
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promoting photocatalytic CO2 reduction through facile electronic modification of N-annulated perylene diimide rhenium bipyridine dyads†
Josh D. B.
Koenig
*,
Warren E.
Piers
 and
Gregory C.
Welch
*
and
Gregory C.
Welch
*
Department of Chemistry, University of Calgary, 2500 University Drive N.W., Calgary, Alberta T2N 1N4, Canada. E-mail: joshua.koenig1@ucalgary.ca; gregory.welch@ucalgary.ca
First published on 28th December 2021
Abstract
The development of CO2 conversion catalysts has become paramount in the effort to close the carbon loop. Herein, we report the synthesis, characterization, and photocatalytic CO2 reduction performance for a series of N-annulated perylene diimide (NPDI) tethered Re(bpy) supramolecular dyads [Re(bpy-C2-NPDI-R)], where R = –H, –Br, –CN, –NO2, –OPh, –NH2, or pyrrolidine (–NR2). The optoelectronic properties of these Re(bpy-C2-NPDI-R) dyads were heavily influenced by the nature of the R-group, resulting in significant differences in photocatalytic CO2 reduction performance. Although some R-groups (i.e. –Br and –OPh) did not influence the performance of CO2 photocatalysis (relative to –H; TONco ∼60), the use of an electron-withdrawing –CN was found to completely deactivate the catalyst (TONco < 1) while the use of an electron-donating –NH2 improved CO2 photocatalysis four-fold (TONco = 234). Despite being the strongest EWG, the –NO2 derivative exhibited good photocatalytic CO2 reduction abilities (TONco = 137). Using a combination of CV and UV-vis-nIR SEC, it was elucidated that the –NO2 derivative undergoes an in situ transformation to –NH2 under reducing conditions, thereby generating a more active catalyst that would account for the unexpected activity. A photocatalytic CO2 mechanism was proposed for these Re(bpy-C2-NPDI-R) dyads (based on molecular orbital descriptions), where it is rationalized that the photoexcitation pathway, as well as the electronic driving-force for NPDI2− to Re(bpy) electron-transfer both significantly influence photocatalytic CO2 reduction. These results help provide rational design principles for the future development of related supramolecular dyads.
Introduction
The adverse effects on climate change related to increased anthropogenic CO2 emissions has inspired the utilization of excess CO2 as a sustainable feedstock for value-added chemicals and fuels.1,2 While the activation of CO2 is kinetically unfavorable, it can be readily accomplished electro-/photocatalytically via proton-couple multielectron chemical reductions.3,4 Consequently, the development of capable molecular electro-/photocatalysts has mainly focused on improving the efficiency and selectivity of the CO2 conversion process.5–9 Among the many comprehensively studied molecular catalyst systems, Re(2,2′-bipyridine)(CO)3Cl [Re(bpy)] is notable for its highly selective CO2-to-CO conversion.10 The versatile bpy ligand has been modified with a variety of substituents to change both the electronic properties and/or the second-sphere H-bonding character of the catalyst.11–18 And while Re(bpy) alone can be used as an effective CO2 reduction photocatalyst,19–24 the photocatalytic CO2 reduction performance is greatly enhanced via the direct functionalization of Re(bpy) with photosensitizing (PS) units.25–31The development of ruthenium(II) diimine photosensitized Re(bpy) supramolecular dyads has been extensively reported by the Ishitani group.5,25–31 These RuII–ReI dyads make use of a Z-scheme architecture whereby the photoexcited electrons of the RuII-moiety are reductively quenched and subsequently transferred to the ReI catalyst center to enable CO2 reduction. To facilitate efficient electron-transfer (eT) and CO2 photocatalysis, several supra-molecular dyad design principles have been established. First, the photoexcited electron should be localized near the tethering portion between the PS and the catalyst.5 Second, the tether between the PS and catalyst moieties should be as short as possible (without being through-conjugated) to enable rapid intramolecular eT.27–30 Third, increasing the molar absorptivity of the PS-moiety (i.e. by incorporating multiple PS units) can improve the quantum efficiency capabilities and the ensuing eT dynamics of the supramolecular dyad.31 Restricted by the first two design principles, attempts to improve the quantum efficiency of these Re(bpy) dyads has been made by using more strongly absorbing PS units, such as porphyrins,32–37 naphthalimide,38,39 naphthalene diimide,40–42 and perylene diimide (PDI).43,44 Although the photophysical dynamics of these dyads appear fundamentally well-understood, only a handful have been properly evaluated as CO2 reduction photocatalysts (see ESI, Table S1†).35–39
Recently, we reported on four N-annulated perylene diimide (NPDI) functionalized Re(bpy) dyads as CO2 reduction electro-catalysts.45 Our investigation of these Re(bpy)–NPDI dyads revealed that the PS unit (NPDI) functions as an electron-reservoir for Re(bpy), enabling efficient CO2 reduction at an overpotential 300 mV lower than conventional Re(bpy)-type electrocatalysts. Moreover, it was also elucidated that the tether length between Re(bpy) and NPDI governs which CO2 reduction mechanism is preferred for the supramolecular dyad(s), where the ethyl-linked Re(bpy)–NPDI dyad possessed the greatest degree of electronic communication. These promising results from our initial Re(bpy)–NPDI dyads led us to hypothesize that eT from the electron-reservoir to the Re(bpy) catalyst could be improved by electronically modifying NPDI in two different ways. It was theorized that the introduction of electron withdrawing groups (EWGs) on NPDI may inductively stabilize the entire dyad, thus enabling more efficient eT by increasing the overall electron affinity of Re(bpy)-moiety. Alternatively, the use of electron donating groups (EDGs) on NPDI could make the electron-reservoir more electron-rich and thus more willing to transfer electrons to the Re(bpy)-moiety. To determine which of these two opposing hypotheses was correct, a series of electronically modified ethyl-linked Re(bpy)–NPDI dyads [Re(bpy-C2-NPDI-R)] (where R = –H, –Br, –CN, –NO2, –OPh, –NH2, or –NR2) were designed (Fig. 1, left) and, for the first time, their photocatalytic CO2-to-CO reduction performance was evaluated. It was revealed that installing an EDG, such as –NH2, led to a four-fold enhancement in turnover numbers of CO (TONco), with respect to the benchmark Re(bpy-C2-NPDI-H) dyad. A mechanism based on molecular orbital (MO) energy levels is proposed to explain the observed differences in photocatalytic CO2 reduction performance for these dyads caused by the installation of EWGs and EDGs on NPDI.
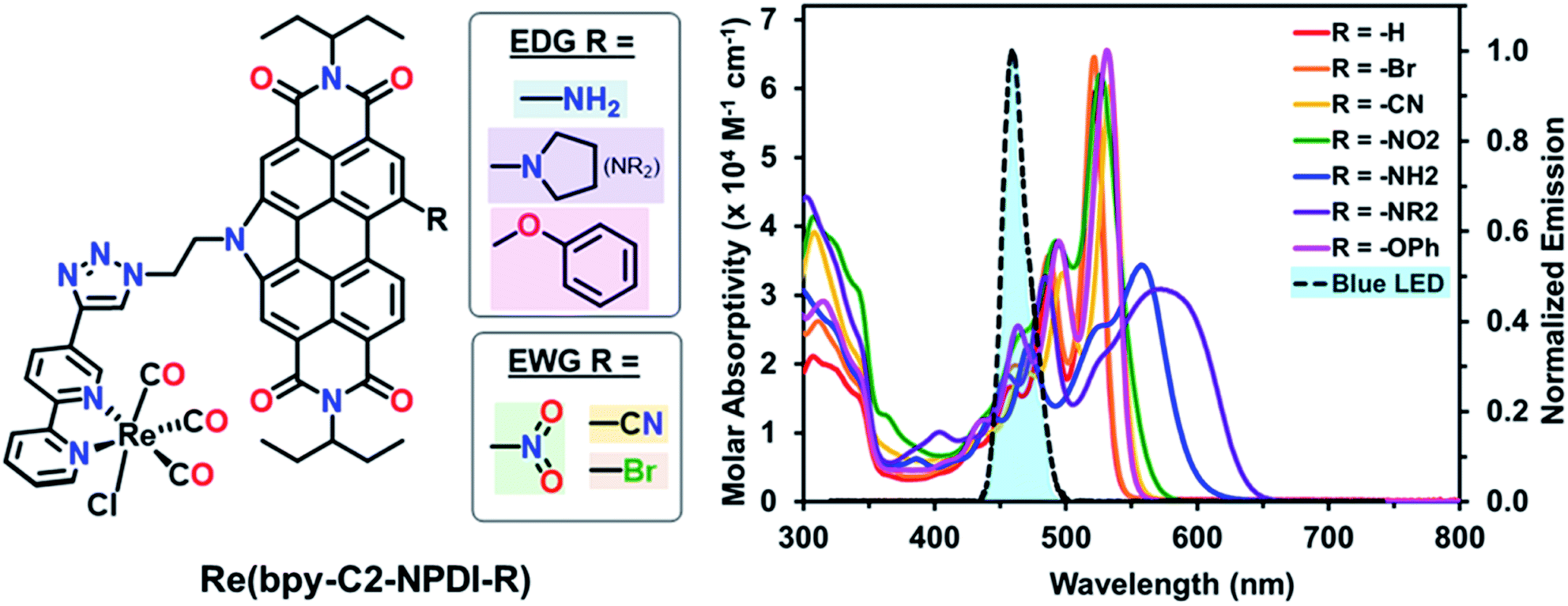 | ||
| Fig. 1 (Left) chemical structures and (right) UV-vis absorption spectra of Re(bpy-C2-NPDI-R) dyads in CHCl3 (∼10−5 M), where R = –H (red), R = –Br (orange), R = –CN (yellow), R = –NO2 (green), R = –NH2 (blue), R = –NR2 (purple), and R = –OPh (magenta). The optical profiles of all Re(bpy-C2-NPDI-R) dyads overlap with the emission spectrum of the blue LED (λ = 470 ± 30 nm; 4 mW cm−2) used for photocatalytic CO2 reduction testing. | ||
Results & discussion
Synthesis & characterization
The synthesis of all azide-ethyl-NPDI precursors (N3-C2-NPDI-R) starts from the HNPDI synthon (Scheme 1).47–49 The pyrrolic nitrogen was alkylated with 1,2-dibromoethane and a terminal azide was installed using SN2 chemistry to give N3-C2-NPDI-H.45,47 Treatment of N3-C2-NPDI-H with either fuming HNO3 (at −78 °C) or Br2 (at 20 °C) affords the N3-C2-NPDI-NO2 and N3-C2-NPDI-Br precursors, respectively. When refluxed with pyrrolidine, the N3-C2-NPDI-Br precursor can be converted to N3-C2-NPDI-NR2. Alternatively, reacting N3-C2-NPDI-Br with an excess of phenol and K2CO3 in N,N-dimethylformamide (DMF; at 80 °C) generates N3-C2-NPDI-OPh. Attempts to cyano-functionalize N3-C2-NPDI using the Rosenmund–von Braun reaction conditions resulted in the elimination of the terminal azide. Consequently, N3-C2-NPDI-CN had to be synthesized starting from HNPDI. After selectively brominating HNPDI,50 HNPDI-Br was reacted with excess CuCN in refluxing DMF to yield HNPDI-CN. This intermediate was then alkylated and azide-functionalized to give the N3-C2-NPDI-CN precursor.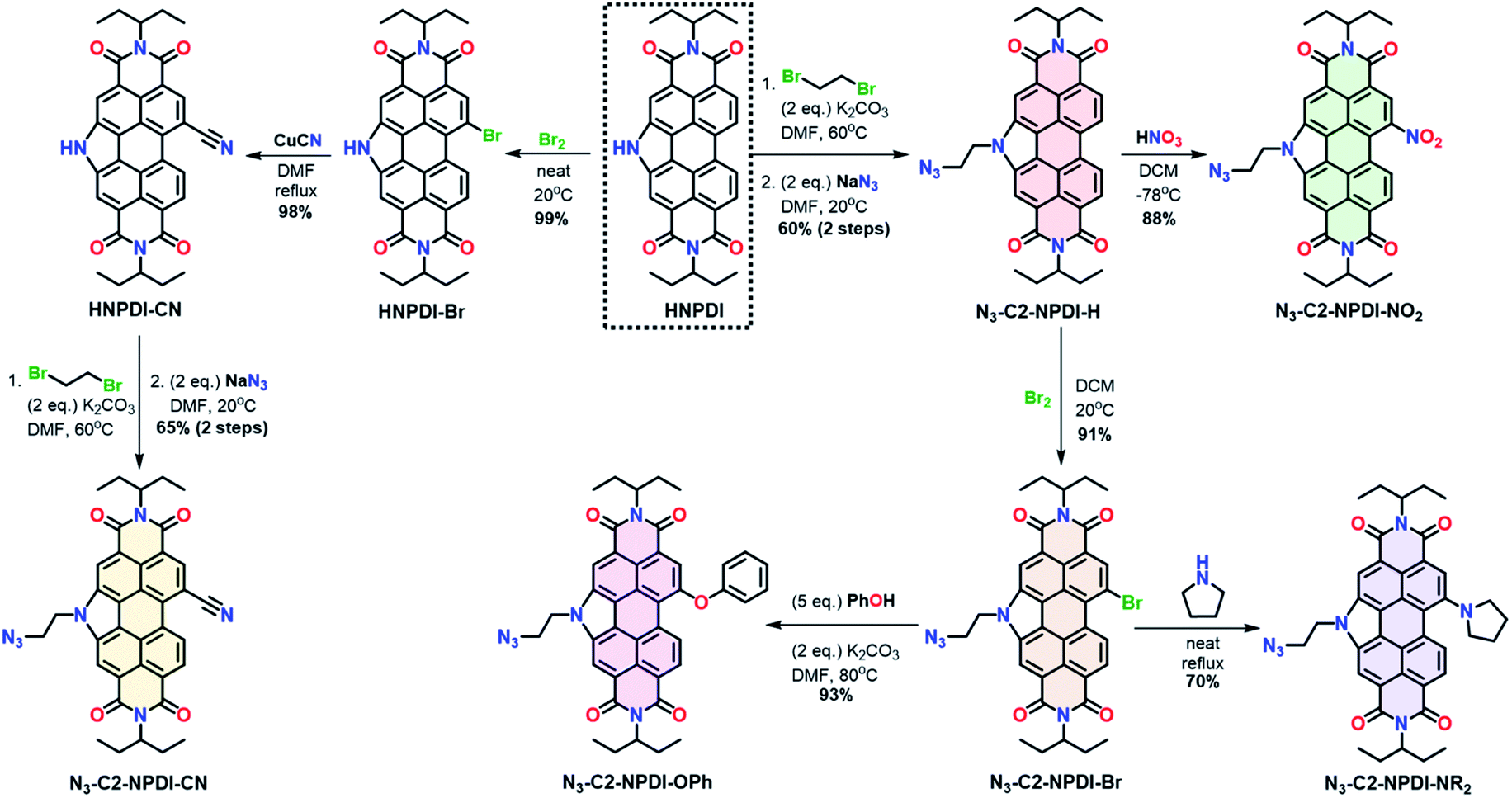 | ||
| Scheme 1 Synthesis of N3-C2-NPDI-R precursors that are converted into Re(bpy-C2-NPDI-R) dyads. | ||
Next, these six N3-C2-NPDI-R precursors were linked to Re(5-ethynyl-2,2′-bipyridine)(CO)3Cl [Re(ethynyl-bpy)] using a standard copper catalyzed azide–alkyne cycloaddition (CuAAC) procedure (see ESI Section II for more details†).45,47 While most Re(bpy-C2-NPDI-R) dyads (where R = –H, –Br, –CN, –NR2, and –OPh) were obtained in excellent yields as the exclusive product, the CuAAC reaction between N3-C2-NPDI-NO2 and Re(ethynyl-bpy) generated both Re(bpy-C2-NPDI-NO2) (major product) and Re(bpy-C2-NPDI-NH2) (minor product). To obtain each catalyst selectively, two additional CuAAC protocols were developed. The selective synthesis of Re(bpy-C2-NPDI-NO2) was accomplished by changing the catalyst from CuSO4 (with sodium ascorbate) to CuI, suggesting that the substoichiometric sodium ascorbate used under our standard CuAAC conditions acted as a reducing agent.51–53 With this insight, Re(bpy-C2-NPDI-NH2) was afforded exclusively using catalytic CuSO4 and a stoichiometric excess of sodium ascorbate. The identity of all Re(bpy-C2-NPDI-R) dyads was confirmed using 1H and 13C NMR spectroscopies, as well as MALDI-TOF mass spectrometry and CHN elemental analysis (Fig. S1–S54†). Note, each specific Re(bpy-C2-NPDI-R) dyad will henceforth be referred to by their R-group only (where R = –H, –Br, –CN, –NO2, –NH2, –NR2, and –OPh).
The optical properties of these Re(bpy-C2-NPDI-R) dyads were probed using both UV-visible-near-infrared (UV-vis-nIR) absorption and Fourier transform infrared (FTIR) spectroscopies. Visually, the optical properties of all Re(bpy-C2-NPDI-R) dyads were dominated by the NPDI-moiety (Fig. 1 and S55–S56†). Relative to –H (λmax = 521), the installation of –Br, –CN, –NO2, and –OPh groups on NPDI all caused a minor bathochromic shift of the absorption profile (Δλmax = 1–13 nm) while maintaining most of the vibronic transitions of the parent complex. As for the –NH2 and –NR2 dyads, the optical spectra were significantly red-shifted (λmax = 558 and 572 nm, respectively) and the molar absorptivity also decreased (from ∼60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to ∼40000 M−1 cm−1). The broadened absorption of –NH2 and –NR2 near the λmax is consistent with the donation of electron-density from the lone-pair of the amino nitrogen into the NPDI π-system.54–56 Notably, all dyads exhibit an absorption peak around λ = 341–343 nm, characteristic of a Re(bpy)-based metal-to-ligand charge transfer band.57 By FTIR, all Re(bpy-C2-NPDI-R) dyads possessed three carbonyl stretching frequencies (vco ∼1900, 1925, and 2025 cm−1), which is consistent with other Re(bpy) catalysts (Fig. S57†).11–18,58 Together, these data confirm successful Re(bpy-C2-NPDI-R) dyad formation.
000 to ∼40000 M−1 cm−1). The broadened absorption of –NH2 and –NR2 near the λmax is consistent with the donation of electron-density from the lone-pair of the amino nitrogen into the NPDI π-system.54–56 Notably, all dyads exhibit an absorption peak around λ = 341–343 nm, characteristic of a Re(bpy)-based metal-to-ligand charge transfer band.57 By FTIR, all Re(bpy-C2-NPDI-R) dyads possessed three carbonyl stretching frequencies (vco ∼1900, 1925, and 2025 cm−1), which is consistent with other Re(bpy) catalysts (Fig. S57†).11–18,58 Together, these data confirm successful Re(bpy-C2-NPDI-R) dyad formation.
The electrochemical properties of these Re(bpy-C2-NPDI-R) dyads were next evaluated using cyclic voltammetry (CV). CV analysis was first performed in CH2Cl2 (Fig. 2A and S65†), with all reported redox events being referenced to the Fc+/0 internal standard. Under an atmosphere of argon, all Re(bpy-C2-NPDI-R) dyads exhibited four reduction and two oxidation redox processes. The first two reversible reductions may be assigned to the NPDI˙−/0 and NPDI2−/˙− redox couples while the third and fourth reductions correspond to the quasi-reversible bpy˙−/0 and the irreversible Re0/I redox events, respectively.11–18,45 With respect to the oxidation events, the irreversible ReII/I redox process remains consistently near Ep ≈ +1.0 V. The quasi-reversible NPDI-based oxidation event, on the other hand, underwent dramatic shifts in potential depending on the nature of the electronic substituent.
Based on the presented CV data (Fig. 2A, S64 and S65†), it was observed that the installation of EWGs (i.e. –Br, –CN, and –NO2) on NPDI caused both the redox events assigned to the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) energy levels of NPDI to shift to more positive potentials. In all cases, the electronic bandgap was narrowed because the LUMO energy level was more significantly perturbed than the HOMO energy level. Conversely, the installation of EDGs (i.e. –OPh, –NH2, and –NR2) on NPDI caused the redox events associated with NPDI HOMO and LUMO energy levels to shift to more negative potentials. Once again, the electronic bandgap was decreased mainly due to the more substantial effects experienced by the HOMO energy level. The observed shifts in E1/2 for first NPDI-R based reduction and oxidation events (Fig. 2B and C), relative to –H, show good Hammett parameter correlation.46 Similar correlations were previously reported for electronically-substituted Re(4,4′-R-bpy) complexes by Kubiak et al.,11 where EWGs shifted redox events more positively and EDGs shifted redox events more negatively. In principle, combining these two relationships could assist with proper energy level matching when designing future supramolecular dyads based on the Re(bpy-C2-NPDI-R) architecture.
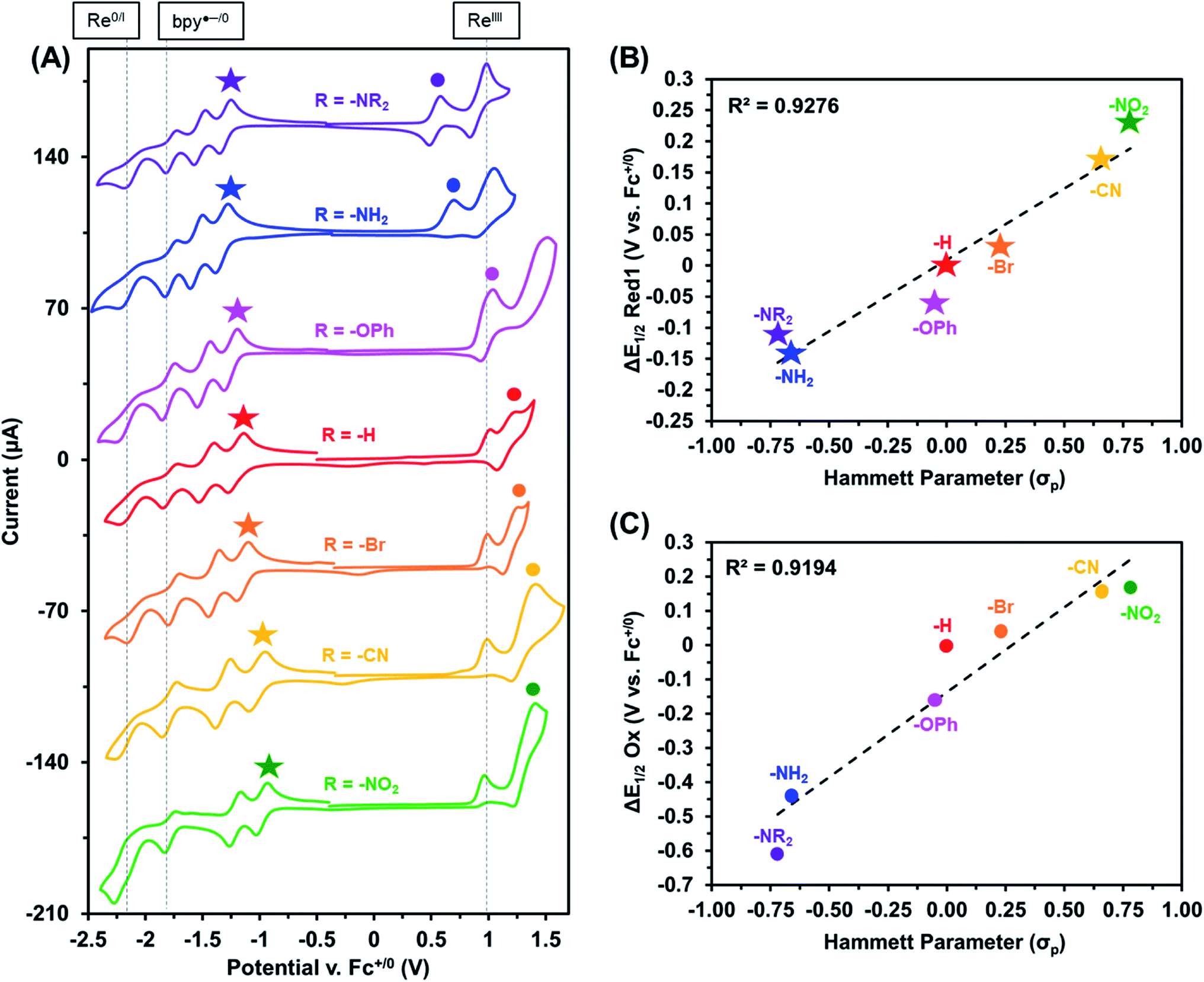 | ||
| Fig. 2 Cyclic voltammograms (A) of Re(bpy-C2-NPDI-R) dyads where R = –NR2 (purple), –NH2 (blue), –OPh (magenta), –H (red), –Br (orange), –CN (yellow), and –NO2 (green). All measurements were recorded at 100 mV s−1, under argon in CH2Cl2 with 0.1 M TBAPF6 supporting electrolyte (WE = glassy carbon, CE = Pt-wire, RE = Ag/AgCl, and Fc+/0 as internal reference standard). Correlation diagrams plot the observed shifts in E1/2 (relative to R = –H) for the first reduction (B) and oxidation (C) as a function of R-group Hammett parameter.46 | ||
Next, the electrochemistry of these Re(bpy-C2-NPDI-R) dyads were assessed in DMF, where only the reduction processes were measured due to solvent window effects.59 Under argon, all Re(bpy-C2-NPDI-R) dyads exhibited the same four previously assigned reduction events (vide supra), where solvent effects caused the potential of most redox events to shift positively by ∼50–100 mV, relative to CH2Cl2 (Fig. S66†).60 Upon measuring CVs at variable scan rates, each dyad displayed a diffusion-limited current response when fitted to the Randles–Sevcik equation (see ESI, eqn (i)†), with a calculated diffusion coefficient between D = 2.3–4.4 × 10−6 cm2 s−1 (Fig. S69–S74†). Upon subjecting these Re(bpy-C2-NPDI-R) dyads to an atmosphere of CO2 (in DMF), a moderate CV current enhancement was observed underneath the fourth redox couple (Fig. 3, red traces with Ecat/2 ≈ −2.1 V). When a catalyst is supplied with the appropriate combination of substrates at a sufficient applied potential, a CV current enhancement is often observed as a result of initiating an electron-consuming catalytic process.61,62 Moreover, these CV current enhancements may be monitored as a function of catalyst or proton-source concentration to gather preliminary mechanistic data on the catalytic process (see ESI, eqn (ii)†).63,64
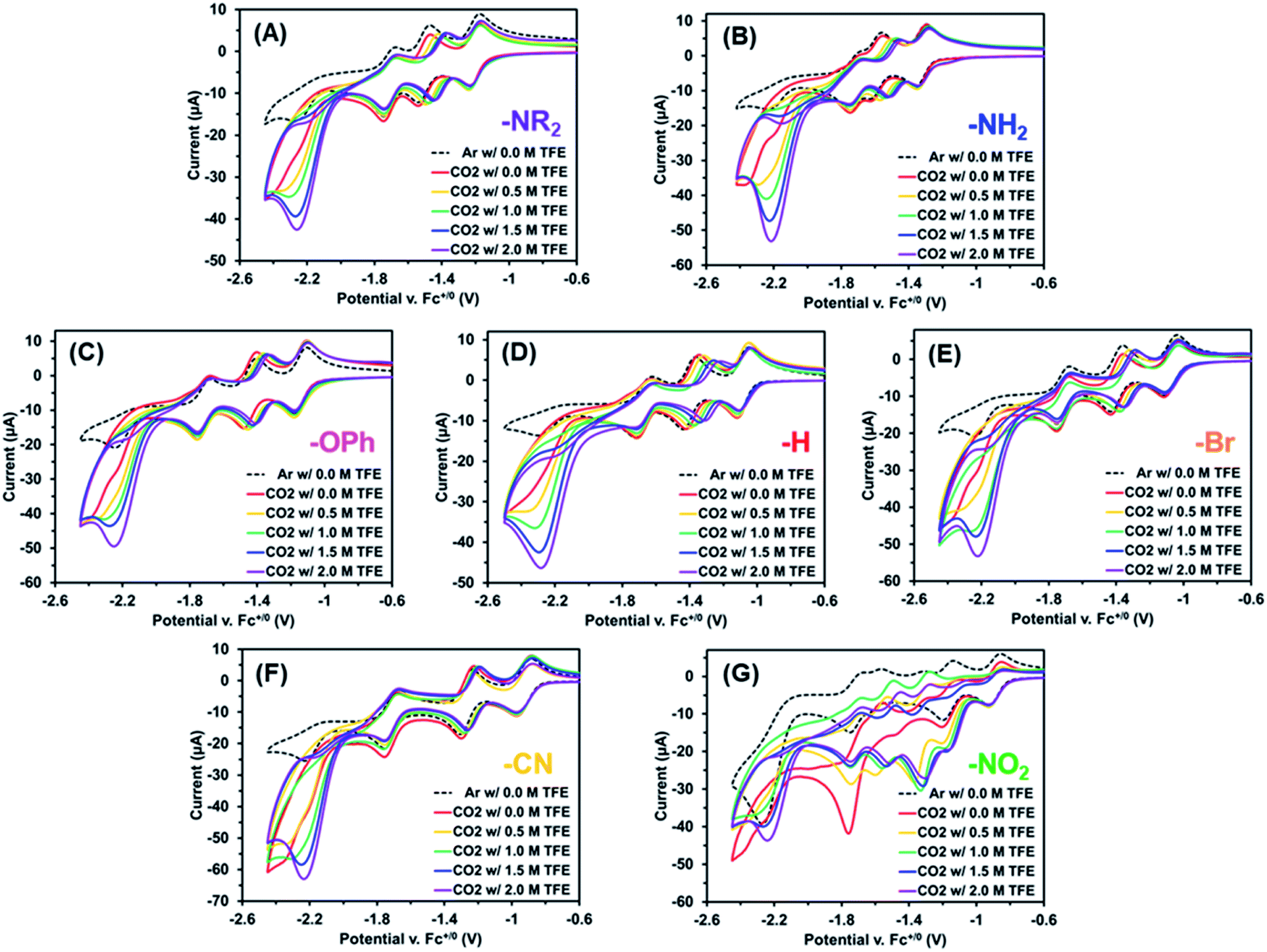 | ||
| Fig. 3 CV current enhancement plots as a function of 2,2,2-trifluoroethanol (TFE) concentration for Re(bpy-C2-NPDI-R) derivatives, where R = –NR2 (A), R = –NH2 (B), R = –OPh (C), R = –H (D), R = –Br (E), R = –CN (F), and R = –NO2 (G). TFE was incrementally added at 0 M (red), 0.5 M (yellow), 1.0 M (green), 1.5 M (blue), and 2 M (purple). All measurements were recorded at 100 mV s−1, under an atmosphere of CO2, in DMF with 0.1 M TBAPF6 supporting electrolyte (WE = glassy carbon, CE = Pt-wire, RE = Ag/AgCl, and Fc+/0 as internal reference standard). Note, the observed current enhancement at Ep = −1.35 V (vs. Fc+/0) for R = –NO2 (G) results from an in situ electrochemical conversion to R = –NH2. | ||
When the Re(bpy-C2-NPDI-R) concentration was varied, a linear CV current enhancement response was also observed for most dyads (Fig. S68†). Similarly, when proton-source 2,2,2-trifluoroethanol (TFE) was incrementally added to a CO2-saturated Re(bpy-C2-NPDI-R) dyad solution, it induced a CV current enhancement increase under the fourth reduction event (Fig. 3). We further note that incremental addition of TFE also caused the NPDI2−/˙− redox couple to gradually shift to more positive potentials. This behavior has been modeled for similar rylene diimide materials as a 2H+/2e− proton-coupled electron-transfer process,65 whereby the NPDI imide oxygens are protonated by the proton-source.66,67 While the collected CV data was not obtained under steady-state conditions (i.e. plateau current),68 modelling these measured CV current enhancements as CO2 reduction variables can qualitatively describe the effects of catalyst and proton-source as first-order and second-order rate-dependent variables, respectively. In other words, these data imply that one Re(bpy-C2-NPDI-R) dyad, with the assistance of two proton-source molecules, can enable electrocatalytic CO2 conversion.45 The obvious exception to these generalized electrochemical trends is the –NO2 dyad (Fig. 3G). An in-depth evaluation of the electrochemical behaviour of –NO2 under argon and CO2 is provided in the ESI (see Section VIII, Fig. S84–S86†). Thorough analysis of this data strongly suggests that –NO2 is converted in situ to –NH2 under reducing conditions; as such, it is difficult to establish meaningful trends for –NO2 (vide infra).
CO2 electro-/photocatalysis
The electrocatalytic CO2 reduction abilities of these Re(bpy-C2-NPDI-R) dyads were evaluated using controlled potential electrolysis (CPE). All experiments were performed in DMF (with 2 M TFE) using our previously described two-compartment H-cell.45,69 At an applied potential (Eappl) of −1.8 V (Fig. S75†), all Re(bpy-C2-NPDI-R) dyads (except –CN) achieved comparable turnover numbers of CO (TONco = 21–25) and faradaic efficiencies (FEco = 87–99%) after 6 hours of electrocatalysis. The –CN derivative, on the other hand, attained about half the performance (TONco = 13) at a slightly lower FEco (87%). This drop in performance is consistent with decreased efficacy of the electron-reservoir effect due to the increased electron-affinity of NPDI-CN (Fig. S77†). Unlike the electronically modified Re(4,4′-R-bpy) series reported by Kubiak et al., where electrocatalytic CO2-to-CO conversion efficiency was highly dependent on the nature of the R-group,11 all Re(bpy-C2-NPDI-R) dyads achieved high FEco at an overpotential that is ∼300 mV lower than the measured Ecat/2 ≈ −2.1 V (Fig. S67†). It should be noted, however, that when an Eappl = −1.7 V was used for CPE (Fig. S76†), electrocatalytic CO2 reduction was essentially shut-off for all Re(bpy-C2-NPDI-R) dyads (TONco ≤ 6). This could indicate that altering the electronic properties of the electron-reservoir may not be the most effective strategy towards further lowering the overpotentials required to enable electrocatalytic CO2 reduction.In our previous report, we noticed that the use of blue light during CPE experiments significantly increased the rates of CO production for –H.45 To determine the origin of this TONco enhancement,28,70,71 we next turned our attention towards the photocatalytic CO2 reduction capabilities of these Re(bpy-C2-NPDI-R) dyads. Following established literature protocols,31,34–37 each dyad (30 μM) was dissolved in a (5:1) DMF: triethanolamine (TEOA) mixture containing sacrificial reducing agent 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH; 3 mM). The glass vials were sealed with rubber septa, sparged with CO2, and then irradiated with blue light (λ = 470 ± 30 nm; 4 mW cm−2) for 24 hours (Table 1).
|
|
|||
|---|---|---|---|
| R = | TONCOa | TONH2a | CO:H2b |
| a Calculated based on bulk catalyst concentration from quadruplicate trials. b No other gaseous (i.e. CH4) or liquid (i.e. HCOO–) products were detected. c Re(bpy-C2-NPDI-NO2) is in situ converted to Re(bpy-C2-NPDI-NH2). | |||
| –H | 57 ± 1 | 1.8 ± 1 | 97:3 |
| –CN | <1 | <1 | — |
| –NO2c | 137 ± 15 | 3 ± 0.6 | 98:2 |
| –Br | 61 ± 5 | 2.5 ± 1 | 96:4 |
| –NR2 | 86 ± 8 | 1.4 ± 1 | 98:2 |
| –OPh | 59 ± 6 | 3.6 ± 2 | 95:5 |
| –NH2 | 234 ± 13 | 2.0 ± 0.1 | >99:1 |

Over the 24 h testing period, the Re(bpy-C2-NPDI-R) dyads all showed good activity for ∼9 hours, after which CO production would level-off for the remainder of the experiment (Fig. S78†). The benchmark dyad, –H, achieved a TONco of 57 ± 1 with a selectivity for CO of 97%. The –Br (TONco = 61 ± 5) and –OPh (TONco = 59 ± 6) dyads achieved the same performance with roughly the same CO selectivity (≥95%). The –CN derivative was completely inactive for CO2 photocatalysis under these conditions. Interestingly, despite being the most EWG, the –NO2 dyad achieved the second best TONco (134 ± 15) with a very high CO selectivity of 98%. When EDGs were functionalized on NPDI, the CO2-to-CO production and selectivity was improved to (TONco = 86 ± 8) for –NR2 and (TONco = 234 ± 13) for –NH2. We note that the photocatalytic performance of –NH2 was not greatly improved over the 24 h testing period by replenishing both BIH and CO2 in 6 h intervals (TONco = 294; Fig. S79†), suggesting that depletion of substrate was not the limiting factor for TONco.
To confirm the importance of each component in the photocatalytic CO2 reduction setup, various control experiments were conducted. As expected, the omission of Re(bpy-C2-NPDI-R) dyads or CO2 from the setup stopped the production of CO. When sacrificial reducing agent BIH was excluded (Table S2†), the TONco was decreased by at least two-fold for all dyads. This result implies that while TEOA alone can simultaneously act as the proton-source and the sacrificial electron-donor,20 BIH is more efficient at reductively quenching the photoexcited dyads. When TEOA was replaced by TFE (Table S3†), the production of CO for all dyads was decreased almost four-fold, except for –NH2 (TONco = 143). Overall, this result points towards the utility of TEOA as a sacrificial electron-donor, as well as the importance of forming Re(bpy)-adducts during CO2 reduction, as seen in previous literature examples.5,26–31 When the irradiation source was switched to a green light LED array (λ = 525 ± 32 nm; 1.9 mW cm−2), the measured TONco decreased at least five-fold for all Re(bpy-C2-NPDI-R) dyads (Table S4†). This significant drop in performance could be the result of either inefficient photoexcitation pathways in the dyad40–44 and/or the elimination of a photo-assisted CO cleavage process.70,71 Lastly, the importance of tethering the NPDI-moiety to the Re(bpy)-moiety was demonstrated by combining N3-C2-NPDI-R with Re(bpy), where it was observed that all samples obtained the same performance as Re(bpy) alone (Table S5†). We further showed that all N3-C2-NPDI-R precursors were essentially inactive for CO2 conversion under the optimized photocatalysis conditions (TONco < 3; Table S6†).
While these control experiments clearly highlight the necessity of each component in the photocatalytic CO2 reduction process, it does not fully account for the performance differences of each Re(bpy-C2-NPDI-R) dyad. Therefore, based on the photocatalytic CO2 reduction results, the Re(bpy-C2-NPDI-R) dyads may be grouped together in three categories: (i) standard catalysts (weak EWGs/EDGs = –H, –Br, and –OPh), (ii) inactive catalysts (strong EWGs = –CN), and (iii) top-performing catalysts (strong EDGs = –NR2 and –NH2). Note, the –NO2 dyad can also be classified as a top-performing catalyst because –NO2 undergoes an in situ transition to –NH2 under photocatalytic CO2 reduction conditions (see ESI Section VIII for more details†).
Mechanistic investigation
During electro-/photocatalytic CO2 reduction testing of these Re(bpy-C2-NPDI-R) dyads, a series of dramatic colour changes were observed. Prior to light irradiation, the absorption profile of all Re(bpy-C2-NPDI-R) dyads were unchanged by the addition of both TEOA and BIH, confirming that neither reagent reduces Re(bpy-C2-NPDI-R) immediately. After sparging with CO2, the samples were then subjected to blue light and the progression of colour changes was monitored periodically by UV-vis-nIR spectroscopy (Fig. S80†). Each Re(bpy-C2-NPDI-R) dyad underwent a transition from their initial colour to either a green (–H, –Br, and –OPh), a dark blue (–CN), or a beige (–NO2, –NR2, and –NH2) colour that was found to persist throughout the remainder of catalysis. Photoluminescence spectroscopy revealed that the photoexcited state of all Re(bpy-C2-NPDI-R) dyads can be reductively quenched by both TEOA and BIH (Fig. S81†). Thus, to gain further insight into the photoelectrochemical processes that were occurring during CO2 reduction catalysis, UV-vis-nIR and FTIR spectroelectrochemistry (SEC) experimentation was conducted. UV-vis-nIR and FTIR SEC data was collected by monitoring an air-free Re(bpy-C2-NPDI-R) dyad solution that was held at a constant Eappl (where Red1, Red2, and Red3 correspond to the NPDI˙−/0, NPDI2−/˙−, and bpy˙−/0 reductions, respectively).The UV-vis-nIR SEC data of each Re(bpy-C2-NPDI-R) dyad (Fig. 4) correlates very well with what was observed when we periodically monitored our photocatalytic experiments. When using an Eappl = Red1, the λmax of all Re(bpy-C2-NPDI-R) dyads was bathochromically shifted, the relative molar absorptivity of this λmax was stronger, and some new vibrational fine-structure was also observed between 800–1000 nm. While the nature of installed R-group influences the position of the λmax and the absorption fine-structure, all these absorption features are consistent with selective formation of NPDI˙−.43,44 When the Eappl is switched to Red2, the spectral features of NPDI˙− are rapidly depleted and replaced by shifted broad-band absorption peak(s). These spectral features are commonly associated with NPDI2−,67 where the vibronic structure of the NPDI2− absorption profile is once again influenced by the nature of installed R-group. With respect to –H (Fig. 4D), the incorporation of HOMO-modifying EDGs (i.e. –NR2, –NH2, and –OPh) resulted in minimal changes to the NPDI2− absorption profile shape (Fig. 4A–C). Conversely, when LUMO-modifying EWGs (i.e. –Br and –CN) are used, the vibronic structure of NPDI2− is significantly different (Fig. 4E and F). When the Eappl was changed to Red3, no other significant spectral changes were observed. This result is unsurprising given the differences in molar absorptivity of the NPDI and Re(bpy)-moieties.57,72 Except for –NO2 (Fig. 4G), all Re(bpy-C2-NPDI-R) dyads displayed very similar spectral transitions. In-depth analysis of the UV-vis-nIR SEC data for –NO2 (Fig. S86†) shows that –NO2 is in situ converted to –NH2 under the conditions necessary for CO2 reduction catalysis. These results help confirm why –NO2 served as an efficient CO2 reduction photocatalyst despite having a stronger EWG than the totally inactive –CN dyad derivative.
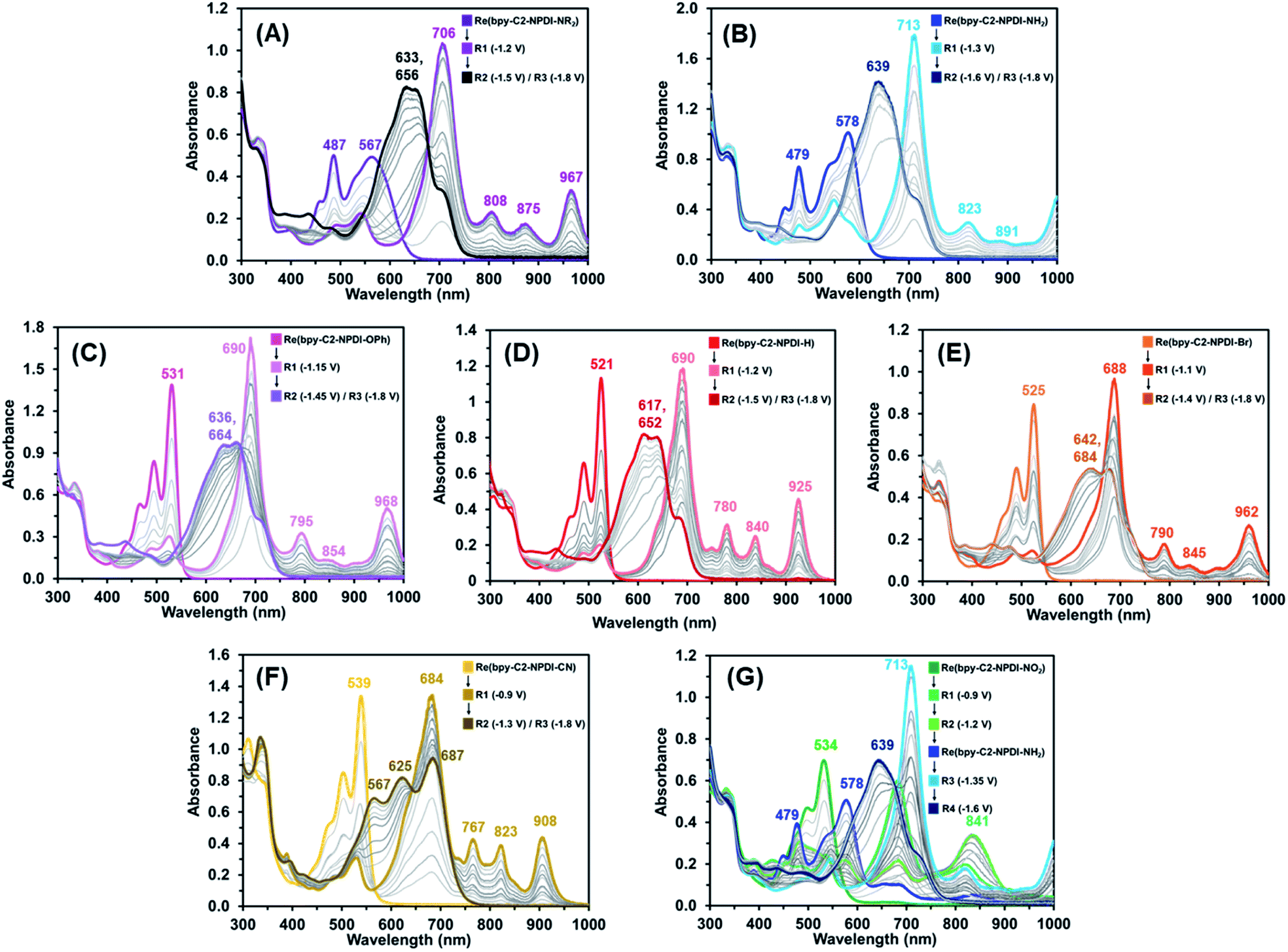 | ||
| Fig. 4 UV-vis-nIR SEC absorbance spectra of Re(bpy-C2-NPDI-R) derivatives, where R = –NR2 (A), R = –NH2 (B), R = –OPh (C), R = –H (D), R = -Br (E), R = –CN (F), and R = –NO2 (G). All experiments were performed in DMF with 0.1 M TBAPF6 supporting electrolyte (WE = Pt-mesh, CE = Pt-wire, pseudo-RE = Ag-wire). | ||
Moving onto the FTIR SEC data, the behavior of all Re(bpy-C2-NPDI-R) dyads were nearly identical (Fig. S58–S63†). While no spectral changes were detected at Eappl = Red1 and Red2, significant shifts in vco were observed at Eappl = Red3. At Red3, the Re(bpy)-moiety of these dyads is formally reduced by one-electron [ReI(bpy˙−-C2-NPDI2−-R)]. The added electron density at the Re(bpy)- moiety results in a lowering of vco from 1895, 1915, and 2019 cm−1 to roughly 1865, 1885, and 1995 cm−1, respectively. Over time, an equilibration process occurs whereby electron-density is shifted from bpy˙− to Re, and leads to Re–Cl dissociation [Re0(bpy-C2-NPDI2−-R)].58 This crucial process generates a 5-coordinate Re metal-center and can be characterized by a Δvco to 1843, 1862, and 1978 cm−1. Although this Cl-dissociation process was more readily observed for dyads bearing EWGs (–CN, –NO2, and –Br) than it was for dyads bearing EDGs (–NR2, –NH2, and –OPh), it was still detected to some degree for all Re(bpy-C2-NPDI-R) catalysts.
The similarities between our SEC data and the results reported for other Re(bpy) and RuII–ReI catalysts suggests that our Re(bpy-C2-NPDI-R) systems likely operate via similar photocatalytic CO2 reduction mechanisms.5,25–31 It should be noted, however, that the photocatalytic CO2 reduction mechanism for Re(bpy) is still heavily debated.18–31,73 As such, the goal of our proposed mechanism was not to precisely determine the exact identity, electron-spin configuration (singlet vs. triplet), and/or the rate dynamics of eT for all photocatalytic CO2 reduction intermediates. Instead, we focus on developing a molecular orbital (MO) description of these Re(bpy-C2-NPDI-R) dyads that helps account for the observed differences in photocatalytic CO2 reduction performance (Fig. 5). The presented mechanism was modeled after the benchmark dyad, –H.
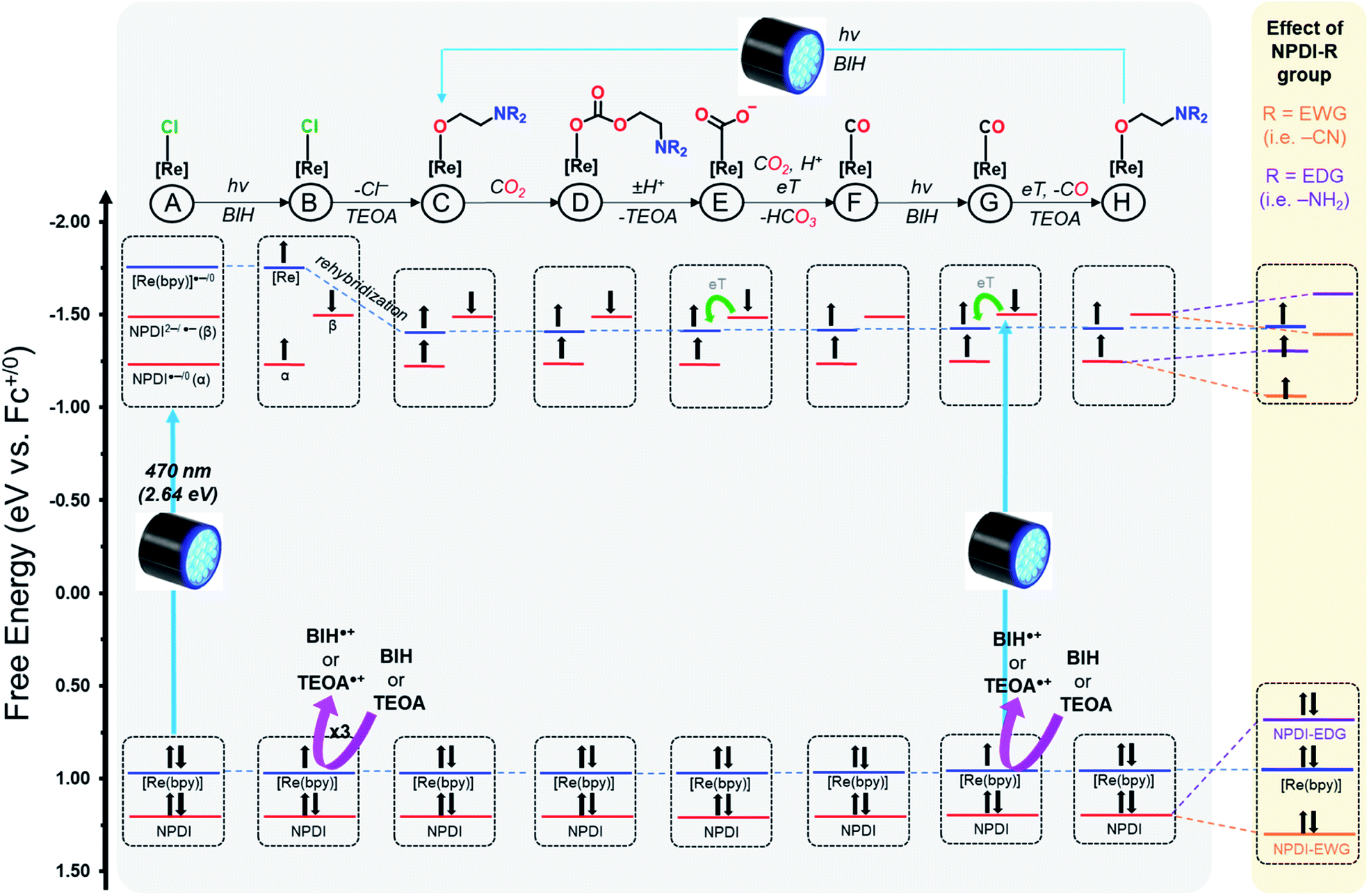 | ||
| Fig. 5 Molecular orbital description for the proposed photocatalytic CO2 reduction mechanism of Re(bpy-C2-NPDI-R), where Re(bpy) orbitals are shown in blue and NPDI-R orbitals are shown in red. The influence of electron withdrawing groups (EWGs, orange) and electron donating groups (EDGs, purple) on NPDI-R molecular orbitals is highlighted (right). Both BIH and TEOA are depicted as sacrificial electron donors that reductively quench the photoexcited electrons. | ||
By CV, it was elucidated for all Re(bpy-C2-NPDI-R) dyads (except –NH2 and –NR2) that the HOMO is Re(bpy)-based and the LUMO is NPDI-based (Fig. 5A). Consequently, the first two photoreductions most likely result from a Re–π* intersystem crossing (ISC) process; however, a direct NPDI-based π–π* transition may also be possible.43,44 The third photoreduction likely occurs either via direct Re(bpy) 3MLCT19 or eT from a photoexcited state of NPDI2−.43,44 Following the formal three electron reduction of the Re(bpy-C2-NPDI-R) dyads (Fig. 5B), the next step is Re–Cl dissociation which generates a 5-coordinate Re metal-center whose axial position subsequently forms an adduct with TEOA.20,74 Another consequence of the chloro-dissociation step is that the Re metal-center undergoes a rehybridization process that lowers the overall energy of the Re(bpy)-based MOs.45,75 Following the formation of the TEOA-Re(bpy) adduct, it is possible for CO2 insertion to occur without direct eT from the catalyst center (Fig. 5D).20,27–30 Protonation of the resulting carbonate intermediate induces a reorganization process that releases TEOA and forms a Re-CO2˙− species (Fig. 5E).22 Due to the presence of 13CO and H13CO3− in the 13C{1H} NMR spectrum after blue light irradiation (Fi. S83†), it is postulated that these Re(bpy-C2-NPDI-R) dyads operate via a BIH-mediated disproportionation reaction between Re–CO2˙− and another equivalent of CO2 that liberates HCO3−,25 rather than a proton-coupled electron transfer process from NPDI2− to the Re(bpy)-moiety that liberates OH−.18 From there, reductive quenching of a photoexcited electron restores NPDI2− (Fig. 5G) and the ensuing transfer of this electron to the Re(bpy)-moiety produces CO, as well as opens a coordination site for TEOA (Fig. 5H).21 The photocatalytic cycle is completed by the photoexcitation and reductive quenching of the electron to regenerate NPDI2−.
Based on photocatalytic CO2 reduction performance, the catalysts were loosely grouped into three categories: (i) standard catalysts (–H, –Br, and –OPh), (ii) inactive catalysts (–CN), and (iii) top-performing catalysts (–NR2 and –NH2). Looking at the proposed photocatalytic CO2 reduction mechanism, it is also possible to map out the effects of EWGs and EDGs on the provided MO description of these Re(bpy-C2-NPDI-R) dyads (Fig. 5, highlighted in yellow). In the case of –Br and –OPh, the overall influence of these R-group does not appear to change the eT dynamics of the Re(bpy-C2-NPDI-R) dyad (with respect to –H). The HOMO–LUMO transition (Re–π*) is identical and the relative shift(s) of the NPDI2− energy level does not significantly alter the electronic driving-force of eT between NPDI2− and Re(bpy). In the case of –CN, the fixation of that EWG on NPDI served to lower the energy of the NPDI-based HOMO and LUMOs. While the net result of this transformation retains the original HOMO–LUMO transition (Re–π*), it appears to lower the energy level of NPDI2− enough to effectively prevent eT from NPDI2− to the Re(bpy)-moiety, thus shutting down catalysis. Conversely, the installation of strong EDGs (–NR2 and –NH2) causes the energy levels of the NPDI-based HOMO and LUMOs to increase. The overall result of this transformation not only changes the HOMO–LUMO transition to an exclusively NPDI-based process (π–π*), but it also increases the driving-force for eT from NPDI2− to Re(bpy). The sum of these two effects together lead to improved dyad eT dynamics, thereby enhancing photocatalytic CO2 reduction (with respect to –H).
It should be noted that another feasible explanation for the improved performance of the amino-functionalized dyads, in particular –NH2, is the possibility of second-sphere H-bonding effects.15,18 Previously we calculated the optimized geometries of various intermediates during CO2 catalysis for the –H dyad.45 It was shown in this study that, although the NPDI was initially folded over the Re(bpy)-moiety, reduction of the –H dyad caused the two moieties to extend away from one another (likely due to coulombic repulsion effects). By analogy, the amino-functionalized NPDI-R bay position would most likely also be extended away from the Re(bpy)-moiety during CO2 photocatalysis. While in this case the distance between the catalyst center and the amino-groups of –NR2 and –NH2 make it unlikely that second-sphere H-bonding effects are involved in CO2 photocatalysis, they can't be conclusively ruled out at this time. If nothing else, the synthetic versatility of the NPDI chromophore means that future iterations of the Re(bpy-C2-NPDI-R) motif could incorporate proximal second-sphere H-bonding groups as a means to further improve CO2 conversion performance.
Conclusions
In conclusion, we present the synthesis and full characterization of six new Re(bpy-C2-NPDI-R) supramolecular dyad materials (where R = –Br, –CN, –NO2, –OPh, –NH2, or –NR2). The installation of R-groups on NPDI altered the optoelectronic properties of these dyads, as well as impacted the photocatalytic CO2 reduction performance. Relative to the benchmark Re(bpy-C2-NPDI-H) dyad (TONco = 57), the incorporation of EDGs (i.e. –NH2) led to an over four-fold improvement in photocatalytic CO2 reduction performance (TONco = 234) while strong EWGs (i.e. –CN) resulted in complete deactivation of the dyads. Despite being the most electron-withdrawing, the –NO2 functionalized NPDI was among the top performing CO2 reduction photocatalysts (TONco = 137), making it an outlier to the proposed trend. Through CV and UV-vis-nIR SEC experimentation, it was elucidated that –NO2 undergoes an in situ conversion to –NH2, thereby forming a different dyad that is responsible for catalysis. A photocatalytic CO2 reduction mechanism is proposed for these dyads, where EDGs served to accelerate CO2 reduction rates by simultaneously changing the HOMO–LUMO excitation pathway and by increasing the electronic driving-force of intramolecular electron transfer from NPDI2− to Re(bpy). Conversely, EWGs shifted the LUMO energy levels of NPDI to the point where photocatalysis is shut down because there is no electronic driving force for eT between NPDI2− and Re(bpy). This study clearly highlights the importance of evaluating structure–property relationships to develop and optimize the future design of new supramolecular dyad photocatalysts.Author contributions
JDBK performed all experimental work and data analysis and prepared the manuscript. GCW directed the project and provided resources. WEP co-directed the project and provided resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GCW acknowledges funding from the NSERC DG program (2019-04392), the Canada Foundation for Innovation, and the University of Calgary. WEP acknowledges the Canada Research Chairs Program. JK acknowledges NSERC CGS-D scholarship program. This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund (CFREF).Notes and references
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. R. Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, ACS Catal., 2018, 8, 2021–2029 CrossRef CAS.
- S. Sung, D. Kumar, M. Gil-Sepulcre and M. Nippe, J. Am. Chem. Soc., 2017, 139, 13993–13996 CrossRef CAS PubMed.
- E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2018, 140, 12451–12456 CrossRef CAS PubMed.
- A. N. Hellman, R. Haiges and S. C. Marinescu, Dalton Trans., 2019, 48, 14251–14255 RSC.
- K. Talukdar, S. Sinha Roy, E. Amatya, E. A. Sleeper, P. Le Magueres and J. W. Jurss, Inorg. Chem., 2020, 59, 6087–6099 CrossRef CAS PubMed.
- M. R. Madsen, J. B. Jakobsen, M. H. Rønne, H. Liang, H. C. D. Hammershøj, P. Nørby, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Organometallics, 2020, 39, 1480–1490 CrossRef CAS.
- S. Lense, K. A. Grice, K. Gillette, L. M. Wolf, G. Robertson, D. McKeon, C. Saucedo, P. J. Carroll and M. Gau, Organometallics, 2020, 39, 2425–2437 CrossRef CAS.
- J. Mukherjee and I. Siewert, Eur. J. Inorg. Chem., 2020, 2020, 4319–4333 CrossRef CAS.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed.
- T. W. Schneider, M. Z. Ertem, J. T. Muckerman and A. M. Angeles-Boza, ACS Catal., 2016, 6, 5473–5481 CrossRef CAS.
- P. Lang, R. Giereth, S. Tschierlei and M. Schwalbe, Chem. Commun., 2019, 55, 600–603 RSC.
- J. H. Jo, S. Choi, H.-Y. Cheong, J. Y. Shin, C. H. Kim, D. W. Cho, H.-J. Son, C. Pac and S. O. Kang, Chem.–Eur. J., 2020, 26, 16733–16754 CrossRef CAS PubMed.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- K. Koike, D. C. Grills, Y. Tamaki, E. Fujita, K. Okubo, Y. Yamazaki, M. Saigo, T. Mukuta, K. Onda and O. Ishitani, Chem. Sci., 2018, 9, 2961–2974 RSC.
- R. Kamata, H. Kumagai, Y. Yamazaki, G. Sahara and O. Ishitani, ACS Appl. Mater. Interfaces, 2019, 11, 5632–5641 CrossRef CAS PubMed.
- Y. Yamazaki, K. Ohkubo, D. Saito, T. Yatsu, Y. Tamaki, S. Tanaka, K. Koike, K. Onda and O. Ishitani, Inorg. Chem., 2019, 58, 11480–11492 CrossRef CAS PubMed.
- K. Kamogawa, Y. Shimoda, K. Miyata, K. Onda, Y. Yamazaki, Y. Tamaki and O. Ishitani, Chem. Sci., 2021, 12, 9682–9693 RSC.
- A. M. Cancelliere, F. Puntoriero, S. Serroni, S. Campagna, Y. Tamaki, D. Saito and O. Ishitani, Chem. Sci., 2020, 11, 1556–1563 RSC.
- A. Gabrielsson, F. Hartl, H. Zhang, J. R. Lindsay Smith, M. Towrie, V. Antonín and R. N. Perutz, J. Am. Chem. Soc., 2006, 128, 4253–4266 CrossRef CAS PubMed.
- K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, J. Phys. Chem. C, 2009, 113, 11667–11673 CrossRef CAS.
- C. D. Windle, M. W. George, R. N. Perutz, P. A. Summers, X. Z. Sun and A. C. Whitwood, Chem. Sci., 2015, 6, 6847–6864 RSC.
- P. Lang, M. Pfrunder, G. Quach, B. Braun-Cula, E. G. Moore and M. Schwalbe, Chem.–Eur. J., 2019, 25, 4509–4519 CrossRef CAS PubMed.
- Y. Kuramochi, Y. Fujisawa and A. Satake, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef CAS PubMed.
- Y. Kuramochi and A. Satake, Chem.–Eur. J., 2020, 26, 16365–16373 CrossRef CAS PubMed.
- F. Franco, C. Cometto, C. Garino, C. Minero, F. Sordello, C. Nervi and R. Gobetto, Eur. J. Inorg. Chem., 2015, 2015, 296–304 CrossRef CAS.
- D. R. Case, A. Spear, A. F. Henwood, M. Nanao, S. Dampf, T. M. Korter, T. Gunnlaugsson, J. Zubieta and R. P. Doyle, Dalton Trans., 2021, 50, 3479–3486 RSC.
- J. F. Martinez, N. T. La Porte and M. R. Wasielewski, J. Phys. Chem. C, 2018, 122, 2608–2617 CrossRef CAS.
- J. F. Martinez, N. T. La Porte, S. Chaudhuri, A. Sinopoli, Y. J. Bae, M. Sohail, V. S. Batista and M. R. Wasielewski, J. Phys. Chem. C, 2019, 123, 10178–10190 CrossRef CAS.
- J. F. Martinez, N. T. La Porte and M. R. Wasielewski, J. Photochem. Photobiol., A, 2019, 372, 21–28 CrossRef CAS.
- S. Hedström, S. Chaudhuri, N. T. La Porte, B. Rudshteyn, J. F. Martinez, M. R. Wasielewski and V. S. Batista, J. Am. Chem. Soc., 2017, 139, 16466–16469 CrossRef PubMed.
- N. T. L. Porte, J. F. Martinez, S. Hedström, B. Rudshteyn, B. T. Phelan, C. M. Mauck, R. M. Young, V. S. Batista and M. R. Wasielewski, Chem. Sci., 2017, 8, 3821–3831 RSC.
- J. D. B. Koenig, Z. S. Dubrawski, K. R. Rao, J. Willkomm, B. S. Gelfand, C. Risko, W. E. Piers and G. C. Welch, J. Am. Chem. Soc., 2021, 143, 16849–16864 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. R. Cann, C. Cabanetos and G. C. Welch, Eur. J. Org. Chem., 2018, 2018, 6933–6943 CrossRef CAS.
- A. D. Hendsbee, J.-P. Sun, W. K. Law, H. Yan, I. G. Hill, D. M. Spasyuk and G. C. Welch, Chem. Mater., 2016, 28, 7098–7109 CrossRef CAS.
- C. R. Harding, J. Cann, A. Laventure, M. Sadeghianlemraski, M. Abd-Ellah, K. R. Rao, B. S. Gelfand, H. Aziz, L. Kaake, C. Risko and G. C. Welch, Mater. Horiz., 2020, 7, 2959–2969 RSC.
- M. Vespa, J. R. Cann, S. V. Dayneko, O. A. Melville, A. D. Hendsbee, Y. Zou, B. H. Lessard and G. C. Welch, Eur. J. Org. Chem., 2018, 2018, 4592–4599 CrossRef CAS.
- A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, ACS Catal., 2019, 4867–4874 CrossRef CAS.
- X. Zhang, K. Yamauchi and K. Sakai, ACS Catal., 2021, 10436–10449 CrossRef CAS.
- C. D. Sahm, G. M. Ucoski, S. Roy and E. Reisner, ACS Catal., 2021, 11266–11277 CrossRef CAS.
- L. Hao, W. Jiang and Z. Wang, Tetrahedron, 2012, 68, 9234–9239 CrossRef CAS.
- G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 196–203 CrossRef CAS PubMed.
- D. H. Harris, S. Brixi, B. S. Gelfand, B. H. Lessard and G. C. Welch, J. Mater. Chem. C, 2020, 8, 9811–9815 RSC.
- S. Sato, Y. Matubara, K. Koike, M. Falkenström, T. Katayama, Y. Ishibashi, H. Miyasaka, S. Taniguchi, H. Chosrowjan, N. Mataga, N. Fukazawa, S. Koshihara, K. Onda and O. Ishitani, Chemistry, 2012, 18, 15722–15734 CrossRef CAS PubMed.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2008 Search PubMed.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- C. Costentin and J.-M. Savéant, ChemElectroChem, 2014, 1, 1226–1236 CrossRef CAS.
- S. Dey, M. E. Ahmed and A. Dey, Inorg. Chem., 2018, 57, 5939–5947 CrossRef CAS PubMed.
- M. E. Ahmed, A. Rana, R. Saha, S. Dey and A. Dey, Inorg. Chem., 2020, 59, 5292–5302 CrossRef CAS PubMed.
- C. Wiberg, M. Busch, L. Evenäs and E. Ahlberg, Electrochim. Acta, 2021, 367, 137480 CrossRef CAS.
- E. Shirman, A. Ustinov, N. Ben-Shitrit, H. Weissman, M. A. Iron, R. Cohen and B. Rybtchinski, J. Phys. Chem. B, 2008, 112, 8855–8858 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt and F. Würthner, Chem. Sci., 2015, 6, 1663–1667 RSC.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- J. D. B. Koenig, J. Willkomm, R. Roesler, W. E. Piers and G. C. Welch, ACS Appl. Energy Mater., 2019, 2, 4022–4026 CrossRef CAS.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, ACS Catal., 2018, 3411–3417 CrossRef CAS.
- S. Fernández, F. Franco, C. Casadevall, V. Martin-Diaconescu, J. M. Luis and J. Lloret-Fillol, J. Am. Chem. Soc., 2020, 142, 120–133 CrossRef PubMed.
- S. S. Saund, M. A. Siegler and V. S. Thoi, Inorg. Chem., 2021, 60, 13011–13020 CrossRef CAS PubMed.
- A. C. Tsipis and A. A. Sarantou, Dalton Trans., 2021, 50, 14797–14809 RSC.
- H. Koizumi, H. Chiba, A. Sugihara, M. Iwamura, K. Nozaki and O. Ishitani, Chem. Sci., 2019, 10, 3080–3088 RSC.
- R. N. Schaugaard, K. Raghavachari and L. Li, Inorg. Chem., 2018, 57, 10548–10556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05465a |
| This journal is © The Royal Society of Chemistry 2022 |