouraged by these results, we next tested saccharide derivatives which are important components in medicines that contain multiple sensitive functional groups. The asymmetric cycloaddition of imino esters derived from fructose and glucose was readily achieved, leading to the corresponding products 5e–5g in high yields (69–78%) and with high stereoselectivities (18:1–>20:1 dr). Finally, L-menthol is undoubtedly one of the most important chiral precursors for the synthesis of agrochemicals and pharmaceuticals. Herein, we easily incorporated this unit with a difluoropyrrolidinyl motif to give product 5h in 79% yield and with >20:1 dr.
Table 3 Modification of natural compounds and pharmaceuticals.a
1,3-Dipolar cycloaddition of 1,1,2-trifluorostyrenes
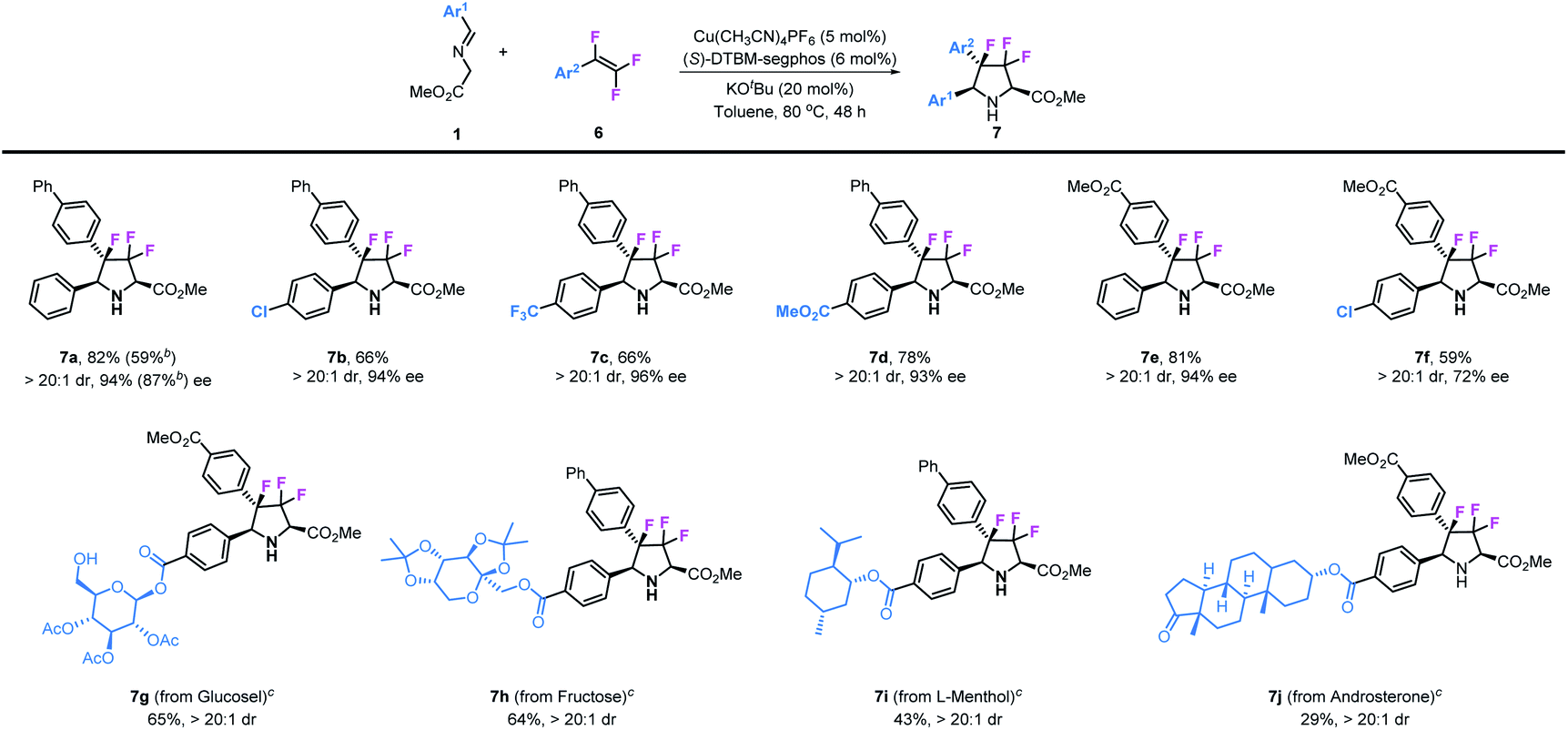
Besides 3,3-difluoropyrrolidines, chiral 3,3,4-trifluoropyrrolidinyl derivatives were also prepared by using 1,1,2-trifluorostyrenes as substrates in this transformation. As shown in Table 4, various substituted 1,1,2-trifluorostyrenes could readily participate in this reaction to access corresponding chiral 3,3,4-trifluoropyrrolidines. Imino esters and trifluorostyrenes with simple structures can be applied as compatible reactants, leading to the desired 3,3,4-trifluoro-pyrrolidines 7a–7f in moderate to good yields (59–82%) and with excellent stereoselectivities (>20:1 dr and 72–96% ee). Similar to the synthesis of 3,3-difluoropyrrolidines, we introduced these unique structural motifs into some kinds of biologically active natural compounds such as glucose (7g), fructose (7h), L-menthol (7i) and androsterone (7j).
Table 4 Synthesis of trifluoropyrrolidine derivatives from trifluorostyrenesa
These reactions should be conducted at 60 °C to prevent decomposition of the obtained products.
Mechanism studies
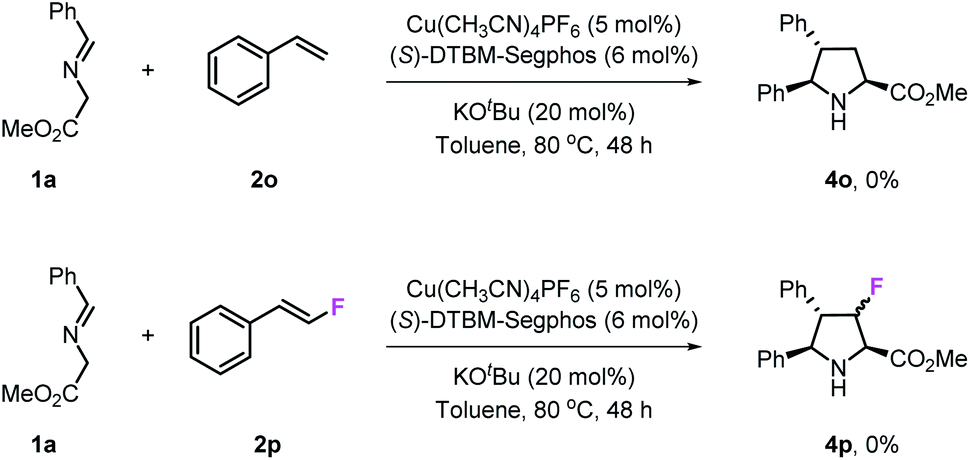
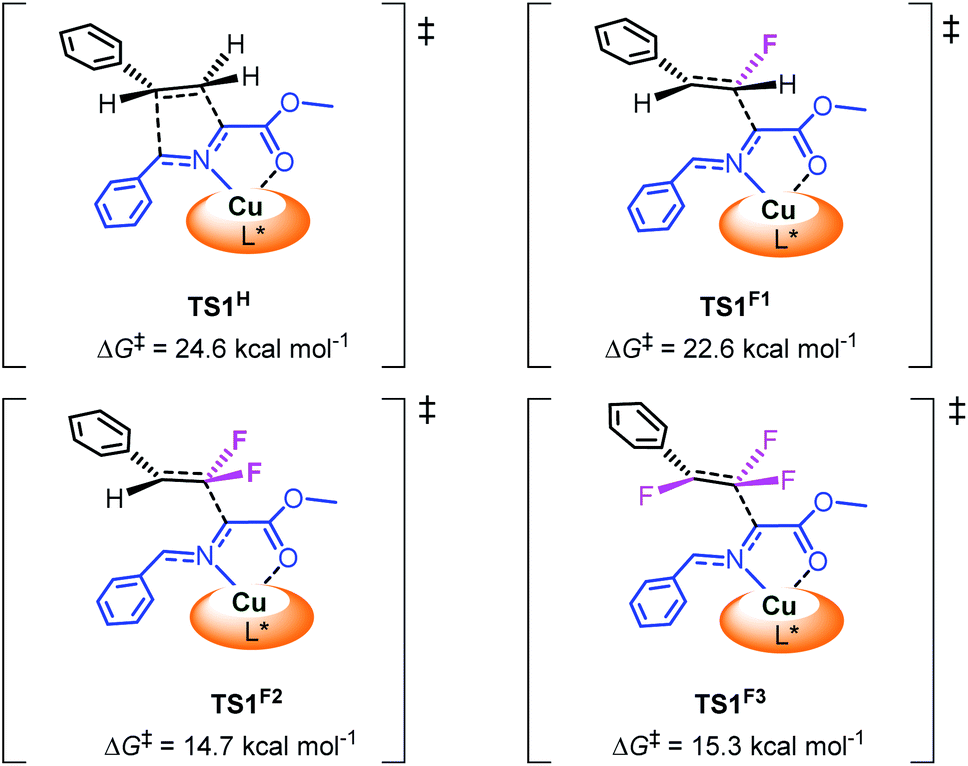
To investigate the influence of fluorine atoms on the reactivity, the reactions of iminoester 1a with styrene 2o and monofluorostyrene 2p were tested. As expected, neither of these reactions happened under the optimal conditions (Scheme 3), which proved that the strong electron withdrawing inductive effect plays a key role in the activation of double bonds. Next, density functional theory (DFT) calculations were performed on the reaction of iminoester 1a with styrene, monofluorostyrene, difluorostyrene and trifluorostyrene. As shown in Scheme 4, in the case of styrene, the reaction tends to proceed via a concerted process, and the activation energy ΔG‡ of TS1H is approximately 24.6 kcal mol−1. In contrast, the 1,3-dipolar cycloaddition of fluorostyrenes underwent stepwise routes that needed to overcome two energy barriers TS1 and TS2 (see the ESI†). However, the activation energy ΔG‡ values of key transition states TS1 for the three reactions are quite different. In the case of monofluorostyrene, the activation energy ΔG‡ of TS1F1 is calculated to be 22.6 kcal mol−1 which is similar to that of styrene. However, with difluorostyrene and trifluorostyrene as substrates, the activation energy ΔG‡ values are much lower (TS1F2 = 14.7 kcal mol−1 and TS1F2 = 15.3 kcal mol−1). These theoretical data are consistent with the experimental results perfectly and provide strong evidence for the crucial role of fluorine atoms in the activation of dipoles.
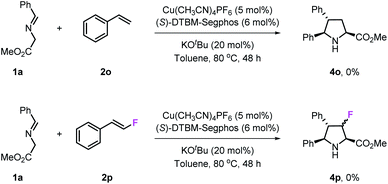 |
| | Scheme 3 Reactions of styrene 2o and monofluorostyrene 2p. | |
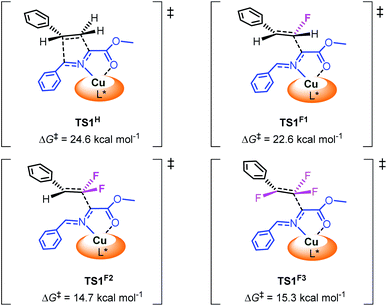 |
| | Scheme 4 Relative free energy barrier for cycloaddition of azomethine ylide with styrene and fluorostyrenes. | |
Scale-up reactions
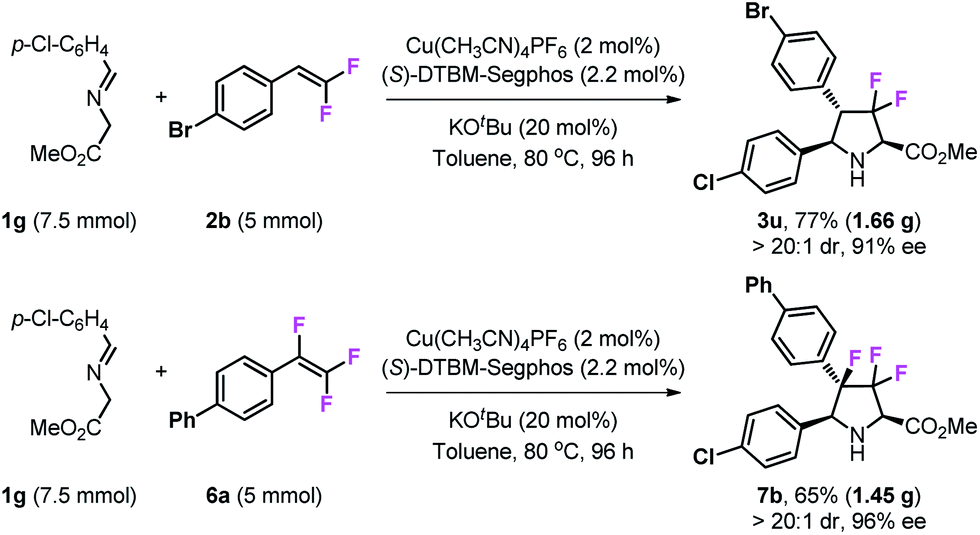
To test the practicability of this methodology, gram-scale syntheses of 3u and 7b were examined under optimal conditions (Scheme 5). The reaction of 7.5 mmol of imino ester 1g with 5 mmol of gem-difluorostyrene 2b performed very well and gave 1.66 g of product 3u in 77% yield, and with high stereoselectivity (>20:1 dr and 91% ee). In addition, 1.45 g of 3,3,4-trifluoropyrrolidine 7b can be prepared by the reaction of 7.5 mmol of imino ester 1g with 5 mmol of 1,1,2-trifluorostyrene 6a.
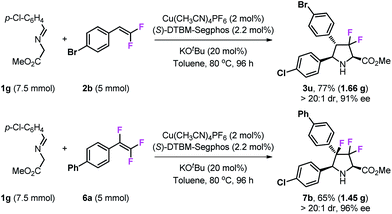 |
| | Scheme 5 Gram scale reactions. | |
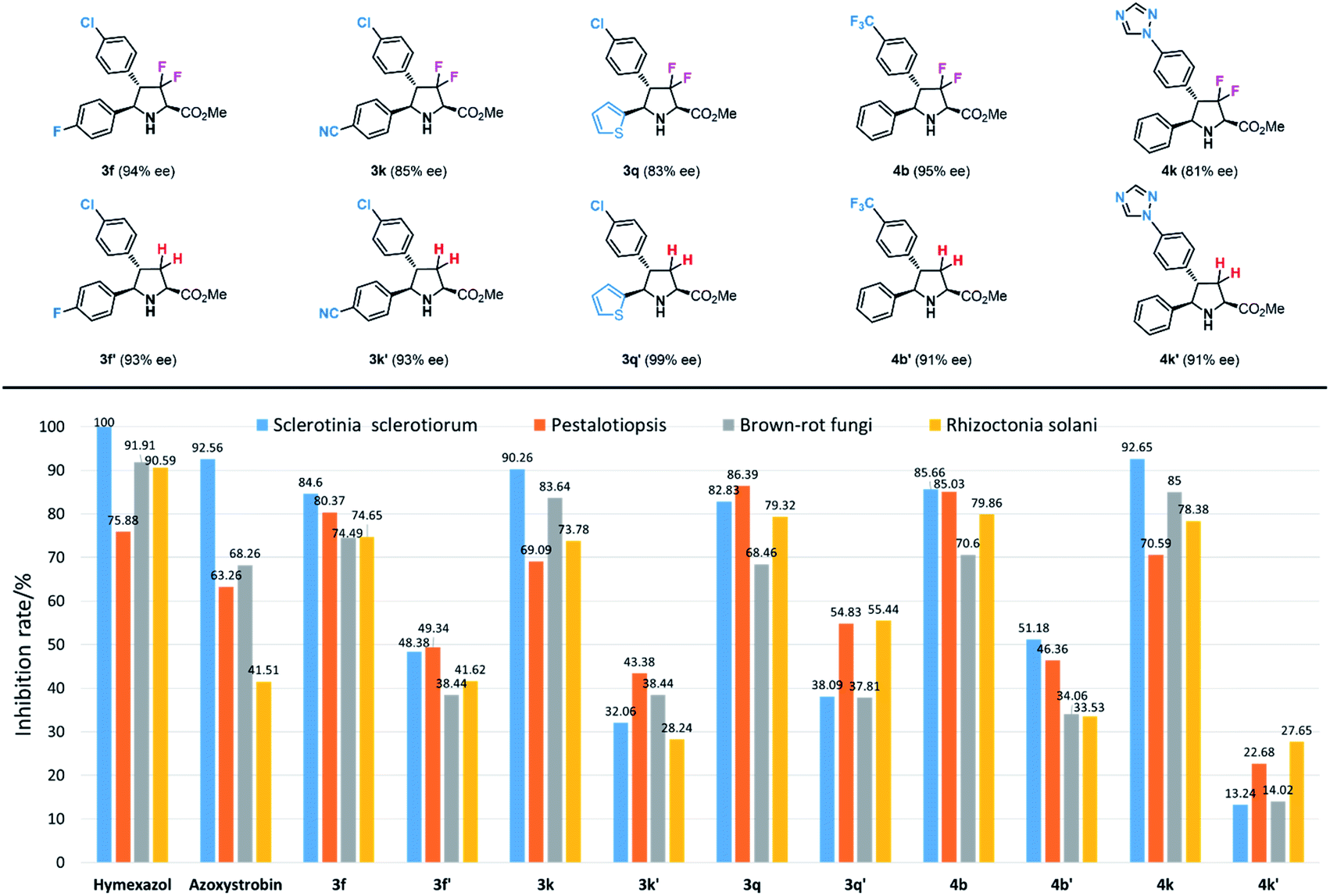
Antifungal activity evaluation
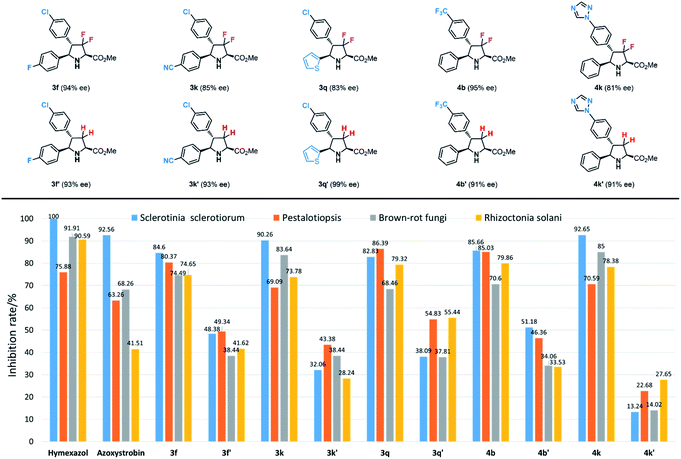
With these unique fluorinated compounds in hand, we next tested their antifungal activities. In this part, four common fungi, Sclerotinia sclerotiorum, Pestalotiopsis, brown-rot fungi and Rhizoctonia solani were chosen as experimental inhibitory subjects. Meanwhile, two popular and commercial antifungal insecticides hymexazol and azoxystrobin were used as contrapositions. As shown in Table S1,† most 3,3-difluorinated products showed general antifungal activities against the four fungi, however, in contrast, most 3,3,4-trifluoro-compounds gave poorer results. Among all biologically active difluoropyrrolidines, products 3f, 3k, 3q, 4b and 4k provided similar and even better antifungal inhibition effects than hymexazol and azoxystrobin (Scheme 6). It seems that additional fluorine groups contribute greatly to antifungal inhibition. Product 3f with an additional fluorine atom at the para-position of the phenyl group gave 84.60%, 80.37%, 74.49% and 74.65% inhibition rates against Sclerotinia sclerotiorum, Pestalotiopsis, brown-rot fungi and Rhizoctonia solani respectively, and similarly, compound 4b which has a trifluoromethyl (CF3) substituent also showed great results (85.66%, 85.03%, 70.62% and 79.86%). Meanwhile, product 3k bearing a cyano-group (CN) also performed very well towards all fungi (90.26%, 69.09%, 83.64% and 73.78%). Heterocycles, which are important building blocks in agricultural compounds, often provide a key contribution to bioactivity; for example, thienyl-substituted product 3q can inhibit these fungi effectively (82.83%, 86.39%, 68.46% and 79.32%), and the same results were obtained by compound 4k with 1,2,4- triazolyl unit (92.65%, 70.59%, 85.05% and 78.38%). These experimental results reveal that the combination of certain functional groups is necessary to enhance biological activities.
 |
| | Scheme 6 Investigation of “fluorine” influence on antifungal activities. | |
As mentioned in the beginning, fluorine factors play a crucial role in biological activity, therefore, to confirm the contribution of fluorine atoms in these structures to the antifungal inhibitory reactivity, corresponding chiral pyrrolidinyl derivatives 3f′, 3k′, 3q′, 4b′ and 4k′ without fluorine atoms at the heterocyclic rings were prepared according to Carretero's methods.55 The antifungal inhibition results of these contrapositive compounds are summarized in Tables 5 and S2,† and the antifungal activities between fluorinated compounds and their contrasts are quite different; as expected, all the contrapositive samples gave poor results in antifungal experiments. 3f′ showed only approximately half of the inhibition rate of 3f against Sclerotinia sclerotiorum, Pestalotiopsis brown-rot fungi, and Rhizoctonia solani (48.38%, 49.34%, 38.44% and 41.62%). 3k′ and 3q′ led to even lower biological reactivities (32.06%, 43.38%, 38.44%, 28.24% and 38.09%, 54.83%, 37.81%, 55.44%). Compound 4b′ bearing a trifluoromethyl (CF3) group also provided a moderate inhibition rate towards the four fungi (51.18%, 46.36%, 34.06%, 33.53%). Furthermore, the nonfluorinated trizolyl-substituted contrast compound 4k′ showed almost no effect on the antifungal inhibition (13.24%, 22.68%, 14.02% and 27.65%). Through these control experiments, we can see that the two fluorine atoms at the pyrrolidine ring play a crucial role in biological activities.
Table 5 Inhibitory effect on Sclerotinia sclerotiorum at different concentrations
| Concentration (mg L−1) |
6.25 |
12.50 |
25.00 |
50.00 |
100.00 |
EC50 |
|
3f
|
40.00 |
44.56 |
64.41 |
83.24 |
84.60 |
23.8 |
|
3k
|
38.53 |
50.15 |
69.16 |
78.82 |
90.26 |
17.96 |
|
3q
|
0.00 |
28.68 |
62.35 |
71.62 |
82.83 |
13.60 |
|
4b
|
42.30 |
54.20 |
64.30 |
70.00 |
85.66 |
10.17 |
|
4k
|
0.00 |
13.18 |
47.95 |
70.62 |
92.65 |
25.78 |
| Hymexazol |
45.72 |
49.06 |
65.10 |
83.25 |
100.00 |
10.08 |
| Azoxystrobin |
45.32 |
55.30 |
92.55 |
95.38 |
92.56 |
15.14 |
Meanwhile, antifungal experiments were carried out with racemic samples under the same conditions. As shown in Table S2,† (±)-3f, (±)-3q and (±)-4b gave slightly poorer results than chiral samples, but (±)-3k and (±)-4k showed very poor antifungal inhibition. This revealed that stereoselectivity is also a key factor in biological activity. Considering the requirement for low pesticide doses in modern practical applications, we finally examined the antifungal inhibition effects of these active compounds at different concentrations (6.25, 12.50, 25.00, 50.00 and 100.00 mg L−1) with Sclerotinia sclerotiorum as the research subject (Table 5). All inhibition rates decreased as the concentration decreased from 100.00 to 6.25 mg L−1, but their rates of decline were different. The inhibitory effects of 3q and 4k dropped sharply, and no effects were observed at 6.25 mg L−1 concentration, while those of 3f and 3k declined slower and still possessed similar inhibition rates to the commercial pesticides hymexazol and azoxystrobin at 6.25 mg L−1. The best result was obtained for 4b, which gave 42.3% inhibition rate even with 6.25 mg L−1 concentration. And EC50 (concentration for 50% of maximal effect) values of compounds 3q (13.60 mg L−1) and 4b (10.17 mg L−1) maintained at the same level of commercial pesticides hymexazol (10.08 mg L−1) and azoxystrobin (15.14 mg L−1). This result illustrates that this structure has some potential for applications in the creation of agricultural compounds.
Conclusion
In conclusion, we developed a Cu(I)-catalysed asymmetric 1,3-dipolar cycloaddition of azomethine ylides with less active 1,1-gem-difluorostyrenes and 1,1,2-trifluorostyrenes, leading to a series of novel chiral 3,3-difluoro- and 3,3,4-trifluoropyrrolidinyl derivatives. This methodology features high yields (up to 96% yield), stereoselectivity (up to >20:1 dr and 97% ee) and broad substrate scope (55 cases). The biological activity of these newly prepared compounds was evaluated via antifungal experiments, and some of these fluorinated pyrrolidines performed very well in antifungal experiments against Sclerotinia sclerotiorum, Pestalotiopsis, brown-rot fungi and Rhizoctonia solani. Control experiments between fluorinated and nonfluorinated compounds proved that the fluorine atoms on heterocyclic rings are crucial to the biological activity.
Experimental section
General procedure for the synthesis of 3,3-difluoro-and 3,3,4-trifluoropyrrolidines
In a N2 atmosphere, Cu(CH3CN)4PF6 (3.7 mg, 0.01 mmol) and (S)-DTBM-segphos (14.2 mg, 0.012 mmol) were dissolved in 2.0 mL of toluene and stirred at room temperature for 1 h. Then, KOtBu (0.04 mmol, 0.2 eq.), imino esters 1 (0.4 mmol, 2.0 eq.), 1,1-gem-difluorostyrenes 2 or 1,1,2-trifluorostyrenes 6 (0.2 mmol, 1.0 eq.) were added successively, the reaction was kept at 80 °C for 48 h. After completion, determined by TLC, the solvent was removed under vacuum and the diastereoselectivity was determined by crude 1H NMR, then the reaction mixture was purified by flash chromatography on silica gel to give the corresponding fluorinated pyrrolidines, whose enantiomeric excess was analyzed directly by chiral HPLC.
Methods for antifungal activity investigation
All tested compounds (10 mg) were dissolved in 1.0 mL acetone, then the solution was added to 100 mL of Potato Dextrose Agar medium to prepare a drug-loaded PDA medium plate having a concentration of 100 mg L−1, and 1.0 mL acetone was added to the PDA as a blank sample. Azoxystrobin and hymexazol were selected as control samples. Sclerotinia sclerotiorum, Pestalotiopsis, brown-rot fungi and Rhizoctonia solani were activated beforehand (prepared by a punch with φ = 0.50 cm), and then they were cut from the edge of the colony and inoculated into the above PDA plate, each strain and blank control experiment were repeated three times. After sealing with a sealing film to prevent contamination of other bacteria, the poisonous PDA culture dishes were placed in a constant temperature incubator at the temperature of 24 ± 1 °C. After the blank control group culture was grown to about two-thirds of the culture dish, the colony diameter was measured by the cross method in millimeters (mm) with a cross and a vertical method, and the average value was taken, and the inhibition rate of each compound against the fungus was calculated.
Author contributions
X. X. developed the synthetic methods. L. B. performed the antifungal activity investigations. L. R and Z. Y. participated in the synthetic work. D. Y. took charge of the characterization of compounds. C.-J. W. and H. T. wrote the manuscript with revisions provided by the other authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21772147, 21801086, 22071072, 220711186) and the Fundamental Research Funds for the Central Universities of China (2662018PY052) and Hubei Province NSF (2020CFA036). We thank Prof. Jiasen Cheng and Guotian Li for providing funguses. Special thanks to Prof. Gen Luo for DFT calculation studies.
Notes and references
- D. B. Harper and D. O'Hagan, Nat. Prod. Rep., 1994, 11, 123–133 RSC.
-
T. Hiyama, Organofluorine compounds: chemistry and applications, 2000, Springer Science & Business Media Search PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
-
P. Kirsch, Modern fluoroorganic chemistry: synthesis, reactivity, applications, 2013, John Wiley & Sons Search PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed.
- B. E. Smart, J. Fluorine Chem., 2001, 109, 3–11 CrossRef CAS.
-
R. D. Chambers, Fluorine in organic chemistry, 2004, CRC press Search PubMed.
-
V. A. Soloshonok, Fluorine-containing synthons, 2005, ACS Publications Search PubMed.
-
K. Uneyama, Organofluorine chemistry, 2008, John wiley & Sons Search PubMed.
-
J.-P. Bégué and D. Bonnet-Delpon, Bioorganic and medicinal chemistry of fluorine, 2008, John Wiley & Sons Search PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
-
A. Tressaud and G. Haufe, Fluorine and health: molecular imaging, biomedical materials and pharmaceuticals, 2008, Elsevier Search PubMed.
- D. O'Hagan and H. Deng, Chem. Rev., 2015, 115, 634–649 CrossRef PubMed.
- A. J. Sicard and R. T. Baker, Chem. Rev., 2020, 120, 9164–9303 CrossRef CAS PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed.
- J. Charpentier, N. Fruh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed.
- T. Koike and M. Akita, Chem, 2018, 4, 409–437 CAS.
- S. M. Khake and N. Chatani, Chem, 2020, 6, 1056–1081 CAS.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed.
- J. P. Michael, Nat. Prod. Rep., 2008, 25, 139–165 RSC.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- M. Kuhnert, A. Blum, H. Steuber and W. E. Diederich, J. Med. Chem., 2015, 58, 4845–4850 CrossRef CAS PubMed.
- J. Hartung, S. N. Greszler, R. C. Klix and J. M. Kallemeyn, Org. Process Res. Dev., 2018, 23, 2532–2537 CrossRef.
- M. J. C. Scanio, X. B. Searle, B. Liu, J. R. Koenig, R. Altenbach, G. A. Gfesser, A. Bogdan, S. Greszler, G. Zhao, A. Singh, Y. Fan, A. M. Swensen, T. Vortherms, A. Manelli, C. Balut, Y. Jia, W. Gao, H. Yong, M. Schrimpf, C. Tse, P. Kym and X. Wang, ACS Med. Chem. Lett., 2019, 10, 1543–1548 CrossRef CAS PubMed.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
-
A. Padwa and W. H. Pearson, Synthetic applications of 1,3-dipolar cycloaddition chemistry toward heterocycles and natural products, 2002, Wiley-VCH, New York Search PubMed.
- T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- J. Adrio and J. C. Carretero, Chem. Commun., 2019, 55, 11979–11991 RSC.
- L. Wei, X. Chang and C.-J. Wang, Acc. Chem. Res., 2020, 53, 1084–1100 CrossRef CAS PubMed.
- X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS.
- J. M. Percy and M. H. Rock, Tetrahedron Lett., 1992, 33, 6177–6180 CrossRef CAS.
- M. P. Jonathan and H. R. Michael, Tetrahedron Lett., 1992, 33, 6177–6180 CrossRef.
- T. P. Suzanne and S. Kuen-Wang, Tetrahedron Lett., 1992, 33, 3289–3292 CrossRef.
- J. M. Percy, K. Stansfield, P. J. Crowley and K. Stansfield, Chem. Commun., 1997, 997, 2033–2034 RSC.
-
W. Gao, T. Lau, S. Pan, D. P. Phillips, X. Wang, Y. Yang, European Patent, 2012, WO2012082947(A1). Search PubMed.
- W.-R. Hanna and K. Henryk, J. Fluorine Chem., 2012, 135, 225–230 CrossRef.
- I. McAlpine, M. T. Dubé, F. Wang, S. Scales, J. Matthews, M. R. Collins, S. K. Nair, M. Nguyen, J. W. Bian, L. M. Alsina, J. M. Sun, J. Y. Zhong, J. S. Warmus and B. T. O'Neill, J. Org. Chem., 2015, 80, 7266–7274 CrossRef CAS PubMed.
- S. Yamada, M. Higashi, T. Konno and T. Ishihara, Eur. J. Org. Chem., 2016, 4561–4568 CrossRef CAS.
- D. L. Orsi, B. J. Easley, A. M. Lick and R. A. Altman, Org. Lett., 2017, 19, 1570–1573 CrossRef CAS PubMed.
- X. Liu, E. E. Lin, G. Chen, J.-L. Li, P. Liu and H. Wang, Org. Lett., 2019, 21, 8454–8458 CrossRef CAS PubMed.
- B. Gao, Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2015, 54, 638–642 CAS.
- P. Tian, C. Feng and T.-P. Loh, Nat. Commun., 2015, 6, 7472–7478 CrossRef PubMed.
- J. Xie, J. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416–9421 CrossRef CAS PubMed.
- R. T. Thornbury and F. D. Toste, Angew. Chem., Int. Ed., 2016, 55, 11629–11632 CrossRef CAS PubMed.
- D. Zell, U. Dhawa, V. Müller, M. Bursch, S. Grimme and L. Ackermann, ACS Catal., 2017, 7, 4209–4213 CrossRef CAS.
- X. Lu, Y. Wang, B. Zhang, J.-J. Pi, X.-X. Wang, T.-J. Gong, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2017, 139, 12632–12637 CrossRef CAS PubMed.
- H. Sakaguchi, Y. Uetake, M. Ohashi, T. Niwa, S. Ogoshi and T. Hosoya, J. Am. Chem. Soc., 2017, 139, 12855–12862 CrossRef CAS PubMed.
- J. Liu, J. Yang, F. Ferretti, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 4690–4694 CrossRef CAS PubMed.
- L. Zhou, C. Zhu, P. Bi and C. Feng, Chem. Sci., 2019, 10, 1144–1149 RSC.
- J. Li, M. Liu, Y. Li, D.-D. Sun, Z. Shu, Q. Tan, S. Guo, R. Xie, L. Gao and H. Ru, J. Med. Chem., 2020, 63, 12748–12772 CrossRef CAS PubMed.
- P. Tian, C.-Q. Wang, S.-H. Cai, S. Song, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2016, 138, 15869–15872 CrossRef CAS PubMed.
- H. J. Tang, L. Z. Lin, C. Feng and T. P. Loh, Angew. Chem., Int. Ed., 2017, 56, 9872–9876 CrossRef CAS PubMed.
- P. E. Daniel, C. I. Onyeagusi, A. A. Ribeiro, K. Li and S. J. Malcolmson, ACS Catal., 2018, 9, 205–210 CrossRef PubMed.
- W. J. Yoo, J. Kondo, J. A. Rodríguez-Santamaría, T. V. Nguyen and S. Kobayashi, Angew. Chem., Int. Ed., 2019, 58, 6772–6775 CrossRef CAS PubMed.
- H. Liu, L. Ge, D. X. Wang, N. Chen and C. Feng, Angew. Chem., Int. Ed., 2019, 58, 3918–3922 CrossRef CAS PubMed.
- T. Y. Lin, Z. Pan, Y. Tu, S. Zhu, H. H. Wu, Y. Liu, Z. Li and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 22957–22982 CrossRef CAS PubMed.
- A. L'Heureux, F. Beaulieu, C. Bennett, D. R. Bill, S. Clayton, F. Laflamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, J. Org. Chem., 2010, 75, 3401–3411 CrossRef PubMed.
- G. Courchesne, M. Couturier, F. Laflamme, A. L'Heureux, F. Beaulieu and L. P. Beauregard, Org. Lett., 2009, 11, 5050–5053 CrossRef PubMed.
- F. Rossi, F. Corcella, F. S. Caldarelli, F. Heidempergher, C. Marchionni, M. Auguadro, M. Cattaneo, L. Ceriani, G. Visentin, G. Ventrella, V. Pinciroli, G. Ramella, I. Candiani, A. Bedeschi, A. Tomasi, B. J. Kline, C. A. Martinez, D. Yazbeck and D. J. Kucera, Org. Process Res. Dev., 2008, 12, 322–338 CrossRef CAS.
- S. H. Hu, C. A. Martinez, B. Kline, D. Yazbeck, J. H. Tao and D. J. Kucera, Org. Process Res. Dev., 2006, 10, 650–654 CrossRef CAS.
- L. Chen, Y. Kim, D. Kucera, K. Harrison, S. Bahmanyar, J. Scott and D. Yazbeck, J. Org. Chem., 2006, 71, 5468–5473 CrossRef CAS PubMed.
- L. Wei, T. M. Makowski and J. L. Rutherford, J. Fluorine Chem., 2012, 135, 354–357 CrossRef CAS.
- X. Feng, B. Simmons, J. A. Iii and J. Murry, J. Org. Chem., 2005, 70, 6105–6107 CrossRef PubMed.
- S. Filippone, E. E. Maroto, Á. Martín-Domenech, M. Suarez and N. Martín, Nat. Chem., 2009, 1, 578–582 CrossRef CAS PubMed.
- A. Pascual-Escudero, A. de Cózar, F. P. Cossío, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2016, 55, 15334–15338 CrossRef CAS PubMed.
- K. Liu, Y. Xiong, Z.-F. Wang, H.-Y. Tao and C.-J. Wang, Chem. Commun., 2016, 52, 9458–9461 RSC.
- X. Chang, Y. Yang, C. Shen, K.-S. Xue, Z.-F. Wang, H. Cong, H.-Y. Tao, L. W. Chung and C.-J. Wang, J. Am. Chem. Soc., 2021, 143, 3519–3535 CrossRef CAS PubMed.
- C. D. McCune, M. L. Beio, J. M. Sturdivant, R. de la Salud-Bea, B. M. Darnell and D. B. Berkowitz, J. Am. Chem. Soc., 2017, 139, 14077–14089 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2064529. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04595d |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Huailong
Teng
*b and
Huailong
Teng