
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed selective C–C bond cleavage and stereoselective alkenylation between cyclopropanol and 1,3-diyne: one-step synthesis of diverse conjugated enynes†
Bedadyuti Vedvyas
Pati
 ,
Asit
Ghosh
,
Komal
Yadav
,
Shyam Kumar
Banjare
,
Shalini
Pandey
,
Upakarasamy
Lourderaj
* and
Ponneri C.
Ravikumar
*
,
Asit
Ghosh
,
Komal
Yadav
,
Shyam Kumar
Banjare
,
Shalini
Pandey
,
Upakarasamy
Lourderaj
* and
Ponneri C.
Ravikumar
*
School of Chemical Sciences, National Institute of Science Education and Research (NISER) Bhubaneswar, HBNI, Jatani, Khurda, 752050 Odisha, India. E-mail: u.lourderaj@niser.ac.in; pcr@niser.ac.in
First published on 8th February 2022
Abstract
The stereoselective synthesis of 1,3-enynes from 1,3-diynes is demonstrated by palladium-catalyzed selective C–C bond cleavage of cyclopropanol. Exclusive formation of mono-alkenylated adducts was achieved by eliminating the possibility of di-functionalization with high stereoselectivity. Indeed, this protocol worked very well with electronically and sterically diverse substrates. Several studies, including deuterium labeling experiments and intermolecular competitive experiments, were carried out to understand the mechanistic details. The atomic-level mechanism followed in the catalytic process was also validated using DFT calculations, and the rate-controlling states in the catalytic cycle were identified. Furthermore, preliminary mechanistic investigations with radical scavengers revealed the non-involvement of the radical pathway in this transformation.
Introduction
Transition metal-catalysed selective cleavage of C–H and C–C bonds has gained enormous significance in recent years owing to its potential application in chemical transformations.1,2 Especially, the strategy involving selective cleavage of a C–H bond followed by subsequent insertion of a 2π-unsaturated unit has been recognized as one of the most attractive approaches to access diverse molecular entities from the readily available feedstock.3 This strategy also helps in building rapid complexity following the high atom-economical process. In this context, the one-step synthesis of conjugated 1,3-enynes through this process is of great importance due to their vast abundance in various natural products and pharmaceuticals (Scheme 1).4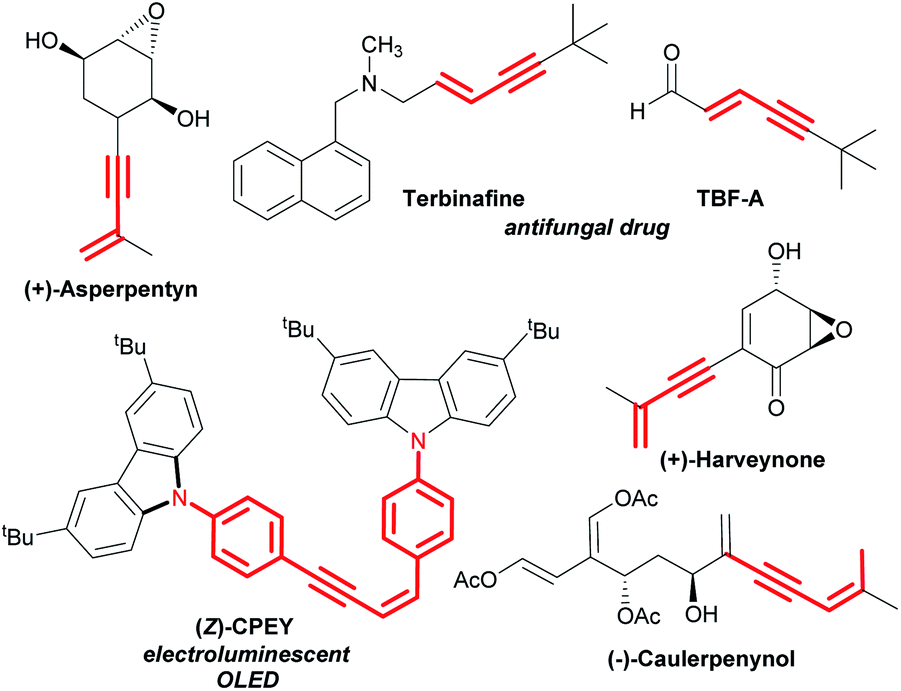 | ||
| Scheme 1 Representative examples of natural products and drug molecules bearing conjugated 1,3-enyne scaffolds. | ||
Consequently, they have been well recognized as multifaceted building units in organic synthesis.5 Moreover, these structural units also received considerable interest in materials science and medicinal chemistry.6
Considering their profound synthetic usefulness, 1,3-diynes have been recently introduced as a coupling partner for transition-metal catalysed reactions to access conjugated 1,3-enynes.7 Nevertheless, the use of 1,3-diynes in these reactions is always associated with particular challenges. The most commonly faced challenges are: (a) difficulty in controlling stereo- and regio-selectivity and (b) difficulty in controlling mono-functionalization over di-functionalization.3b,7 As a result, only a few protocols for the synthesis of 1,3-enynes using 1,3-diynes have been described to date.7 Ge and co-workers developed a cobalt-catalysed regiodivergent and stereoselective hydroboration of 1,3-diynes to afford boryl-functionalized enynes (Scheme 2a(i)).7a In another report, Beller and co-workers also disclosed palladium-catalysed selective synthesis of conjugated enynes employing alkoxycarbonylation of 1,3-diynes (Scheme 2a(ii)).7b Interestingly, highly selective synthesis of 1,3-enynes was also achieved by Glorius and co-workers via manganese-catalysed C–H activation-alkenylation of arenes and heteroarenes with 1,3-diynes (Scheme 2a(iii)).7c Because of the challenges mentioned above, reports on using 1,3-diynes to synthesize conjugated enynes are minimal. Although palladium-catalysed hydroalkylation of alkynes with cyclopropanols was developed by Yao and co-workers,11h the possibility of selective C–C bond cleavage and alkenylation sequence for 1,3-dynes has never been attempted.
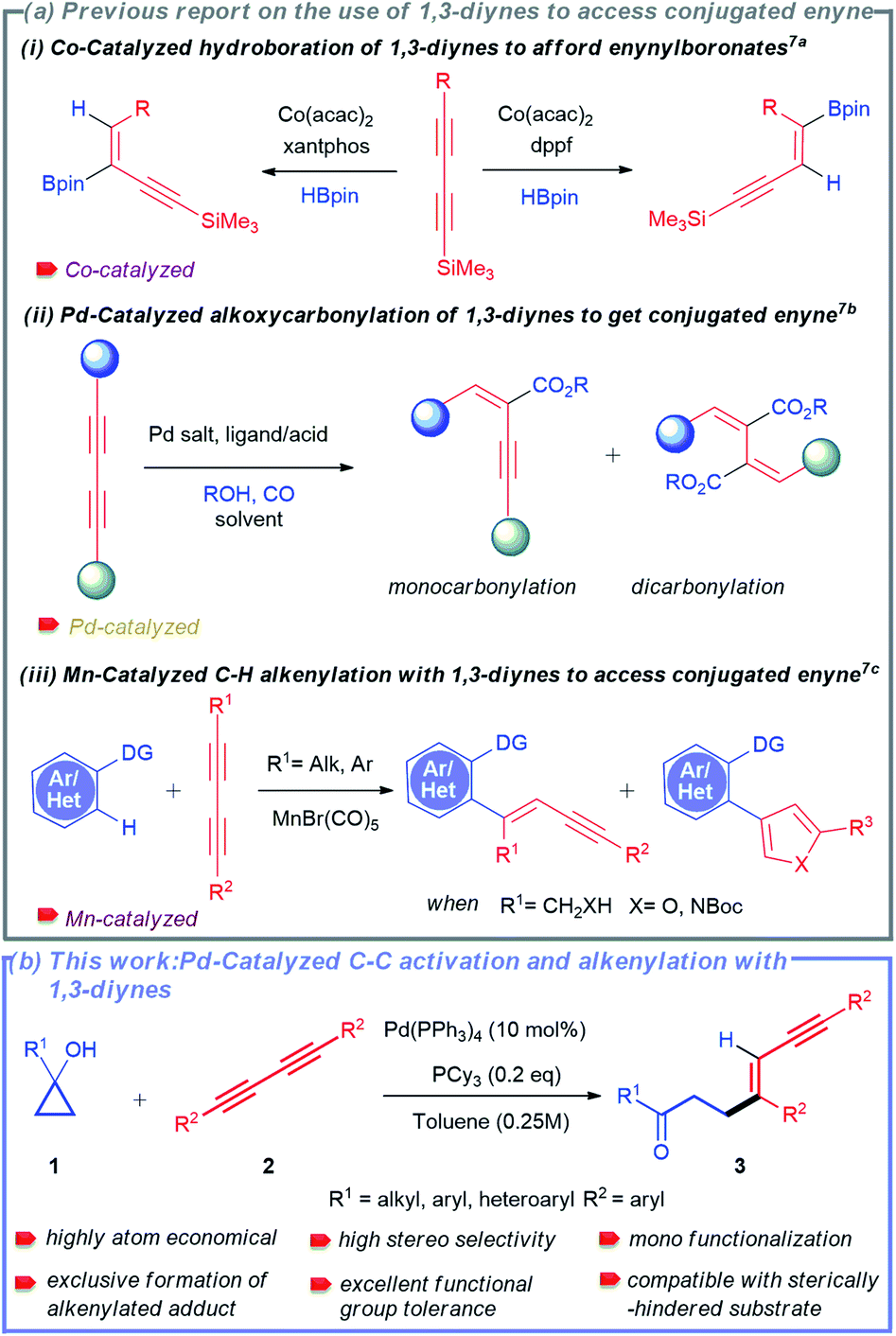 | ||
| Scheme 2 Use of 1,3-diynes to access conjugated enynes. (a) Previous report and (b) this work. | ||
In this context, the tandem cleavage and functionalization of the C–C bond is quite challenging. It is mainly owing to the thermodynamic stability and kinetic inertness of C–C bonds. In addition, poor accessibility and poor orbital directionality of C–C bonds by the catalyst as compared to C–H bonds make them less favorable for effective interactions with transition metal complexes. Despite these difficulties, substantial efforts have been made over the last two decades to functionalize different types of C–C bonds.2,8 Some valuable strategies employed include β-carbon elimination,8a,c oxidative addition,2d and aromatization driven processes.8d In this regard, one of the recent and attractive processes includes the cleavage of the C–C bond of small rings by taking advantage of their intrinsic ring strain.9 The thermodynamic barrier is largely offset with the aid of release of ring strain. The synthetic potential of this method was successfully demonstrated by several research groups such as the Dong, Jun, Murakami, Bower, Yu, Loh, and Marek groups to access an array of useful structural motifs.10 Cyclopropanol obtained from Kulinkovich protocol is one of the smallest ring molecules whose ring strain has been exploited to synthesize various molecular architectures utilizing metal-homoenolates.11–13 Our group has also developed the strain-driven C–C bond cleavage of cyclopropanol and cyclopropenone to furnish diverse functionalized molecular units.14
Owing to the synthetic value of conjugated enynes and the significance of developing new methodology using 1,3-diynes, we envisioned that the strain-driven C–C bond cleavage-subsequent alkenylation of cyclopropanol would effectively result in the formation of the desired 1,3-enynes. Herein, we report a palladium-catalysed selective C–C bond cleavage and stereo-selective mono-alkenylation of readily accessible cyclopropanol to rapidly synthesize divergent 1,3-enynes (Scheme 2b).
Results and discussion
To establish our methodology, we started by identifying the suitable reaction conditions for the palladium-catalysed C–C bond activation of cyclopropanol and the subsequent alkenylation with 1,3-diyne. Subsequently, 1-benzylcyclopropan-1-ol 1a and 1,4-diphenylbuta-1,3-diyne 2a were chosen as the model substrates in the presence of 10 mol% of a palladium catalyst. Initially, different palladium catalysts were screened in the absence of a ligand in toluene at 100 °C (Table 1, entries 1–5). The desired alkenylated adduct 3aa was obtained in 19% and 56% yields with Pd(dba)2 and Pd(PPh3)4 respectively (Table 1, entries 3 and 5). Other palladium catalysts such as PdCl2, PdCl2(PPh3)2, and Pd(OCOCF3)2 remained ineffective (Table 1, entries 1, 2 and 4). The use of other solvents such as tetrahydrofuran, acetonitrile, and dimethylformamide, with Pd(PPh3)4 as the catalyst, did not improve the product yield (Table 1, entries 6–8). Increasing or decreasing the reaction temperature (120 °C and 80 °C) had a deleterious effect on the reaction furnishing the desired product 3aa in 34% and 42% yields, respectively (Table 1, entries 9 and 10). Hence, we chose Pd(PPh3)4 as the catalyst in toluene at 100 °C and varied the other reaction parameters. It is worth mentioning here that the reactive metal-homoenolate generated after the β-carbon elimination from 1a may undergo protodemetalation and β-hydride elimination to give a ring-opened isomerized product and α,β-unsaturated ketone respectively resulting in a significant loss of the product yield. Therefore, we increased the equivalence of 1a. To our delight, 69% of 3aa was obtained using 2 equivalents of 1a (Table 1, entry 11). Next, we investigated the effects of different electron-rich N-heterocyclic and phosphine ligands (Table 1, entries 12–20). No improvement in the yield of 3aa was noticed using electron-rich N-heterocyclic ligands (Table 1, entries 12–15). Gratifyingly, product yield was further enhanced in the cases of P(tBu)3.HBF4 (77%) and PCy3 (89%) (Table 1, entries 18, 19). However, a reduced yield of 3aa was observed with the PCy3-Pd(PPh3)4 catalyst at lower temperatures (Table 1, entries 21 and 22). Furthermore, varying the molar percentage of Pd(PPh3)4 and PCy3 did not increase the product yield (Table 1, entries 23 and 24). We then performed a control experiment without the Pd(PPh3)4 catalyst. As expected, it did not produce the product, thus, confirming the key role of the catalyst in this reaction (entry 25). Hence, the use of 10 mol% of Pd(PPh3)4, 0.2 equivalents of PCy3 in toluene (0.25 M) at 100 °C is the best optimized condition for the synthesis of 3aa (Table 1, entry 19).| a Unless otherwise specified, all reactions were carried out using catalyst (10 mol%), ligand (0.2 equiv.), 1a (0.10 mmol, 1.0 equiv.), and 2a (0.10 mmol, 1.0 equiv.) in a solvent (0.25 M) for 16 h. b Yields determined by NMR, using 1,3,5-trimethoxy benzene as the internal reference. c nd = not detected. d Using 1a (0.20 mmol, 2.0 equiv.) and 2a (0.10 mmol, 1.0 equiv.) while the other conditions remained the same. e L = 3-(tert-butyl)-1-(2,6-diisopropylphenyl)-1H-imidazol-3-ium hexafluorophosphate (V). f Using a catalyst (5 mol%) while the other conditions remained the same. g Using catalyst (10 mol%) and ligand (0.1 equiv.) while the other conditions remained the same. Isolated yield is mentioned in the parenthesis. |
|---|
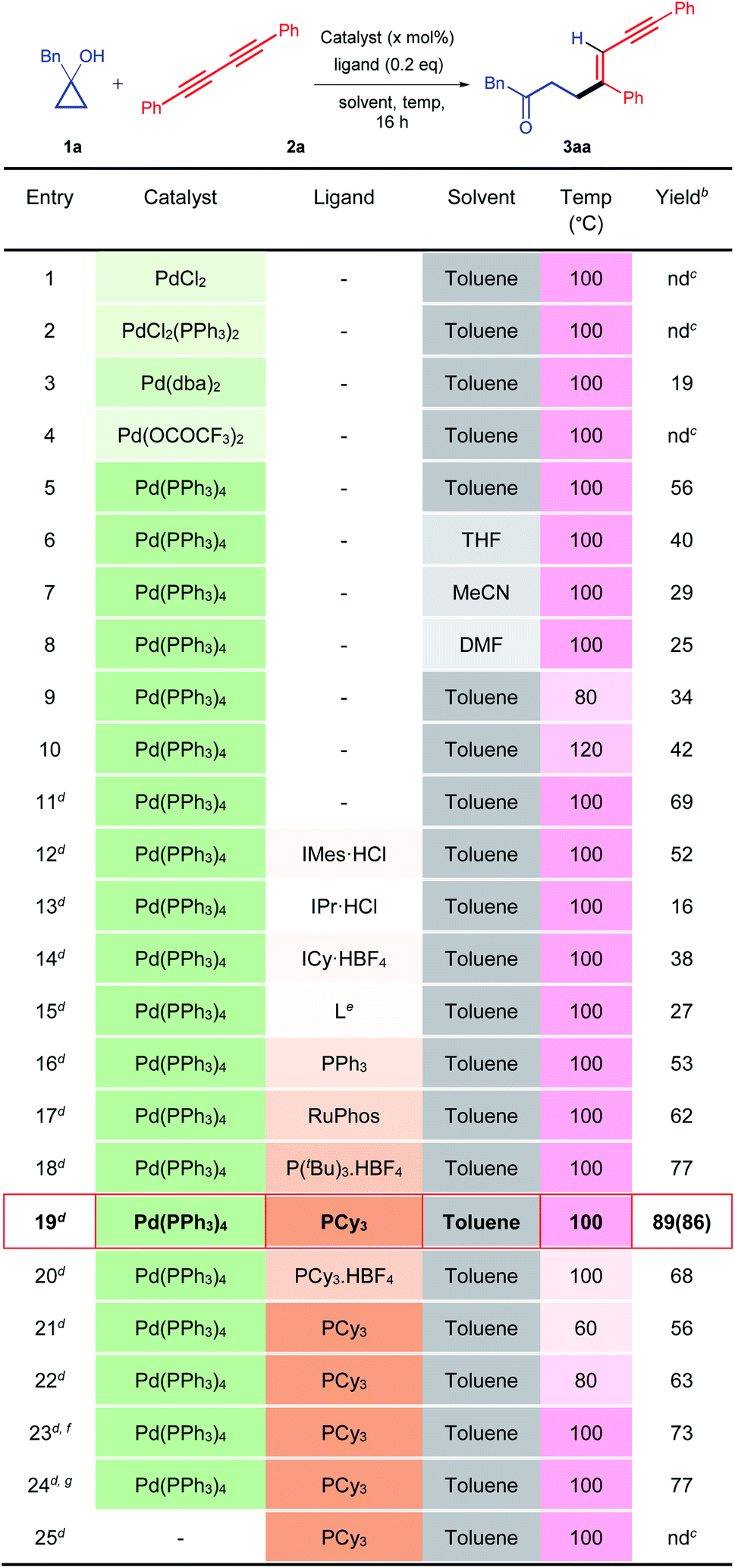
|
Having established the optimized reaction conditions, we next moved to assess the robustness of this highly stereo-selective alkenylation of cyclopropanols (Table 2). To showcase the diversity of this protocol, we subjected various benzyl-, alkyl-, aryl-, heteroaryl-substituted cyclopropanols 1 and aryl-substituted 1,3-diynes 2 to the standard reaction conditions. Initially, we examined the feasibility of different benzyl-substituted cyclopropanols. Cyclopropanols bearing Me, OMe, F, Cl, and CF3 substituents worked efficiently, furnishing 61–86% yield of their respective alkenylated adducts 3aa–3fa. Furthermore, we also found that the alkenylation reaction was viable with disubstituted cyclopropanols affording their respective adducts 3ga and 3ha in 91% and 50% yields respectively. It is worth mentioning that various alkyl (linear-chain and alicyclic) substituted cyclopropanols and sterically hindered cyclopropanol 1j reacted smoothly to produce the desired alkenylated adducts 3ia–3na in 64–80% yield.
| a All reactions were carried out using Pd(PPh3)4 (10 mol%), PCy3 (0.2 equiv.), 1 (0.20 mmol, 2.0 equiv.), and 2a (0.10 mmol, 1.0 equiv.) in toluene (0.25 M) at 100 °C for 16 h. |
|---|
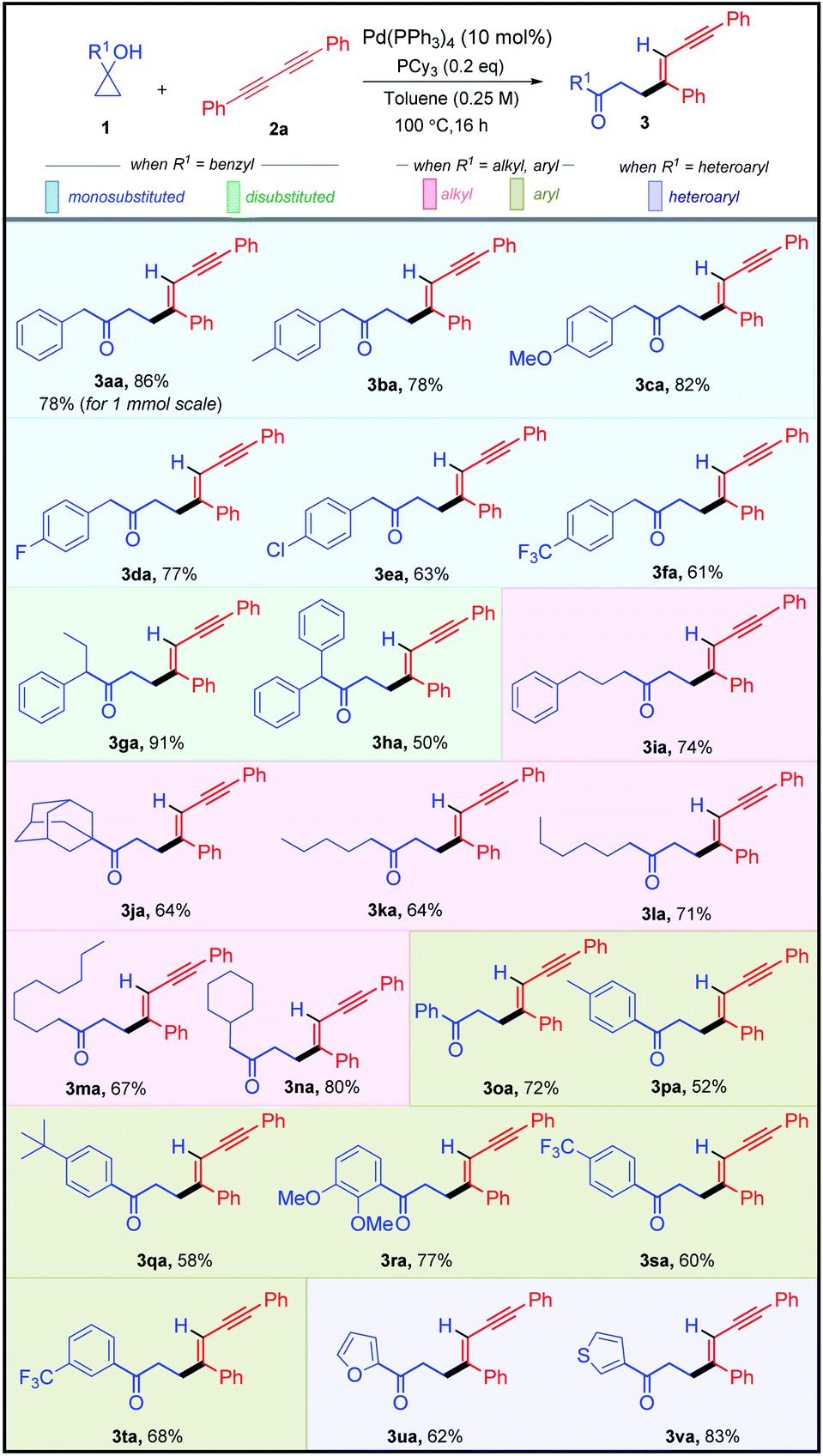
|
After successfully demonstrating the scope of alkenylation with benzyl- and alkyl-substituted cyclopropanols, we focused our attention on unraveling the scope of different aryl- and heteroaryl-substituted cyclopropanols. Similar to the benzyl-substituted cyclopropanols, Me, tBu, 2,3-di-OMe, and CF3 substituted aryl cyclopropanols were facile, resulting in good yields of their corresponding alkenylated adducts 3oa–3ta. Pleasingly, cyclopropanols bearing furan- and thiophene-substituents underwent the transformation efficiently, yielding 3ua and 3va in 62% and 83% yield, respectively.
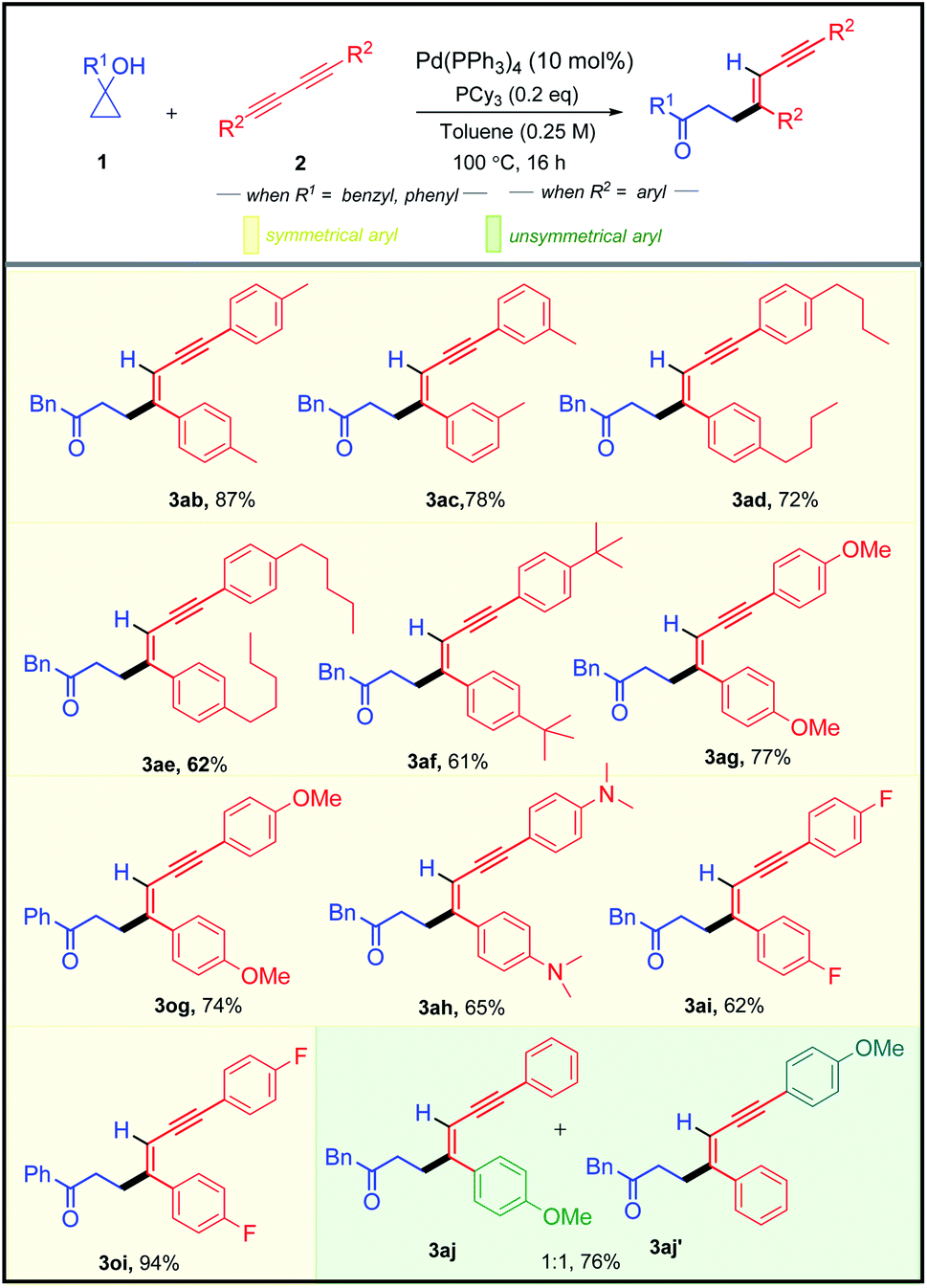
After successfully exploring the scope of structurally and electronically distinct cyclopropanols for the developed methodology, we further investigated the scope with different 1,3-diynes (Table 3). Both electronically rich and poor 1,3-diynes furnished their corresponding alkenylated adducts 3ab–3aj in 61–94% yields. The regio- and the stereo-selectivity of the product molecule was confirmed unambiguously from the NOE experiment of 3aa. To test the synthetic practicality of the developed C–C activation protocol, we performed a 1 mmol scale reaction with 1a and 2a to produce 3aa in 78% yield.
| a All reactions were carried out using Pd(PPh3)4 (10 mol%), PCy3 (0.2 equiv.), 1 (0.20 mmol, 2.0 equiv.), and 2a (0.10 mmol, 1.0 equiv.) in toluene (0.25 M) at 100 °C for 16 h. |
|---|
|
Encouraged by the versatility of the developed transformation, we performed a series of experiments to get insight into the mechanism of the catalytic cycle. Initially, we conducted a set of deuterium incorporation studies using the deuterated substrate 1a-[d] and deuterated solvent CD3OD (Scheme 3a). The reaction of 1a-[d] (100% D) and 2a afforded 3aa-[d] in 81% yield with 48% deuteration at the olefinic proton, 6% deuteration at the carbonyl α-position and 27% deuteration at the benzylic proton of 3aa-[d] (Scheme 3aii). When we used CD3OD as a co-solvent, 57% deuteration at the olefinic proton, 75% deuteration at the benzylic proton, and 7% deuteration at the carbonyl α-position of 3aa-[d] were observed with 77% yield (Scheme 3aiii). These results reveal that not only the alcoholic protons but also the carbonyl α-protons are also the proton source for the olefinic position. Furthermore, the treatment of the isolated product 3aa with CD3OD under the optimized conditions resulted in no deuteration at the olefinic position of 3aa-[d] (Scheme 3aiv). To investigate the involvement of a radical pathway, we carried out two reactions in the presence of radical scavengers such as 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) and 2,6-di-tert-butyl-4-methyl-phenol (BHT) (Scheme 3b). The 3aa yields of 51% and 73% in the presence of TEMPO and BHT respectively refuted the possibility of a radical pathway.
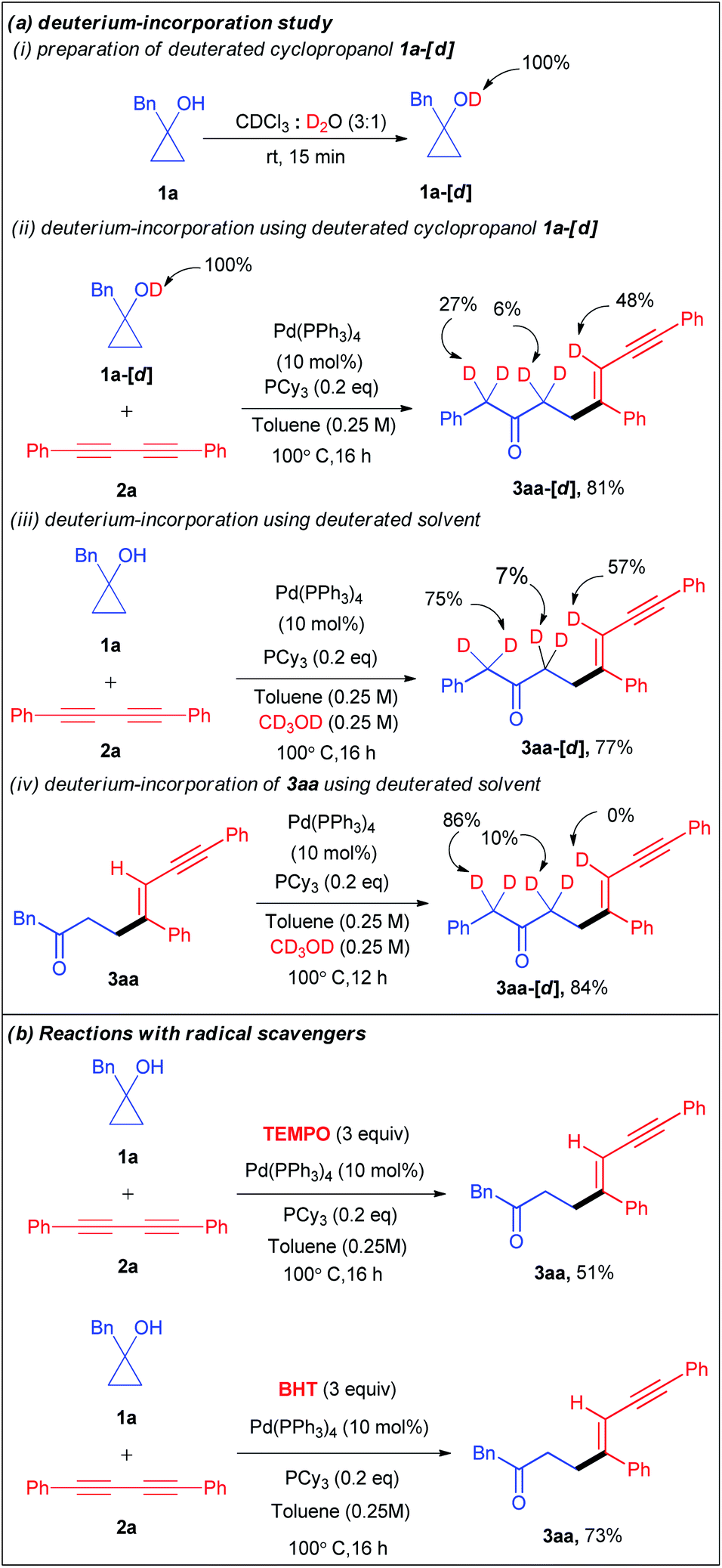 | ||
| Scheme 3 Mechanistic studies. | ||
Moreover, for further mechanistic insights and to understand the electronic effects, we conducted two intermolecular competitive experiments. First, a one pot competition reaction between two electronically different aryl cyclopropanols 1p and 1s was performed under the standard reaction conditions with diyne 2a, affording 3pa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3sa in a 6.14:1 ratio (see ESI section 4.1.1-page S16† for details). In a similar vein, the competition reaction between the cyclopropanols in a parallel set of reactions resulted in 3pa:3sa in a 3.3:1 ratio (Scheme 4a). Similarly, another set of one pot competition reactions between two different cyclopropanols 1b and 1f (benzyl substituents) when reacted with diyne 2a, produced 3ba:3fa in a 1:0.90 ratio (see ESI section 4.1.3-page S21† for details). Clearly, these experimental observations indicate the role of direct relay of electronic effects in the aryl substrates which are absent in the benzyl substrates. Next, the one pot competitive experiment between two different diynes 2g and 2i with cyclopropanol 1a afforded 3ag:3ai in a 1:1.68 ratio (see ESI section 4.2.1-page S23† for details). Similarly, the competitive one pot reaction between diynes 2g and 2i with cyclopropanol 1o le to 3og:3oi in a 1:2.16 ratio (see ESI section 4.2.2-page S25† for details). Likewise, the competition reaction between the diynes 2g and 2i with cyclopropanol 1o in a parallel set of reactions resulted in 3og:3oi in a 1:1.2 (Scheme 4a). These results reveal that alkenylation is more favourable for electronically rich cyclopropanols and electronically poor diynes.
:3sa in a 6.14:1 ratio (see ESI section 4.1.1-page S16† for details). In a similar vein, the competition reaction between the cyclopropanols in a parallel set of reactions resulted in 3pa:3sa in a 3.3:1 ratio (Scheme 4a). Similarly, another set of one pot competition reactions between two different cyclopropanols 1b and 1f (benzyl substituents) when reacted with diyne 2a, produced 3ba:3fa in a 1:0.90 ratio (see ESI section 4.1.3-page S21† for details). Clearly, these experimental observations indicate the role of direct relay of electronic effects in the aryl substrates which are absent in the benzyl substrates. Next, the one pot competitive experiment between two different diynes 2g and 2i with cyclopropanol 1a afforded 3ag:3ai in a 1:1.68 ratio (see ESI section 4.2.1-page S23† for details). Similarly, the competitive one pot reaction between diynes 2g and 2i with cyclopropanol 1o le to 3og:3oi in a 1:2.16 ratio (see ESI section 4.2.2-page S25† for details). Likewise, the competition reaction between the diynes 2g and 2i with cyclopropanol 1o in a parallel set of reactions resulted in 3og:3oi in a 1:1.2 (Scheme 4a). These results reveal that alkenylation is more favourable for electronically rich cyclopropanols and electronically poor diynes.
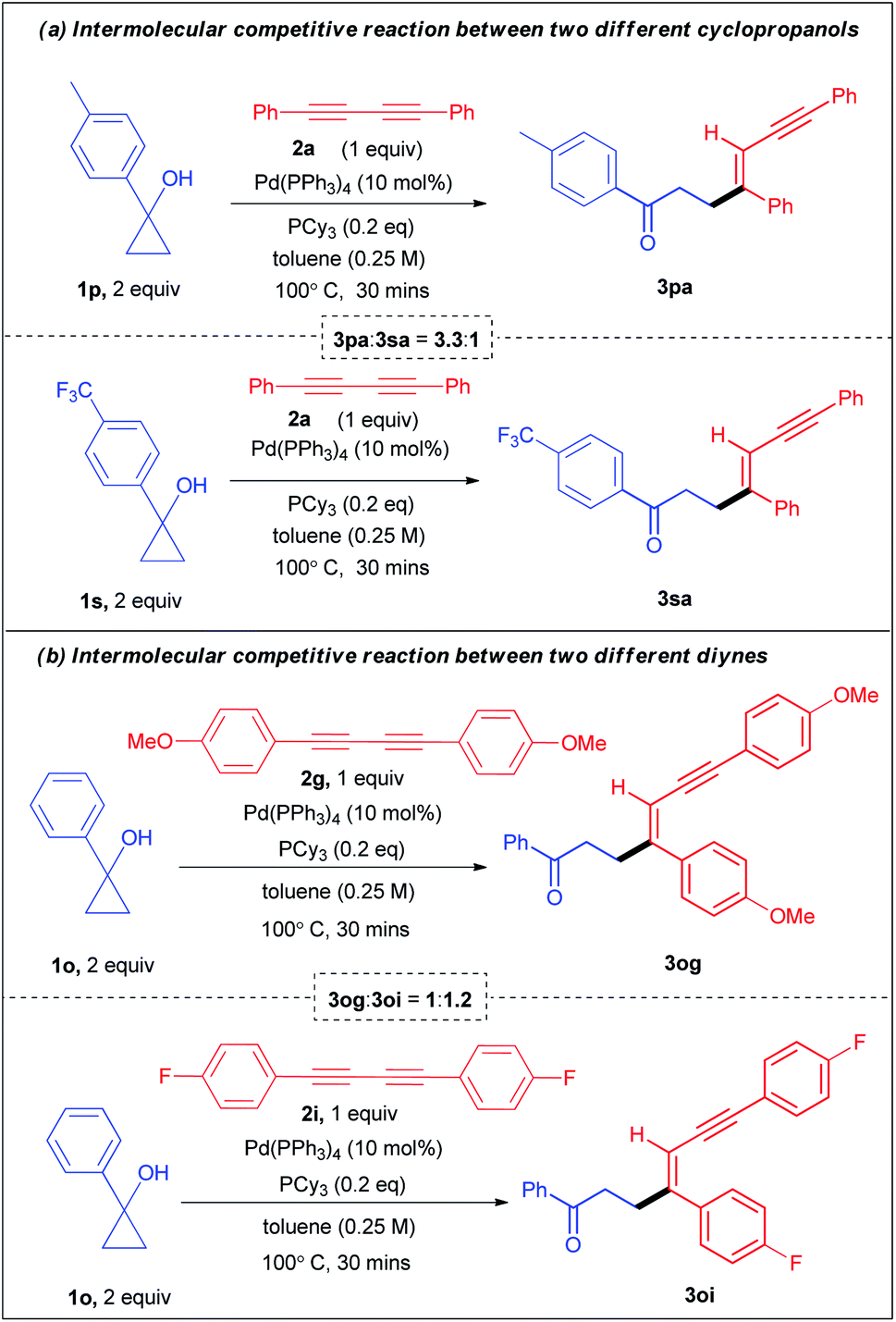 | ||
| Scheme 4 Competitive experiments. | ||
To gain insight into the atomic-level mechanism followed in the reaction between 1a and 2a catalyzed by Pd(PPh3)4, we performed density functional theory (DFT) calculations using the B3LYP-D3 (ref. 15–17) functional with 6-31G(d,p)18 basis sets and the SDD19 and LANL2DZ20 pseudopotentials to represent the core electrons of Pd. The LANL2DZ and SDD pseudopotentials have successfully been used in several studies to represent the core electrons of Pd.21,22 We performed all calculations in the presence of toluene as the solvent using the SMD23 solvation model and mapped the (Gibbs) free energy profiles along the reaction paths. The free energies were calculated by considering different pathways at the experimental temperature of 373.15 K. See the ESI† for a detailed description of the methods and the different reaction pathways considered in the DFT study. We found that the overall nature of the reaction profiles for different pathways was similar when using the SDD and LANL2DZ pseudopotentials. However, the SDD gave comparatively lower barriers for the various pathways considered and hence we discuss below the mechanisms obtained using the B3LYP-D3/6-31G(d,p)/SDD level of theory.
In cross-coupling reactions involving Pd catalysts, PdL4 dissociates to PdL3, which is in equilibrium with PdL2.24 PdL2 has been observed to behave as the active species and thus controls the oxidative addition step. The energy of the active species, PdL2 was found to be 6.4 kcal mol−1 lower as compared to that of PdL4, where L = PPh3. The relative energies were computed with respect to PdL4. Due to the complexity of the reaction, we considered different types of mechanisms for the catalytic cycle. Four different pathways were investigated: (i) Path I: oxidative addition → 1,3-diyne coordination → β-carbon elimination → syn-addition → reductive elimination (Fig. 1 and Scheme 5), (ii) Path II: 1,3-diyne coordination → ligand exchange with cyclopropanol → syn-addition → β-carbon elimination → reductive elimination (Fig. S3 and S4†), (iii) Path III: oxidative addition → β-carbon elimination → 1,3-diyne coordination → syn-addition → reductive elimination (Fig. S5 and S6†), and (iv) Path IV: oxidative addition → 1,3-diyne coordination → syn-addition → β-carbon elimination → reductive elimination (Fig. S7 and S8†). The effective barriers calculated using the energy span model25 for Paths I–IV were 39.0, 49.1, 36.1, and 43.9 kcal mol−1 respectively indicating that Path III could be the favorable mechanism with oxidative addition as the rate-determining step. However, the deuterium substituted reaction resulted in a KIE value of 1.70 (refer to the ESI† for details). This KIE value was found to be in sharp contrast to the theoretically calculated KIE (3.77, refer to the ESI† for details) computed by considering the oxidative addition barrier of 36.1 kcal mol−1 for Path III. These observations clearly revealed the non-involvement of the O–H oxidative-addition as the rate-limiting-step and hence Path III was eliminated as the possible reaction pathway. Moreover, the electronic effect seen in Scheme 4 cannot be explained if O–H activation is the rate-limiting step (Path III). Of the remaining three reaction pathways considered, Path I had the lowest effective barrier and was found to be consistent with the experimental observations as discussed below. Fig. 1 gives the complete free energy profile for the reaction path (Path I) involving various stationary points. The molecular structures and important geometrical parameters of the stationary points are given in Fig. S1 and S9 in the ESI.† The catalytic reaction cycle begins with the approach of the active catalyst Pd(PPh3)2 and 1a towards each other to form the complex int1 having an energy of −4.7 kcal mol−1 with the H atom of the OH group of 1a pointing towards Pd (H–Pd distance = 2.28 Å). The next step is the oxidative addition of the alcoholic –OH group11f,g to the Pd center to result in int2. The oxidative addition is achieved by the concerted dissociation of the O–H bond and the formation of Pd–O and Pd–H coordinate bonds via a three-membered cyclic transition state ts1 with a barrier of 36.1 kcal mol−1 with respect to int1. In int2, the Pd center is tetracoordinated with Pd–O (2.06 Å) and Pd–H (1.57 Å) bonds and has a square planar geometry. Then, the addition of 2a to palladium takes place to result in int3 preceded by the elimination of PPh3 which is crucial to lower the steric hindrance and accommodate incoming 2a. It should be noted that the free rotation of the O–C bond in the intermediates (int1 and int2) leads to the formation of different conformational isomers. However, the energy differences between the conformational isomers were less than 1 kcal mol−1. The intermediate int3 then undergoes β-carbon elimination to form int4. During this process, a PPh3 group is first eliminated to give int3′. Then, the Pd–Cβ bond is formed which is accompanied by the simultaneous breaking of the Cα–Cβ and O–Pd bonds via a four-membered cyclic transition state ts2 with a barrier of 4.5 kcal mol−1 with respect to int3 to form int3′′. A PPh3 group is then added to int3′′ to give int4. The intermediates int3′ and int3′′are not shown in Fig. 1 for clarity. What ensues is the hydrogenation of the unsaturated alkyne bond by the syn addition of Pd–H to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond to form the intermediate int5. This is a concerted step via a four-membered cyclic transition state ts3 with a barrier of 11.3 kcal mol−1. When going from int4 to ts3, the hydrogen atom of the Pd–H bond is stretched, and the CC bond is aligned coplanar to the square planar orientation of the Pd center to facilitate the H⋯CC interaction. From ts3, the H migration results in int5 with the Pd center maintaining a planar geometry.
C bond to form the intermediate int5. This is a concerted step via a four-membered cyclic transition state ts3 with a barrier of 11.3 kcal mol−1. When going from int4 to ts3, the hydrogen atom of the Pd–H bond is stretched, and the CC bond is aligned coplanar to the square planar orientation of the Pd center to facilitate the H⋯CC interaction. From ts3, the H migration results in int5 with the Pd center maintaining a planar geometry.
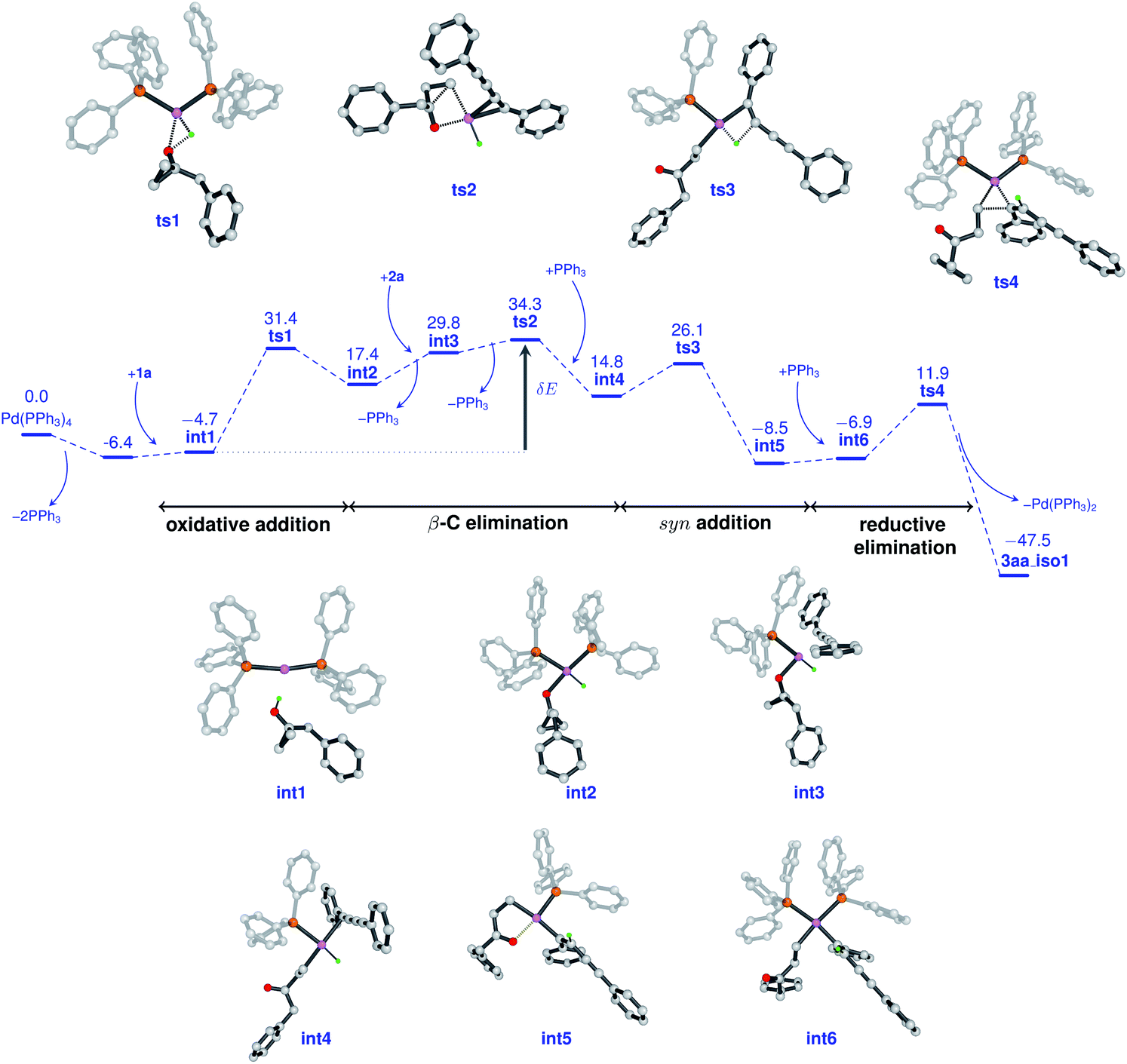 | ||
| Fig. 1 Free energy profile for the reaction between 1a and 2a catalyzed by Pd(PPh3)4, calculated at the B3LYP-D3/6-31G(d,p)/SDD level of theory. The energies are reported in kcal mol−1. The effective free energy barrier (δE) for the reaction is shown by the dark gray arrow. | ||
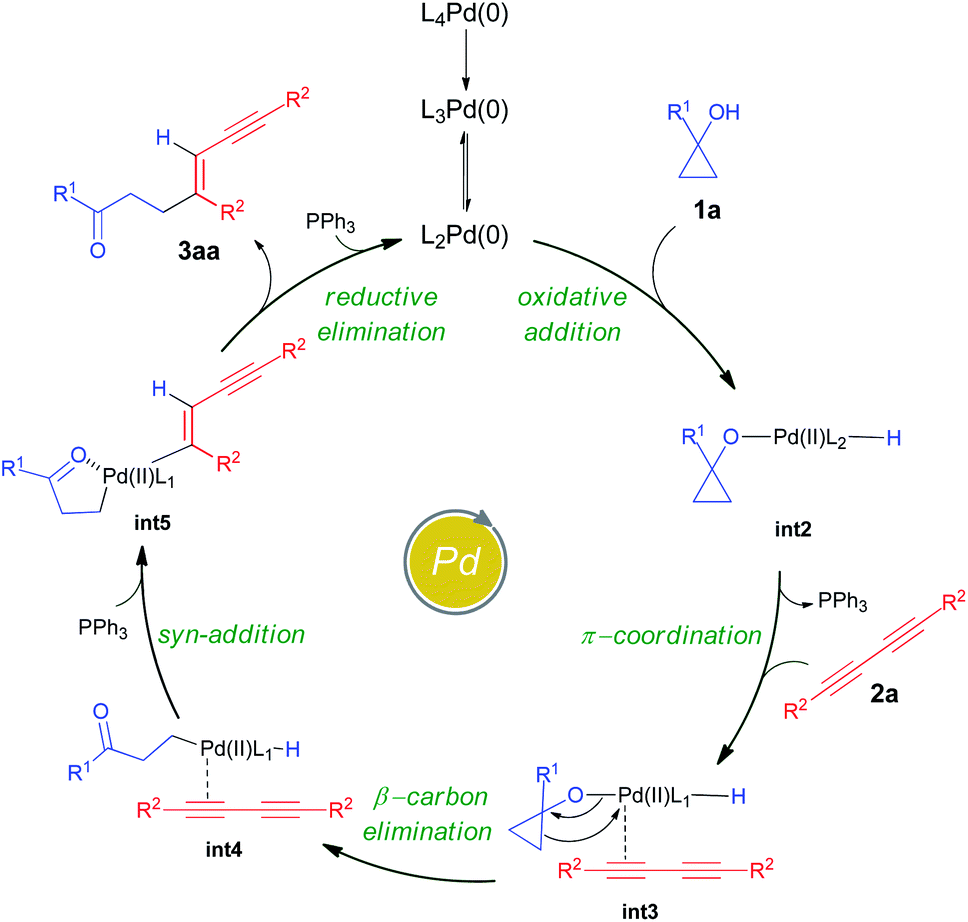 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Now, the addition of a PPh3 group to int5 leads to the formation of the intermediate int6 with a tetra-coordinated Pd center. The final step in the cycle is the reductive elimination process at the Pd center from int6 to the product 3aa_iso1. Here, the two Pd–C bonds in int6 are broken with the concomitant bond formation between the associated carbon atoms via a cyclic three-membered transition state ts4 with a 18.8 kcal mol−1 barrier. The product 3aa_iso1 is stabilized by −47.5 kcal mol−1 with respect to the reactants. The product 3aa_iso1 can exist in different isomeric forms (3aa_iso2, 3aa_iso3, and 3aa_iso4, Fig. S2†) due to rotation about C–C sigma bonds that are within 4 kcal mol−1 in energies.
It is of interest to identify the rate-controlling states involved in the overall reaction. To this end, we computed the turnover frequency (TOF) for the reaction using the energetic span model.25 From the energetics of the reaction, we can see that the TOF determining transition state (TDTS) and the TOF determining intermediate (TDI) are ts2 and int1, respectively. Hence, the effective free energy barrier (δE) for the catalytic cycle is 39.0 kcal mol−1 which is lower than that of the Paths II and IV, as noted in the energy profile (Fig. 1).
It should be noted that the competitive experiments (Scheme 4a) of the electron rich and poor cyclopropanols with diynes (2a) resulted in a ratio of 3.3:1 (3pa:3sa). Moreover, reactions of the electron rich (2g) and poor (2i) diynes with cyclopropanol (1) resulted in a ratio of 1:1.2 (3og:3oi). To understand this distribution in the products where electron rich cyclopropanols and electron poor diynes are favoured, we calculated the thermal rate constants using the Eyring-Polanyi equation26,27 at 373.15 K. For the reactions involving p-Me-cyclopropanol and p-CF3-cyclopropanol, the kTST(3pa)/kTST(3sa) was found to be 2.3:1, whereas the kTST(3og)/kTST(3oi) was found to be 1:1.42, in close agreement with the experimentally observed product ratios. In addition, the KIE (kH/kD) calculated from the DFT free energies of 2.56 was found to be in qualitative agreement with the experimentally determined value of 1.70, indicating the absence of any primary kinetic isotope effect in the reaction. Thus, the agreement of the calculated KIE and product ratios from the DFT energies with the experimental KIE and product ratios obtained in the electronic effect experiments, further supports Path I as the possible mechanism.
In accordance with the aforementioned mechanistic outcomes from the experiments supported by the DFT calculations at the B3LYP-D3/6-31G(d,p)/SDD level of theory and literature precedents,11 a plausible catalytic cycle is depicted in Scheme 5. The catalytic cycle starts with the oxidative addition of cyclopropanol11f,g1a to the active PdL2(0) catalyst to form the intermediate int2. The intermediate int2 then undergoes π-coordination with 1,3-diyne 2a to afford the intermediate int3. β-Carbon elimination from int3 leads to the formation of the intermediate int4. This intermediate int4 then undergoes syn addition with hydrogen atom transfer to furnish the homoenolate intermediate int5. Finally, the reductive elimination of palladium homoenolate intermediate int5 results in the alkenylated adduct 3aa with the regeneration of the Pd(0) catalyst.
Conclusions
In conclusion, we have developed a palladium-catalyzed novel and highly stereo-selective synthetic strategy to get an array of 1,3-enyne derivatives via selective C–C bond cleavage of cyclopropanol followed by alkenylation with 1,3-diynes. Apart from high and stereo-selectivity, excellent functional group tolerance has also been achieved with both cyclopropanol and 1,3-diynes. The protocol was found to be amenable to sterically hindered cyclopropanols such as adamantane cyclopropanol. Importantly, by eliminating the possibility of difunctionalization, exclusive mono-functionalization has also been achieved. Several mechanistic investigations including deuterium labelling experiments have been carried out. In addition, the reaction mechanism of the catalytic process was supported using DFT calculations and it was found that the step corresponding to the ring opening of cyclopropanol acted as the rate-controlling step for the reaction. The preliminary mechanistic findings with radical scavengers revealed non-involvement of the radical pathway in the transformation.Data availability
We have deposited all the experimental and computational data as ESI.†Author contributions
B. V. P. planned, conducted, and analyzed the experiments, and A. G. carried out the substrate scope and contributed in writing the first draft of the manuscript. S. K. B. carried out the mechanistic studies. K. Y. conducted the computational studies. S. P. carried out the NOE experiments. P. C. R. supervised the overall project and U. L. supervised the computational calculations in the project. All authors contributed to discussions and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Dedication
This work is dedicated to Prof. S. Chandrasekaran, IISc, Bangalore, on the occasion of his 75th birthday.Acknowledgements
We acknowledge DAE, Govt. of India, Council of Scientific and Industrial Research (CSIR), New Delhi (Grant 02(0256)/16/EMR II), and the Science and Engineering Research Board (SERB), New Delhi (Grant EMRII/2017/001475) for financial support. B. V. P. thanks DST-INSPIRE, and A. G., K. Y., S. K. B., and S. P. thank the DAE for the research fellowship.Notes and references
- For selected recent reviews on transition-metal-catalyzed C−H activation, see: (a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (b) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (c) V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29–39 RSC; (d) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (e) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS PubMed; (f) C. Sambiagio, D. Schonbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maesa and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (g) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratzand and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (h) U. Dutta, S. Maiti, T. Bhattacharya and D. Maity, Science, 2021, 372, 701 CrossRef PubMed and references cited therein..
- For selected recent reviews on transition-metal-catalyzed C−C activation, see: (a) M. Murakami and T. Matsuda, Chem. Commun., 2011, 47, 1100–1105 RSC; (b) K. Ruhland, Eur. J. Org. Chem., 2012, 2683–2706 CrossRef CAS; (c) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed; (d) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (e) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed; (f) P.-h. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed; (g) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC; (h) T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3–79 CrossRef CAS PubMed and references cited therein..
- For selected examples, see: (a) W. Zhang, H. Li and L. Wang, Adv. Synth. Catal., 2019, 361, 2885–2896 CrossRef CAS; (b) D.-G. Yu, F. de Azambuja, T. Gensch, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 9650–9654 CrossRef CAS PubMed; (c) S. Kathiravan and I. Nicholls, Org. Lett., 2017, 19, 4758–4761 CrossRef CAS PubMed; (d) M. S. R. Mandal, A. Das, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2017, 23, 17454–17457 CrossRef PubMed; (e) Y. Gao, F. F. Zeng, X. D. Sun, M. F. Zeng, Z. Yang, X. Q. Huang, G. D. Shen, Y. S. Tan, R. K. Feng and C. Z. Qi, Adv. Synth. Catal., 2018, 360, 1328–1333 CrossRef CAS; (f) Á. M. Martínez, I. Alonso, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem.–Eur. J., 2019, 25, 5733–5742 CrossRef PubMed; (g) R. Feng, H. Ning, H. Su, Y. Gao, H. Yin, Y. Wang, Z. Yang and C. Qi, J. Org. Chem., 2017, 82, 10408–10417 CrossRef CAS PubMed; (h) R. Mei, W. Ma, Y. Zhang, X. Guo and L. Ackermann, Org. Lett., 2019, 21, 6534–6538 CrossRef CAS PubMed; (i) S. Kumar, A. M. Nair and C. M. R. Volla, Org. Lett., 2020, 22, 2141–2146 CrossRef CAS PubMed; (j) A. Dey and C. M. R. Volla, Org. Lett., 2020, 22, 7480–7485 CrossRef CAS PubMed; (k) B. V. Pati, P. S. Sagara, A. Ghosh, G. K. D. Adhikari and P. C. Ravikumar, J. Org. Chem., 2021, 82, 10408–10417 Search PubMed; (l) F. Zhao, X. Gong, Y. Lu, J. Qiao, X. Jia, H. Ni, X. Wu and X. Zhang, Org. Lett., 2021, 23, 727–733 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Rudi, M. Schleyer and Y. Kashman, J. Nat. Prod., 2000, 63, 1434–1436 CrossRef CAS PubMed; (b) S. L. Iverson and J. P. Uetrecht, Chem. Res. Toxicol., 2001, 14, 175–181 Search PubMed; (c) N. El-Jaber, A. Estévez-Braun, A. G. Ravelo, O. Munoz-Munoz, A. Rodrí-guez Afonso and J. R. Murguia, J. Nat. Prod., 2003, 66, 722–724 CrossRef CAS PubMed; (d) A. Fürstner and L. Turet, Angew. Chem., Int. Ed., 2005, 44, 3462–3466 CrossRef PubMed.
- For selected reviews, see: (a) R. G. Bergman, Acc. Chem. Res., 1973, 6, 25–31 CrossRef CAS; (b) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2916 CrossRef CAS PubMed; (c) R. Chinchilla and C. Njera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed; (d) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed; (e) Y. Xiao and J. Zhang, Angew. Chem., Int. Ed., 2008, 47, 1903–1906 CrossRef CAS PubMed; (f) D. J. Burns, D. Best, M. D. Wieczysty and H. W. Lam, Angew. Chem., Int. Ed., 2015, 54, 9958–9962 CrossRef CAS PubMed; (g) Y. Huang, J. del Pozo, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2018, 140, 2643–2655 CrossRef CAS PubMed; (h) C. Ye, Y. Li, X. Zhu, S. Hu, D. Yuan and H. Bao, Chem. Sci., 2019, 10, 3632–3636 RSC; (i) X. Zhu, W. Deng, M.-F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548–559 CrossRef CAS PubMed.
- For selected examples, see: (a) C.-K. Choi, I. Tomita and T. Endo, Macromolecules, 2000, 33, 1487–1488 CrossRef CAS; (b) K. Campbell, C. J. Kuehl, M. J. Ferguson, P. J. Stang and R. R. Tykwinski, J. Am. Chem. Soc., 2002, 124, 7266–7267 CrossRef CAS PubMed; (c) Y. Liu, M. Nishiura, Y. Wang and Z. Hou, J. Am. Chem. Soc., 2006, 128, 5592–5593 CrossRef CAS PubMed; (d) G. S. Pilzak, K. van Gruijthuijsen, R. H. van Doorn, B. van Lagen, E. J. R. Sudhçlter and H. Zuilhof, Chem.–Eur. J., 2008, 14, 7939–7950 CrossRef CAS PubMed; (e) Z. Cao and T. Ren, Organometallics, 2011, 30, 245–250 CrossRef CAS.
- For selected examples, see: (a) H. L. Sang, C. Wu, G. G. D. Phua and S. Ge, ACS Catal., 2019, 9, 10109–10114 CrossRef CAS; (b) J. Liu, J. Yang, C. Schneider, R. Frank, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2020, 59, 9032–9040 CrossRef CAS PubMed; (c) S. Cembellín, T. Dalton, T. Pinkert, F. Schäfers and F. Glorius, ACS Catal., 2020, 10, 197–202 CrossRef; (d) J. Liu, C. Schneider, J. Yang, Z. Wei, H. Jiao, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2021, 133, 375–383 CrossRef; (e) F. Sun, C. Yang, J. Ni, G. J. Cheng and X. Fang, Org. Lett., 2021, 23, 4045–4050 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Korotvička, I. Císařová, J. Roithová and M. Kotora, Chem.–Eur. J., 2012, 18, 4200–4207 CrossRef PubMed; (b) Z. Gu, G. B. Boursalian, V. Gandon, R. Padilla, H. Shen, T. V. Timofeeva, P. Tongwa, K. P. C. Vollhardt and A. A. Yakovenko, Angew. Chem., Int. Ed., 2011, 50, 9413–9417 CrossRef CAS PubMed; (c) H. Takano, K. S. Kanyiva and T. Shibata, Org. Lett., 2016, 18, 1860–1863 CrossRef CAS PubMed; (d) Y. Xu, X. Qi, P. Zheng, C. C. Berti, P. Liu and G. Dong, Nature, 2019, 567, 373–378 CrossRef CAS PubMed and references cited therein..
- (a) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed; (b) B. Biletskyi, P. Colonna, K. Mason, J. L. Parrain, L. Commeiras and G. Chouraqui, Chem. Soc. Rev., 2021, 50, 7513–7538 RSC.
- For selected examples, see: (a) C.-H. Jun, H. Lee, C. W. Moon and H.-S. Hong, J. Am. Chem. Soc., 2001, 123, 8600–8601 CrossRef CAS PubMed; (b) L. Jiao, C. Yuan and Z.-X. Yu, J. Am. Chem. Soc., 2008, 130, 4421–4430 CrossRef CAS PubMed; (c) L. Deng, M. Chen and G. Dong, J. Am. Chem. Soc., 2018, 140, 9652–9658 CrossRef CAS PubMed; (d) J. Bruffaerts, A. Vasseur, S. Singh, A. Masarwa, D. Didier, L. Oskar, L. Perrin, O. Eisenstein and I. Marek, J. Org. Chem., 2018, 83, 3497–3515 CrossRef CAS PubMed; (e) X. Ma, I. R. Hazelden, T. Langer, R. H. Munday and J. F. Bower, J. Am. Chem. Soc., 2019, 141, 3356–3360 CrossRef CAS PubMed; (f) S. Li, P. Shi, R.-H. Liu, X.-H. Hu and T.-P. Loh, Org. Lett., 2019, 21, 1602–1606 CrossRef CAS PubMed.
- For selected examples, see: (a) D. Rosa and A. Orellana, Chem. Commun., 2013, 49, 5420–5422 RSC; (b) B. B. Parida, P. P. Das, M. Niocel and J. K. Cha, Org. Lett., 2013, 15, 1780–1783 CrossRef CAS PubMed; (c) K. Cheng and P. J. Walsh, Org. Lett., 2013, 15, 2298–2301 CrossRef CAS PubMed; (d) N. Nithiy and A. Orellana, Org. Lett., 2014, 16, 5854–5857 CrossRef CAS PubMed; (e) A. Reding, P. G. Jones and D. B. Werz, Org. Lett., 2018, 20, 7266–7269 CrossRef CAS PubMed; (f) J. Le. Bras and J. Muzart, Tetrahedron, 2019, 76(12), 130879, DOI:10.1016/j.tet.2019.130879; (g) H. Okumoto, T. Jinnai, H. Shimizu, Y. Harada, H. Mishima and A. Suzuki, Synlett, 2000, 629–630 CAS; (h) H. Liu, Z. Fu, S. Gao, Y. Huang, A. Lin and H. Yao, Adv. Synth. Catal., 2018, 360, 3171–3175 CrossRef CAS.
- For selected examples, see: (a) Z. Ye and M. Dai, Org. Lett., 2015, 17, 2190–2193 CrossRef CAS PubMed; (b) Z. Ye, K. E. Gettys, X. Shen and M. Dai, Org. Lett., 2015, 17, 6074–6077 CrossRef CAS PubMed; (c) Y. Li, Z. Ye, T. M. Bellman, T. Chi and M. Dai, Org. Lett., 2015, 17, 2186–2189 CrossRef CAS PubMed; (d) X.-P. He, Y.-J. Shu, J.-J. Dai, W.-M. Zhang, Y.-S. Feng and H.-J. Xu, Org. Biomol. Chem., 2015, 13, 7159–7163 RSC; (e) H. Zhang, G. Wu, H. Yi, T. Sun, B. Wang, Y. Zhang, G. Dong and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 3945–3950 CrossRef CAS PubMed; (f) Y. A. Konik, M. Kudrjashova, N. Konrad, S. Kaabel, I. Järving, M. Lopp and D. G. Kananovich, Org. Biomol. Chem., 2017, 15, 8334–8340 RSC; (g) Z. Ye, X. Cai, J. Li and M. Dai, ACS Catal., 2018, 8, 5907–5914 CrossRef CAS PubMed; (h) E. Gyanchander, S. Ydhyam, N. Tumma, K. Belmore and J. K. Cha, Org. Lett., 2016, 18, 6098–6101 CrossRef CAS PubMed and references cited therein..
- For selected examples, see: (a) J. Yang, Y. Shen, Y. J. Lim and N. Yoshikai, Chem. Sci., 2018, 9, 6928–6934 RSC; (b) J. Yang, Q. Sun and N. Yoshikai, ACS Catal., 2019, 9, 1973–1978 CrossRef CAS; (c) J. Yang, Y. Sekiguchi and N. Yoshikai, ACS Catal., 2019, 9, 5638–5644 CrossRef CAS; (d) L. R. Mills, C. H. Zhou, E. Fung and S. A. L. Rousseaux, Org. Lett., 2019, 21, 8805–8809 CrossRef CAS PubMed; (e) J. Li, Y. Zheng, M. Huang and W. Li, Org. Lett., 2020, 22, 5020–5024 CrossRef CAS PubMed; (f) Y. Sekiguchi and N. Yoshikai, J. Am. Chem. Soc., 2021, 143(12), 4775–4781 CrossRef CAS PubMed; (g) W. Huang and F. Meng, Angew. Chem., Int. Ed., 2021, 60, 2694–2698 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Nanda and P. C. Ravikumar, Org. Lett., 2020, 22, 1368–1374 CrossRef CAS PubMed; (b) B. V. Pati, A. Ghosh and P. C. Ravikumar, Org. Lett., 2020, 22, 2854–2860 CrossRef CAS PubMed; (c) T. Nanda, P. Biswal, B. V. Pati, S. K. Banjare and P. C. Ravikumar, J. Org. Chem., 2021, 86(3), 2682–2695 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- P. Fuentealba, H. Stoll, L. Von Szentpaly, P. Schwerdtfeger and H. Preuss, J. Phys. B Atom. Mol. Phys., 1983, 16, 323–328 CrossRef.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82(1), 284–298 CrossRef CAS.
- S. Bag, A. Mondal, R. Jayarajan, U. Dutta, S. Porey, R. B. Sunoj and D. Maiti, J. Am. Chem. Soc., 2020, 142(28), 12453–12466 CrossRef CAS PubMed.
- F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 50(35), 8192–8195 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- U. W. Maes, S. Verbeeck, T. Verhelst, A. Ekomié, N. von Wolff, G. Lefèvre, E. A. Mitchell and A. Jutand, Chem.–Eur. J., 2015, 21, 7858–7865 CrossRef PubMed.
- S. Kozuch, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 795–815 CAS.
- H. Eyring, J. Chem. Phys., 1935, 3(2), 107–115 CrossRef CAS.
- H. Eyring and M. Polanyi, Z. Phys. Chem., Abt. B, 1931, 12, 279–311 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04780a |
| This journal is © The Royal Society of Chemistry 2022 |