DOI:
10.1039/D1SC05374D
(Edge Article)
Chem. Sci., 2022,
13, 1992-2000
Rh2(II)-catalyzed enantioselective intramolecular Büchner reaction and aromatic substitution of donor–donor carbenes†
Received
29th September 2021
, Accepted 19th January 2022
First published on 19th January 2022
Abstract
The chiral dirhodium(II) tetracarboxylate-catalyzed enantioselective intramolecular Büchner reaction of donor/donor-carbenes was reported and a series of valuable chiral polycyclic products were synthesized. Both aryloxy enynones and diazo compounds were efficient carbene precursors for this reaction. Excellent yields (up to 99%) and outstanding enantioselectivities (up to >99% ee) were achieved under standard conditions. For furyl substituted chiral cyclohepta[b]benzofurans bearing a substituent at the C4 position on cycloheptatrienes, control reactions showed that the chiral Büchner products could slowly racemize either under dark or natural light conditions. A diradical-involved mechanism rather than a zwitterionic intermediate was proposed to explain the racemization. Furthermore, furyl substituted chiral fluorene derivatives were obtained via asymmetric aromatic substitution when biaryl enynones were employed as carbene precursors.
Introduction
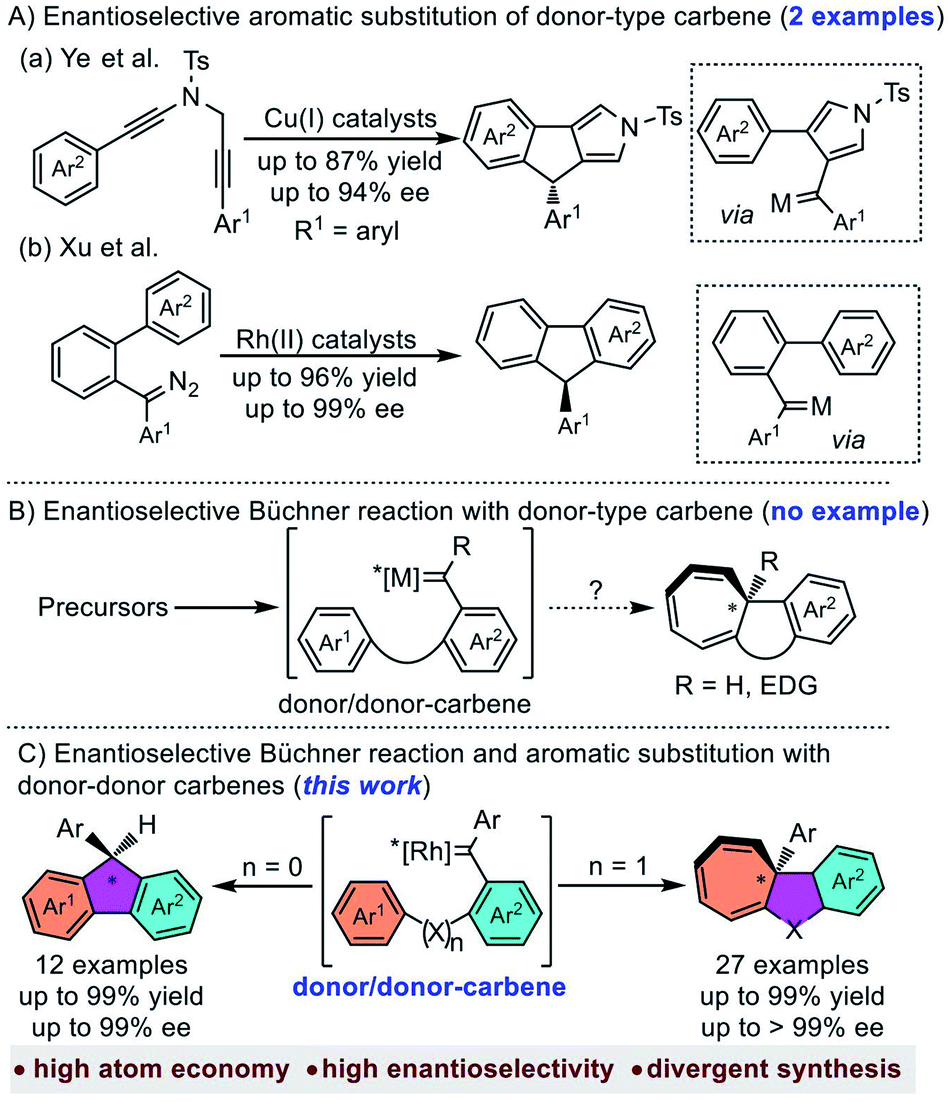
Metal-carbene is one of the most important reactive intermediates in organic synthesis.1 Among the numerous carbene-transfer reactions, the reaction of metal-carbene with arenes has gained much interest in the past few decades.2 The main reaction pathways of metal-carbene with arenes include aromatic substitution and Büchner reaction, which could provide efficient approaches to polycyclic molecules, especially in an intramolecular manner.3 Compared to the well-developed non-asymmetric Büchner reaction4 and aromatic substitution,5 the asymmetric ones were much less explored,6 especially for donor type carbene (donor- and donor/donor-carbenes) involved reactions.7 The main reason is that donor-type carbenes were usually regarded as less efficient for carbene-transfer reactions than acceptor-type carbenes (acceptor-, donor/acceptor-, and acceptor/acceptor-carbenes).8 Recently, Ye et al. disclosed the first enantioselective aromatic substitution with donor–donor copper carbenes through diyne cyclization.9 Xu et al. also reported a rhodium catalysed asymmetric carbene arylation reaction with diazo compounds as donor–donor carbene precursors (Scheme 1A).10 These achievements indicated the tunability of donor or donor–donor type carbenes in asymmetric synthesis. However, to the best of our knowledge, there is still no example of donor-type carbene involved asymmetric Büchner reaction (Scheme 1B), which could be partially attributed to (1) the dual electron donating aryl groups making the carbene carbon less electrophilic;11 (2) the commonly-used diazo precursors are highly unstable, potentially explosive and easily dimerized.12
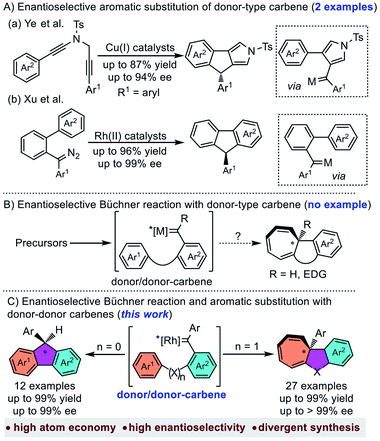 |
| | Scheme 1 Donor-type carbene mediated enantioselective aromatic substitution and Büchner reactions. | |
Over the past two decades, carbene chemistry with non-diazo precursors has experienced tremendous growth due to the emerging gold chemistry.13 Despite that these newly developed carbene precursors provide several useful means in the transformation of donor type metal-carbenes, the commonly used gold catalyst usually suffered inefficient chiral induction due to the linear geometry.14 As a reliable and safe donor-type metal-carbene source, enynones have received much attention due to its versatile activities.15 In our previous work, we realized the asymmetric intramolecular C–H insertion and cyclopropanation with enynones as carbene precursors.16 With our continuous interest in the development of donor type metal-carbenes,17 herein we would like to report the first example of the chiral Rh2(II)-catalyzed enantioselective Büchner reaction with donor–donor carbenes (Scheme 1C, right). In addition, the asymmetric aromatic substitution is also achieved using the same catalyst as well (Scheme 1C, left).
Results and discussion
Rh2(II)-catalyzed asymmetric Büchner reaction of enynones
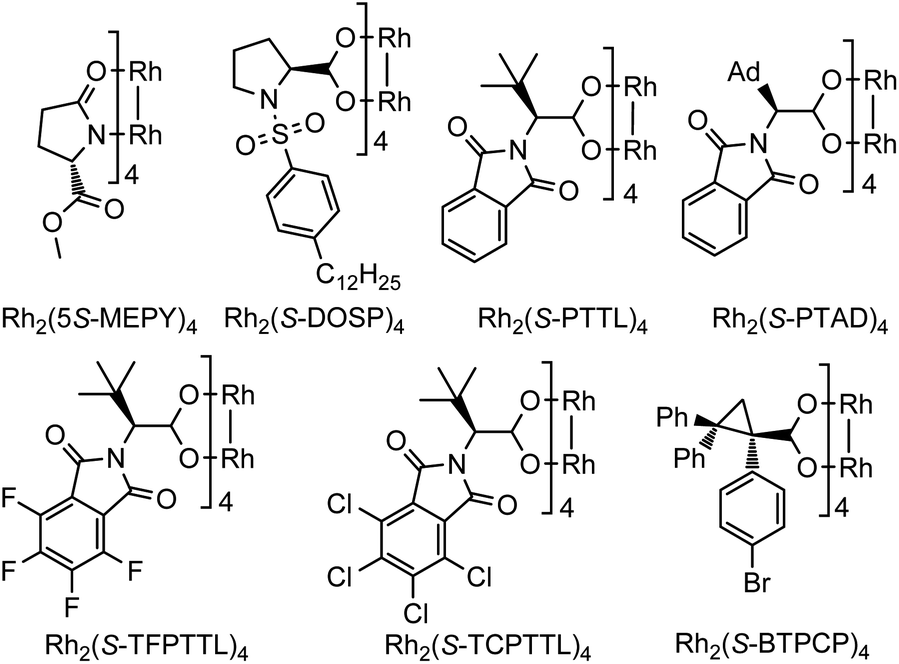
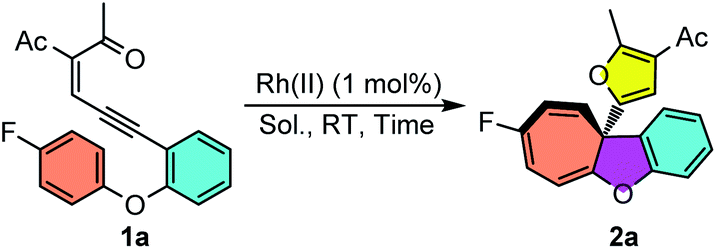
To start this investigation, enynone 1a was chosen as the model substrate to screen the asymmetric Büchner reaction conditions. As shown in Table 1, the desired furyl substituted cyclohepta[b]benzofuran 2a was obtained in 94% yield under the catalysis of 1 mol% Rh2(OPiv)4 at room temperature (Table 1, entry 1). In line with our speculation, the electron-rich chiral dirhodium(II) carboxamide catalyst [Rh2(5S-MEPY)4] proved to have no catalytic activity for this reaction, which indicated that the more d-π donating catalyst would further decrease the electrophilicity of the donor–donor carbene carbon (entry 2). Dirhodium N-sulfonylprolinate [Rh2(S-DOSP)4] resulted in a quantitative yield, but without asymmetric induction (entry 3). The phthalimide-based catalysts showed better selectivity, giving the desired product 2a in 91–99% yields and 6–73% ee (entries 4–7). For example, the enantioselectivity was only 6% ee when [Rh2(S-PTTL)4] was used as the catalyst (entry 4). When t-butyl was replaced with the adamantly group, catalyst [Rh2(S-PTAD)4] gave 29% ee and 91% yield (entry 5). The enantioselectivity of 2a can be increased dramatically to 73% ee when more electron–deficient complexes [Rh2(S-TFPTTL)4] and [Rh2(S-TCPTTL)4] were applied (entries 6 and 7). To our delight, when dirhodium triarylcyclopropane carboxylate [Rh2(S-BTPCP)4] was used as the catalyst, the highest enantioselectivity of 83% ee was obtained with 99% yield (entry 8). The solvents were screened as well and DCE proved to be the optimal one, which gave the desired product in 92% ee and quantitative yield (entry 11). Lowering the reaction temperature to −20 °C further improved the enantioselectivity to 96% ee (entry 12), which was then used as the optimal reaction condition. In addition, the catalyst loading could be reduced to 0.5 mol%, and both the reaction yield and enantioselectivity were unaffected, albeit with a longer reaction time (entry 13).
Table 1 Optimization of the reaction conditionsa
|
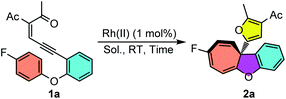
|
| Entry |
Cat. |
Sol. |
Time |
Yieldb |
eec |
|
The reaction was performed in DCE under N2; 1a = 0.2 mmol and [1a] = 0.1 M.
Isolated yield.
The ee value of 2a was determined by HPLC using a chiral stationary phase.
−20 °C.
−20 °C, 0.5 mol% Rh2(S-BTPCP)4.
|
| 1 |
Rh2(OPiv)4 |
DCM |
3 h |
94% |
— |
| 2 |
Rh2(5S-MEPY)4 |
DCM |
4 h |
Trace |
— |
| 3 |
Rh2(R-DOSP)4 |
DCM |
4 h |
99% |
Racemic |
| 4 |
Rh2(S-PTTL)4 |
DCM |
12 h |
97% |
6% |
| 5 |
Rh2(S-PTAD)4 |
DCM |
12 h |
91% |
29% |
| 6 |
Rh2(S-TFPTTL)4 |
DCM |
4 h |
99% |
−20% |
| 7 |
Rh2(S-TCPTTL)4 |
DCM |
4 h |
99% |
73% |
| 8d |
Rh2(S-BTPCP)4 |
DCM |
2 h |
99% |
83% |
| 9 |
Rh2(S-BTPCP)4 |
PhMe |
4 h |
99% |
60% |
| 10 |
Rh2(S-BTPCP)4 |
CHCl3 |
8 h |
99% |
70% |
| 11 |
Rh2(S-BTPCP)4 |
DCE |
2 h |
99% |
92% |
|
12
|
Rh
2
(S-BTPCP)
4
|
DCE
|
30 h
|
99%
|
96%
|
| 13e |
Rh2(S-BTPCP)4 |
DCE |
53 h |
99% |
96% |
|
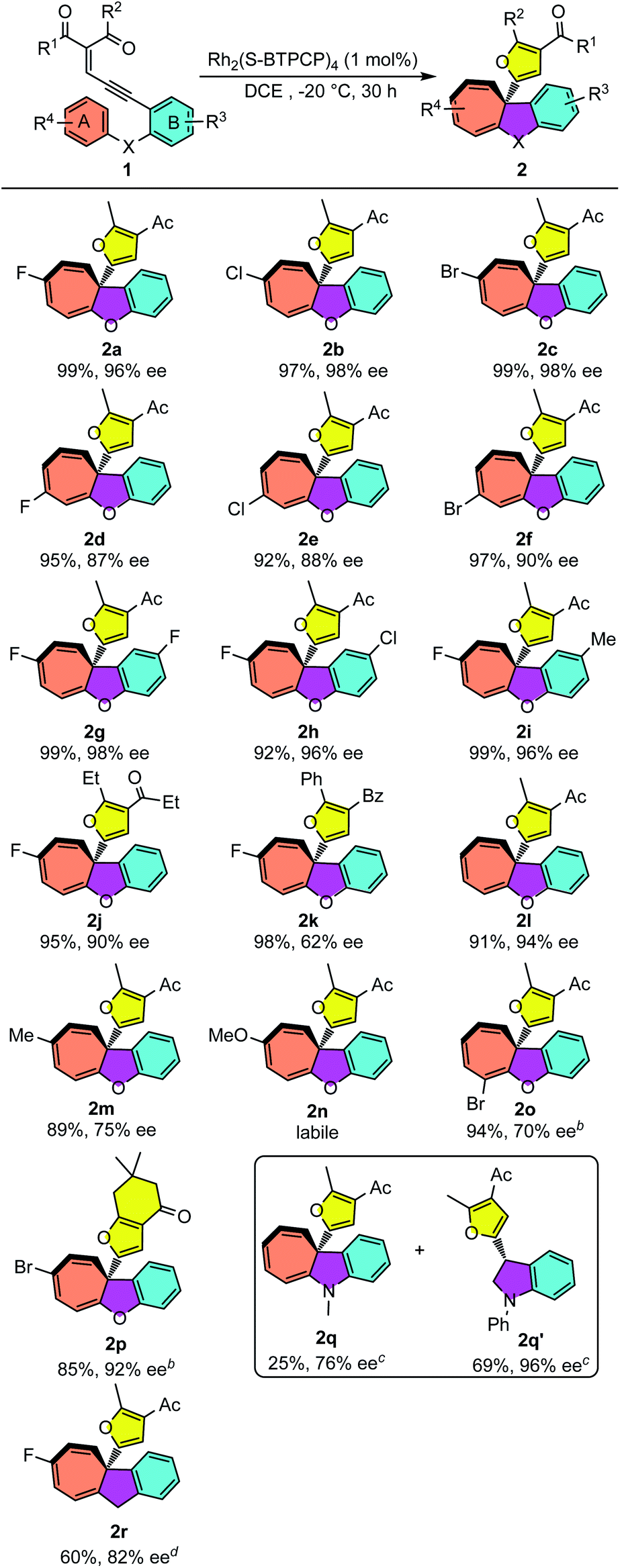
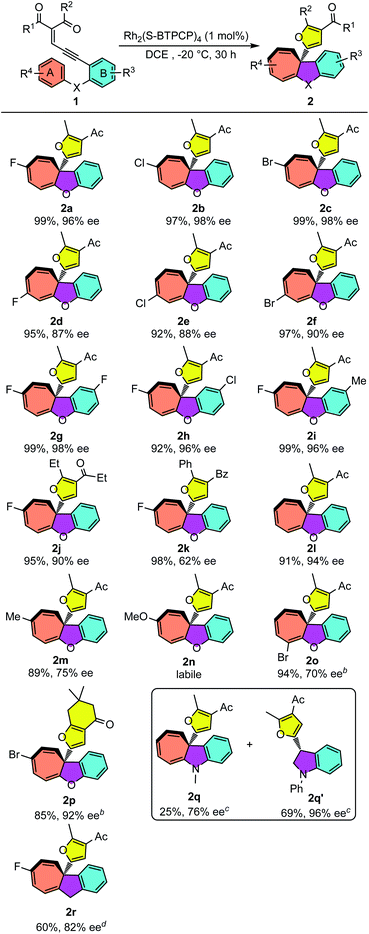
With the optimized reaction conditions for the Büchner reaction in hand (Table 1, entry 12), we next examined the reaction scope with different enynones. As shown in Table 2, enynones tethered with para-halogen-substituted aryloxy reacted smoothly, giving the desired polycyclic products 2a–c in 97–99% yields with excellent enantioselectivities (96–98% ee). The reactions of meta-halogenated enynones (R4 is halogen) proceeded smoothly as well, giving the corresponding products 2d–f in good enantioselectivities (87–90% ee) and excellent yields (92–97%). Variations in the R3 group of enynones were also investigated, which led to the desired Büchner ring-expansion products in high yields and excellent enantioselectivities with both electron-withdrawing and donating groups (2g–i). The enynones with different carbonyl substituents showed good reactivities as well. Moderate to good enantioselectivity of products 2j and 2k was achieved. It seems that the enantioselectivity of this Büchner reaction was sensitive to the electronic properties of aromatic ring A; the ee values for cyclohepta[b]benzofurans 2l and 2m dropped to 94% and 75% when phenoxyl- and 4-methylphenoxyl-enynones were utilized as substrates. When electronically richer 4-methoxy-substituted substrate 1n was applied, the product 2n could be observed from the 1H NMR spectrum but decomposed quickly after purification on the silica gel. Ortho-halogenated enynones 1o was tested as well, giving the desired cyclohepta[b]benzofuran 2o in 94% yield and 70% ee. It should be pointed out that the 1,3-diketone derived substrate 1p performed well in this annulation and gave 2p in 85% yield and 92% ee under −10 °C conditions. Lastly, N–Me phenyl substituted enynone 1q and CH2 tethered enynone 1r were also tested under these catalytic conditions. Interestingly, the Büchner product 2q was obtained only in 25% yield and 76% ee, accompanied by the C–H insertion product dihydroindole 2q′ (69%, 96% ee).18 Indan fused cycloheptatriene 2r was achieved with both moderate yield and good enantioselectivity.
Table 2 Scope of the asymmetric Büchner reactiona
|
Reaction conditions: 1 = 0.2 mmol, [1] = 0.1 M, isolated yield.
−10 °C, 48 h.
69% N–Me C–H insertion product 2q′ was isolated.
40 °C, 24 h.
|
|
Rh2(II)-catalyzed Büchner reaction of diazo compounds
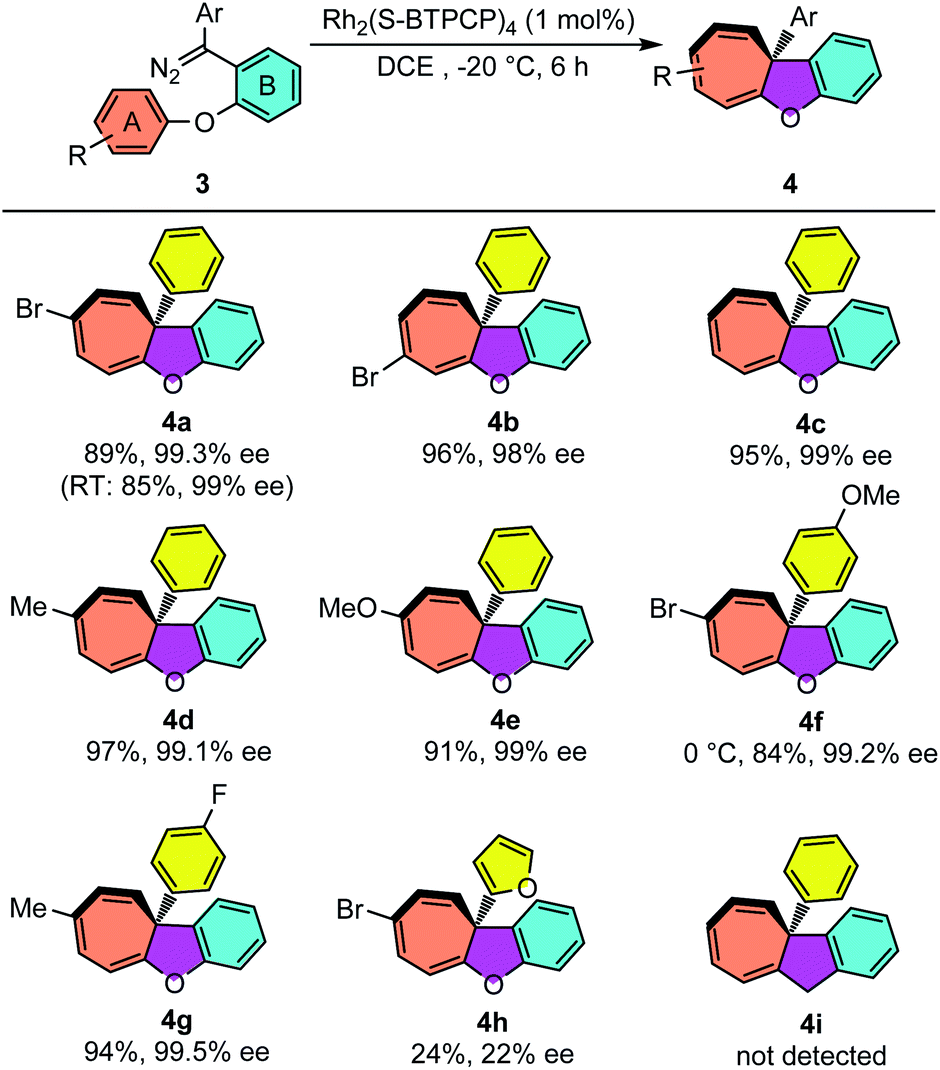
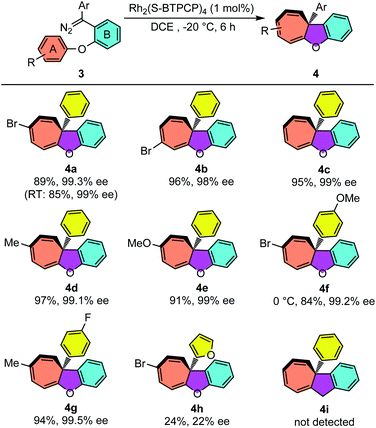
The reaction conditions of Rh2(S-BTPCP)4-catalyzed enantioselective intramolecular Büchner reaction were not only applied well for enynones, but also showed excellent performance when diazo compounds were used as donor–donor carbene sources. As shown in Table 3, aryl substituted cyclohepta[b]benzofurans were produced from aryloxy substituted diphenyl diazo compounds 3. Unlike the enynone-based system, the diazo-based system was found to be almost not noticeably affected by the electronic properties of the substituents on aromatic ring A. The phenyl substituted cyclohepta[b]benzofurans 4a–e were obtained with high yields (84–97%) and outstanding enantioselectivity (98–99.5% ee). The cyclohepta[b]benzofuran 4e derived from the diazo compound with electron-rich aromatic ring A could also be obtained in 91% yield and 99% ee. The product 4e was stable and no decomposition was observed when purified on silica gel, which is different with the enynone-based system (see 2n). Furthermore, the introduction of electron-donating and -withdrawing groups on the Ar group of diazo compounds also provided the desired products 4f–g with excellent enantioselectivities. Furyl substituted diazo compound 3h was synthesized and reacted under the standard conditions, and only Büchner product 4h was isolated with 24% yield and 22% ee. Interestingly, an unexpected furan ring-opening product enynal 4h′ was obtained in 55%.15a Lastly, CH2-tethered diazo compound 3i was not an effective substrate for this Büchner reaction and only vinyl product 4i′via carbene dimerization was detected (see the ESI†).
Table 3 Scope of the asymmetric Büchner reaction with diazo compounds as carbene sourcesa
|
Reaction conditions: 3 = 0.1 mmol, [3] = 0.025 M, isolated yield; the diazo compound 3 was added dropwise over 1 h.
|
|
Rh2(II)-catalyzed asymmetric aromatic substitution reaction of enyones
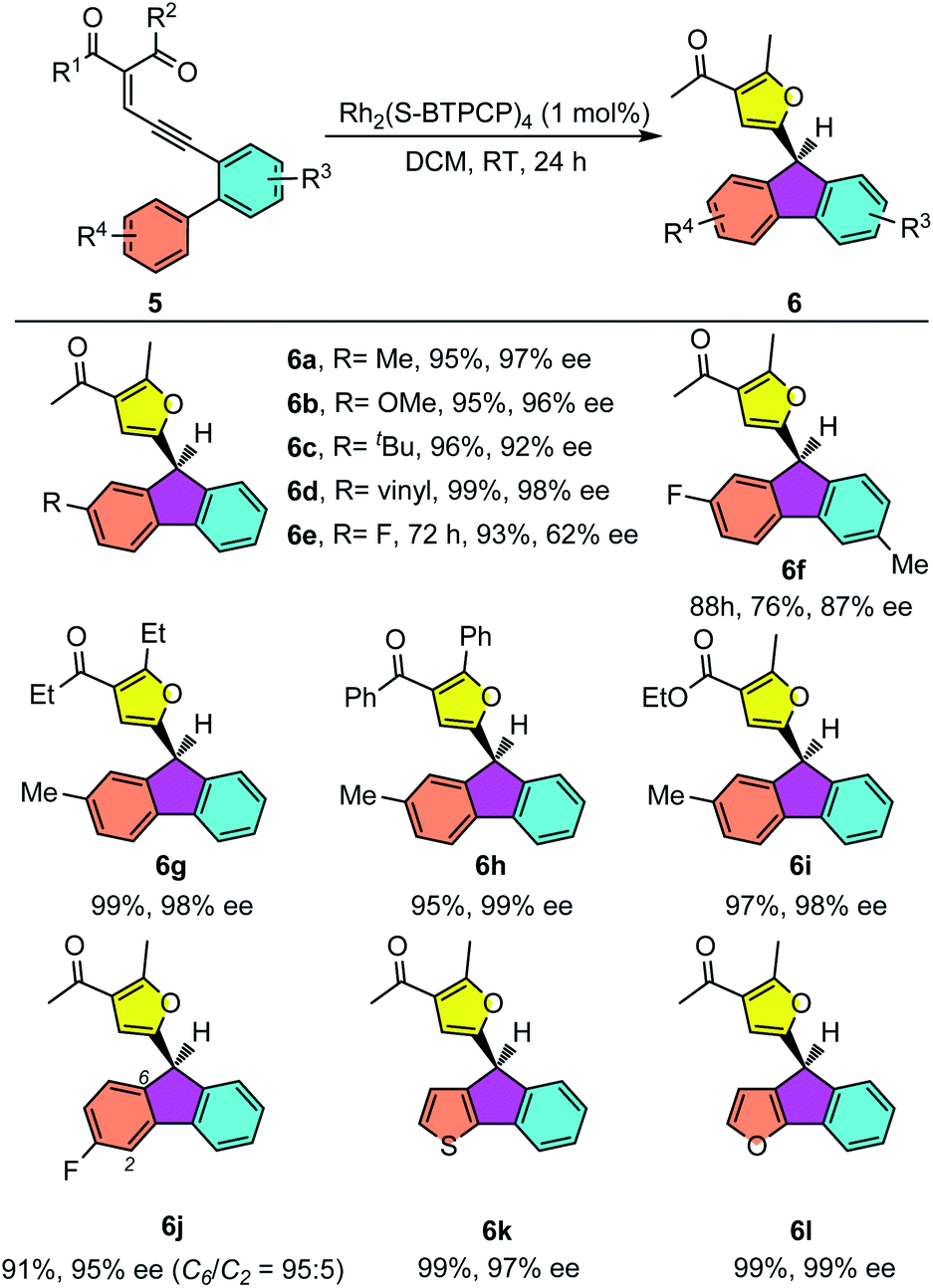
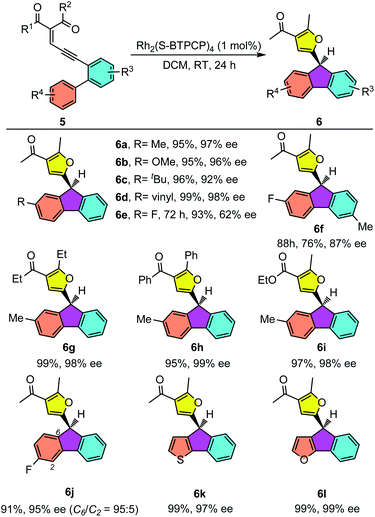
Having established the eantioselective intramolecular Büchner reaction as a reliable and efficient synthetic protocol, biaryl enynones 5 were then employed as donor carbene precursors to test the asymmetric aromatic substitution reaction. As shown in Table 4, the intramolecular aromatic substitution reactions proceeded smoothly and gave the desired furyl substituted fluorenes with high yields and excellent enantioselectivities under slightly modified reaction conditions (see the ESI†). It was shown that the catalytic process could be successfully applied to different enynone substrates bearing different R4 groups. For example, in addition to enynone 5a, various enynone derivatives 5 with para-substituted groups could be efficiently converted into the desired fluorenes 6 in good enantioselectivities (62–98% ee) (6a–e). The yields were typically higher than 90% for most substrates. The substrates with electron-rich aryl groups have higher reactivity, furnishing the fluorene products 6a–e in almost quantitative yields. Moreover, enynones with different substituents on carbonyl groups were also suitable for this transformation, leading to fluorenes 6g–i in excellent yields (95–99%) and enantioselectivities (98–99% ee). In addition to the enynones with para-substituted phenyl groups, the meta-substituted enynone was also investigated. The reaction took place smoothly to give the desired product 6j in excellent yield and enantioselectivity. More importantly, a good regioselectivity was observed (C6/C2 = 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). This result indicated that the aromatic substitution reaction is sensitive to the steric effects of the substrates. Notably, thienyl and furyl substituted enynones were also suitable substrates to provide the desired aromatic substation products 6k and 6l with excellent results. It is worthy of note that a similar non-asymmetric divergent Büchner reaction and aromatic substitution reaction were reported by Wang and co-workers in a metal free manner with hydrazones as donor carbene precursors.19
:5). This result indicated that the aromatic substitution reaction is sensitive to the steric effects of the substrates. Notably, thienyl and furyl substituted enynones were also suitable substrates to provide the desired aromatic substation products 6k and 6l with excellent results. It is worthy of note that a similar non-asymmetric divergent Büchner reaction and aromatic substitution reaction were reported by Wang and co-workers in a metal free manner with hydrazones as donor carbene precursors.19
Table 4 Scope of the aromatic substitution reactiona
|
Reaction conditions: 5 = 0.2 mmol, [5] = 0.1 M, isolated yield.
|
|
DFT studies on the reaction mechanism and chemoselectivity
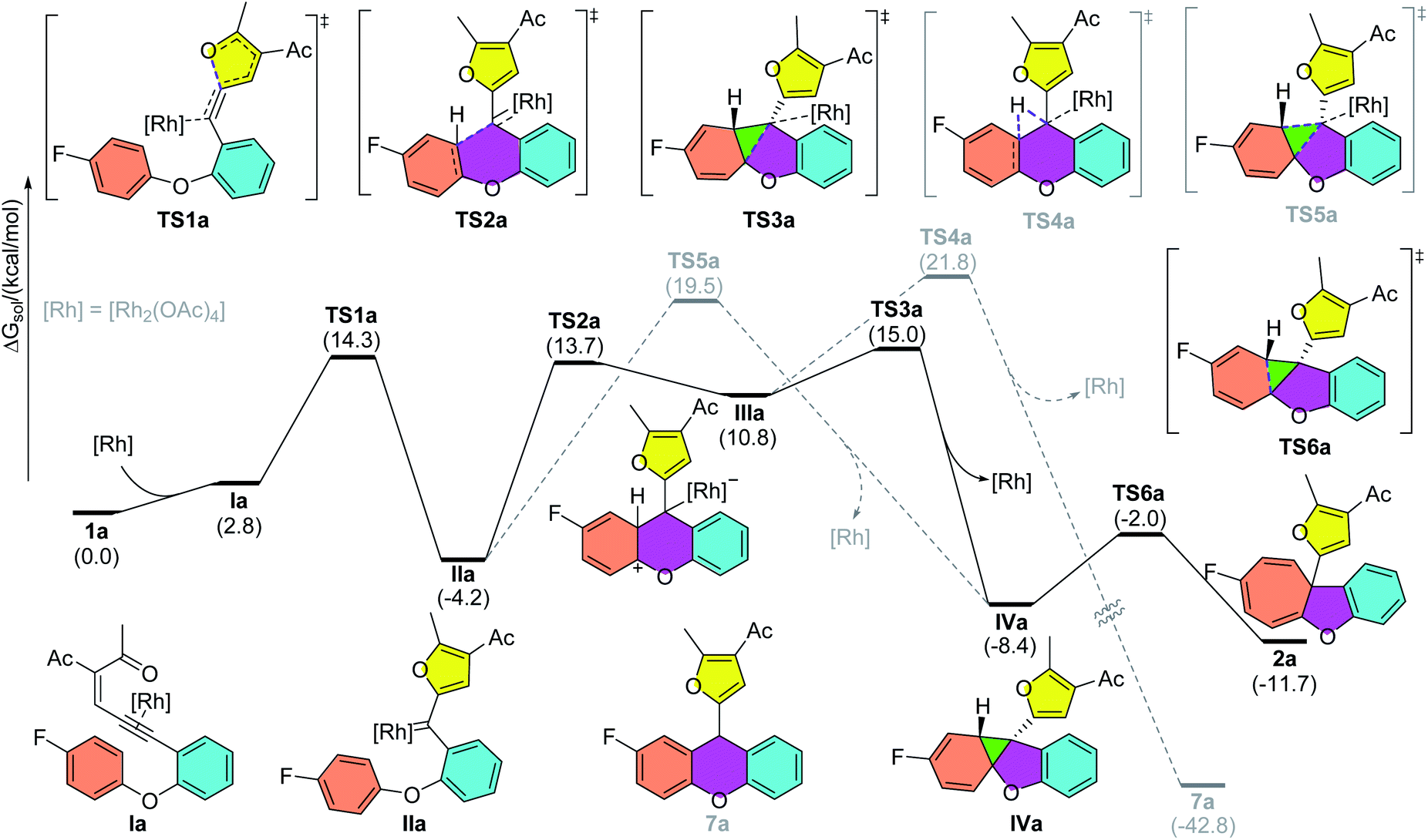
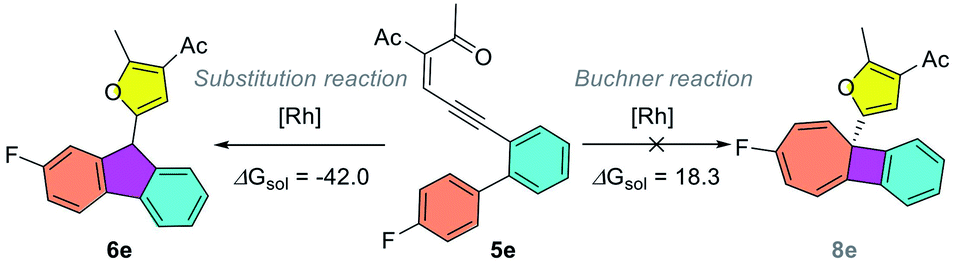
To gain insights into the reaction mechanism, DFT calculations were carried out using Gaussian16 at the M06/def2TZVP-SMD(dichloroethane)//B3LYP-D3(BJ)/def2SVP theoretical level.20–22 As shown in Scheme 2, the reaction starts with the coordination of the rhodium catalyst with the alkyne moiety, followed by a 5-exo-dig cyclization to afford dirhodium carbene intermediate IIa. This is supported by our experiments, as diazo compounds could also undergo a similar Büchner reaction under standard conditions. Then, a stepwise cyclopropanation process involving electrophilic addition of carbene to the phenyl ring and a subsequent ring-closure step will lead to the formation of the cyclopropane intermediate IVa. Moreover, a concerted cyclopropanation step viaTS5a is also considered, which is disfavored over the stepwise pathway (viaTS3a) by 4.5 kcal mol−1. Finally, an electrocyclic step viaTS6a opens the cyclopropane ring to form the Büchner reaction product 2a. The whole potential energy surface shows that the ring-closure step is the rate-determining step, with an activation barrier about 19.2 kcal mol−1 in terms of the Gibbs free energy. In addition, intermediate IIIa could also undergo a [1,2]-H shift viaTS4a, with the release of the rhodium catalyst, to give the aromatic substitution product 7a. DFT calculations indicated that the Büchner reaction pathway via ring-closure transition state TS3a is favored over the aromatic substitution pathway via [1,2]-H shift transition state TS4a by 6.8 kcal mol−1. In contrast, for biaryl enynone 5, a similar Büchner reaction was not observed, possibly due to the high ring strain of the fused cyclic butene moiety in 8 (Scheme 3). Thermochemistry calculations show that the Büchner process of 5e is endergonic (ΔGsol = 18.3 kcal mol−1), and thus could not happen spontaneously. However, the overall aromatic substitution reaction of biaryl enynone 5e is exergonic by 42.0 kcal mol−1, leading to the formation of stable 9H-fluorene 6e (see the ESI†).
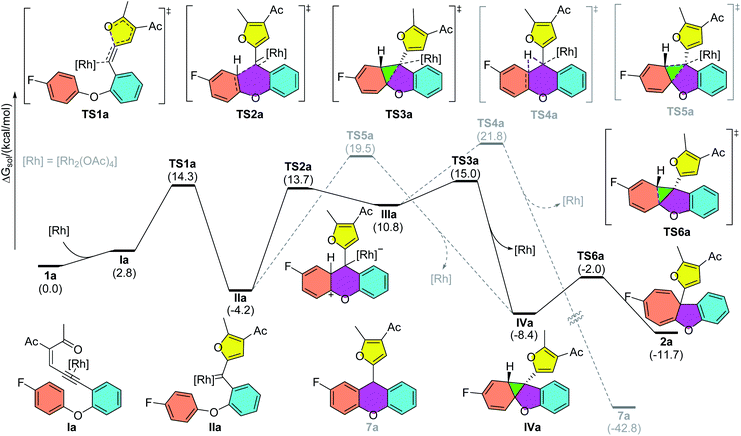 |
| | Scheme 2 Free energy profiles of Rh2(II)-catalyzed reaction pathways of enynone 1a. If not otherwise noted, relative Gibbs free energies computed at the M06/def2TZVP-SMD(dichloroethane)//B3LYP-D3(BJ)/def2SVP theoretical level are reported in kcal mol−1. | |
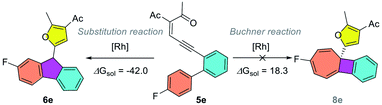 |
| | Scheme 3 Competition between the Büchner reaction and aromatic substitution pathways for biaryl enynone 5e. | |
Control experiments and mechanism for the rationalization of the chiral Büchner products
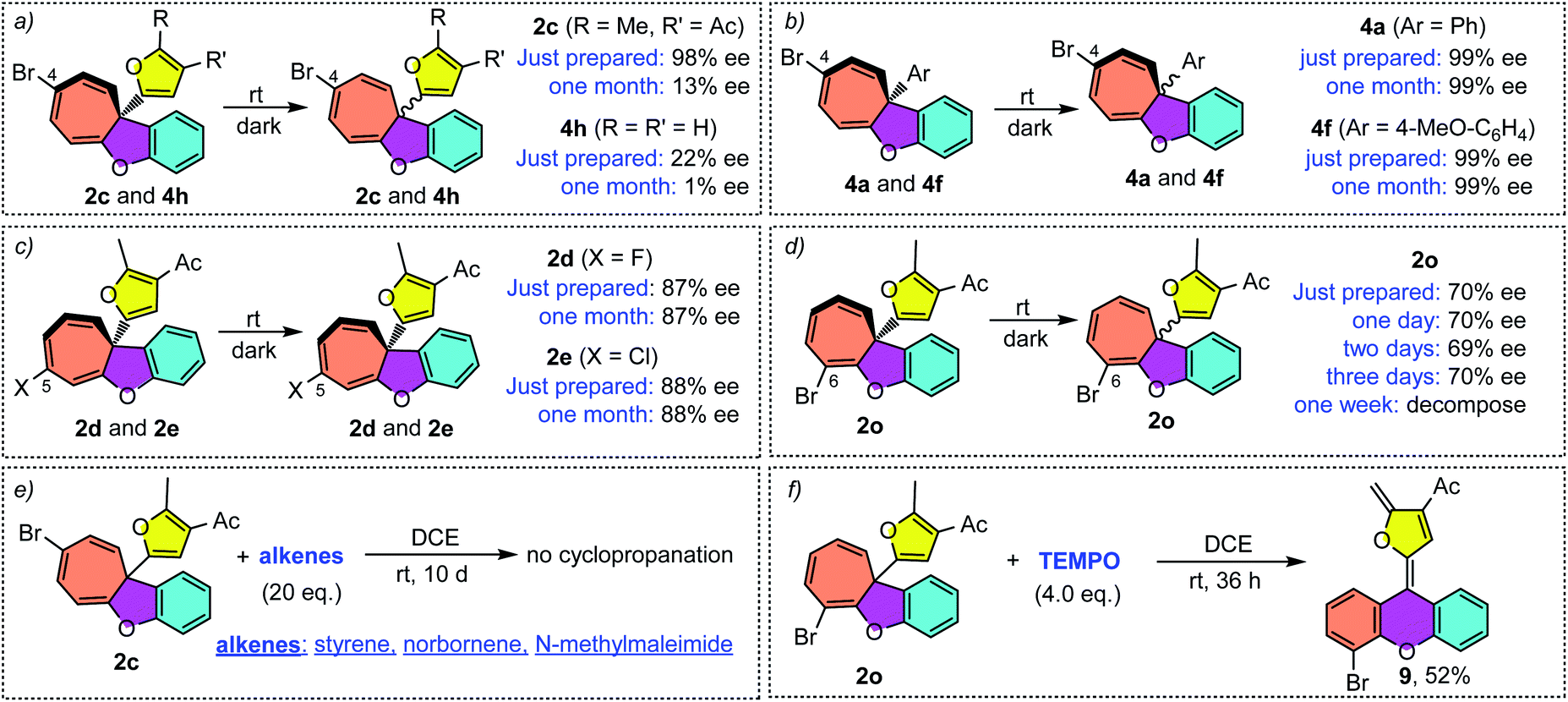
It should be noted that the ee value of furyl substituted Büchner products 2c and 4h was found to slowly decrease when preserved in i-PrOH at room temperature (Scheme 4a). Similar racemization was also found for other C4-substituted chiral cyclohepta[b]benzofurans (see the ESI†). This interesting phenomenon drove us to figure out the reason for racemization. Controlled experiments indicated that this racemization is independent of the solvent (Table S3†) but both temperature and natural light will accelerate the racemization rate. Interestingly, the phenyl substituted Büchner products (4a and 4f) derived from diazo compounds maintained good enantioselectivity. Surprisingly, the chiral cyclohepta[b]benzofuran products derived from ortho- and meta-substituted phenoxy enynones (such as 2d, 2e and 2o) did not racemize (Scheme 4c and d). These results implied that both the type of substituents attached to cyclohepta[b]benzofurans (2c and 4a) and the position of the functional groups on cycloheptatriene (2c and 2f) had a great influence on racemization. In view of this racemization, several following possible mechanisms were proposed. First, we speculated that furyl substituted chiral cyclohepta[b]benzofurans might racemize via a free donor–donor carbene intermediate through a retro-Büchner reaction. To validate this conjecture, various alkenes were added into the solution of 2c to trap the proposed free carbene intermediate (Scheme 4e). However, no desired cyclopropanation product was detected, which suggested that the racemization process might not proceed through a reversible Büchner reaction. Furthermore, the results that only C4-substituted Büchner products (such as 2c and 4h) were observed to racemize also exclude the free carbene process because cycloheptatriene bearing functional groups at other positions (such as 2d and 2o) will racemize through free carbene intermediates as well. In addition, studies by Dolye, Moody and Maguire suggested that a zwitterionic intermediate is also operative for the racemization of the Büchner products, wherein the C–C bond of cyclopropane could break and form reversibly.23–25 Similarly, Houk et al. also suggested a diradical species to explain the “walk rearrangement” substituted norcaradiene.26 Compared to the zwitterionic intermediate, we preferred a diradical mediated racemization process in our system because no electron-withdrawing group could stabilize the electronic mismatched zwitterionic species. Thus, a control experiment intended to intercept the postulated diradical species was then designed. Interestingly, a ring isomerization product 9 was obtained in 52% when TEMPO was used as the trapping reagent (Scheme 4f), which makes the reaction more likely to go through a diradical mechanism.
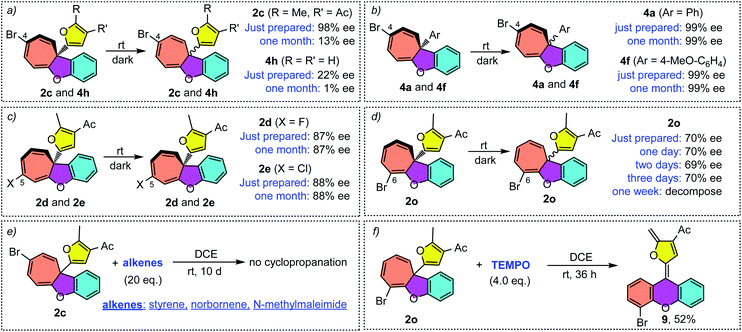 |
| | Scheme 4 Control experiments and mechanistic investigation. | |
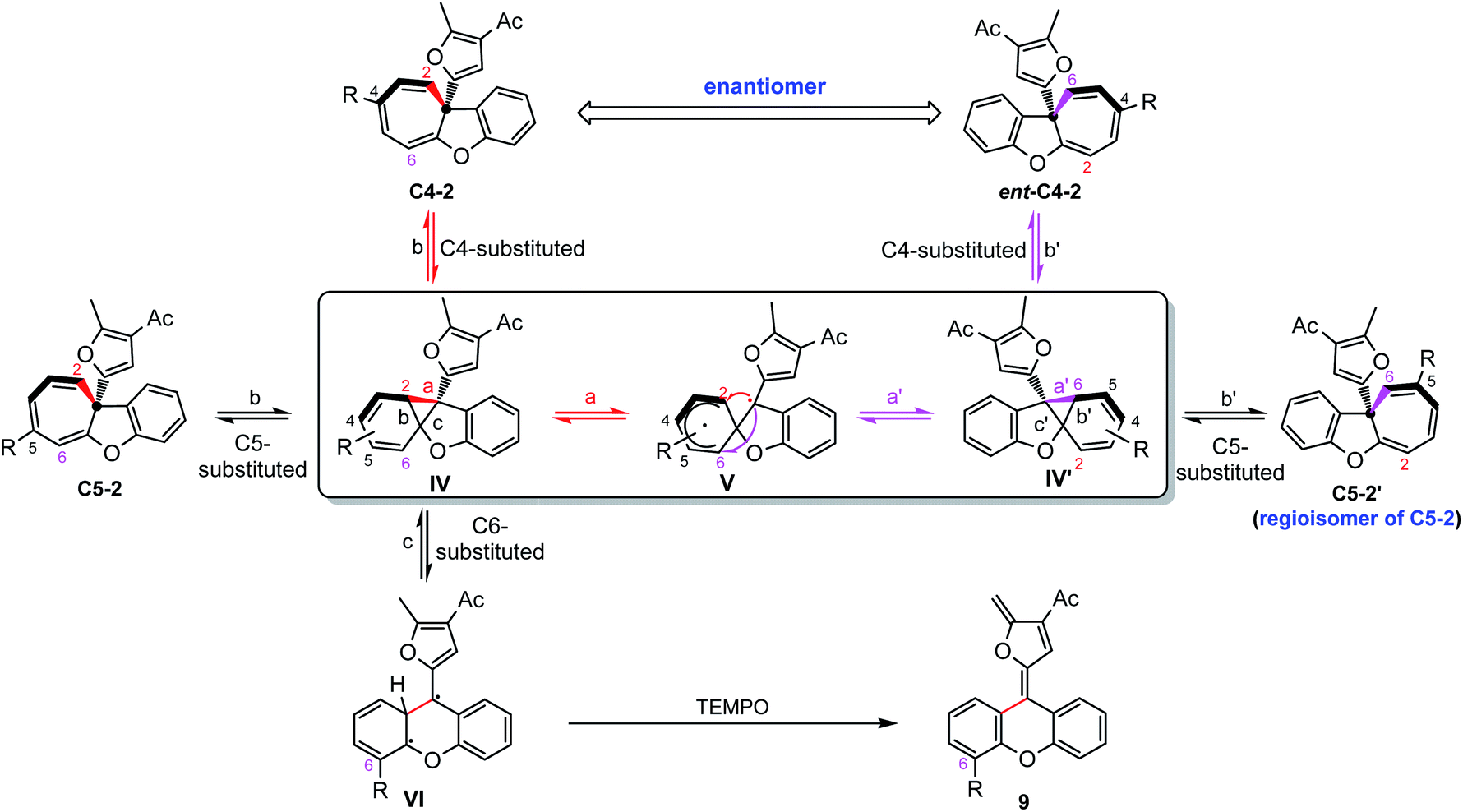
Taken together, a diradical process is the probable mechanism for the racemization of furyl substituted chiral cyclohepta[b]benzofurans. As shown in Scheme 5, the chiral cyclohepta[b]benzofuran C4-2 bearing a substituent at the C4 position first generates norcaradiene IV and then leads to the diradical tautomer V through homolytic cleavage of the cyclopropane (bond a). The diradical V has two possible pathways to evolve, one is to norcaradiene IV through the reversible process by attacking the C2 position, and another is attacking the C6 position to give the dissymmetric norcaradiene IV' which leads to the enantiomer ent-C4-2 after ring expansion, which eventually results in the racemization of chiral cyclohepta[b]benzofuran 2. It's also possible that the diradical VI could be generated via cleavage of bond c of intermediate IV due to the increasing steric hindrance when a substituent was introduced at the C6-position. The diradical VI could be oxidized by TEMPO to give ring isomerization product 9. It is noteworthy that when it comes to the chiral cyclohepta[b]benzofuran products derived from ortho- and meta-substituted phenoxy enynones, a similar process will take place to generate a regioisomer, rather than an enantiomer, without loss of the enantiomeric excess. Numerous efforts to isolate or detect the proposed regioisomer failed, which might be ascribed to that the “walking rearrangement” to the sterically more congested isomer was highly unfavourable (see the ESI for details†). Thus, the cyclopropane would prefer to break bond c to release the steric torsion to give diradical intermediate VI, which could be intercepted by TEMPO to give the ring isomerization product 9.27
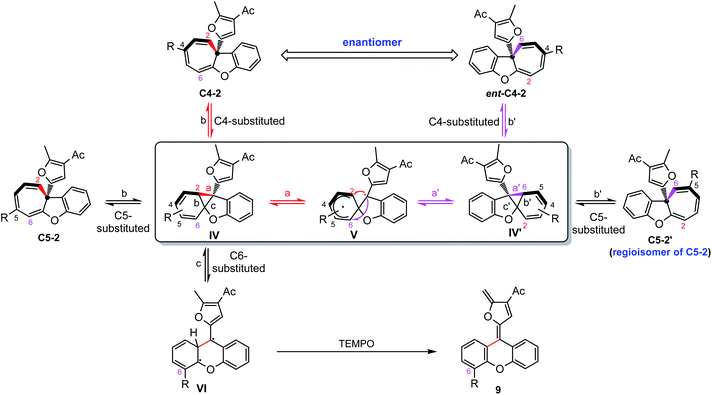 |
| | Scheme 5 Plausible mechanism for the racemization through diradical intermediates. | |
Derivatization of the cycloheptatriene skeleton
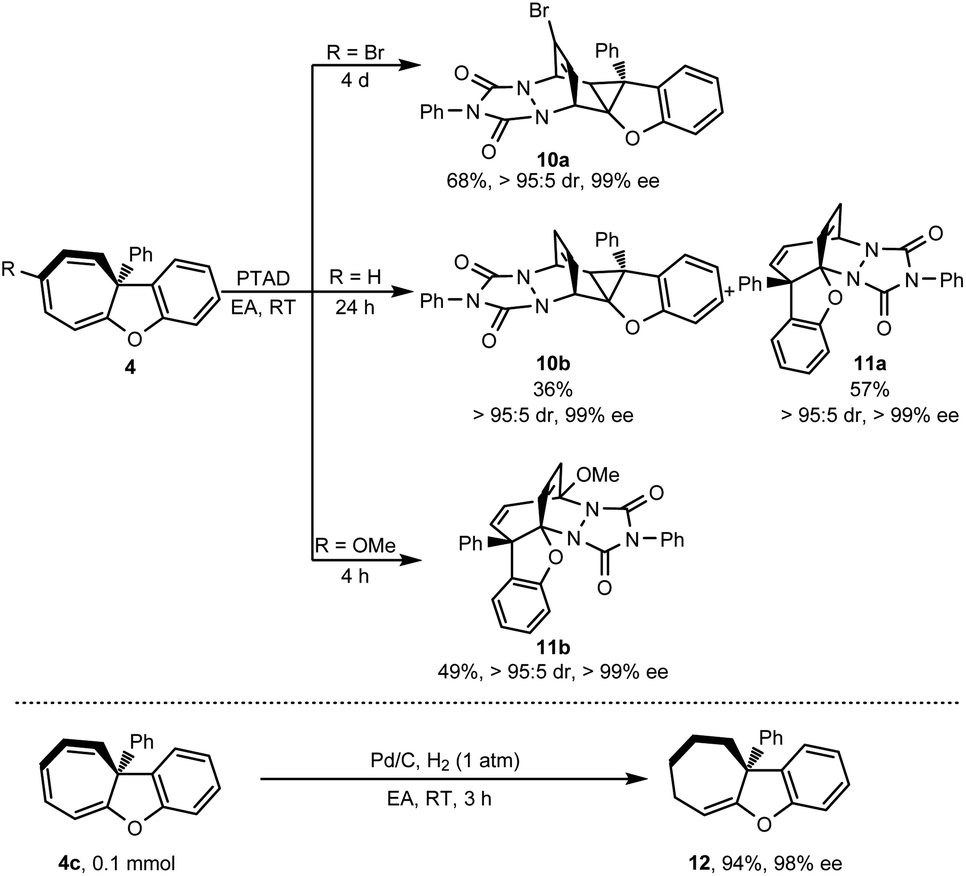
Cycloheptatriene is usually found as a useful skeleton in natural products, such as colchicine,28 hinokitiol29 and other series of tropolones and azaheptafulvenes.30 These kinds of compounds are also flexible to transform into other diverse structures. To demonstrate the synthetic potential of this methodology, [4 + 2] cycloaddition was conducted with PTAD (4-phenyl-1,2,4-triazoline-3,5-dione) as a dienophile. As shown in Scheme 6, when the R group was the Br atom, [4 + 2] cycloaddition proceeded via a norcaradiene intermediate because of the cycloheptatriene–norcaradiene (CHT–NCD) equilibrium.31 Only highly fused cyclopropane 10a was obtained as a single regioisomer in 68% yield with high stereoselectivity (>95:5 dr, 99% ee).
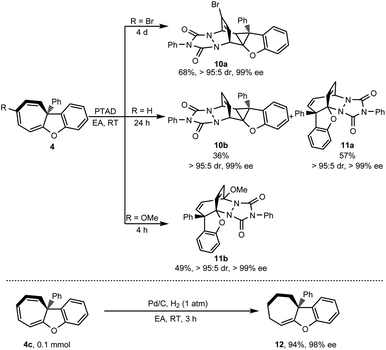 |
| | Scheme 6 Transformation of cycloheptatriene 4a. aThe reaction was conducted on 0.1 mmol scale, PTAD (1.05 equivalent). | |
Interestingly, when the R group was the H atom, cyclopropane 10b (36% yield) via norcaradiene addition competed with the bridged polycyclic product 11a (57% yield) which was derived from direct [4 + 2] cycloaddition.32 Both the addition products were achieved with high diastereoselectivity (>95:5 dr) and without loss of chiral integrity (99% ee). When a more electron-donating group such as OMe substituted cycloheptatriene was used, only direct [4 + 2] cycloaddition product 11b was isolated in 49% yield with excellent stereoselectivity (>95:5 dr, >99% ee). Besides the [4 + 2] cycloaddition, cycloheptatriene 4c was further hydrogenated in the presence of a catalytic amount of Pd/C under a hydrogen atmosphere (Scheme 6). This reaction selectively reduced two of three double bonds of cycloheptatriene and provided tetrahydro-7H-cyclohepta[b]benzofuran 12 in 94% yield while retaining the chiral integrity (98% ee).
Conclusion
In summary, we have developed a chiral dirhodium(II) tetracarboxylate-catalyzed enantioselective intramolecular reaction of arenes with donor–donor carbenes. This reaction represents the first enantioselective intramolecular Büchner reaction of donor–donor carbenes. When enynone tethered aryloxy substrates were employed as carbene precursors, the Büchner reaction took place smoothly. The desired furyl substituted cyclohepta[b]benzofuran polycyclic products were synthesised in good yields and moderate to excellent enantioselectivity. Aryloxy-substituted diazo compounds were also efficient donor–donor carbene precursors for this asymmetric Büchner reaction and phenyl substituted cyclohepta[b]benzofurans were obtained with high yields and excellent enantioselectivities. Moreover, when biaryl enynones were used as substrates, the aromatic substitution reaction rather than the Büchner reaction occurred to produce the furyl substituted fluorene derivatives with quantitative yields and excellent enantioselectivities. For furyl substituted chiral cyclohepta[b]benzofurans from para-substituted phenoxy enynones and diazo compounds, the loss of enantioselectivity was observed. A diradical mechanism was proposed to account for the racemization which was supported by the controlled experiments. The cycloheptatriene products could be further transformed into other polycyclic compounds by [4 + 2] cycloaddition and hydrogenation.
Data availability
All the data have been included in the ESI.†
Author contributions
D. Zhu performed all the experiments. T. Cao is appreciated for his help with English language revision. K. Chen performed the DFT calculations to indicate the mechanistic difference for the Büchner reaction and the aromatic substitution reaction. D. Zhu and S. Zhu contributed to the conception of the experiments, discussion of the results and preparation of manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We appreciate financial support from the National Natural Science Foundation of China (22071062, 21871096, and 22003077), Guangdong Science and Technology Department (2018B030308007), and the China Postdoctoral Science Foundation (2021M701244).
Notes and references
- Selected reviews:
(a) M. P. Doyle, Chem. Rev., 1986, 86, 919–939 CrossRef CAS;
(b) A. Padwa and S. F. Hornbuckle, Chem. Rev., 1991, 91, 263–309 CrossRef CAS;
(c) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–935 CrossRef CAS PubMed;
(d) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- Y.-P. Li, Z.-Q. Li and S.-F. Zhu, Tetrahedron Lett., 2018, 59, 2307–2316 CrossRef CAS.
-
(a) C. P. Park, A. Nagle, C. H. Yoon and C. Chen, J. Org. Chem., 2009, 74, 6231–6236 CrossRef CAS PubMed;
(b) J. Kim, Y. Ohk, S. H. Park, Y. Jung and S. Chang, Chem.–Asian J., 2011, 6, 2040–2047 CrossRef CAS PubMed;
(c) S. E. Reisman, R. R. Nani and S. Levin, Synlett, 2011, 2437–2442 CrossRef CAS;
(d) R. R. Nani and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7304–7311 CrossRef CAS PubMed;
(e) R. A. Brawn, K. Zhu and J. S. Panek, Org. Lett., 2014, 16, 74–77 CrossRef CAS PubMed.
- Selected examples:
(a) E. Büchner and T. Curtius, Ber. Dtsch. Chem. Ges., 1885, 18, 2377–2379 CrossRef;
(b) M. A. McKervey, S. M. Tuladhar and M. F. Twohig, J. Chem. Soc., Chem. Commun., 1984, 129–130 RSC;
(c) M. P. Doyle, M. S. Shanklin and H. Q. Pho, Tetrahedron Lett., 1988, 29, 2639–2642 CrossRef CAS;
(d) A. R. Maguire, N. R. Buckley, P. O'Leary and G. Ferguson, Chem. Commun., 1996, 2595–2596 RSC;
(e) O. A. McNamara, N. R. Buckley, P. O'Leary, F. Harrington, N. Kelly, S. O'Keeffe, A. Stack, S. O'Neill, S. E. Lawrence, C. N. Slattery and A. R. Maguire, Tetrahedron, 2014, 70, 6870–6878 CrossRef CAS;
(f) S. Mo and J. Xu, ChemCatChem, 2014, 6, 1679–1683 CrossRef CAS;
(g) S. Mo, X. Li and J. Xu, J. Org. Chem., 2014, 79, 9186–9196 CrossRef CAS PubMed;
(h) M. R. Rodríguez, Á. Beltrán, Á. L. Mudarra, E. Álvarez, F. Maseras, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2017, 56, 12842–12847 CrossRef PubMed;
(i) K. Ji and L. Zhang, Adv. Synth. Catal., 2018, 360, 647–651 CrossRef CAS PubMed.
- Selected examples:
(a) M. P. Doyle, M. S. Shanklin, H. Q. Pho and S. N. Mahapatro, J. Org. Chem., 1988, 53, 1017–1022 CrossRef CAS;
(b) F. Pan, S. Liu, C. Shu, R.-K. Lin, Y.-F. Yu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2014, 50, 10726–10729 RSC;
(c) L. Li, B. Zhou, Y.-H. Wang, C. Shu, Y.-F. Pan, X. Lu and L.-W. Ye, Angew. Chem., Int. Ed., 2015, 54, 8245–8249 CrossRef CAS PubMed;
(d) B. Seo, W. H. Jeon, J. Kim, S. Kim and P. H. Lee, J. Org. Chem., 2015, 80, 722–732 CrossRef CAS PubMed;
(e) A. Conde, G. Sabenya, M. Rodríguez, V. Postils, J. M. Luis, M. M. D.-Requejo, M. Costas and P. J. Pérez, Angew. Chem., Int. Ed., 2016, 55, 6530–6534 CrossRef CAS PubMed.
-
(a) M. Kennedy, M. A. McKervey, A. R. Maguire and G. H. P. Roos, J. Chem. Soc., Chem. Commun., 1990, 361–362 RSC;
(b) M. P. Doyle, D. G. Ene, D. C. Forbes and T. H. Pillow, Chem. Commun., 1999, 1691–1692 RSC;
(c) S. O'Keeffe, F. Harrington and A. R. Maguire, Synlett, 2007, 2367–2370 Search PubMed;
(d) S. O'Neill, S. O'Keeffe, F. Harrington and A. R. Maguire, Synlett, 2009, 2312–2314 Search PubMed;
(e) C. N. Slattery, L.-A. Clarke, S. O'Neill, A. Ring, A. Ford and A. R. Maguire, Synlett, 2012, 765–767 CAS;
(f) D. C. Crowley, D. Lynch and A. R. Maguire, J. Org. Chem., 2018, 83, 3794–3805 CrossRef CAS PubMed;
(g) T. Hoshi, E. Ota, Y. Inokuma and J. Yamaguchi, Org. Lett., 2019, 21, 10081–10084 CrossRef CAS PubMed;
(h) N. P. T. Thanh, M. Tone, H. Inoue, I. Fujisawa and S. Iwasa, Chem. Commun., 2019, 55, 13398–13401 RSC;
(i) N. Watanabe, Y. Ohtake, S. Hashimoto, M. Shiro and S. Ikegami, Tetrahedron Lett., 1995, 36, 1491–1494 CrossRef CAS;
(j) N. Watanabe, T. Ogawa, Y. Ohtake, S. Ikegami and S. Hashimoto, Synlett, 1996, 85–86 CrossRef CAS;
(k) H. Tsutsui, Y. Yamaguchi, S. Kitagaki, S. Nakamura, M. Anada and S. Hashimoto, Tetrahedron: Asymmetry, 2003, 14, 817–821 CrossRef CAS.
-
(a) Y. Wang, P. R. McGonigal, B. Herlé, M. Besora and A. M. Echavarren, J. Am. Chem. Soc., 2014, 136, 801–809 CrossRef CAS PubMed;
(b) H. Wang, C.-Y. Zhou and C.-M. Che, Adv. Synth. Catal., 2017, 359, 2253–2258 CrossRef CAS;
(c) Q. Zeng, K. Dong, J. Huang, L. Qiu and X. Xu, Org. Biomol. Chem., 2019, 17, 2326–2330 RSC.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed.
- F.-L. Hong, Z.-S. Wang, D.-D. Wei, T.-Y. Zhai, G.-C. Deng, X. Lu, R.-S. Liu and L.-W. Ye, J. Am. Chem. Soc., 2019, 141, 16961–16970 CrossRef CAS PubMed.
- K. Dong, X. Fan, C. Pei, Y. Zheng, S. Chang, J. Cai, L. Qiu, Z.-X. Yu and X. Xu, Nat. Commun., 2020, 11, 2363–2372 CrossRef CAS PubMed.
- B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2021, 60, 6864–6878 CrossRef CAS PubMed.
- D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Chem. Soc. Rev., 2020, 49, 908–950 RSC.
-
(a) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef PubMed;
(b) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902–912 CrossRef CAS PubMed;
(c) L. Zhang, Acc. Chem. Res., 2014, 47, 877–888 CrossRef CAS PubMed;
(d) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS PubMed.
- R. Jouhannet, S. Dagorne, A. Blanc and P. de Frémont, Chem. - Eur. J., 2021, 27, 9218–9240 CrossRef CAS PubMed.
-
(a) J. Ma, L. Zhang and S. Zhu, Curr. Org. Chem., 2016, 20, 102–118 CrossRef CAS;
(b) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1208–1262 CrossRef CAS;
(c) R. Vicente, J. González, L. Riesgo, J. González and L. A. López, Angew. Chem., Int. Ed., 2012, 51, 8063–8067 CrossRef CAS PubMed;
(d) J.-M. Yang, Z.-Q. Li, M.-L. Li, Q. He, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 3784–3789 CrossRef CAS PubMed.
-
(a) D. Zhu, J. Ma, K. Luo, H. Fu, L. Zhang and S. Zhu, Angew. Chem. Int. Ed., 2016, 55, 8452–8456 CrossRef CAS PubMed;
(b) D. Zhu, L. Chen, H. Zhang, Z. Ma, H. Jiang and S. Zhu, Angew. Chem., Int. Ed., 2018, 57, 12405–12409 CrossRef CAS PubMed.
-
(a) J. Ma, H. Jiang and S. Zhu, Org. Lett., 2014, 16, 4472–4475 CrossRef CAS PubMed;
(b) S. Zhu, X. Huang, T.-Q. Zhao, T. Ma and H. Jiang, Org. Biomol. Chem., 2015, 13, 1225–1233 RSC;
(c) J. Ma, K. Chen, H. Fu, L. Zhang, W. Wu, H. Jiang and S. Zhu, Org. Lett., 2016, 18, 1322–1325 CrossRef CAS PubMed;
(d) H. Luo, K. Chen, H. Jiang and S. Zhu, Org. Lett., 2016, 18, 5208–5211 CrossRef CAS PubMed;
(e) T. Cao, K. Chen and S. Zhu, Org. Chem. Front., 2017, 4, 450–454 RSC;
(f) R. Wu, K. Chen, J. Ma, Z.-X. Yu and S. Zhu, Sci. China: Chem., 2020, 63, 1230–1239 CrossRef CAS;
(g) R. Wu, J. Lu, T. Cao, J. Ma, K. Chen and S. Zhu, J. Am. Chem. Soc., 2021, 143, 14916–14925 CrossRef CAS PubMed;
(h) S. Qiu, X. Gao and S. Zhu, Chem. Sci., 2021, 12, 13730–13736 RSC;
(i) Y. Chen, P. Shen, T. Cao, H. Chen, Z. Zhao and S. Zhu, Nat. Commun., 2021, 12, 6165–6177 CrossRef CAS PubMed;
(j) X. Liu, X. Xu and W. Hu, Chin. J. Org. Chem., 2020, 40, 4370–4371 CrossRef CAS.
- When using 1q as the substrate under Rh(ii)-catalyzed conditions, N-Me C-H insertion gave 2q′ as the major product with 69% isolated yield and 96% ee. The desired cyclohepta[b]benzofuran 2q was only isolated with 25% yield and 76% ee.
- Z. Liu, H. Tan, L. Wang, T. Fu, Y. Xia, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 3056–3060 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, R. C. 01, Gaussian, Inc., Wallingford C. T., 2016 Search PubMed.
-
(a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed;
(d) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
-
(a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed;
(b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC;
(c) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- C. J. Moody, S. Miah, A. M. Z. Slawin, D. J. Mansfield and I. C. Richards, J. Chem. Soc., Perkin Trans. 1, 1998, 4067–4075 RSC.
- A. R. Maguire, P. O'Leary, F. Harrington, S. E. Lawrence and A. J. Blake, J. Org. Chem., 2001, 66, 7166–7177 CrossRef CAS PubMed.
- X. Xu, X. Wang, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2015, 17, 790–793 CrossRef CAS PubMed.
- A. Kless, M. Nendel, S. Wilsey and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 4524–4525 CrossRef CAS.
-
(a) W. S. Trahanovsky and T. J. Cassady, J. Am. Chem. Soc., 1984, 106, 8197–8201 CrossRef CAS;
(b) W. S. Trahanovsky, D. L. Miller and Y. Wang, J. Org. Chem., 1997, 62, 8980–8986 CrossRef CAS;
(c) C.-S. Huang, C.-C. Peng and C.-H. Chou, Tetrahedron Lett., 1994, 35, 4175–4176 CrossRef CAS.
- R. S. Nett, W. Lau and E. S. Sattely, Nature, 2020, 584, 148–153 CrossRef CAS PubMed.
- M. Miyashita, S. Hara and A. Yoshikoshi, J. Org. Chem., 1987, 52, 2602–2604 CrossRef CAS.
-
(a) J.-X. Zhao, Y.-Y. Fan, J.-B. Xu, L.-S. Gan, C.-H. Xu, J. Ding and J.-M. Yue, J. Nat. Prod., 2017, 80, 356–362 CrossRef CAS PubMed;
(b) D. R. Hirsch, D. V. Schiavone, A. J. Berkowitz, L. A. Morrison, T. Masaoka, J. A. Wilson, E. Lomonosova, H. Zhao, B. S. Patel, S. H. Datla, S. G. Hoft, S. J. Majidi, R. K. Pal, E. Gallicchio, L. Tang, J. E. Tavis, S. F. J. Le Grice, J. A. Beutler and R. P. Murelli, Org. Biomol. Chem., 2018, 16, 62–69 RSC;
(c) W. E. Truce and J. P. Shepherd, J. Am. Chem. Soc., 1977, 99, 6453–6454 CrossRef CAS;
(d) C. Zhao, S. A. Blaszczyk and J. Wang, Green Synth. Catal., 2021, 2, 198–215 CrossRef.
-
(a) T. Ito, S. Harada, H. Homma, H. Takenaka, S. Hirose and T. Nemoto, J. Am. Chem. Soc., 2021, 143, 604–611 CrossRef CAS PubMed;
(b) W. D. Mackay and J. S. Johnson, Org. Lett., 2016, 18, 536–539 CrossRef CAS PubMed.
- S. Wang, C. Rodríguez-Escrich and M. A. Pericàs, Angew. Chem., Int. Ed., 2017, 56, 15068–15072 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05374d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Shifa
Zhu
*b and
Shifa
Zhu