
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencewSDTNBI: a novel network-based inference method for virtual screening†
Zengrui
Wu‡
 ,
Hui
Ma‡
,
Zehui
Liu‡
,
Lulu
Zheng
,
Zhuohang
Yu
,
Shuying
Cao
,
Wenqing
Fang
,
Lili
Wu
,
Weihua
Li
,
Guixia
Liu
,
Jin
Huang
* and
Yun
Tang
*
,
Hui
Ma‡
,
Zehui
Liu‡
,
Lulu
Zheng
,
Zhuohang
Yu
,
Shuying
Cao
,
Wenqing
Fang
,
Lili
Wu
,
Weihua
Li
,
Guixia
Liu
,
Jin
Huang
* and
Yun
Tang
*
Shanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: ytang234@ecust.edu.cn; huangjin@ecust.edu.cn
First published on 21st December 2021
Abstract
In recent years, the rapid development of network-based methods for the prediction of drug–target interactions (DTIs) provides an opportunity for the emergence of a new type of virtual screening (VS), namely, network-based VS. Herein, we reported a novel network-based inference method named wSDTNBI. Compared with previous network-based methods that use unweighted DTI networks, wSDTNBI uses weighted DTI networks whose edge weights are correlated with binding affinities. A two-pronged approach based on weighted DTI and drug–substructure association networks was employed to calculate prediction scores. To show the practical value of wSDTNBI, we performed network-based VS on retinoid-related orphan receptor γt (RORγt), and purchased 72 compounds for experimental validation. Seven of the purchased compounds were confirmed to be novel RORγt inverse agonists by in vitro experiments, including ursonic acid and oleanonic acid with IC50 values of 10 nM and 0.28 μM, respectively. Moreover, the direct contact between ursonic acid and RORγt was confirmed using the X-ray crystal structure, and in vivo experiments demonstrated that ursonic acid and oleanonic acid have therapeutic effects on multiple sclerosis. These results indicate that wSDTNBI might be a powerful tool for network-based VS in drug discovery.
1 Introduction
Lead discovery is an essential step in the pipeline of drug discovery.1 However, identification of lead compounds by in vitro and in vivo experiments is often expensive and time-consuming. To improve efficiency and reduce costs, virtual screening (VS), a computer-aided technology aiming to identify promising compounds for targets of interest from chemical libraries, has been widely used in lead discovery.2,3 Currently, there are two principal types of VS, namely, structure-based and ligand-based VS.3,4 Ligand-based VS can be further divided into several subtypes, such as pharmacophore-based,5 similarity-based,6 and machine learning-based.7,8 As their names imply, structure-based VS uses the information of three-dimensional (3D) structures of targets to identify new ligands, while ligand-based VS uses the information of ligands. To date, these two types of VS have achieved great success in drug discovery.3,9,10 Meanwhile, with the rapid development of network pharmacology, the drug discovery paradigm has been shifting from the linear mode of “one drug → one target → one disease” to the network mode of “multiple drugs → multiple targets → multiple diseases”.11,12 A series of network-based methods have been proposed for the prediction of drug–target interactions (DTIs),13 which provides an opportunity for the emergence of a new type of VS, namely, network-based VS.Representative network-based methods, including network-based inference (NBI) methods,14–16 random walk-based methods,17 and local community-based methods,18 are derived from link prediction algorithms in complex networks.19 Compared with structure-based methods, network-based methods do not rely on 3D structures of targets.13 Moreover, compared with supervised machine learning-based methods, network-based methods do not rely on experimentally determined inactive DTIs, namely, negative samples.13 For example, our previously developed NBI only uses a known DTI network to predict potential targets for drugs in the DTI network.14 No other information such as 3D structures of targets and negative samples is required. The two improved versions of NBI, substructure–drug–target NBI (SDTNBI) and balanced SDTNBI (bSDTNBI),15,16 use both DTI and drug–substructure association (DSA) networks as inputs, and hence can predict potential targets for not only drugs within the DTI network, but also various compounds outside the DTI network. Based on the predicted DTIs, it is possible to explore the molecular mechanisms of the therapeutic and adverse effects of approved drugs and natural products,15,20,21 and identify active compounds for targets of interest.15 Using bSDTNBI in combination with in vitro experiments, we discovered new compounds targeting estrogen receptor α,15 prostaglandin E2 receptor EP4 subtype,21 and NAD(P)H:quinone oxidoreductase 1 (NQO1).22 These applications indicate that network-based methods, especially our NBI methods, have the potential to be applied in VS.
Despite the successful applications of network-based methods, there is still a weakness, namely, prediction scores are not correlated with binding affinities. For each predicted DTI between a drug and a target, the prediction score only measures the probability that the drug can interact with the target, rather than the potency of the drug against the target.14–16 A higher score does not mean a stronger interaction. This weakness makes it hard to quantitatively assess the activities of the screened compounds in network-based VS, just like scoring functions do in structure-based VS.23 The possible reason for this weakness is that network-based methods are usually based on unweighted DTI networks, whose edge weights are always equal to a constant (e.g., 1). Binding affinities have not been considered in network construction and network-based prediction yet. Hence, it is necessary to improve network-based methods by introducing binding affinity data in order to make them more suitable for VS.
In this study, we developed a novel network-based method named weighted substructure-drug–target NBI (wSDTNBI). Compared with previous network-based methods that use unweighted DTI networks, wSDTNBI uses weighted DTI networks whose edge weights are correlated with binding affinities. As shown in Fig. 1, a two-pronged approach based on DTI and DSA networks was employed to calculate prediction scores for all possible DTIs. This improved method can output prediction scores correlated with binding affinities, and help to find more active compounds with higher activities for targets of interest. To show the practical value of wSDTNBI, we performed network-based VS on retinoid-related orphan receptor γt (RORγt, a specific isoform of RORγ), and purchased 72 compounds for experimental validation. Seven of the purchased compounds were confirmed to be novel RORγt inverse agonists by in vitro experiments, including ursonic acid and oleanonic acid with IC50 values of 10 nM and 0.28 μM, respectively. Moreover, the direct contact between the best compound ursonic acid and RORγt was confirmed using the X-ray crystal structure, and in vivo experiments demonstrated that ursonic acid and oleanonic acid have therapeutic effects on multiple sclerosis (MS). The success rate of our network-based VS on RORγt (7/72 ≈ 9.7%) is higher than the success rates of recent structure-based and deep learning-based VS on RORγt (only about 5%).24,25 These results indicate that wSDTNBI might be a powerful tool for network-based VS in drug discovery.
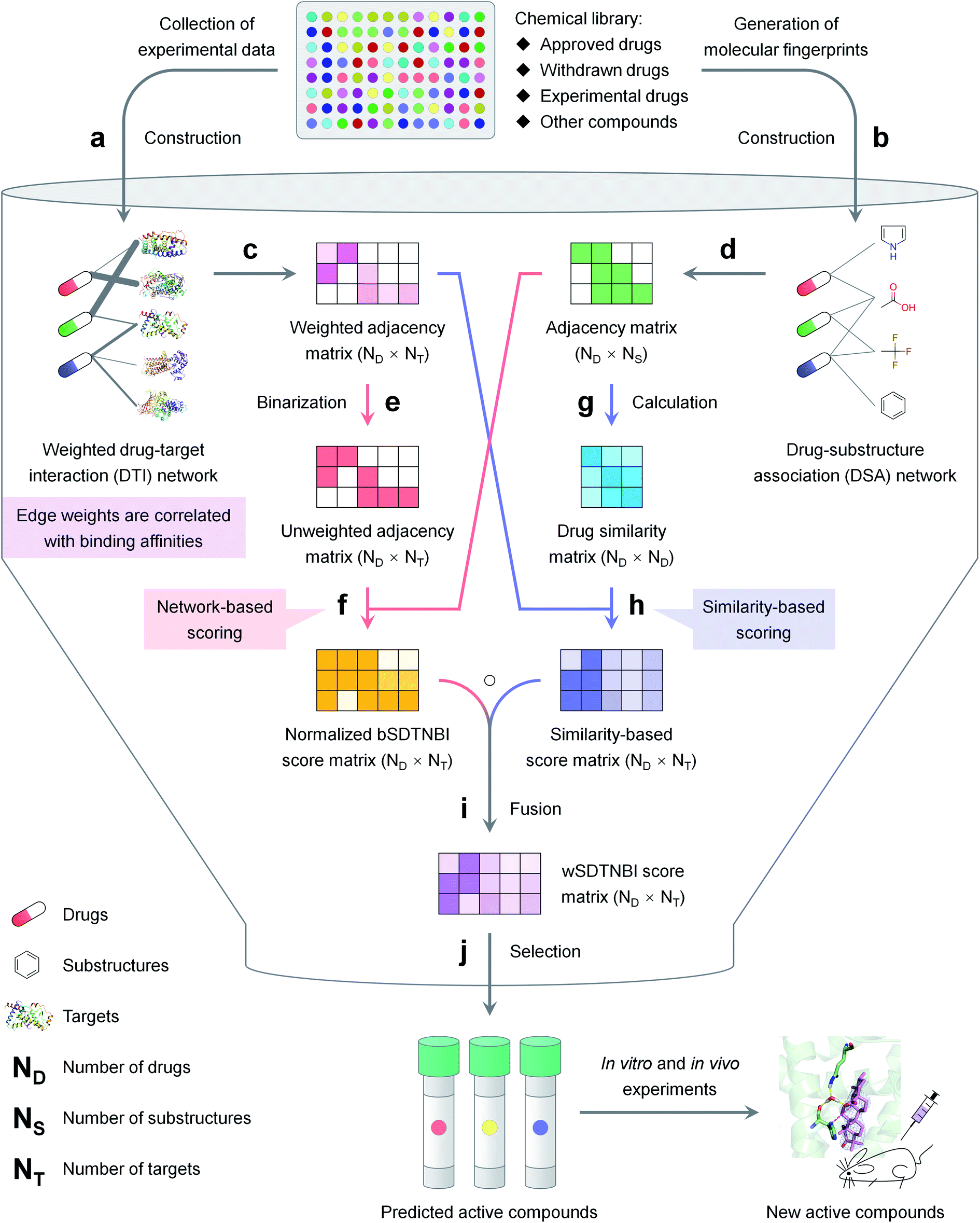 | ||
| Fig. 1 The schematic diagram of network-based VS with wSDTNBI, including (a) construction of a weighted DTI network, (b) construction of a DSA network, (c) representing the weighted DTI network as an adjacency matrix, (d) representing the DSA network as an adjacency matrix, (e) converting the weighted DTI network into an unweighted DTI network, (f) calculation and normalization of bSDTNBI scores using the unweighted DTI network and the DSA network, (g) generation of a drug similarity matrix, (h) calculation of similarity-based scores using the weighted DTI network and the drug similarity matrix, (i) calculation of wSDTNBI scores by fusing normalized bSDTNBI scores and similarity-based scores, (j) selection of potential active compounds according to the calculated wSDTNBI scores. The details are described in the Methods and ESI† methods. | ||
2 Methods
2.1 Description of wSDTNBI
The schematic diagram of network-based VS with wSDTNBI is illustrated in Fig. 1. Just as its name implies, wSDTNBI uses a weighted DTI network and a DSA network as inputs to predict potential DTIs. In the weighted DTI network, nodes represent drugs and targets, and edges represent DTIs (Fig. 1a). A drug node and a target node are connected by an edge if the drug is known to interact with the target. The edge weights are not always equal to 1, but set to be positively correlated with binding affinities. In the DSA network, nodes represent drugs and substructures, and edges represent DSAs (Fig. 1b). A drug node and a substructure node are connected by an edge if the chemical structure of the drug contains the substructure. The DSA network generally contains DSAs for not only all drugs in the DTI network, but also novel compounds without any known targets, such as newly discovered natural products and newly synthesized compounds.Assuming that there are a total of ND drugs and novel compounds in the DTI and DSA networks, and a total of NT targets in the DTI network, the weighted DTI network can be represented as a ND × NT adjacency matrix WDTI (Fig. 1c). In this matrix, WDTI(i, j) = the weight of the edge between drug Di and target Tj if they are connected in the DTI network, otherwise = 0. Assuming that there are a total of NS substructures in the DSA network, the DSA network can be represented as a ND × NS adjacency matrix ADSA (Fig. 1d). In this matrix, ADSA(i, j) = 1 if drug Di and substructure Sj are connected in the DSA network, otherwise = 0. Based on the two networks represented by adjacency matrices WDTI and ADSA, prediction scores are calculated for all possible DTIs by the following two-pronged approach.
On one prong (Fig. 1, red arrows), our previously developed bSDTNBI is employed to calculate prediction scores. At first, the weighted DTI network is converted into an unweighted DTI network (Fig. 1e), whose adjacency matrix ADTI can be obtained using the following equation:
| ADTI(i, j) = |sgn(WDTI(i, j))| |
Then, bSDTNBI scores are calculated for all possible DTIs, based on a substructure-drug–target network constructed by integrating the unweighted DTI network and the DSA network. In the calculation of bSDTNBI scores, three tuneable parameters α, β and γ are used to address potential imbalances.15 These bSDTNBI scores are subsequently normalized to lie between 0 and 1, and stored in a ND × NT matrix Snorm (Fig. 1f). A new parameter δ is introduced to adjust the distribution of normalized scores. Specifically, assuming that SbSDTNBI(i, j) is the bSDTNBI score of the DTI between drug Di and target Tj, normalized bSDTNBI scores can be calculated using the following equations:
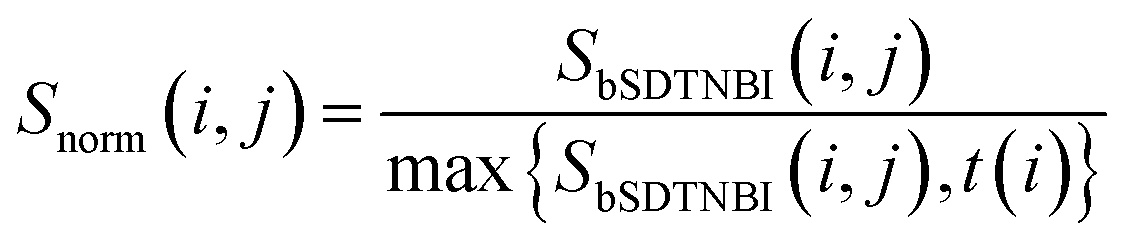
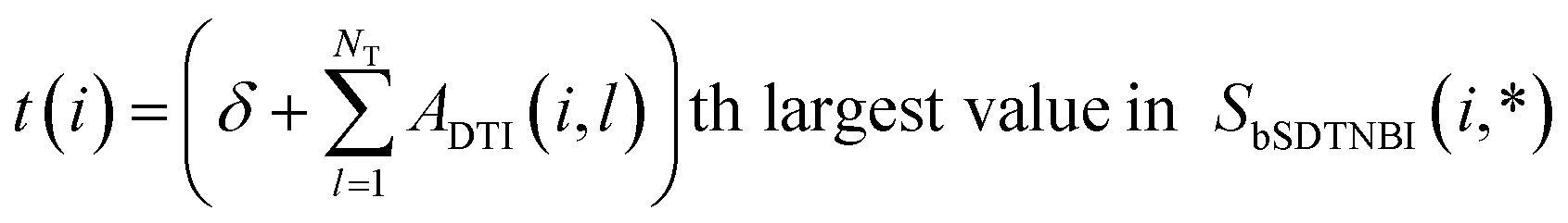
On the other prong (Fig. 1, blue arrows), a similarity-based method is used to calculate prediction scores. At first, a ND × ND drug similarity matrix is obtained by calculating similarity values for all possible drug pairs (Fig. 1g). Specifically, for each pair of drugs, two rows in ADSA corresponding to the two drugs are extracted as two vectors, and the similarity between the two drugs is measured using the Tanimoto coefficient:26
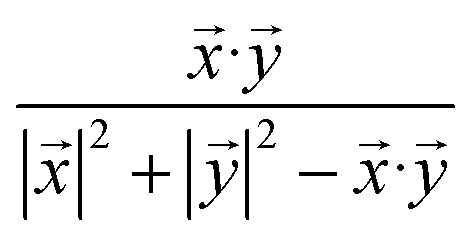
Then, similarity-based scores are calculated for all possible DTIs, and stored in a ND × NT matrix Ssim (Fig. 1h). Specifically, for each DTI between drug Di and target Tj, Ssim(i, j) is set to the average of the edge weights of the DTIs between Tj and its ε known ligands most similar to Di in the weighted DTI network. Compared with normalized bSDTNBI scores, these similarity-based scores may be more correlated with binding affinities.
The final prediction scores, which we called wSDTNBI scores, are obtained by fusing normalized bSDTNBI scores and similarity-based scores (Fig. 1i). Mathematically, the wSDTNBI score matrix SwSDTNBI is the Hadamard product of the aforementioned two score matrices Snorm and Ssim:
| SwSDTNBI = Snorm○Ssim |
In general, only those DTIs having both high normalized bSDTNBI scores and high similarity-based scores will have high wSDTNBI scores. The wSDTNBI scores may help to identify active compounds with high binding affinities (Fig. 1j). The pseudo codes of wSDTNBI and previous network-based methods NBI, SDTNBI and bSDTNBI are provided in ESI† methods.
2.2 Construction of network-based models
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and the number of carbon atoms. Only those drugs that have MW <800 and more than one carbon atoms were retained.
P and the number of carbon atoms. Only those drugs that have MW <800 and more than one carbon atoms were retained.
Using the canonical SMILES of the retained drugs, DTI data with experimentally determined Ki, Kd, half maximal inhibitory concentration (IC50), half maximal effective concentration (EC50), half maximal activity concentration (AC50) or potency values were extracted from ChEMBL32 (version 25), BindingDB33 (version 2019m4), IUPHAR/BPS Guide to PHARMACOLOGY28 (accessed in May 2019) and PDSP Ki database34 (accessed in May 2019). The extracted data were then filtered. Specifically, a DTI data item was retained if it met the following criteria: (i) the target is a protein from Homo sapiens; (ii) the target protein can be represented as an unique UniProt accession number; (iii) the target protein was marked as “reviewed” in UniProt Knowledgebase35 (accessed in June 2019); (iv) the interaction was not explicitly remarked as “inconclusive”, “inactive” or “not active” in source databases.
Based on the processed DTI data, a local DTI network was constructed only using DTI data with Ki values <10 μM. The edge weights in the local network were set as −log10(Ki/10 μM). Meanwhile, a global DTI network was constructed using not only DTI data with Ki values <10 μM, but also DTI data with Kd, IC50, EC50, AC50 or potency values <10 μM. The way of assigning edge weights in the global network was similar to that in the local network. When lacking Ki values, Kd, IC50, EC50, AC50 or potency values were used as alternatives.
2.3 Evaluation of network-based models
At first, all possible DTIs are predicted based on the training set. To avoid meaningless values, only those drugs that have DTIs in the test set were considered in the calculation of evaluation indicators. For each drug Di, its newly predicted DTIs were sorted by prediction scores in descending order, and divided into two classes. Specifically, the ones ranked in the top L were considered as positive, while the others were considered as negative. After comparing the predicted positives and negatives with the test set, four numbers were calculated for drug Di, including the number of true positives TPi(L), the number of false positives FPi(L), the number of true negatives TNi(L) and the number of false negatives FNi(L). Denoting N’D and N’T as the numbers of drugs and targets considered in the calculation of evaluation indicators, recall R(L) and enhancement recall eR(L), two evaluation indicators depending on L, were calculated using the following equations:
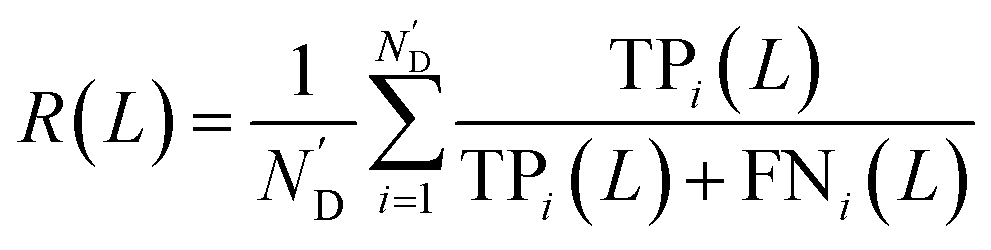
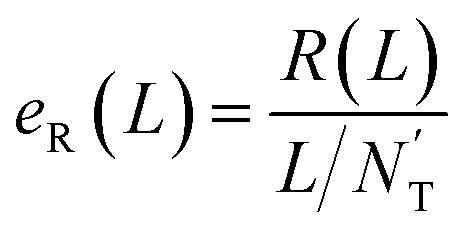
Meanwhile, area under receiver operating characteristic curve (AUROC), an evaluation indicator independent of L, was calculated. The curve was obtained by plotting true positive rates (TPRs) against false positive rates (FPRs), after calculating a series of TPRs and FPRs at different L values using the following equations:

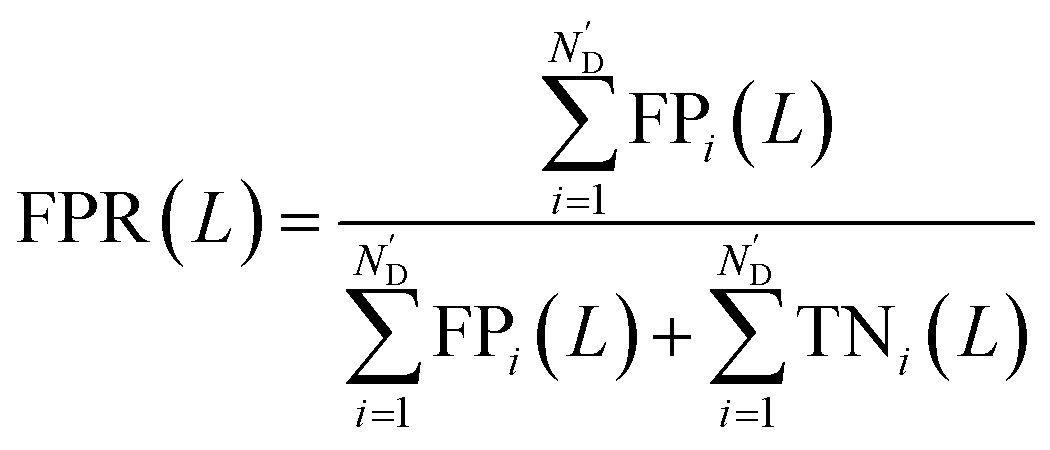
In addition, Pearson correlation coefficient (r), mean absolute error (MAE) and root mean square error (RMSE) were calculated by comparing the known edge weights of the DTIs in the test set with the prediction scores of these DTIs.
For each model, the aforementioned 10-fold cross validation process was repeated ten times, and each of the evaluation indicators was expressed as mean ± standard deviation (SD).
In addition to network-based methods, molecular docking methods were also used to calculate prediction scores. The crystal structures of the five nuclear receptors were chosen according to DUD-E,37 and downloaded from RCSB Protein Data Bank38 (PDB codes: 2AM9 for AR, 1SJ0 for ESR1, 2P54 for PPARA, 2GTK for PPARG, and 1MV9 for RXRA). Each of the crystal structures was prepared by Protein Preparation Wizard in Schrödinger (version 2016; Schrödinger, LLC) with the following three steps. Firstly, the crystal structure was preprocessed using the default settings, and missing side chains were added by Prime (version 4.3; Schrödinger, LLC) if needed. Secondly, redundant chains and water molecules were deleted. Thirdly, the protonation states of residues were optimized at pH 7.0. Based on the prepared crystal structures, receptor grids were generated by Receptor Grid Generation in Schrödinger (version 2016; Schrödinger, LLC). In grid generation, the size of the enclosing box was set to maximum (36 Å) to ensure that most of the ligands in external validation sets can be docked into the prepared receptors. Then, the ligands of the five nuclear receptors were prepared by LigPrep in Schrödinger (version 2016; Schrödinger, LLC), and docked into the prepared receptors by Glide39 (version 7.0; Schrödinger, LLC) in both standard precision (SP) and extra precision (XP) modes. The absolute values of docking scores were used as prediction scores.
Finally, the correlation between prediction scores and pKi values was evaluated by the Pearson correlation test in R (version 3.3.3).
2.4 Construction of drug–target–immunological process network
The best wSDTNBI model Global-wSDTNBI-FCFP_4 was used to predict 20 new targets for all drugs in the global DTI network. Known and predicted DTIs for the drugs marked as “approved” in DrugBank27 and IUPHAR/BPS Guide to PHARMACOLOGY28 were retained. Meanwhile, targets associated with immunological processes were collected from IUPHAR Guide to IMMUNOPHARMACOLOGY.28 Then, a drug–target–immunological process network was constructed by integrating the DTIs for approved drugs and the target–immunological process associations. This network was visualized by Cytoscape40 (version 3.4.0).2.5 Prediction and validation of RORγt ligands
000 rpm for 30 min at 4 °C and the supernatant was loaded on a HisTrap nickel affinity column (GE Healthcare) pre-equilibrated with Buffer A. Protein was eluted with a gradient of 40–500 mM imidazole in buffer A. Fractions containing the protein of interest were pooled and dialyzed overnight against buffer B (20 mM Tris, pH = 7.0, 200 mM NaCl, 4% glycerol, 5 mM DTT). The RORγt-LBD protein was then concentrated using 10 kDa centrifugal filters from Merck Millipore (KGaA of Darmstadt, Germany) and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). All the purification operations were performed at 4 °C.
For luciferase activity assay, HEK293T cells were co-transfected with 2 μg ROR-GAL4, 2 μg pGL4.31 and 100 ng pRL-SV40 in a 6 cm dish using Lipofectamine 2000 (Invitrogen, USA). After 24 h of transfection, the cells were incubated with a variety of concentrations of compounds. After 24 h treatment, the cells were lysed with passive lysis buffer and luciferase activities were measured using a Dual-Glo Luciferase Assay System (Promega, Madison, WI) using an EnVision Multilabel Plate Reader (PerkinElmer, Waltham, MA).
lnK, where ΔG is the change in free energy, ΔH is the change in enthalpy, ΔS is the change in entropy, T is the experimental temperature, R is the gas constant, and K is the binding constant.
000 rpm at 4 °C for 20 min. The supernatant samples were resolved by SDS-PAGE, and then transferred to a PVDF membrane. After incubation with the RORγt antibody and the horseradish peroxidase (HRP)-conjugated secondary antibody, the protein bands were analyzed using the ECL Western Blotting Substrate Kit (Thermo Fisher, USA).
000 rpm for 15 min prior to crystallization experiments. Diffraction-quality crystals of ligand-bound RORγt-LBD-SRC2.2 were grown by the sitting drop vapor diffusion technique. The reservoir solution contained 20% (w/v) PEG3350, 0.2 M CaCl2, and 5 μM EDTA-2Na. Each 10 μL sitting drop consisted of 5 μL sample and 5 μL reservoir solution and was equilibrated against 0.5 ml reservoir solution at 20 °C. Crystals were flash-frozen in liquid nitrogen with cryoprotecant using corresponding reservoir condition supplemented with 30% glycerol. X-ray diffraction data were collected at 100 K on the synchrotron beamline BL19U1 at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China). Diffraction data were processed using XDS. The initial structure was determined by molecular replacement with CCP4 and PDB entry 4XT9 as an initial model. Multiple rounds of positional isotropic B-factor refinement were performed using phenix.refine followed by re-building in Coot. The coordinates and structure factors of the final model of RORγt in complex with ursonic acid were deposited in PDB (PDB code: 6J3N).
2.6 Effects on experimental autoimmune encephalomyelitis (EAE)
3 Results
3.1 Overview of network-based models
DTI networks are the basis of network-based models for DTI prediction. In this study, we constructed two comprehensive DTI networks by collecting experimental data from public databases. One is a local DTI network with 18249 edges connecting 5560 drugs and 968 human target proteins, which was constructed only using DTI data with Ki values <10 μM. The other is a global DTI network with 45018 edges connecting 12751 drugs and 1844 human target proteins, which was constructed using DTI data with Ki, Kd, IC50, EC50, AC50 or potency values <10 μM. These two DTI networks contain various types of drugs and target proteins. Drugs include not only approved drugs, but also withdrawn drugs, clinical drug candidates and experimental drug-like compounds. Target proteins include G protein-coupled receptors (GPCRs), nuclear receptors, enzymes, ion channels and transporters. In previous studies of network-based DTI prediction,14–16,21 edge weights in DTI networks were usually set to a constant (e.g., 1). Compared with them, edge weights in the two weighted DTI networks were greater than zero and positively correlated with binding affinities. In simple terms, the higher the edge weight is, the higher the binding affinity is. The detailed information of drugs (e.g., canonical SMILES, names, types, CAS registry numbers and Anatomical Therapeutic Chemical classification codes), targets (e.g., UniProt accession numbers, names and families) and DTIs (e.g., activity values and references) in the two DTI networks is provided in ESI data 1.†
We then constructed 18 DSA networks by generating nine types of fingerprints for all drugs in the two DTI networks. The nine types of fingerprints include four common fingerprints (FP4, KR, MACCS and PubChem) and five extended-connectivity fingerprints (ECFP_0, ECFP_2, FCFP_0, FCFP_2 and FCFP_4). The statistics of the 18 DSA networks are given in ESI Table S1.† Based on the DTI and DSA networks, we constructed 18 wSDTNBI models, and optimized the parameters of these models by grid search. The optimized parameters are listed in ESI Table S2.† For the purpose of comparison, we also constructed models using previous network-based methods. Specifically, two NBI models were constructed based only on the DTI networks, while 18 SDTNBI and 18 bSDTNBI models were constructed based on the DTI and DSA networks. Some of the models are freely available in our web server NetInfer51 (http://lmmd.ecust.edu.cn/netinfer).
3.2 Performance of network-based models
To compare wSDTNBI with previous network-based methods and find the best model, we evaluated the performance of all 56 models in this study by 10-fold cross validation. The evaluation indicators of local and global models, expressed as mean ± SD, are given in ESI Tables S3 and S4,† respectively. At first, we focused on the performance of recovering missing DTIs, which was measured using three evaluation indicators: R, eR and AUROC. Under the condition that 20 new DTIs were predicted for each drug (i.e., L = 20), 48 of the 56 models have R values >0.5 and eR values >24.3. For a model, the R value >0.5 means that the missing DTIs of each drug can be successfully recovered more than 50% in average, and the eR value >24.3 means that the observed R value is at least 24.3 times larger than the theoretical R value in random prediction. Meanwhile, all models have AUROC values >0.9, further indicating their good performance in recovering missing DTIs. Moreover, we found that the models constructed using FCFP_4 fingerprints usually outperform the models constructed using other types of fingerprints, which is consistent with the finding in our previous study.22We next focused on the correlation and difference between prediction scores and binding affinities, which were measured using the other three evaluation indicators: r, MAE and RMSE. As shown in Fig. 2, all NBI, SDTNBI and bSDTNBI models have r values very close to 0, which means that they cannot output prediction scores correlated with binding affinities. In contrast, the r values of local-wSDTNBI-FCFP_4 and global-wSDTNBI-FCFP_4, the best local and global models, are higher than 0.55 and comparable with those of molecular docking methods.23 The high r values of local-wSDTNBI-FCFP_4 and global-wSDTNBI-FCFP_4 mean that they can output prediction scores positively correlated with binding affinities. Moreover, these two wSDTNBI models have MAE ≤0.80 and RMSE ≤1.07, indicating the small difference between prediction scores and binding affinities.
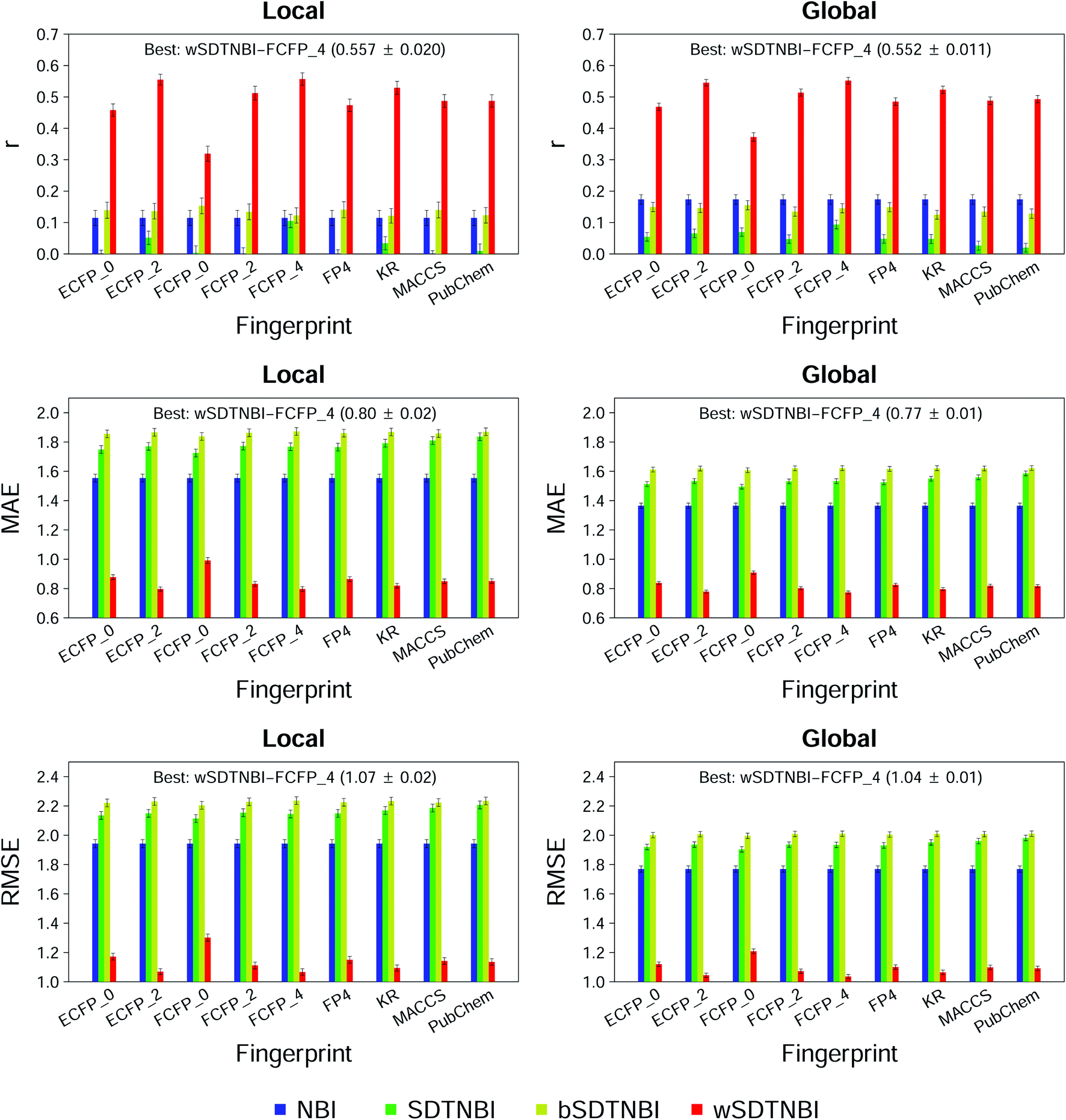 | ||
| Fig. 2 Three evaluation indicators (r, MAE and RMSE) of network-based models in 10-fold cross validation. Results of the models constructed using NBI, SDTNBI, bSDTNBI and wSDTNBI are represented by blue, green, yellow and red bars. | ||
To further evaluate the correlation between prediction scores and binding affinities, we collected hundreds of nuclear receptor ligands outside the local and global DTI networks, including 717 AR ligands, 277 ESR1 ligands, 104 PPARA ligands, 345 PPARG ligands and 147 RXRA ligands. Using these nuclear receptor ligands, we further evaluated the performance of global-wSDTNBI-FCFP_4 and the other two models constructed using FCFP_4 fingerprints, global-bSDTNBI-FCFP_4 and global-SDTNBI-FCFP_4. Besides previous network-based methods, wSDTNBI was also compared with two commonly used molecular docking methods, Glide SP and XP.39 The correlation between prediction scores and pKi values, measured using r and P values, are provided in ESI Fig. S1.† From the P values, we found that wSDTNBI performed well on all five nuclear receptors, while the other four methods only performed well on part of the nuclear receptors. From the r values, we found that wSDTNBI outperforms not only SDTNBI and bSDTNBI, but also Glide SP and XP.
In addition to evaluation indicators, we also focused on application ranges. Global models such as global-wSDTNBI-FCFP_4 were constructed based on the global DTI network, and hence can predict active compounds for 1844 target proteins in the global network. Local models such as local-wSDTNBI-FCFP_4 were constructed based on the local DTI network, and hence can only predict active compounds for 968 target proteins in the local network. It is obvious that the application ranges of global models are wider than those of local models.
Taken together, global-wSDTNBI-FCFP_4 can output prediction scores correlated with binding affinities, and has a wide application range. Hence, we recognized this wSDTNBI model as the best model. It is promising to apply global-wSDTNBI-FCFP_4 in practical applications, such as network analysis and network-based VS.
3.3 Analysis of predicted drug–target interactions
Using the best model global-wSDTNBI-FCFP_4, we predicted 20 new targets for all 12751 drugs in the global DTI network. Interestingly, several newly predicted DTIs can be validated using recently published articles. For example, abiraterone is known as an inhibitor of cytochrome P450 17A1 (UniProt accession number: P05093, gene symbol: CYP17A1) and an approved drug for the treatment of castration-resistant prostate cancer.52 Herein, it was predicted to interact with AR. In a recent study, abiraterone showed IC50 = 7.6 and 9.4 μM in AR competitive binding and transcriptional assays, which means that abiraterone can directly bind to AR and inhibit AR transcriptional activity.52 Antimalarial drugs chloroquine and hydroxychloroquine were predicted to target sigma 1 receptor (UniProt accession number: Q99720, gene symbol: SIGMAR1). In a recent study, chloroquine and hydroxychloroquine were found to be sigma 1 receptor ligands with pKi = 7.1 and 6.9, respectively.53 Besides these DTIs predicted for approved drugs, some DTIs predicted for natural products can also be validated using the literature. For example, resveratrol and piceatannol were predicted to target 5-lipoxygenase (UniProt accession number: P09917, gene symbol: ALOX5). In a recent study, resveratrol and piceatannol were found to be 5-lipoxygenase inhibitors with IC50 = 4.9 and 0.24 μM, respectively.54 These results further indicate the reliability of global-wSDTNBI-FCFP_4.
Our predicted DTIs may facilitate the discovery of new drugs for the treatment of complex diseases. Previous studies have demonstrated that innate immunity and inflammation play important roles in many age-related chronic diseases, such as neurodegenerative diseases, diabetes and cancer.28,55 Meanwhile, autoimmune diseases, such as psoriasis, rheumatoid arthritis, inflammatory bowel disease and MS, are estimated to affect approximately 8–9% of the world population.56 To explore the relationships among drugs, targets and immunological processes, we constructed a drug–target–immunological process network for approved drugs (Fig. 3). This network contains 11 immunological processes [including barrier integrity, inflammation, antigen presentation, T cell (activation), B cell (activation), immune regulation, tissue repair, immune system development, cytokine production and signaling, chemotaxis and migration, and cellular signaling], 247 human target proteins associated with these immunological processes, and 1297 approved drugs that have known or predicted interactions with these targets. Ten of the targets, such as RORγt (UniProt accession number: P51449, gene symbol: RORC), were connected with more than 100 approved drugs. Our drug–target–immunological process network may help to explain the molecular mechanisms of the effects of approved drugs on immunological processes, and discover new drugs for the treatment of inflammatory and immune diseases.
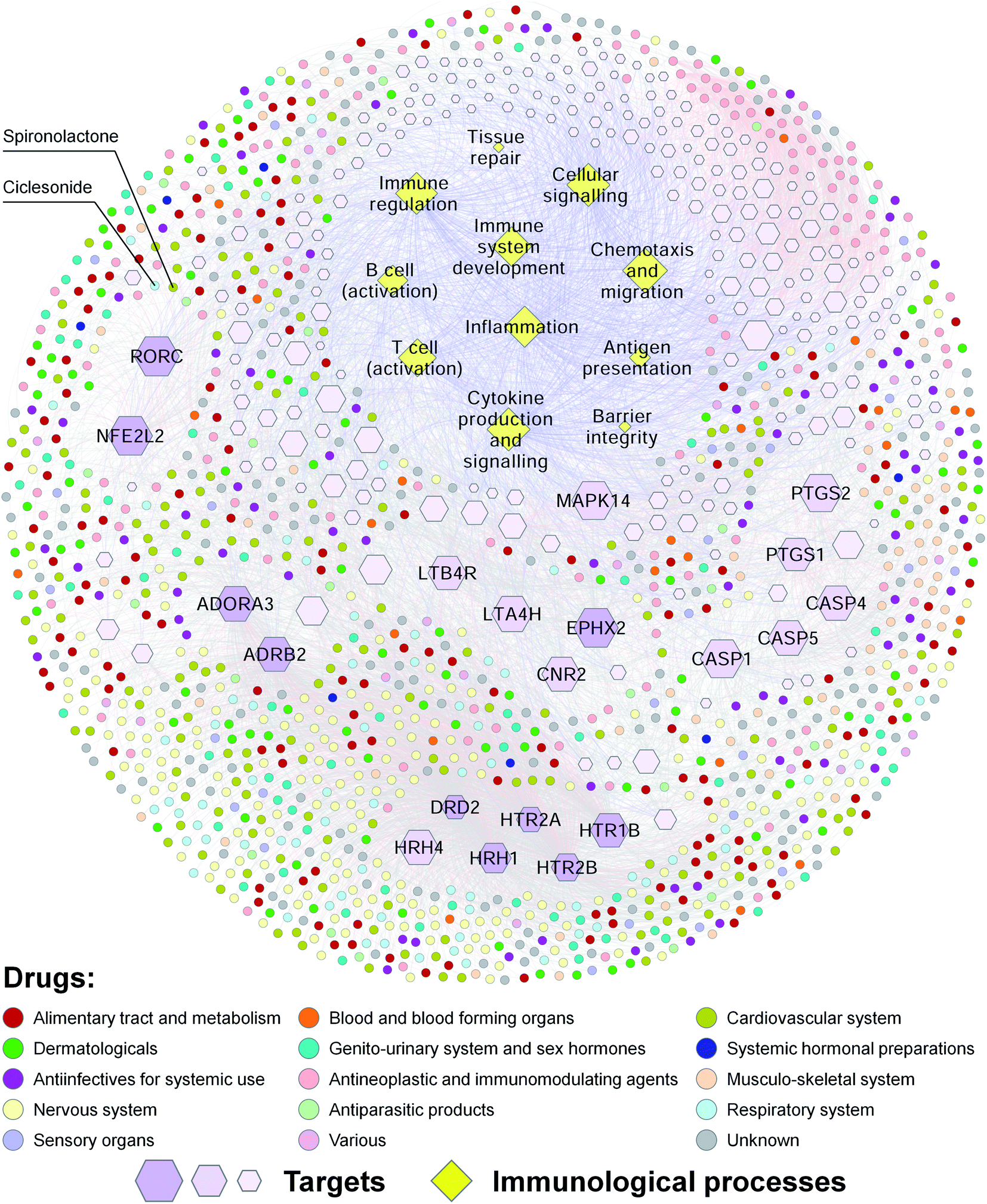 | ||
| Fig. 3 The drug–target–immunological process network for approved drugs. Drugs are represented by circles and coloured according to their first-level Anatomical Therapeutic Chemical classification codes. Targets are represented by purple hexagons. Immunological processes are represented by yellow diamonds. Known and predicted DTIs are represented by light red and gray lines, respectively. Target-immunological process associations are represented by light blue lines. | ||
To confirm this view, we used RORγt as an example. Previous studies have shown that targeting T helper 17 (TH17) cells is a promising strategy for the treatment of various autoimmune diseases.57 As the “master” transcriptional factor of TH17 cells, RORγt orchestrates the differentiation program of TH17 cells, and has emerged as an attractive target for autoimmune diseases.58 In our drug–target–immunological process network, several approved drugs such as spironolactone were predicted to target RORγ and associated with immunological processes such as the activation of T cells. A previous study has shown that mineralocorticoids such as aldosterone and deoxycorticosterone acetate (DOCA) promote autoimmune damage by enhancing TH17-mediated immunity.59 A more recent study has shown that spironolactone can decrease DOCA-salt-induced organ damage in rats by blocking the activation of TH17 cells and the downregulation of regulatory T lymphocytes.60 These studies suggest that some approved drugs such as spironolactone may target RORγt and have therapeutic effects on autoimmune diseases.
3.4 Discovery of new RORγt ligands
Using the best model Global-wSDTNBI-FCFP_4, we performed network-based VS on RORγt, the aforementioned nuclear receptor associated with autoimmune diseases. Considering that natural products are important sources of new drugs over the past few decades,61,62 we predict 20 new targets for not only all 12751 drugs within the global DTI network, but also 468 natural products outside the global DTI network. The prediction results of the 12751 + 468 = 13219 compounds are provided in ESI data 2,† which may provide useful information for researchers interested in VS and network pharmacology. According to the prediction results, we obtained 512 potential RORγt ligands and found that approximately 262 of them were purchasable from TargetMol. After considering the price and availability, we purchased 72 compounds from TargetMol for experimental validation. Most of the purchased compounds are approved drugs and natural products.
A cell-based nuclear receptor RORγt-GAL4 reporter assay was used to confirm the activity of these compounds affecting the transcriptional activity of RORγt. Seven of the 72 purchased compounds (7/72 ≈ 9.7%) showed inhibition of the transcriptional regulation of RORγt with IC50 values ranging from 0.10 μM to 4.97 μM (Fig. 4a), including ursonic acid, oleanonic acid, ciclesonide, AKT inhibitor VIII, BX-471, spironolactone and veratramine. To validate the ability of these compounds to interact with RORγt directly, co-activator peptide SRC1-2 with a biotin label was used to evaluate the affinity of the seven compounds to RORγt by HTRF technology. All seven compounds showed dose dependent inhibition of RORγt binding to SRC1-2 with IC50 values ranging from 10 nM to 19.81 μM (Fig. 4a), further indicating that they are potent RORγt inverse agonists.
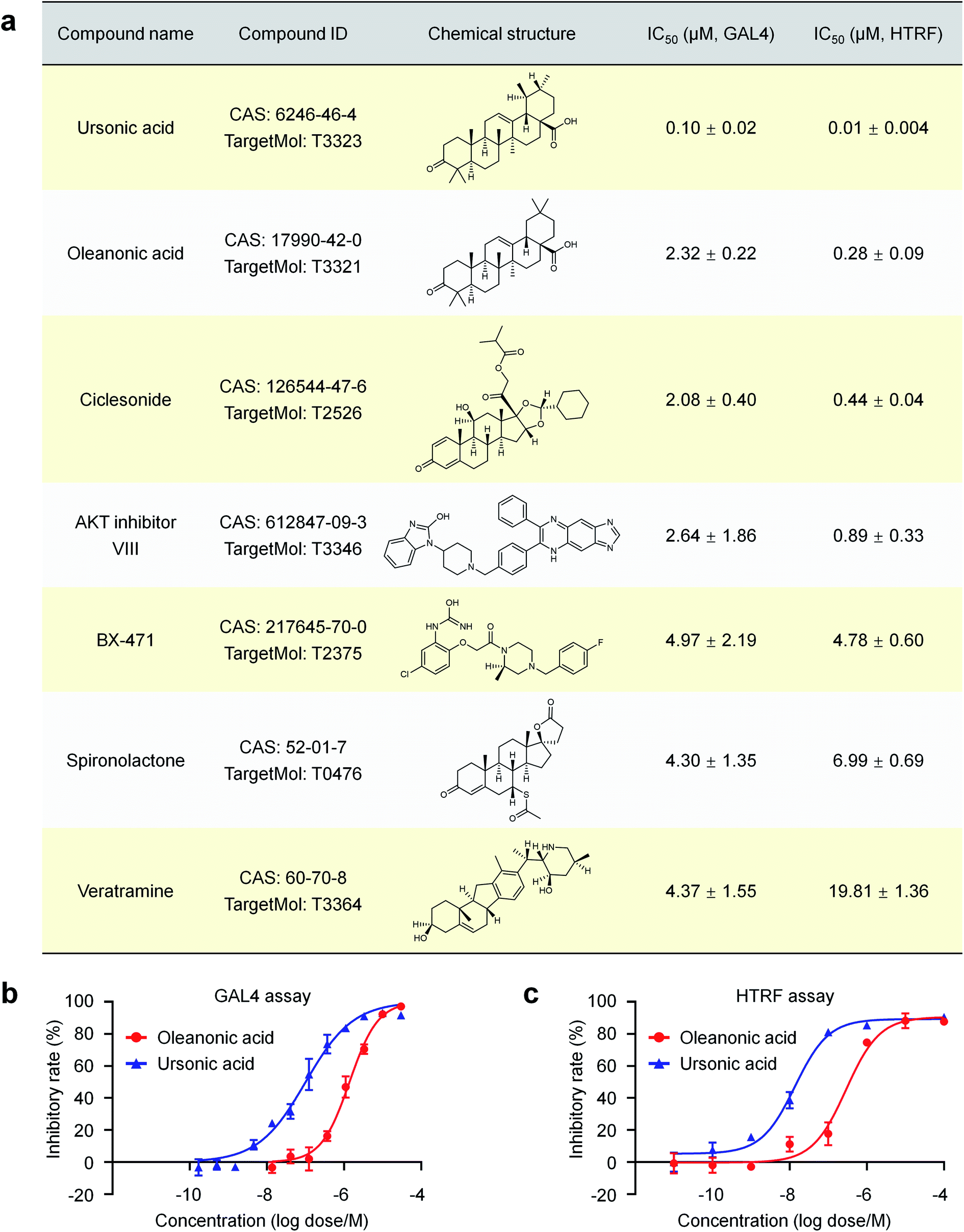 | ||
| Fig. 4 Seven newly discovered RORγt inverse agonists. (a) The names, structures and IC50 values of these RORγt inverse agonists. (b) The dose–response curves of ursonic acid and oleanonic acid in GAL4 assay. (c) The dose–response curves of ursonic acid and oleanonic acid in HTRF assay. | ||
3.5 Further investigation of ursonic acid and oleanonic acid
We turned to focus on ursonic acid and oleanonic acid, and the two natural products showed high activities in both dual-luciferase reporter and HTRF assays (Fig. 4), which may provide useful information for further development.Considering the presence of tryptophan residues in RORγt-LBD (Trp 314 and Trp 317), an intrinsic fluorescence-quenching based assay was used to further verify the direct interactions between the two compounds and RORγt-LBD.63 The intrinsic fluorescence spectra were measured after RORγt-LBD was treated with increasing concentrations of compounds. Fig. 5a showed that ursonic acid and oleanonic acid dose-dependently induced fluorescence quenching of RORγt-LBD, indicating that they are capable of binding to RORγt-LBD.
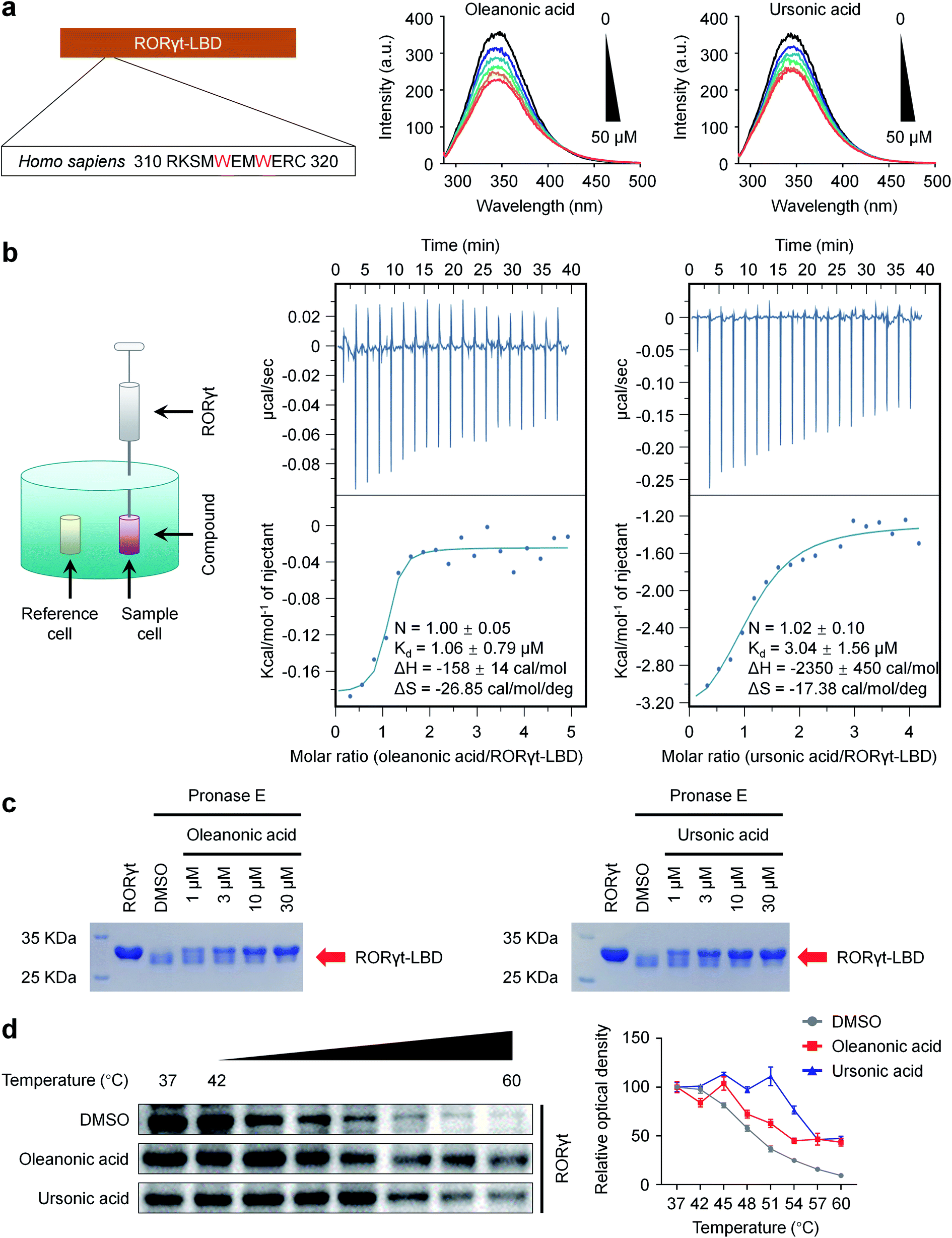 | ||
| Fig. 5 The further experimental validation for ursonic and oleanonic acid binding to RORγt directly. (a) Binding of ursonic acid and oleanonic acid to RORγt-LBD was determined by intrinsic fluorescence quenching assay. RORγt-LBD (30 μM) was incubated with increasing concentrations of ursonic acid or oleanonic acid (0 μM, 10 μM, 20 μM, 30 μM, 40 μM, and 50 μM) for 30 min. The fluorescence emission (285–500 nm) was measured with an excitation wavelength of 280 nm. (b) Thermodynamic characterization of the interaction between RORγt-LBD and ursonic acid or oleanonic acid by ITC. Compounds were titrated with RORγt-LBD in ITC buffer as mentioned in the Methods. Binding curves were fitted as a single binding event. (c) Purified RORγt-LBD-SRC2.2 protein (60 μM) was incubated with indicative concentrations of compounds at 40 °C for 10 min and then subjected to SDS-PAGE and Coomassie (SimpleBlue)-staining. (d) CETSA showed that ursonic acid and oleanonic acid stabilized their target protein RORγt in HEK293T cells. Cells were treated with compounds for 2 h. Quantitative analysis according to the western blotting is shown in the right. | ||
The thermodynamic parameters of the interaction between RORγt-LBD and ursonic acid or oleanonic acid were examined by ITC. As shown in Fig. 5b, ursonic acid and oleanonic acid bound to RORγt-LBD with Kd values of 3.04 μM and 1.06 μM, respectively. According to the changes in free energy and enthalpy (ΔG and ΔH), the binding of ursonic acid and oleanonic acid to RORγt-LBD is based on hydrogen bonds, van der Waals bonds and hydrophobic effects as indicated by the negative binding enthalpy (ΔH) and entropy factor (TΔS). Although the Kd values of the two compounds are similar, ursonic acid has more strong hydrogen bonds and van der Waals bonds as indicated by the bigger enthalpy change. The ITC results indicated that ursonic acid and oleanonic acid could interact with RORγt-LBD directly.
Binding of small compounds may stabilize its target protein and reduce the protease sensitivity of its target protein.44 We also run DARTS44 assay to identify the binding target of ursonic acid and oleanonic acid. As shown in Fig. 5c, proteolysis of RORγt-LBD-SRC2.2 by the pronase E was clearly decreased by the presence of ursonic acid or oleanonic acid in a dose-dependent manner, indicating that the binding of ursonic acid or oleanonic acid reduced the pronase E sensitivity of RORγt-LBD-SRC2.2.
To further validate the interaction between ursonic acid or oleanonic acid and RORγt in a cellular context, we performed CETSA64 in the HEK293T cell line exogenously expressing RORγt. As shown in Fig. 5d, both ursonic acid and oleanonic acid significantly increased the Tm values of RORγt, suggesting that both of them stabilized RORγt through the direct interactions. All the above findings indicated that ursonic acid and oleanonic acid indeed bound with their target protein RORγt.
3.6 Crystal structure of RORγt in complex with ursonic acid
In addition, we obtained the crystal structure of RORγt-LBD-SRC2.2 in complex with ursonic acid at a resolution of 1.99 Å (PDB code: 6J3N) (Fig. 6a and ESI Table S5†), further confirming the direct interaction between ursonic acid and RORγt. The RORγt-LBD-SRC2.2 structure was solved in the space group P41212 with a polypeptide chain per symmetric unit. Common to most nuclear receptor LBDs, this RORγt-LBD-SRC2.2 structure presented a canonical three-layered helix sandwich folded by 12 α-helices and three β-strands (Fig. 6a). X-ray results showed that ursonic acid was buried in the canonical orthosteric ligand pocket (Fig. 6a) and formed van der Waals interactions with residues located in the orthosteric binding site, including Gln 286, Leu 292, Leu 324, Arg 367, Phe 377, Leu 391 and Ile 400 (Fig. 6b). Besides, Gln 286 and His 323 formed a network of hydrogen bonds with ursonic acid mediated by a water molecule (Fig. 6b). Ursonic acid failed to form hydrogen bonds with His 479 in H11 and Tyr 502 and Phe 506 in AF2, and hence behaved as an “agonist lock untouched” inverse agonist of RORγt.65 The crystal results provide useful information on structural modification of ursonic acid and help find new RORγt inverse agonists with higher activities. | ||
| Fig. 6 X-ray structure of ursonic acid with RORγt-LBD-SRC2.2 (PDB code: 6J3N). (a) The overview of the crystal structure of RORγt-LBD-SRC2.2 with ursonic acid. (b) The binding mode of ursonic acid in the ligand binding pocket. RORγt-LBD-SRC2.2 is shown as surface or cartoon with cyan α-helices and purple β-strands. Ursonic acid is drawn as yellow sticks. Key residues and water molecules are shown as grey sticks and red spheres respectively. Hydrogen bonds are indicated by yellow dotted lines. Oxygen and nitrogen are colored by red and blue, respectively. | ||
3.7 RORγt inhibition ameliorated MOG-induced chronic progressive EAE model
MS is a chronic inflammatory disease of the central nervous system that affects more than 2 million people worldwide,66 and one of the common causes of serious physical disability in young adults.67 As mentioned above, RORγt is a potential therapeutic target for autoimmune diseases such as MS. Our newly discovered RORγt inverse agonists may have therapeutic effects on MS. Hence, we investigated the in vivo effects of ursonic acid and oleanonic acid on the disease process of EAE, a most commonly used animal model of human MS.68As shown in Fig. 7a, we induced EAE in female C57BL/6 mice with MOG35-55 and PTX, and the mice were orally administrated usronic acid and oleanonic acid every day from day 12. We observed that EAE mice started to exhibit clinical symptoms at day 13 after immunization (n = 8) (Fig. 7b). Compared with the EAE group, a significant delay of the disease onset was found in oleanonic acid-treated group mice with a mean disease onset date of 15.3 ± 0.2 days (n = 8). Surprisingly, better therapeutic effects were found in ursonic acid-treated group mice with an average onset date of 17.3 ± 0.5 days (n = 8), which may be attributed to its better inhibitory activity. In the course of EAE, body weight variation also reflects disease severity. As expected, the ursonic acid and oleanonic acid administration significantly reversed EAE induced mice body weight loss (Fig. 7c).
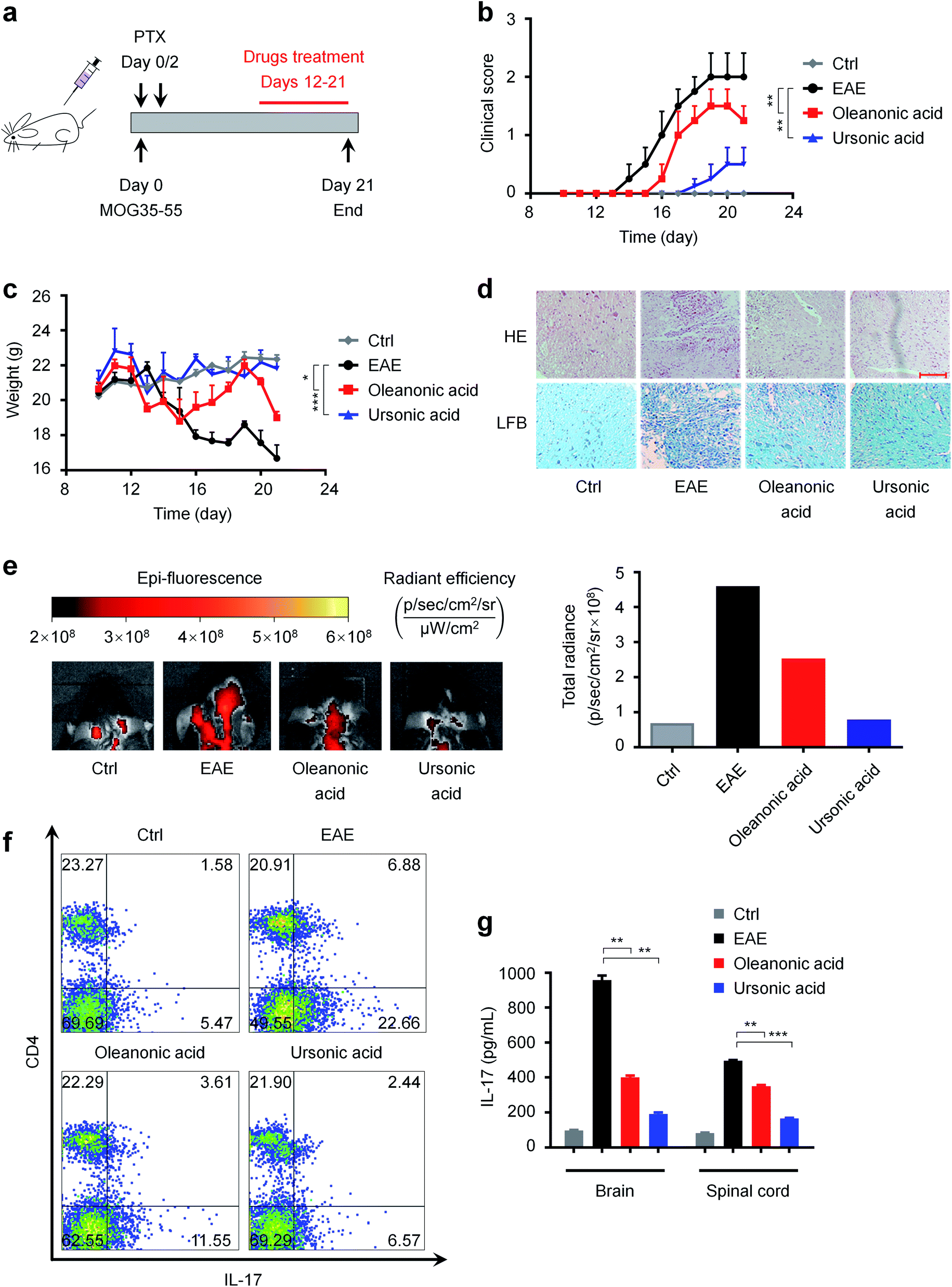 | ||
| Fig. 7 In vivo effects of ursonic acid and oleanonic acid on the disease process of EAE. (a) EAE disease induction and treatment. (b) Disease severity assessed by the clinical scoring system ranging from 0 (no signs) to 5 (death). (c) Disease severity assessed by body weight variation. (d) Histochemical analysis of spinal cords of mice from different groups (scale bar = 100 μm). (e) EAE-induced BBB disruption reflected by BSA-Cy5.5 images in the brain. (f) Frequency of pro-inflammatory TH17 cells detected by CD4 and IL-17 dual-staining in the spleen of mice. (g) IL-17 expression in spinal cords and brains measured by ELISA. | ||
Demyelinated lesions and inflammation in the peripheral white matter of spinal cord are the main pathological features of the EAE model.69 To further assess the disease condition of EAE, representative sections of spinal cord from each group mice were stained with LFB and H&E. Consistent with decreased clinical symptoms, immune cell infiltrations were reduced in white matter of spinal cords from ursonic acid and oleanonic acid treated-group mice (Fig. 7d). Moreover, bovine serum albumin coupled with Cy5.5 (BSA-Cy5.5) was used to assess the degree of blood–brain barrier (BBB) damage, which is one of the early pathophysiological hallmarks of EAE.49,70 Compared with the EAE group, the visualization of brain BSA-Cy5.5 accumulation imaging exhibited that ursonic acid and oleanonic acid remitted the EAE-induced BBB disruption obviously (Fig. 7e), and ursonic acid showed better therapeutic effects than oleanonic acid, which is consistent with their inhibitory activities reflected in vitro.
Accumulating evidence has established that IL-17-producing CD4+ T cells, also known as TH17 cells, play a critical role in the pathogenesis of EAE.71 To examine whether RORγt inhibition by compounds affects the differentiation of TH17 cells in vivo, we thereby performed intracellular cytokine staining to determine the population of TH17 cells in the spleen of mice. As demonstrated in Fig. 7f, ursonic acid and oleanonic acid treatment resulted in a reduced frequency of pro-inflammatory TH17 cells as detected by CD4 and IL-17 dual-staining. Meanwhile, we also detected the IL-17 content in the spinal cord and brain supernatant by ELISA.72,73 Consistent with decreased TH17 population, clinical, ELISA analysis of mice tissue samples revealed that ursonic acid and oleanonic acid effectively reduced IL-17A protein expression in brains and spinal cords (Fig. 7g).
Taken together, ursonic acid and oleanonic acid ameliorated the MOG-induced chronic progressive EAE model by targeting pro-inflammatory TH17 cells, especially ursonic acid. Therefore, they may serve as lead compounds for the treatment of MS.
4 Discussion
In this study, we proposed wSDTNBI, a novel network-based method integrating weighted DTI and DSA networks for DTI prediction. wSDTNBI inherits the good features of previous network-based methods, including independent of 3D crystal structures and negative DTI samples. Furthermore, wSDTNBI can output prediction scores correlated with binding affinities, overcoming the limitation of previous network-based methods. This improvement makes it more suitable for VS, and lead to a new type of VS, namely, network-based VS. Compared with traditional methods, wSDTNBI have several obvious advantages.Firstly, wSDTNBI can help to identify more active compounds with higher activities. In a previous study, Zhang et al. performed structure-based VS on RORγt.24 Only one of 24 purchased compounds (1/24 ≈ 4.2%) was validated by GAL4 reporter assay (IC50 = 11.84 μM). More recently, Zeng et al. developed a deep learning-based method for DTI prediction, and used the method to identify potential RORγt ligands.25 Only one of 18 purchased compounds (1/18 ≈ 5.6%) was validated by GAL4 reporter assay (IC50 = 0.43 μM). In this study, we performed network-based VS on RORγt using Global-wSDTNBI-FCFP_4, and purchased 72 compounds for experimental validation. Seven of them (7/72 ≈ 9.7%) showed IC50 <10 μM in the same GAL4 reporter assay, and the best compound ursonic acid even showed IC50 = 0.10 μM in GAL4 assay and IC50 = 10 nM in HTRF assay. Compared with the two previous VS studies on RORγt,24,25 we obtained not only the highest success rate, but also the compound with the highest activity.
Secondly, wSDTNBI can help to identify some active compounds that other methods cannot. To show this, we inputted the chemical structures of the seven newly discovered RORγt ligands into six web servers for DTI prediction, and then investigated whether these web servers can correctly predict the interactions between these compounds and RORγt. The six web servers include one pharmacophore-based, PharmMapper;74 three similarity-based, SEA,75 ChemMapper,76 and SwissTargetPrediction;77 and two machine learning-based, TargetHunter,78 and TargetNet.79 After trying different parameter settings, still only three of the web servers, ChemMapper, SwissTargetPrediction and TargetHunter, reproduced a small number of the interactions, while the others totally failed (ESI Table S6†). Meanwhile, we also tried previous network-based methods. Unsurprisingly, only one, one and four compounds were predicted as potential RORγt ligands by NBI, SDTNBI and bSDTNBI, respectively (ESI Table S6†).
Thirdly, wSDTNBI can output comprehensive prediction results in a short time. In this study, we used the NVIDIA cuBLAS library, an GPU-accelerated implementation of the basic linear algebra subroutines (BLAS), to accelerate the matrix computation such as matrix multiplication. In three workstations with the hardware support of NVIDIA GeForce RTX 2080 Ti, RTX 2080 Super and GTX 1070, Global-wSDTNBI-FCFP_4 only costs about 48, 53 and 125 seconds to calculate prediction scores for all possible interactions between 13219 compounds and 1844 target proteins, which means that it can simultaneously perform VS on the 1844 targets in one or two minutes. As a comparison, we docked the seven newly discovered RORγt ligands into a crystal structure of RORγt by Glide SP and XP. However, the two docking experiments between seven compounds and only one target cost approximately 7 and 50 minutes, respectively. The high speed allows large-scale VS to be performed using wSDTNBI. The comprehensive prediction results covering hundreds of targets may not only help to discard those compounds binding to the proteins closely associated with toxic or side effects [e.g., human ether-à-go-go-related gene (hERG) potassium channel], but also provide opportunities for multi-target drug discovery.
Despite the aforementioned advantages, there is still room for improvement of our method. wSDTNBI cannot be used for a protein without any known ligands, because this protein cannot be interlinked with the known DTI network. In addition, interaction types (e.g., agonism or antagonism, activation or inhibition) are not considered yet. We are actively developing novel network-based methods to overcome these limitations. For example, we are trying to extend the prediction results to more proteins by introducing different types of protein sequence similarity and structure similarity data.
Besides the methodology we developed, the active compounds we discovered can also facilitate drug discovery and development. In this study, seven compounds were confirmed to be novel RORγt inverse agonists by in vitro experiments, and in vivo experiments demonstrated that two of them (ursonic acid and oleanonic acid) have therapeutic effects on MS, indicating that they may serve as lead compounds for the treatment of autoimmune diseases including MS. Considering that RORγt is a specific isoform of RORγ, the newly discovered RORγt inverse agonists may be applied in other diseases associated with RORγ. For example, RORγ was found to drive the expression of AR and represent a potential therapeutic target for the treatment of prostate cancer in a previous study.80 As one of the newly discovered RORγt inverse agonists, spironolactone was found to reduce the incidence of prostate cancer in a recent study.81 The possible reason of this chemoprevention of prostate cancer is that spironolactone suppresses the expression of AR by inhibiting the transcriptional activity of RORγ. These studies indicate that the newly discovered RORγt inverse agonists may have therapeutic effects on prostate cancer. Moreover, we predicted absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of the newly discovered RORγt inverse agonists, and provided the prediction results in ESI data 2.† Interestingly, most of the newly discovered RORγt inverse agonists, especially ursonic acid and oleanonic acid, were predicted to have not only good human intestinal absorption, but also low carcinogenicity, cardiotoxicity and hepatotoxicity. These predicted ADMET properties further suggest that our newly discovered RORγt inverse agonists are promising lead compounds for the treatment of autoimmune diseases and cancer.
5 Conclusions
We developed a novel network-based method named wSDTNBI, which employed weighted DTI networks instead of unweighted DTI networks for the prediction of potential DTIs, and hence can help to find more active compounds with higher activities for targets of interest. In a network-based VS on RORγt, seven of the 72 purchased compounds including ursonic acid and oleanonic acid were confirmed to be novel RORγt inverse agonists by in vitro experiments. In addition, an X-ray crystal structure of RORγt in complex with ursonic acid (PDB code: 6J3N) was obtained, and in vivo experiments showed the therapeutic effects of ursonic acid and oleanonic acid on MS. The success rate of our network-based VS on RORγt is higher than the success rates of recent structure-based and deep learning-based VS on RORγt. Our newly discovered RORγt inverse agonists and the cocrystal structure may facilitate the discovery of more effective drugs for the treatment of autoimmune diseases including MS. Our methods and workflow can be easily extended to other targets for other diseases. We believe that wSDTNBI would become a powerful tool for network-based VS in drug discovery.Data availability
The detailed information of drugs, targets and DTIs for model construction is provided in ESI data 1.† The prediction results of the best wSDTNBI model and the detailed information of the purchased compounds are provided in ESI data 2.† Some of the wSDTNBI models constructed in this study are freely available in our web server NetInfer (http://lmmd.ecust.edu.cn/netinfer). All other data are available upon reasonable request.Author contributions
Yun Tang and Jin Huang supervised the research project. Zengrui Wu designed and implemented the wSDTNBI method. Zengrui Wu, Lulu Zheng, Zhuohang Yu, Weihua Li and Guixia Liu constructed network-based models, evaluated the performance of network-based models, performed network-based VS on RORγt and analyzed the prediction results. Hui Ma, Zehui Liu, Shuying Cao, Wenqing Fang, Lili Wu performed in vitro and in vivo experiments. Zengrui Wu, Hui Ma, Zehui Liu, Jin Huang and Yun Tang wrote the manuscript. All authors read and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Junhao Li (Uppsala University, Sweden) and Dr Hongbin Yang (University of Cambridge, United Kingdom) for helpful discussions. This work was supported by the National Key Research and Development Program of China (Grants 2016YFA0502304 and 2019YFA0904800), the National Natural Science Foundation of China (Grants 81872800, 81773775, 81973362, 82173746 and 82104066), the Shanghai Frontiers Science Center of Optogenetic Techniques for Cell Metabolism (Shanghai Municipal Education Commission, Grant 2021 Sci & Tech 03-28), the Shanghai Post-doctoral Excellence Program (Grant 2018199), the Shanghai Sailing Program (Grant 19YF1412700), the China Postdoctoral Science Foundation (Grant 2019M661413), and the 111 Project (Grant BP0719034).Notes and references
- Y. Tang, W. L. Zhu, K. X. Chen and H. L. Jiang, Drug Discovery Today, 2006, 3, 307–313 CrossRef PubMed.
- G. Klebe, Drug Discovery Today, 2006, 11, 580–594 CrossRef CAS PubMed.
- A. Lavecchia and C. Di Giovanni, Curr. Med. Chem., 2013, 20, 2839–2860 CrossRef CAS PubMed.
- P. D. Lyne, Drug Discovery Today, 2002, 7, 1047–1055 CrossRef CAS PubMed.
- K. H. Kim, N. D. Kim and B. L. Seong, Expert Opin. Drug Discovery, 2010, 5, 205–222 CrossRef CAS PubMed.
- P. Willett, Drug Discovery Today, 2006, 11, 1046–1053 CrossRef CAS PubMed.
- R. Kurczab, S. Smusz and A. J. Bojarski, J. Cheminf., 2014, 6, 32 Search PubMed.
- A. Lavecchia, Drug Discovery Today, 2015, 20, 318–331 CrossRef PubMed.
- S. Rocha, C. G. Olanda, H. H. Fokoue and C. M. R. Sant'Anna, Curr. Top. Med. Chem., 2019, 19, 1751–1767 CrossRef PubMed.
- N. Singh, L. Chaput and B. O. Villoutreix, Briefings Bioinf., 2021, 22, 1790–1818 CrossRef PubMed.
- A. L. Hopkins, Nat. Chem. Biol., 2008, 4, 682–690 CrossRef CAS PubMed.
- J. L. Medina-Franco, M. A. Giulianotti, G. S. Welmaker and R. A. Houghten, Drug Discovery Today, 2013, 18, 495–501 CrossRef PubMed.
- Z. R. Wu, W. H. Li, G. X. Liu and Y. Tang, Front. Pharmacol., 2018, 9, 1134 CrossRef CAS PubMed.
- F. X. Cheng, C. Liu, J. Jiang, W. Q. Lu, W. H. Li, G. X. Liu, W. X. Zhou, J. Huang and Y. Tang, PLoS Comput. Biol., 2012, 8, e1002503 CrossRef CAS PubMed.
- Z. R. Wu, W. Q. Lu, D. Wu, A. Q. Luo, H. P. Bian, J. Li, W. H. Li, G. X. Liu, J. Huang, F. X. Cheng and Y. Tang, Br. J. Pharmacol., 2016, 173, 3372–3385 CrossRef CAS PubMed.
- Z. R. Wu, F. X. Cheng, J. Li, W. H. Li, G. X. Liu and Y. Tang, Briefings Bioinf., 2017, 18, 333–347 CAS.
- X. Chen, M. X. Liu and G. Y. Yan, Mol. BioSyst., 2012, 8, 1970–1978 RSC.
- C. Duran, S. Daminelli, J. M. Thomas, V. J. Haupt, M. Schroeder and C. V. Cannistraci, Briefings Bioinf., 2018, 19, 1183–1202 CrossRef PubMed.
- L. Y. Lu and T. Zhou, Phys. A, 2011, 390, 1150–1170 CrossRef.
- J. S. Fang, Z. R. Wu, C. P. Cai, Q. Wang, Y. Tang and F. X. Cheng, J. Chem. Inf. Model., 2017, 57, 2657–2671 CrossRef CAS PubMed.
- Z. R. Wu, W. Q. Lu, W. W. Yu, T. D. Y. Wang, W. H. Li, G. X. Liu, H. K. Zhang, X. F. Pang, J. Huang, M. Y. Liu, F. X. Cheng and Y. Tang, Pharmacol. Res., 2018, 129, 400–413 CrossRef CAS PubMed.
- Z. R. Wu, Q. H. Wang, H. B. Yang, J. Y. Wang, W. H. Li, G. X. Liu, Y. Yang, Y. Z. Zhao and Y. Tang, J. Chem. Inf. Model., 2021, 61, 2486–2498 CrossRef CAS PubMed.
- M. Y. Su, Q. F. Yang, Y. Du, G. Q. Feng, Z. H. Liu, Y. Li and R. X. Wang, J. Chem. Inf. Model., 2019, 59, 895–913 CrossRef CAS PubMed.
- Y. Zhang, X. Q. Xue, X. Y. Jin, Y. Song, J. Li, X. Y. Luo, M. Song, W. Q. Yan, H. R. Song and Y. Xu, Eur. J. Med. Chem., 2014, 78, 431–441 CrossRef CAS PubMed.
- X. X. Zeng, S. Y. Zhu, W. Q. Lu, Z. H. Liu, J. Huang, Y. D. Zhou, J. S. Fang, Y. Huang, H. M. Guo, L. Li, B. D. Trapp, R. Nussinov, C. Eng, J. Loscalzo and F. X. Cheng, Chem. Sci., 2020, 11, 1775–1797 RSC.
- P. Willett, J. M. Barnard and G. M. Downs, J. Chem. Inf. Model., 1998, 38, 983–996 CrossRef CAS.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li, Z. Sayeeda, N. Assempour, I. Iynkkaran, Y. F. Liu, A. Maciejewski, N. Gale, A. Wilson, L. Chin, R. Cummings, D. Le, A. Pon, C. Knox and M. Wilson, Nucleic Acids Res., 2018, 46, D1074–D1082 CrossRef CAS PubMed.
- S. D. Harding, J. L. Sharman, E. Faccenda, C. Southan, A. J. Pawson, S. Ireland, A. J. G. Gray, L. Bruce, S. P. H. Alexander, S. Anderton, C. Bryant, A. P. Davenport, C. Doerig, D. Fabbro, F. Levi-Schaffer, M. Spedding, J. A. Davies and I. Nc, Nucleic Acids Res., 2018, 46, D1091–D1106 CrossRef CAS PubMed.
- M. Kanehisa, M. Furumichi, M. Tanabe, Y. Sato and K. Morishima, Nucleic Acids Res., 2017, 45, D353–D361 CrossRef CAS PubMed.
- Y. H. Li, C. Y. Yu, X. X. Li, P. Zhang, J. Tang, Q. X. Yang, T. T. Fu, X. Y. Zhang, X. J. Cui, G. Tu, Y. Zhang, S. Li, F. Y. Yang, Q. Sun, C. Qin, X. Zeng, Z. Chen, Y. Z. Chen and F. Zhu, Nucleic Acids Res., 2018, 46, D1121–D1127 CrossRef CAS PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez, P. Mutowo, F. Atkinson, L. J. Bellis, E. Cibrian-Uhalte, M. Davies, N. Dedman, A. Karlsson, M. P. Magarinos, J. P. Overington, G. Papadatos, I. Smit and A. R. Leach, Nucleic Acids Res., 2017, 45, D945–D954 CrossRef CAS PubMed.
- M. K. Gilson, T. Q. Liu, M. Baitaluk, G. Nicola, L. Hwang and J. Chong, Nucleic Acids Res., 2016, 44, D1045–D1053 CrossRef CAS PubMed.
- B. L. Roth, E. Lopez, S. Patel and W. K. Kroeze, Neuroscientist, 2000, 6, 252–262 CrossRef CAS.
- A. Bateman, M. J. Martin, S. Orchard, M. Magrane, E. Alpi, B. Bely, M. Bingley, R. Britto, B. Bursteinas, G. Busiello, H. Bye-A-Jee, A. Da Silva, M. De Giorgi, T. Dogan, L. G. Castro, P. Garmiri, G. Georghiou, D. Gonzales, L. Gonzales, E. Hatton-Ellis, A. Ignatchenko, R. Ishtiaq, P. Jokinen, V. Joshi, D. Jyothi, R. Lopez, J. Luo, Y. Lussi, A. MacDougall, F. Madeira, M. Mahmoudy, M. Menchi, A. Nightingale, J. Onwubiko, B. Palka, K. Pichler, S. Pundir, G. Y. Qi, S. Raj, A. Renaux, M. R. Lopez, R. Saidi, T. Sawford, A. Shypitsyna, E. Speretta, E. Turner, N. Tyagi, P. Vasudev, V. Volynkin, T. Wardell, K. Warner, X. Watkins, R. Zaru, H. Zellner, A. Bridge, I. Xenarios, S. Poux, N. Redaschi, L. Aimo, G. Argoud-Puy, A. Auchincloss, K. Axelsen, P. Bansal, D. Baratin, M. C. Blatter, J. Bolleman, E. Boutet, L. Breuza, C. Casals-Casas, E. de Castro, E. Coudert, B. Cuche, M. Doche, D. Dornevil, A. Estreicher, L. Famiglietti, M. Feuermann, E. Gasteiger, S. Gehant, V. Gerritsen, A. Gos, N. Gruaz, U. Hinz, C. Hulo, N. Hyka-Nouspikel, F. Jungo, G. Keller, A. Kerhornou, V. Lara, P. Lemercier, D. Lieberherr, T. Lombardot, X. Martin, P. Masson, A. Morgat, T. B. Neto, S. Paesano, I. Pedruzzi, S. Pilbout, M. Pozzato, M. Pruess, C. Rivoire, C. Sigrist, K. Sonesson, A. Stutz, S. Sundaram, M. Tognolli, L. Verbregue, C. H. Wu, C. N. Arighi, L. Arminski, C. M. Chen, Y. X. Chen, J. Cowart, J. S. Garavelli, H. Z. Huang, K. Laiho, P. McGarvey, D. A. Natale, K. Ross, C. R. Vinayaka, Q. H. Wang, Y. Q. Wang, L. S. Yeh, J. Zhang and C. UniProt, Nucleic Acids Res., 2019, 47, D506–D515 CrossRef PubMed.
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- M. M. Mysinger, M. Carchia, J. J. Irwin and B. K. Shoichet, J. Med. Chem., 2012, 55, 6582–6594 CrossRef CAS PubMed.
- S. K. Burley, C. Bhikadiya, C. X. Bi, S. Bittrich, L. Chen, G. V. Crichlow, C. H. Christie, K. Dalenberg, L. Di Costanzo, J. M. Duarte, S. Dutta, Z. K. Feng, S. Ganesan, D. S. Goodsell, S. Ghosh, R. K. Green, V. Guranovic, D. Guzenko, B. P. Hudson, C. L. Lawson, Y. H. Liang, R. Lowe, H. Namkoong, E. Peisach, I. Persikova, C. Randle, A. Rose, Y. Rose, A. Sali, J. Segura, M. Sekharan, C. H. Shao, Y. P. Tao, M. Voigt, J. D. Westbrook, J. Y. Young, C. Zardecki and M. Zhuravleva, Nucleic Acids Res., 2021, 49, D437–D451 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- M. E. Smoot, K. Ono, J. Ruscheinski, P. L. Wang and T. Ideker, Bioinformatics, 2011, 27, 431–432 CrossRef CAS PubMed.
- S. Fujita-Sato, S. Ito, T. Isobe, T. Ohyama, K. Wakabayashi, K. Morishita, O. Ando and F. Isono, J. Biol. Chem., 2011, 286, 31409–31417 CrossRef CAS PubMed.
- F. R. Santori, P. X. Huang, S. A. van de Pavert, E. F. Douglass, D. J. Leaver, B. A. Haubrich, R. Keber, G. Lorbek, T. Konijn, B. N. Rosales, D. Rozman, S. Horvat, A. Rahier, R. E. Mebius, F. Rastinejad, W. D. Nes and D. R. Littman, Cell Metab., 2015, 21, 286–297 CrossRef CAS PubMed.
- X. Xu, X. Xu, P. Liu, Z. Y. Zhu, J. Chen, H. A. Fu, L. L. Chen, L. H. Hu and X. Shen, J. Biol. Chem., 2015, 290, 19888–19899 CrossRef CAS PubMed.
- B. Lomenick, R. Hao, N. Jonai, R. M. Chin, M. Aghajan, S. Warburton, J. N. Wang, R. P. Wu, F. Gomez, J. A. Loo, J. A. Wohlschlegel, T. M. Vondriska, J. Pelletier, H. R. Herschman, J. Clardy, C. F. Clarke and J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21984–21989 CrossRef CAS PubMed.
- X. Liu, Q. Hu, W. Y. Wang, H. Ma, J. Q. Pu, J. Y. Cui, T. Gong, Y. Wu, W. Q. Lu and J. Huang, Biochem. Pharmacol., 2021, 188 CAS.
- T. Xu, X. H. Wang, B. Zhong, R. I. Nurieva, S. Ding and C. Dong, J. Biol. Chem., 2011, 286, 22707–22710 CrossRef CAS PubMed.
- J. R. Huh, M. W. L. Leung, P. X. Huang, D. A. Ryan, M. R. Krout, R. R. V. Malapaka, J. Chow, N. Manel, M. Ciofani, S. V. Kim, A. Cuesta, F. R. Santori, J. J. Lafaille, H. E. Xu, D. Y. Gin, F. Rastinejad and D. R. Littman, Nature, 2011, 472, 486–490 CrossRef CAS PubMed.
- J. R. Huh, E. E. Englund, H. Wang, R. L. Huang, P. X. Huang, F. Rastinejad, J. Inglese, C. P. Austin, R. L. Johnson, W. W. Huang and D. R. Littman, ACS Med. Chem. Lett., 2013, 4, 79–84 CrossRef CAS PubMed.
- K. Schmitz, N. de Bruin, P. Bishay, J. Mannich, A. Haussler, C. Altmann, N. Ferreiros, J. Lotsch, A. Ultsch, M. J. Parnham, G. Geisslinger and I. Tegeder, EMBO Mol. Med., 2014, 6, 1398–1422 CrossRef CAS PubMed.
- D. Druzd, O. Matveeva, L. Ince, U. Harrison, W. Y. He, C. Schmal, H. Herzel, A. H. Tsang, N. Kawakami, A. Leliavski, O. Uhl, L. Yao, L. E. Sander, C. S. Chen, K. Kraus, A. de Juan, S. M. Hergenhan, M. Ehlers, B. Koletzko, R. Haas, W. Solbach, H. Oster and C. Scheiermann, Immunity, 2017, 46, 120–132 CrossRef CAS PubMed.
- Z. R. Wu, Y. Y. Peng, Z. H. Yu, W. H. Li, G. X. Liu and Y. Tang, J. Chem. Inf. Model., 2020, 60, 3687–3691 CrossRef CAS PubMed.
- J. D. Norris, S. J. Ellison, J. G. Baker, D. B. Stagg, S. E. Wardell, S. Park, H. M. Alley, R. M. Baldi, A. Yllanes, K. J. Andreano, J. P. Stice, S. A. Lawrence, J. R. Eisner, D. K. Price, W. R. Moore, W. D. Figg and D. P. McDonnell, J. Clin. Invest., 2017, 127, 2326–2338 CrossRef PubMed.
- D. E. Gordon, G. M. Jang, M. Bouhaddou, J. W. Xu, K. Obernier, K. M. White, M. J. O'Meara, V. V. Rezelj, J. F. Z. Guo, D. L. Swaney, T. A. Tummino, R. Huttenhain, R. M. Kaake, A. L. Richards, B. Tutuncuoglu, H. Foussard, J. Batra, K. Haas, M. Modak, M. Kim, P. Haas, B. J. Polacco, H. Braberg, J. M. Fabius, M. Eckhardt, M. Soucheray, M. J. Bennett, M. Cakir, M. J. McGregor, Q. Y. Li, B. Meyer, F. Roesch, T. Vallet, A. Mac Kain, L. Miorin, E. Moreno, Z. Z. C. Naing, Y. Zhou, S. M. Peng, Y. Shi, Z. Y. Zhang, W. Q. Shen, I. T. Kirby, J. E. Melnyk, J. S. Chorba, K. V. Lou, S. Z. A. Dai, I. Barrio-Hernandez, D. Memon, C. Hernandez-Armenta, J. Lyu, C. J. P. Mathy, T. Perica, K. B. Pilla, S. J. Ganesan, D. J. Saltzberg, R. Rakesh, X. Liu, S. B. Rosenthal, L. Calviello, S. Venkataramanan, J. Liboy-Lugo, Y. Z. Lin, X. P. Huang, Y. F. Liu, S. A. Wankowicz, M. Bohn, M. Safari, F. S. Ugur, C. Koh, N. S. Savar, Q. D. Tran, D. Shengjuler, S. J. Fletcher, M. C. O'Neal, Y. M. Cai, J. C. J. Chang, D. J. Broadhurst, S. Klippsten, P. P. Sharp, N. A. Wenzell, D. Kuzuoglu-Ozturk, H. Y. Wang, R. Trenker, J. M. Young, D. A. Cavero, J. Hiatt, T. L. Roth, U. Rathore, A. Subramanian, J. Noack, M. Hubert, R. M. Stroud, A. D. Frankel, O. S. Rosenberg, K. A. Verba, D. A. Agard, M. Ott, M. Emerman, N. Jura, M. von Zastrow, E. Verdin, A. Ashworth, O. Schwartz, C. D'Enfert, S. Mukherjee, M. Jacobson, H. S. Malik, D. G. Fujimori, T. Ideker, C. S. Craik, S. N. Floor, J. S. Fraser, J. D. Gross, A. Sali, B. L. Roth, D. Ruggero, J. Taunton, T. Kortemme, P. Beltrao, M. Vignuzzi, A. Garcia-Sastre, K. M. Shokat, B. K. Shoichet and N. J. Krogan, Nature, 2020, 583, 459–468 CrossRef CAS PubMed.
- M. Boccellino, M. Donniacuo, F. Bruno, B. Rinaldi, L. Quagliuolo, M. Ambruosi, S. Pace, M. De Rosa, A. Olgac, E. Banoglu, N. Alessio, A. Massa, H. Kahn, O. Werz, A. Fiorentino and R. Filosa, Eur. J. Med. Chem., 2019, 180, 637–647 CrossRef CAS PubMed.
- F. Licastro, G. Candore, D. Lio, E. Porcellini, G. Colonna-Romano, C. Franceschi and C. Caruso, Immun. Ageing, 2005, 2, 8 CrossRef PubMed.
- M. Gutierrez-Arcelus, S. S. Rich and S. Raychaudhuri, Nat. Rev. Genet., 2016, 17, 160–174 CrossRef PubMed.
- J. F. Yang, M. S. Sundrud, J. Skepner and T. Yamagata, Trends Pharmacol. Sci., 2014, 35, 493–500 CrossRef CAS PubMed.
- I. I. Ivanov, B. S. McKenzie, L. Zhou, C. E. Tadokoro, A. Lepelley, J. J. Lafaille, D. J. Cua and D. R. Littman, Cell, 2006, 126, 1121–1133 CrossRef CAS PubMed.
- A. A. Herrada, F. J. Contreras, N. P. Marini, C. A. Amador, P. A. Gonzalez, C. M. Cortes, C. A. Riedel, C. A. Carvajal, F. Figueroa, L. F. Michea, C. E. Fardella and A. M. Kalergis, J. Immunol., 2010, 184, 191–202 CrossRef CAS PubMed.
- C. A. Amador, V. Barrientos, J. Pena, A. A. Herrada, M. Gonzalez, S. Valdes, L. Carrasco, R. Alzamora, F. Figueroa, A. M. Kalergis and L. Michea, Hypertension, 2014, 63, 797–803 CrossRef CAS PubMed.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- Y. P. Zhan, X. P. Du, H. Z. Chen, J. J. Liu, B. X. Zhao, D. H. Huang, G. D. Li, Q. Y. Xu, M. Q. Zhang, B. C. Weimer, D. Chen, Z. Cheng, L. R. Zhang, Q. X. Li, S. W. Li, Z. H. Zheng, S. Y. Song, Y. J. Huang, Z. Y. Ye, W. J. Su, S. C. Lin, Y. M. Shen and Q. Wu, Nat. Chem. Biol., 2008, 4, 548–556 CrossRef CAS PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. H. Cao and P. Nordlund, Science, 2013, 341, 84–87 CrossRef CAS PubMed.
- F. A. Meijer, R. G. Doveston, R. de Vries, G. M. Vos, A. A. A. Vos, S. Leysen, M. Scheepstra, C. Ottmann, L. G. Milroy and L. Brunsveld, J. Med. Chem., 2020, 63, 241–259 CrossRef CAS PubMed.
- D. S. Reich, C. F. Lucchinetti and P. A. Calabresi, N. Engl. J. Med., 2018, 378, 169–180 CrossRef CAS PubMed.
- C. A. Dendrou, L. Fugger and M. A. Friese, Nat. Rev. Immunol., 2015, 15, 545–558 CrossRef CAS PubMed.
- H. Kebir, K. Kreymborg, I. Ifergan, A. Dodelet-Devillers, R. Cayrol, M. Bernard, F. Giuliani, N. Arbour, B. Becher and A. Prat, Nat. Med., 2007, 13, 1173–1175 CrossRef CAS PubMed.
- C. S. Constantinescu, N. Farooqi, K. O'Brien and B. Gran, Br. J. Pharmacol., 2011, 164, 1079–1106 CrossRef CAS PubMed.
- B. Aube, S. A. Levesque, A. Pare, E. Chamma, H. Kebir, R. Gorina, M. A. Lecuyer, J. I. Alvarez, Y. De Koninck, B. Engelhardt, A. Prat, D. Cote and S. Lacroix, J. Immunol., 2014, 193, 2438–2454 CrossRef CAS PubMed.
- T. Aranami and T. Yamamura, Allergol. Int., 2008, 57, 115–120 CrossRef CAS PubMed.
- J. S. Tzartos, M. A. Friese, M. J. Craner, J. Palace, J. Newcombe, M. M. Esiri and L. Fugger, Am. J. Pathol., 2008, 172, 146–155 CrossRef CAS PubMed.
- W. Cao, Y. Q. Yang, Z. Y. Wang, A. L. Liu, L. Fang, F. L. Wu, J. Hong, Y. F. Shi, S. Leung, C. Dong and J. W. Z. Zhang, Immunity, 2011, 35, 273–284 CrossRef CAS PubMed.
- X. Wang, Y. H. Shen, S. W. Wang, S. L. Li, W. L. Zhang, X. F. Liu, L. H. Lai, J. F. Pei and H. L. Li, Nucleic Acids Res., 2017, 45, W356–W360 CrossRef CAS PubMed.
- M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin and B. K. Shoichet, Nat. Biotechnol., 2007, 25, 197–206 CrossRef CAS PubMed.
- J. Y. Gong, C. Q. Cai, X. F. Liu, X. Ku, H. L. Jiang, D. Q. Gao and H. L. Li, Bioinformatics, 2013, 29, 1827–1829 CrossRef CAS PubMed.
- D. Gfeller, A. Grosdidier, M. Wirth, A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2014, 42, W32–W38 CrossRef CAS PubMed.
- L. R. Wang, C. Ma, P. Wipf, H. B. Liu, W. W. Su and X. Q. Xie, AAPS J., 2013, 15, 395–406 CrossRef CAS PubMed.
- Z. J. Yao, J. Dong, Y. J. Che, M. F. Zhu, M. Wen, N. N. Wang, S. Wang, A. P. Lu and D. S. Cao, J. Comput.-Aided Mol. Des., 2016, 30, 413–424 CrossRef CAS PubMed.
- J. J. Wang, J. X. Zou, X. Q. Xue, D. M. Cai, Y. Zhang, Z. J. Duan, Q. P. Xiang, J. C. Yang, M. C. Louie, A. D. Borowsky, A. C. Gao, C. P. Evans, K. S. Lam, J. Z. Xu, H. J. Kung, R. M. Evans, Y. Xu and H. W. Chen, Nat. Med., 2016, 22, 488–496 CrossRef CAS PubMed.
- B. M. Hiebert, B. W. Janzen, R. M. Sanjanwala, A. D. Ong, R. D. Feldman and J. O. Kim, Br. J. Clin. Pharmacol., 2021, 87, 1801–1813 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods, figures, tables and data files. See DOI: 10.1039/d1sc05613a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |