
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalytic enantioselective SN1-type dehydrative nucleophilic substitution: access to bis(indolyl)methanes bearing quaternary carbon stereocenters†
Wen-Run
Zhu‡
,
Qiong
Su‡
,
Xiao-Yi
Deng
 ,
Jia-Sheng
Liu
,
Tao
Zhong
,
Shan-Shui
Meng
,
Ji-Tao
Yi
,
Jiang
Weng
* and
Gui
Lu
*
,
Jia-Sheng
Liu
,
Tao
Zhong
,
Shan-Shui
Meng
,
Ji-Tao
Yi
,
Jiang
Weng
* and
Gui
Lu
*
Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, 510006, P. R. China. E-mail: wengj2@mail.sysu.edu.cn; lugui@mail.sysu.edu.cn
First published on 23rd November 2021
Abstract
A highly general and straightforward approach to access chiral bis(indolyl)methanes (BIMs) bearing quaternary stereocenters has been realized via enantioconvergent dehydrative nucleophilic substitution. A broad range of 3,3′-, 3,2′- and 3,1′-BIMs were obtained under mild conditions with excellent efficiency and enantioselectivity (80 examples, up to 98% yield and >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er). By utilizing racemic 3-indolyl tertiary alcohols as precursors of alkyl electrophiles and indoles as C–H nucleophiles, this organocatalytic strategy avoids pre-activation of substrates and produces water as the only by-product. Mechanistic studies suggest a formal SN1-type pathway enabled by chiral phosphoric acid catalysis. The practicability of the obtained enantioenriched BIMs was further demonstrated by versatile transformation and high antimicrobial activities (3al, MIC: 1 μg mL−1).
:1 er). By utilizing racemic 3-indolyl tertiary alcohols as precursors of alkyl electrophiles and indoles as C–H nucleophiles, this organocatalytic strategy avoids pre-activation of substrates and produces water as the only by-product. Mechanistic studies suggest a formal SN1-type pathway enabled by chiral phosphoric acid catalysis. The practicability of the obtained enantioenriched BIMs was further demonstrated by versatile transformation and high antimicrobial activities (3al, MIC: 1 μg mL−1).
Introduction
Quaternary carbon stereocenters are frequently found in numerous natural products, pharmaceuticals and biologically active compounds.1 The catalytic and enantioselective construction of these stereocenters pose a long-standing challenge, especially in acyclic systems.2 In the past decade, various elegant approaches have been developed to address this challenge.3 Among them, catalytic enantioconvergent substitution reactions of racemic tertiary alkyl electrophiles have always been considered as an efficient and versatile approach to access quaternary stereocenters (Fig. 1a, left).4 However, these reactions generally require a good leaving groups (LG = halides, OAc, OTf, etc.) to promote carbon–carbon bond formation. A representative example is the work of Jacobsen, who developed a pioneering enantioconvergent catalytic SN1 reaction of propargyl acetates using allyl silane as a nucleophile.4k From the viewpoint of synthetic simplicity and efficiency, the dehydrative nucleophilic substitution of readily available tertiary alcohols (LG = OH) with C–H nucleophiles represents a straightforward and economical means for constructing quaternary carbon stereocenters (Fig. 1a, right).5 Despite its attractive potential, this type of reaction has been applied less and enantioselective examples of such process remain rare.6–8 Towards this end, Sun8 and coworkers have elegantly developed formal dehydrative SN1 substitutions of 1,1-diaryl tertiary alcohols for the highly enantioselective formation of acyclic all-carbon quaternary stereocenters, but a directing group was needed. | ||
| Fig. 1 Synthetic strategies to all-carbon quaternary stereogenic center. | ||
Bis(indolyl)methanes (BIMs) are privileged structural motifs in natural products and bioactive molecules with significant antibacterial, anticancer, and anti-inflammatory activities (Scheme 1a).9 Therefore, the development of efficient approaches for BIMs have drawn much attention from the synthetic community. However, these established methods are mainly focused on the construction of symmetrical BIMs bearing tertiary stereocenters,10,11 and the asymmetric synthesis of enantioenriched BIMs bearing quaternary carbon centers still remain less explored.12,13 Moreover, existing synthetic methods are largely limited to symmetrical and unsymmetrical 3,3′-BIMs. In contrast, other types of BIMs (such as 3,1′-, 3,2′- and 3,n′-BIMs) received less attention and their asymmetric synthesis using the same strategy has remained unstudied, although these BIMs exhibit potential biological activities and deserve special attention as well. Therefore, a broadly applicable synthetic strategy for a wide variety of chiral BIMs would be of high importance.
 | ||
| Scheme 1 Representative bis(indolyl)methanes (BIMs) in our current work. | ||
Given the significance of both the dehydrative nucleophilic substitution and bis(indolyl)methanes (BIMs), herein we report the direct SN1-type dehydrative nucleophilic substitution of racemic 3′-indolyl tertiary alcohols with various indole nucleophiles catalyzed by chiral phosphoric acids, providing various BIMs bearing quaternary stereogenic carbon centers with good to excellent enantioselectivities; remarkably, we establish a unified strategy for the first time to enable the asymmetric synthesis of a wide range of enantioenriched 3,3′-, 3,2′- and 3,1′-BIMs under mild conditions (Scheme 1b). We further illustrate that the BIM products could be transformed into other valuable enantioenriched molecules bearing all-carbon quaternary centers such as chiral triarylmethanes that are difficult to access. Moreover, preliminary biological activities demonstrate that these chiral BIMs are potential antibacterial agents with MIC (minimum inhibitory concentration) of up to 1 μg mL−1 against Bacillus subtilis.
Results and discussion
To initiate our investigation, racemic α-indolyl-α-trifluoromethyl propargylic alcohol 1a was selected as the model substrate in consideration of the privileged role of the CF3 group in medicinal chemistry14,15 and the versatile alkynyl handle for further diversification.16 Moreover, we envisioned that the Brønsted acid catalyst17 could activate the tertiary alkyl alcohol 1avia dehydration to form the corresponding tertiary carbocation intermediate, which is stabilized by its iminium type resonance form. Furthermore, the chiral Brønsted acid catalyst is expected to achieve efficient chiral induction through ion-pair or hydrogen-bonding with the corresponding carbocation and indole nucleophile simultaneously, to deliver the desired all-carbon quaternary stereocenter. To test this hypothesis, BINOL-derived chiral phosphoric acid (R)-A1 was initially employed as the catalyst to promote the reaction of 1a with 3-methylindole 2a, and the reaction proceeded smoothly to afford 3,2′-bis(indolyl)methane 3a in 90% yield with 64:36 er (Table 1, entry 1). Next, a series of BINOL- and SPINOL-derived chiral phosphoric acid catalysts were examined to improve the enantioselectivities (Table 1, entries 1–11). We were pleased to find that catalyst (R)-A6 bearing a perfluorophenyl substituent at the 3,3′-position of the BINOL backbone showed a significant increase in enantioselectivity (91:9 er) with 90% yield (Table 1, entry 6). Interestingly, the catalyst loading was successfully reduced to 5 mol% without any loss in chemical efficiency and enantioselectivity (entry 12). Various solvents, reaction temperatures, concentrations and additives were further investigated in the presence of 5 mol% (R)-A6 (Table 1, entries 13–16. For details, see the ESI†). Finally, the highest yield (92%) and the best enantioselectivity (96:4 er) were achieved when the reaction was catalyzed by 5 mol% (R)-A6 in DCM (3 mL) at 10 °C with MgSO4 (35 mg) as the additive (Table 1, entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent (× mL) | Time | Yieldb (%) | er |
|
a Unless otherwise specified, all reactions were carried out with the catalyst (10 mol%), 1a (0.10 mmol) and 2a (0.12 mmol) in DCM (× mL) at room temperature.
b Isolated yield of 3a.
c Determined by chiral-phase HPLC analysis.
d Catalyst (5 mol%) was used.
e MgSO4 (35 mg) was used.
f Performed at 10 °C.
g
1a:2a (1:1) was used.
|
|||||
| 1 | (R)-A1 | DCM (1) | 20 h | 90 | 64:36 |
| 2 | (R)-A2 | DCM (1) | 72 h | Trace | — |
| 3 | (R)-A3 | DCM (1) | 20 h | 90 | 56:44 |
| 4 | (R)-A4 | DCM (1) | 20 h | 88 | 61:39 |
| 5 | (R)-A5 | DCM (1) | 72 h | Trace | — |
| 6 | (R)-A6 | DCM (1) | 20 h | 90 | 91:9 |
| 7 | (R)-A7 | DCM (1) | 20 h | 68 | 53:47 |
| 8 | (R)-A8 | DCM (1) | 20 h | 65 | 65:35 |
| 9 | (R)-A9 | DCM (1) | 20 h | 53 | 64:36 |
| 10 | (R)-B1 | DCM (1) | 72 h | Trace | — |
| 11 | (R)-B2 | DCM (1) | 72 h | Trace | — |
| 12d | (R)-A6 | DCM (1) | 20 h | 90 | 91:9 |
| 13d | (R)-A6 | DCM (3) | 24 h | 89 | 93:7 |
| 14d,e | (R)-A6 | DCM (3) | 20 h | 90 | 94:6 |
| 15d,e,f | (R)-A6 | DCM (3) | 5 d | 92 | 95:5 |
| 16d,e,f,g | (R)-A6 | DCM (3) | 5 d | 92 | 96:4 |
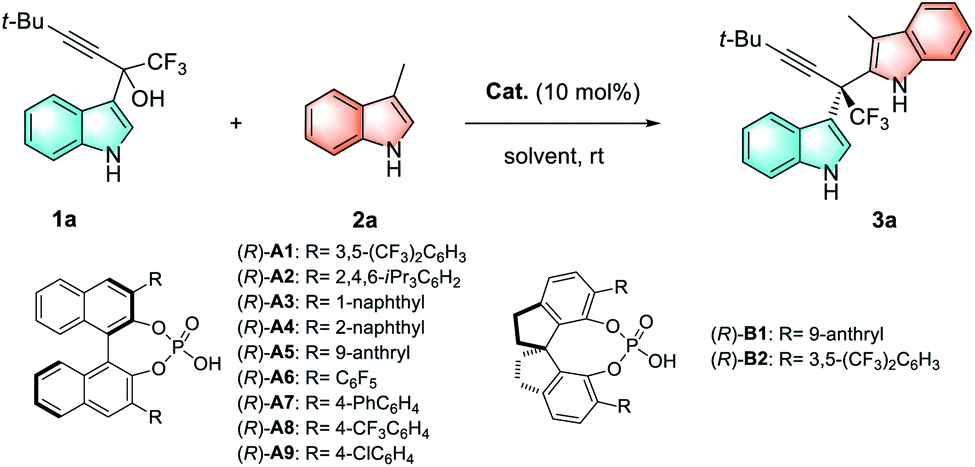
Having established the optimal reaction conditions, we then proceed to examine the reaction scope (Tables 2–4). First, we evaluated the method for the synthesis of chiral 3,2′-BIMs bearing all-carbon quaternary stereocenters. As illustrated in Table 2, a broad range of 3-methylindoles 2 bearing either electron-donating or -withdrawing groups at different positions of the indole ring (C4, C5 and C6) participated in this reaction, furnishing corresponding 3,2′-BIMs (3a–3j) in 62–98% yields with 87:13–98:2 ers. Next, various α-indolyl propargylic alcohols 1 bearing substituents on the indolyl moiety were investigated, and excellent enantioselectivities (95:5–98:2 ers) were obtained for the corresponding 3,2′-BIMs (3k–3ab).
| a Unless otherwise specified, all reactions were carried out with the catalyst (5 mol%), 1 (0.10 mmol), 2 (0.10 mmol) and MgSO4 (35 mg) in DCM (3 mL) at 10 °C for 12–288 h. b Carried out with the catalyst (10 mol%), 1 (0.10 mmol), 2 (0.12 mmol) and MgSO4 (25 mg) in DCM (1 mL) at rt. c Carried out with the catalyst (10 mol%), 1 (0.10 mmol), 2 (0.10 mmol) and MgSO4 (35 mg) in DCM (3 mL) at rt. |
|---|

|
Notably, α-ethoxycarbonyl substituted propargylic alcohol was used to produce 3s in 85% yield and 95:5 er. In addition, a scale-up reaction for the synthesis of 3t was also successfully performed with similar results. To our delight, the R3 group of 1 could be expanded from alkyne to various alkenyl, alkyl and aryl functionalities. When racemic allyl alcohols were employed, alkenyl substituent products 3ac–3ak were afforded in up to 95% yield and 96:4 er, while moderate yields and enantioselectivities were observed for alkyl-substituted tertiary alcohols. Finally, triarylmethane 3an could also be obtained with high yield and excellent enantioselectivity (98:2 er). The absolute configuration of 3t was determined to be R based by X-ray crystallographic analysis after derivatization, and those of other chiral 3,2′-BIMs were assigned analogously.18
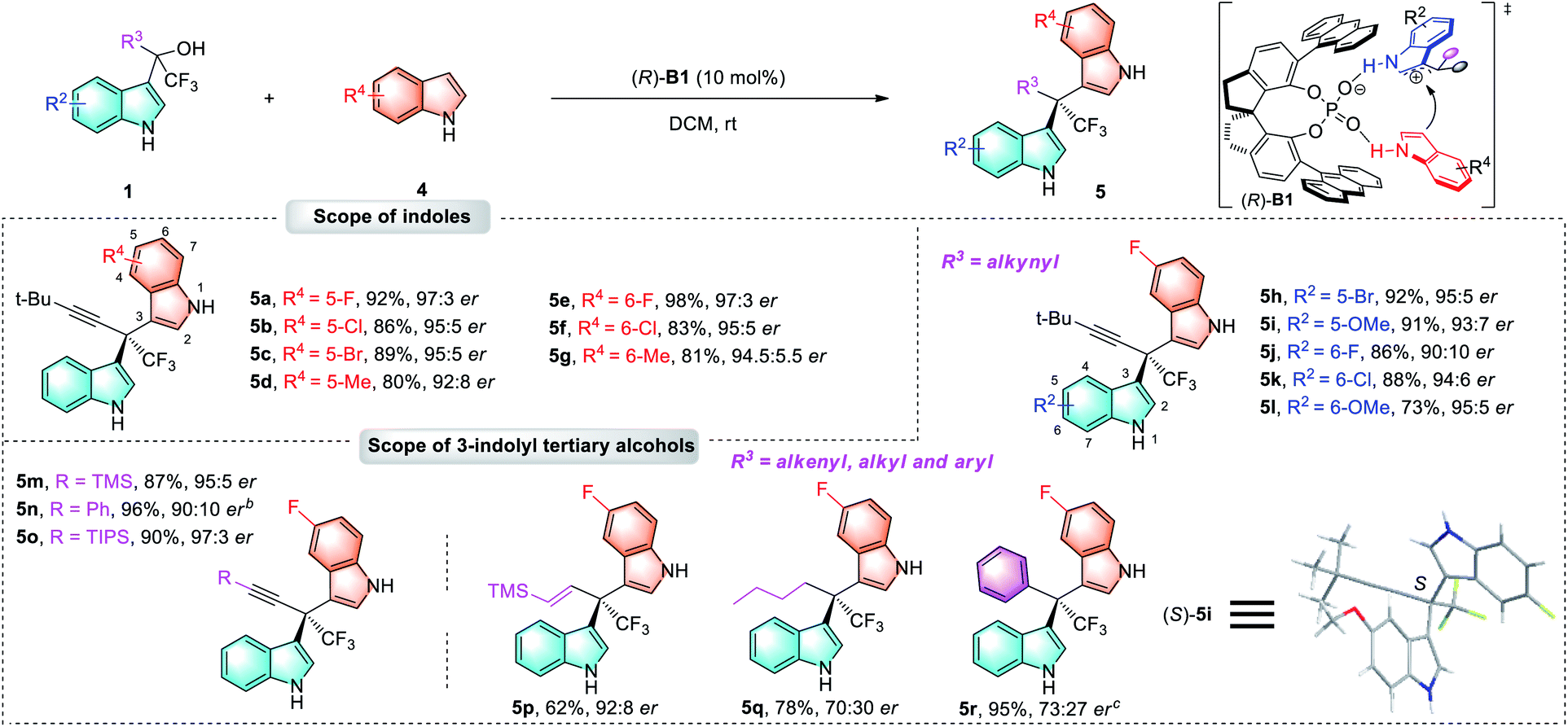
As a significant extension of this asymmetric catalytic method, we then turned our attention to the construction of more challenging chiral 3,3′-BIMs which have two similar indole rings. To our disappointment, the optimal catalyst (R)-A6 for chiral 3,2′-BIMs failed to deliver chiral 3,3′-BIM products with good enantioselectivity (Table S2 in the ESI†). After slight modification of the reaction conditions (see ESI for details†), the highest enantioselectivity of 97:3 er with 92% yield was achieved when the reaction was catalyzed by spirocyclic phosphoric acid (R)-B1 (10 mol%) in DCM at room temperature (5a, Table 3). Next, the scope of the enantioenriched 3,3′-BIMs was investigated. As shown in Table 3, various substituted groups on the indole 4 could be well tolerated (5a–5g, 80%–98% yields, 92:8–97:3 ers). Similarly, this process was applicable to a wide range of tertiary alcohols 1 bearing various R2/R3 substituents, which afforded products 5h–5r in generally high yields (62%–96%), and moderate to excellent enantioselectivities (70:30–97:3 ers). A single crystal X-ray analysis of 5i established the absolute configuration of the 3,3′-BIM products.18
| a Unless otherwise specified, all reactions were carried out with the catalyst (10 mol%), 1 (0.10 mmol) and 4 (0.12 mmol) in DCM (1 mL) at room temperature for 10–240 h. b Carried out with the catalyst (5 mol%), 1 (0.10 mmol) and 4 (0.12 mmol) in DCM (1 mL) at rt. c Carried out with catalyst (R)-A6 (10 mol%), 1 (0.10 mmol) and 4 (0.12 mmol) in toluene (1 mL) at 80 °C. |
|---|
|
During the investigation of the nucleophilic substitution reaction of 3-methylindole 2a with α-indolyl propargylic alcohol 1ae, an unexpected cyclic 3,1′-BIM 7a was isolated in 30% yield with 53:47 er under the standard conditions shown in Table 2, which was proposed to be formed through the intramolecular hydroamination of the allene intermediate 8a (Scheme 2). Encouraged by this interesting result, we next turned our attention to improve the enantioselectivity of this reaction. After optimization (see the ESI for details†), 3,1′-BIM 7a could be afforded in 86% yield and 97:3 er when the reaction was performed at room temperature with (R)-A7 as the catalyst (Table 4, 7a).
 | ||
| Scheme 2 Model reaction employed for condition optimization. | ||
| a Unless otherwise specified, all reactions were carried out with the catalyst (10 mol%), 1 (0.10 mmol) and 2 (0.12 mmol) in DCM (1 mL) at room temperature for 10–96 h. |
|---|

|
Next, the scope of cyclic 3,1′-BIMs was explored. As shown in Table 4, various indoles 2 with different R1/R4 substituents produced the desired 3,1′-BIMs 7a–7j in 45%–96% yields with 95:5–98:2 ers. Different substituents including halogens, methyl and methoxyl on the phenyl ring of the indolyl moiety were all compatible, giving 7k–7w in 68%–98% yields with an excellent stereocontrol (92.5:7.5–>99:1 ers). Besides, when α–CHF2-substituted propargylic alcohol was subjected to this process, the corresponding 3,1′-BIM 7x was obtained in 76% yield with 96:4 er. The absolute configurations of these 3,1′-BIMs were assigned based on the X-ray crystal structure of compound 18 (derivated from 7a, Scheme 4).18
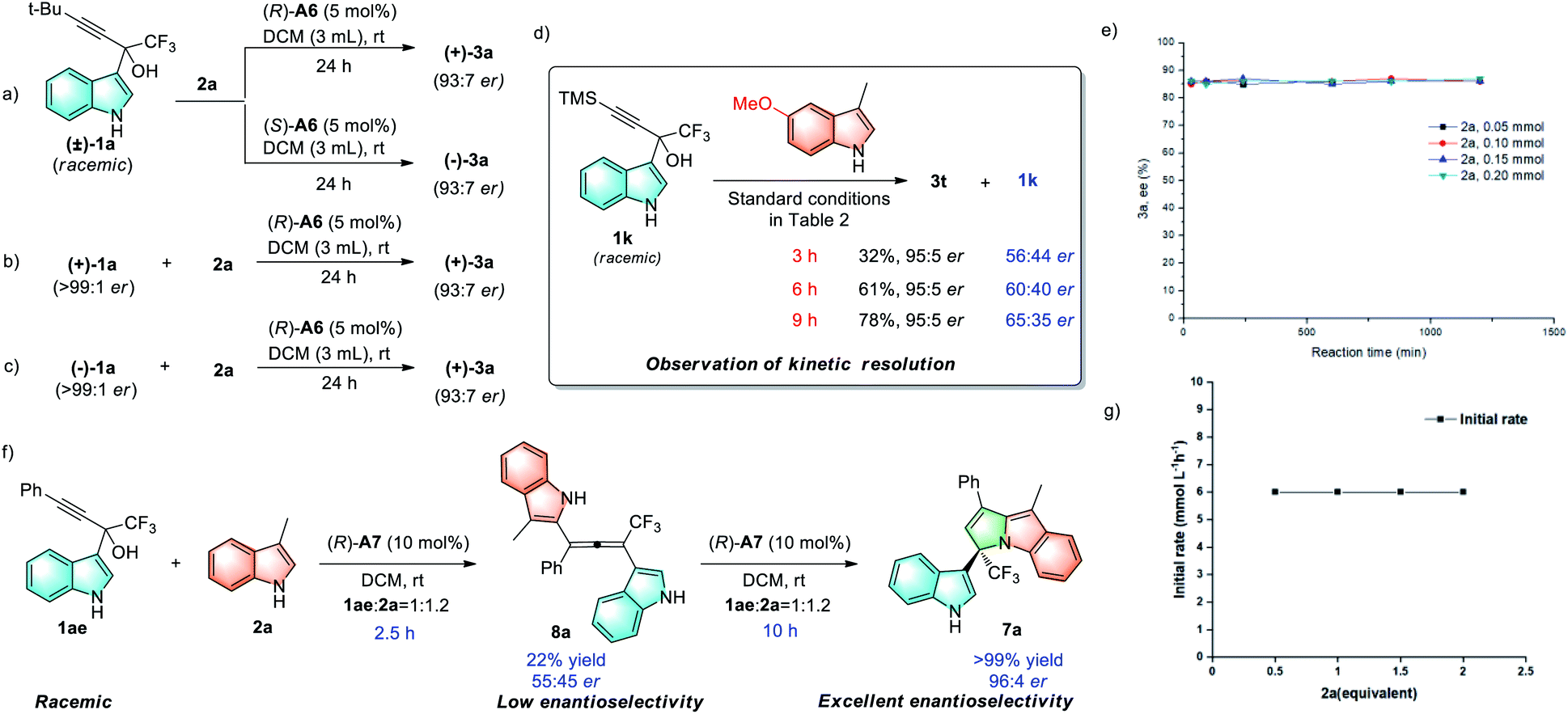
To gain some insights into the mechanism, a series of control experiments were performed. First, the products with opposite chirality can be isolated in a similar yield after subjecting racemic 1a to the chiral (R)- or (S)-phosphoric acid catalyst A6 (Scheme 3a). Using the same (R)-A6 catalyst, both (+)-1a and (−)-1a reacted with 2a to deliver product (+)-3a with the same configuration (Schemes 3b and 3c). Based on these results, an enantiospecific mechanism could be ruled out. Moreover, under the standard conditions, substrate 1k was observed to undergo a partial kinetic resolution process (Scheme 3d). However, variations in the initial concentrations of 2a (with the same initial concentration of 1a) produce good consistency in the reaction rate, which shows that the reaction is zeroth-order for 2a, and it has no rate dependence on the initial concentration of the nucleophile 2a (Scheme 3g). Meanwhile, reactions carried out with different initial concentrations of 2a produce good overlay in the enantiomeric excess data of product 3a, indicating no catalyst decomposition or product inhibition in this process (Scheme 3e). When taken together, these experimental results excluded the SN2 mechanism coupled with dynamic kinetic resolution.
 | ||
| Scheme 3 Control experiments and mechanistic studies. | ||
 | ||
| Scheme 4 Synthetic transformations of compounds 3ac, 3t, 5m and 7a. | ||
To explore the origin of the excellent stereocontrol in the formation of chiral cyclic 3,1′-BIMs, control experiments were carried out using 1ae and 2a under the standard reaction conditions (Scheme 3f). After 2.5 hours, the allene intermediate 8a can be isolated in 22% yield with low enantioselectivity (55:45 er). Subjecting 8a to the standard conditions provided 7a with comparable yield and enantioselectivity (96:4 er) to those observed in the one-pot as shown process in Table 4 (see the ESI for details†), thus indicating that hydroamination of the alkene intermediate is the enantiodetermining step (Scheme 3f, right).
Next, ten randomly selected chiral BIMs (3l, 3o, 3u, 3y, 3ad, 3ah, 3al, 5m, 7a and 7g) were evaluated for their antimicrobial activities with the FDA-approved broad-spectrum antimicrobial drug ampicillin or vancomycin as the positive control. We first screened their antibacterial activities against escherichia coli and bacillus subtilis with ampicillin as the positive control. As shown in Table 5, 3,2′-BIMs 3o and 3u exhibited antibacterial effects towards escherichia coli. Meanwhile, all selected compounds showed remarkable antibacterial activities against bacillus subtilis (MIC: 1–8 μg mL−1). Notably, 3,2′-BIM 3al bearing a butyl group was 2 times more potent than ampicillin with a MIC value of 1 μg mL−1. We further examined their antibacterial potency against methicillin-resistant staphylococcus aureus (MRSA) using vancomycin as the reference. To our delight, most of these compounds showed impressive antibacterial effects against MRSA (MIC: 2–16 μg mL−1), especially compounds 3al and 5m (MIC: 2–4 μg mL−1). These preliminary antibacterial tests indicated that enantioenriched BIMs might be developed as novel antibacterial agents.
| Compound | MIC (μg mL−1) | ||
|---|---|---|---|
| Escherichia coli | Bacillus subtilis | Methicillin-resistant Staphylococcus aureus | |
| a MIC values were obtained by the microdilution method following CLSI standards and are given in μg mL−1. | |||
| 3l | 64 | 4 | 16 |
| 3o | 8 | 4 | 16 |
| 3u | 16 | 2 | 16 |
| 3y | >64 | 8 | >64 |
| 3ad | 64 | 2 | 16 |
| 3ah | 64 | 8 | >64 |
| 3al | >64 | 1 | 2 |
| 5m | >64 | 4 | 4 |
| 7a | 64 | 4 | 16 |
| 7g | 64 | 8 | 16 |
| Ampicillin | 2 | 2 | — |
| Vancomyci | — | — | 1 |
To illustrate the synthetic utilities of this new methodology, we have also tried to transform these 3,n′-BIM products into a variety of useful enantioenriched compounds with quaternary stereocenters. In the case of 3ac, its methoxyl group can be smoothly converted into triflate (9) in good yield without loss of enantiomeric purity, which provides more possibilities for further functionalization. For 3,2′-BIM 3t, regioselective protection of the indole NH moiety provided 10; hydrogenation of the alkyne afforded α-alkyl BIM 11; removal of the TMS group generated terminal alkyne 12, which underwent either a Cu-catalyzed azide–alkyne cycloaddition (CuAAC) to deliver triarylmethanes 13a–13c, or an intramolecular cyclization to give 14 with a pyrrolo[1,2-a]indole scaffold (Scheme 4a). Furthermore, 3,3′-BIM 5m with the TMS-protected alkyne moiety can be transformed into aryl alkyne 15 through desilylation followed by Sonogashira coupling (Scheme 4b). The synthetic utility of 3,1′-BIM 7a was also demonstrated by N–H protection, selective hydrogenation and bromination (Scheme 4c).
Conclusions
In conclusion, we have developed an enantioselective SN1-type dehydration nucleophilic substitution of racemic tertiary alcohols with various substitution patterns (Csp, Csp2 and Csp3). Under chiral phosphoric acid catalysis, a wide range of readily available racemic 3-indolyl tertiary alcohols were attacked by indoles to produce acyclic bis(indolyl)methanes (3,2′-BIMs and 3,3′-BIMs) and cyclic 3,1′-BIMs bearing quaternary stereogenic centers with high efficiencies and enantioselectivities (80 examples, up to 98% yield and >99:1 er). The rich chemistry of the indolyl, alkynyl, and other functional groups in the obtained BIMs allows for facile transformation into valuable chiral compounds bearing quaternary centers that are otherwise difficult to access. Mechanism investigations exclude the SN2 mechanism and suggest a formal SN1-type pathway enabled by CPA catalysts. Preliminary biological evaluation demonstrated that these enantioenriched BIMs exhibited impressive preliminary antibacterial activities, and notably 3al is 2 times more potent than ampicillin against bacillus subtilis with a MIC value of 1 μg mL−1. This strategy not only provides an efficient and unified method for accessing valuable chiral BIMs, but also opens opportunities for asymmetric dehydrative cross-coupling reactions employing tertiary alcohols as competent electrophiles without pre-activation.
Data availability
The ESI† include experimental detail, HPLC data, NMR data and HRMS data.Author contributions
W.-R. Zhu and Q. Su conceived the project, performed the experiments, and wrote the manuscript. X.-Y. Deng, T. Zhong, and J.-T. Yi. Carried out experiments and prepared the ESI.† J.-S. Liu performed the experiments of antimicrobial activities. J. Weng, S.-S. Meng, and G. Lu analyzed the results. G. Lu, and J. Weng conceived and directed the project and wrote the paper. All authors contributed to discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 81972824), Guangdong Basic and Applied Basic Research Foundation (No 2020A1515011513 and 2020A1515010684), Guangdong Provincial Key Laboratory of Chiral Molecule and Drug Discovery (No 2019B030301005), and National Engineering and Technology Research Center for New Drug Druggability Evaluation (Seed Program of Guangdong Province, No 2017B090903004). We are grateful to Prof. Ming Lu and Shu-Ting Li (SYSU) for their help in the single crystal preparation. We are also thankful to Prof. Ge Zhang (SYSU) for her help in the evaluation of antimicrobial activities, and to Prof. Hong-Gen Wang (SYSU) for helpful discussion.Notes and references
- (a) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS; (b) C. Li, S. S. Ragab, G. Liu and W. Tang, Nat. Prod. Rep., 2020, 37, 276–292 RSC.
- For selected reviews, see: (a) K. Fuji, Chem. Rev., 2002, 93, 2037–2066 CrossRef; (b) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC; (c) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS; (d) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef CAS.
- For selected recent reviews, see: (a) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS; (b) X. Zhang and C.-H. Tan, Chem, 2021, 7, 1451–1486 CrossRef CAS; (c) J. Sietmann and J. M. Wiest, Angew. Chem., Int. Ed., 2020, 59, 6964–6974 CrossRef CAS; For some recent examples, see: (d) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340–344 CrossRef CAS; (e) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef CAS; (f) B. W. H. Turnbull and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 6156–6159 CrossRef CAS; (g) Z. Wang, Y. Zhu, X. Pan, G. Wang and L. Liu, Angew. Chem., Int. Ed., 2020, 59, 3053–3057 CrossRef CAS; (h) D. Ma, C. B. Miao and J. Sun, J. Am. Chem. Soc., 2019, 141, 13783–13787 CrossRef CAS PubMed; (i) V. Lanke and I. Marek, J. Am. Chem. Soc., 2020, 142, 5543–5548 CrossRef CAS PubMed; (j) M. Isomura, D. A. Petrone and E. M. Carreira, J. Am. Chem. Soc., 2021, 143, 3323–3329 CrossRef CAS; (k) S. Liu, Y. Tanabe, S. Kuriyama, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2021, 60, 11231–11236 CrossRef CAS; (l) K. Liao, Y. Gong, R.-Y. Zhu, C. Wang, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2021, 60, 8488–8493 CrossRef CAS.
- For selected reviews, see: (a) C. H. Ding and X. L. Hou, Chem. Rev., 2011, 111, 1914–1937 CrossRef CAS; (b) W. Xue, X. Jia, X. Wang, X. Tao, Z. Yin and H. Gong, Chem. Soc. Rev., 2021, 50, 4162–4184 RSC; For selected examples, see: (c) B. M. Trost and C. Jiang, J. Am. Chem. Soc., 2001, 123, 12907–12908 CrossRef CAS PubMed; (d) S. Ma, X. Han, S. Krishnan, S. C. Virgil and B. M. Stoltz, Angew. Chem., Int. Ed., 2009, 48, 8037–8041 CrossRef CAS PubMed; (e) P. Zhang, H. Le, R. E. Kyne and J. P. Morken, J. Am. Chem. Soc., 2011, 133, 9716–9719 CrossRef CAS PubMed; (f) P. A. Evans, S. Oliver and J. Chae, J. Am. Chem. Soc., 2012, 134, 19314–19317 CrossRef CAS PubMed; (g) H. Zhang, L. Hong, H. Kang and R. Wang, J. Am. Chem. Soc., 2013, 135, 14098–14101 CrossRef CAS PubMed; (h) A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257–11260 CrossRef CAS PubMed; (i) K. Tsuchida, Y. Senda, K. Nakajima and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 9728–9732 CrossRef CAS; (j) X. Liu, P. Wang, L. Bai, D. Li, L. Wang, D. Yang and R. Wang, ACS Catal., 2018, 8, 10888–10894 CrossRef CAS; (k) A. E. Wendlandt, P. Vangal and E. N. Jacobsen, Nature, 2018, 556, 447–451 CrossRef CAS; (l) X. Zhang, R. Ren, S. M. Tan, D. Tan, R. Lee and C.-H. Tan, Science, 2019, 363, 400–404 CrossRef CAS PubMed; (m) V. Lanke and I. Marek, Chem. Sci., 2020, 11, 9378–9385 RSC; (n) Z. Wang, Z.-P. Yang and G. C. Fu, Nat. Chem., 2021, 13, 236–242 CrossRef CAS.
- For selected reviews on the dehydrative nucleophilic substitution, see: (a) R. Kumar and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 1121–1146 RSC; (b) J. Moran, M. Dryzhakov and E. Richmond, Synthesis, 2016, 48, 935–959 CrossRef; (c) S. O'Neill, J. Rodriguez and M. A. Walczak, Chem.–Asian J., 2018, 13, 2978–2990 CrossRef; (d) S. Estopina-Duran and J. E. Taylor, Chem.–Eur. J., 2021, 27, 106–120 CrossRef CAS PubMed; (e) A. Gualandi, G. Rodeghiero and P. G. Cozzi, Asian J. Org. Chem., 2018, 7, 1957–1981 CrossRef CAS.
- For selected examples on the synthesis of tertiary stereocenters via asymmetric dehydrative nucleophilic substitution, see: (a) P. G. Cozzi, F. Benfatti and L. Zoli, Angew. Chem., Int. Ed., 2009, 48, 1313–1316 CrossRef CAS; (b) B. Xu, Z.-L. Guo, W.-Y. Jin, Z.-P. Wang, Y.-G. Peng and Q.-X. Guo, Angew. Chem., Int. Ed., 2012, 51, 1059–1062 CrossRef CAS; (c) Z. Lai, Z. Wang and J. Sun, Org. Lett., 2015, 17, 6058–6061 CrossRef CAS PubMed; (d) S. Tanaka and M. Kitamura, Chem. Rec., 2021, 21, 1385–1397 CAS.
- For selected examples on the synthesis of quaternary stereocenters in cyclic systems via asymmetric dehydrative nucleophilic substitution, see: (a) C. Guo, J. Song, J. Z. Huang, P. H. Chen, S. W. Luo and L. Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 1046–1050 CrossRef CAS; (b) Y. Liu, H. H. Zhang, Y. C. Zhang, Y. Jiang, F. Shi and S. J. Tu, Chem. Commun., 2014, 50, 12054–12057 RSC; (c) C. Ma, F. Jiang, F.-T. Sheng, Y. Jiao, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 3014–3020 CrossRef CAS PubMed; (d) L. Song, Q. X. Guo, X. C. Li, J. Tian and Y. G. Peng, Angew. Chem., Int. Ed., 2012, 51, 1899–1902 CrossRef CAS.
- For selected examples on the synthesis of quaternary stereocenters in acyclic systems via asymmetric dehydrative nucleophilic substitution, see: (a) Z. Wang, Y. F. Wong and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 13711–13714 CrossRef CAS; (b) W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910–1913 CrossRef CAS; (c) X. Li, M. Duan, Z. Deng, Q. Shao, M. Chen, G. Zhu, K. N. Houk and J. Sun, Nat. Catal., 2020, 3, 1010–1019 CrossRef CAS.
- (a) X. He, S. Hu, K. Liu, Y. Guo, J. Xu and S. Shao, Org. Lett., 2006, 8, 333–336 CrossRef; (b) H. J. Kim, H. Lee, J. H. Lee, D. H. Choi, J. H. Jung and J. S. Kim, Chem. Commun., 2011, 47, 10918–10920 RSC; (c) R. Bell, S. Carmeli and N. Sar, J. Nat. Prod., 1994, 57, 1587–1590 CrossRef CAS; (d) G. Bifulco, I. Bruno, L. Minale, R. Riccio, A. Calignano and C. Debitus, J. Nat. Prod., 1994, 57, 1294–1299 CrossRef CAS PubMed; (e) G. Bifulco, I. Bruno, R. Riccio, J. Lavayre and G. Bourdy, J. Nat. Prod., 1995, 58, 1254–1260 CrossRef CAS; (f) N. Ichite, M. B. Chougule, T. Jackson, S. V. Fulzele, S. Safe and M. Singh, Clin. Cancer Res., 2009, 15, 543–552 CrossRef CAS; (g) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2010, 110, 2250–2293 CrossRef CAS PubMed; (h) M. Marrelli, X. Cachet, F. Conforti, R. Sirianni, A. Chimento, V. Pezzi, S. Michel, G. A. Statti and F. Menichini, Nat. Prod. Res., 2013, 27, 2039–2045 CrossRef CAS PubMed; (i) S. Roy, R. Gajbhiye, M. Mandal, C. Pal, A. Meyyapan, J. Mukherjee and P. Jaisankar, Med. Chem. Res., 2014, 23, 1371–1377 CrossRef CAS; (j) M. M. Queiroz, E. F. Queiroz, M. L. Zeraik, S. N. Ebrahimi, L. Marcourt, M. Cuendet, I. Castro-Gamboa, M. Hamburger, V. da Silva Bolzani and J.-L. Wolfender, J. Nat. Prod., 2014, 77, 650–656 CrossRef CAS PubMed; (k) I. Syahrul, T. Muhammad and I. Nor Hadiani, Curr. Med. Chem., 2015, 22, 4412–4433 CrossRef.
- For selected examples on the construction of symmetrical BIMs: see: (a) T. Tsuchimoto and M. Kanbara, Org. Lett., 2011, 13, 912–915 CrossRef CAS PubMed; (b) T. Mehrabi and A. Ariafard, Chem. Commun., 2016, 52, 9422–9425 RSC; (c) F. Ling, L. Xiao, L. Fang, C. Feng, Z. Xie, Y. Lv and W. Zhong, Org. Biomol. Chem., 2018, 16, 9274–9278 RSC; (d) E. B. McLean, F. M. Cutolo, O. J. Cassidy, D. J. Burns and A.-L. Lee, Org. Lett., 2020, 22, 6977–6981 CrossRef CAS; (e) J. Nie, G.-W. Zhang, L. Wang, A. Fu, Y. Zheng and J.-A. Ma, Chem. Commun., 2009, 2356–2358 RSC; (f) S. Sasaki, Y. Ikekame, M. Tanayama, T. Yamauchi and K. Higashiyama, Synlett, 2012, 23, 2699–2703 CrossRef CAS; (g) Y. Wang, Y. Yuan, C.-H. Xing and L. Lu, Tetrahedron Lett., 2014, 55, 1045–1048 CrossRef CAS; (h) W. E. Noland, H. V. Kumar, G. C. Flick, C. L. Aspros, J. H. Yoon, A. C. Wilt, N. Dehkordi, S. Thao, A. K. Schneerer, S. Gao and K. J. Tritch, Tetrahedron, 2017, 73, 3913–3922 CrossRef CAS; (i) V. K. Pandey and P. Anbarasan, J. Org. Chem., 2017, 82, 12328–12336 CrossRef CAS PubMed; (j) Y.-M. Ren, M.-D. Xu and X. Wang, Catalysts, 2017, 7, 300–306 CrossRef; (k) A. Muthukumar, G. N. Rao and G. Sekar, Org. Biomol. Chem., 2019, 17, 3921–3933 RSC; (l) Z. Wu, G. Wang, S. Yuan, D. Wu, W. Liu, B. Ma, S. Bi, H. Zhan and X. Chen, Green Chem., 2019, 21, 3542–3546 RSC; (m) J. Qiao, S. Gao, L. Wang, J. Wei, N. Li and X. Xu, J. Organomet. Chem., 2020, 906, 121039–121046 CrossRef CAS; (n) Y. Ling, D. An, Y. Zhou and W. Rao, Org. Lett., 2019, 21, 3396–3401 CrossRef CAS PubMed; (o) A. A. More and A. M. Szpilman, Org. Lett., 2020, 22, 3759–3764 CrossRef CAS PubMed.
- For selected examples on the construction of chiral BIMs bearing tertiary stereocenters, see: (a) M. H. Zhuo, Y. J. Jiang, Y. S. Fan, Y. Gao, S. Liu and S. Zhang, Org. Lett., 2014, 16, 1096–1099 CrossRef CAS; (b) H.-Q. Wang, M.-M. Xu, Y. Wan, Y.-J. Mao, G.-J. Mei and F. Shi, Adv. Synth. Catal., 2018, 360, 1850–1860 CrossRef CAS; (c) C. Yue, F. Na, X. Fang, Y. Cao and J. C. Antilla, Angew. Chem., Int. Ed., 2018, 57, 11004–11008 CrossRef CAS.
- For selected examples on the synthesis of chiral BIMs bearing cyclic quaternary stereogenic carbon centers, see: (a) X.-X. Sun, B.-X. Du, H.-H. Zhang, L. Ji and F. Shi, ChemCatChem, 2015, 7, 1211–1221 CrossRef CAS; (b) M.-H. Zhuo, G.-F. Liu, S.-L. Song, D. An, J. Gao, L. Zheng and S. Zhang, Adv. Synth. Catal., 2016, 358, 808–815 CrossRef CAS.
- For selected examples on the synthesis of chiral BIMs bearing acyclic quaternary stereogenic carbon centers, see: (a) Y. Zhang, S.-X. Zhang, L.-N. Fu and Q.-X. Guo, ChemCatChem, 2017, 9, 3107–3110 CrossRef CAS; (b) X.-K. Guan, H. Zhang, J.-G. Gao, D.-Y. Sun, X.-S. Qin, G.-F. Jiang, G.-L. Zhang and S. Zhang, J. Org. Chem., 2019, 84, 12562–12572 CrossRef CAS PubMed.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) X. H. He, Y. L. Ji, C. Peng and B. Han, Adv. Synth. Catal., 2019, 361, 1923–1957 CrossRef CAS.
- For our recent related work, see: (a) W.-J. Huang, Q. Chen, N. Lin, X.-W. Long, W.-G. Pan, Y.-S. Xiong, J. Weng and G. Lu, Org. Chem. Front., 2017, 4, 472–482 RSC; (b) W.-R. Zhu, Q. Chen, N. Lin, K.-B. Chen, Z.-W. Zhang, G. Fang, J. Weng and G. Lu, Org. Chem. Front., 2018, 5, 1375–1380 RSC; (c) W.-R. Zhu, K. Liu, J. Weng, W.-H. Huang, W.-J. Huang, Q. Chen, N. Lin and G. Lu, Org. Lett., 2020, 22, 5014–5019 CrossRef CAS; (d) W.-R. Zhu, Q. Su, H.-J. Diao, E.-X. Wang, F. Wu, Y.-L. Zhao, J. Weng and G. Lu, Org. Lett., 2020, 22, 6873–6878 CrossRef CAS; (e) W.-R. Zhu, Q. Su, N. Lin, Q. Chen, Z.-W. Zhang, J. Weng and G. Lu, Org. Chem. Front., 2020, 7, 3452–3458 RSC.
- For selected reviews, see: (a) B. M. Trost and C.-J. Li, Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, Wiley, New York, 2014 Search PubMed; (b) I.-T. Trotuş, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761–1782 CrossRef; (c) H. Schobert, Chem. Rev., 2014, 114, 1743–1760 CrossRef CAS; (d) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed.
- For selected reviews, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS; (b) M. Terada, Chem. Commun., 2008, 4097–4112 RSC; (c) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS; (d) T. James, M. van Gemmeren and B. List, Chem. Rev., 2015, 115, 9388–9409 CrossRef CAS. For selected examples, see: (e) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed; (f) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS; (g) F. Jiang, K.-W. Chen, P. Wu, Y.-C. Zhang, Y. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 15104–15110 CrossRef CAS PubMed; (h) S.-S. Meng, P. Yu, Y.-Z. Yu, Y. Liang, K. N. Houk and W.-H. Zheng, J. Am. Chem. Soc., 2020, 142, 8506–8513 CrossRef CAS.
- (a) ESI.†; (b) X. Ou, X. Li, H. Rong, L. Yu and M. Lu, Chem. Commun., 2020, 56, 9950–9953 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2101620, 2101638 and 2101639. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05174a |
| ‡ Authors with equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |