
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective formation of propan-1-ol from propylene via a chemical looping approach†
A. R. P.
Harrison
 * and
E. J.
Marek
*
* and
E. J.
Marek
*
Department of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK. E-mail: arph2@cam.ac.uk; ejm94@cam.ac.uk
First published on 22nd August 2022
Abstract
A novel chemical looping approach for propan-1-ol production from propylene was investigated. Silver- and gold-based catalysts were prepared, with SrFeO3−δ perovskite as a catalyst support, and characterised using X-ray diffraction (XRD), electron microscopy (SEM and STEM-EDS), and X-ray photoelectron spectroscopy (XPS). Particles of the catalysts were used in experiments carried out in a packed bed reactor, by passing 5 vol% propylene over the material to generate oxygenated products, with oxygen provided via reduction of the catalyst support, SrFeO3−δ. The solid particles were subsequently re-oxidised with air in a separate step, closing the chemical loop. Catalysts composed of AgCl and AgAu alloy supported on SrFeO3−δ gave up to 70–80% stable selectivity towards propan-1-ol over the temperature range 260–300 °C, with little to no formation of the secondary alcohol, propan-2-ol, albeit at low propylene conversion (∼0.5–1.5%). Postulated mechanisms for propan-1-ol formation under chemical looping conditions are discussed, via propylene oxide or allyl alcohol intermediates.
1. Introduction
Selective oxidation of propylene to form value-added C3 products, such as propylene oxide (PO), allyl alcohol (AA), and propan-1-ol, is a significant challenge in heterogeneous catalysis.1–4 In particular, catalysts for producing propylene oxide from propylene using air as the oxidising agent have been studied extensively,5,6 but are not yet commercially competitive, despite the environmental and economic problems of the incumbent chlorohydrin and hydroperoxide processes.1 Production of propan-1-ol directly from propylene is also challenging due to the preferential formation of the secondary alcohol isomer, propan-2-ol, under most reaction conditions.4Silver-based catalysts on metal oxide supports have been widely investigated for selective oxidation of propylene,6 but have generally shown relatively poor selectivity towards C3 oxygenates. For propylene reacting with oxygen over Ag, the γ-carbon atom in propylene preferentially reacts with atoms of oxygen adsorbed at the Ag surface, Oa, to form CO2via complete combustion. The complete combustion reaction can be partially suppressed by the addition of chloride species, either on the surface of the catalyst or in the feed gas,7–11 but at the penalty of decreased propylene conversion.
Selectivity towards C3 oxygenates via oxidation of propylene is further limited by isomerisation reactions. Barteau and co-workers12 investigated the reactions of PO over Ag surfaces under aerobic and anaerobic conditions. They found that PO can undergo H-transfer reactions to form a broad distribution of products, including propanal, acetone, allyl alcohol, and acrolein.
The use of Ag–Au bimetallic catalysts in selective oxidation reactions has been investigated by Geenen et al.13 Increasing the proportion of Au to Ag reduced the extent of complete combustion, with acrolein as the favoured oxygenate product. The increase in selectivity towards acrolein was attributed to the catalysts having isolated Ag sites (i.e. the separation between the catalyst's active sites greatly exceeded the size of propylene molecules), which inhibit the dissociation of molecular oxygen from forming two neighbouring Oa–Ag species. Therefore, the favoured surface species was adsorbed molecular (O2)a,13,14 which is selective towards acrolein.
Previous studies have also investigated using solid oxides (e.g. PdOx, VOx) as oxidising agents for propylene oxidation.15 Unsupported PdOx catalysts produced PO at up to 28% selectivity, with acrolein and propanal forming via isomerisation of PO at 38% and 33% selectivity, respectively. The overall conversion of propylene was low, at ∼0.4%. However, the PdOx catalysts used did not show a consistent product distribution over multiple redox cycles, with ∼98% selectivity towards acrolein at ∼0.01% propylene conversion after regenerating the catalyst in air. The broad distribution of oxygenate products was ascribed to the ability of Pd to act as a catalyst for hydrogen transfer reactions, which aid isomerisation of PO to other oxygenates. In contrast, near 100% selectivity for PO was achieved using unsupported V2O5 as a solid oxidant, but at very low (∼0.03%) conversion of propylene. The use of V2O5 supported on SiO2 or γ-Al2O3 allowed for a modest improvement in the conversion of propylene, up to ∼0.1%, at ∼70% selectivity towards PO over a single reduction step. The use of Bi2O3–MoO3 and Fe2SbOx for oxidising propylene at elevated temperatures (>450 °C) has also been investigated.16,17 Acrolein was found to be the dominant C3 oxygenate product over the catalysts MoO3, 2 Bi2O3·MoO3, and Fe2SbOx, with C6 hydrocarbons (benzene and hexadiene) being the favoured products over Bi2O3 and Sb2O4.
Recently, a chemical looping approach has been proposed18 for selective oxidation reactions, including epoxidation of olefins,19 oxidative dehydrogenation of alkanes,20 or conversion of methane to methanol.21 Here, lattice oxygen is provided from a metal oxide support (termed the oxygen carrier) to react at the surface of a catalyst, in the absence of gaseous oxygen. The oxygen carrier is then re-oxidised in air, in a separate step (a schematic of the process is shown in Fig. 1), giving the chemical looping approach the advantage of inherent separation between the organic species and the air, thereby decreasing the risk of forming explosive mixtures.
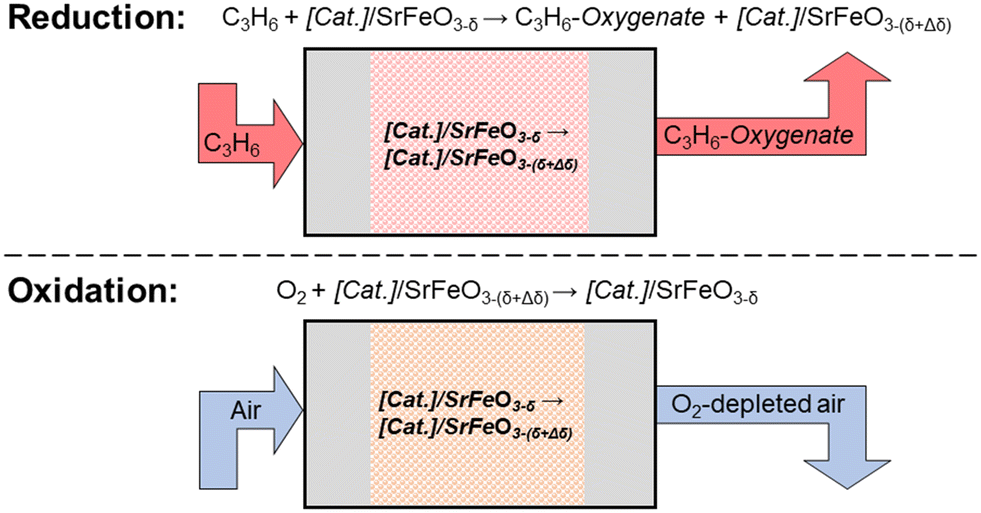 | ||
| Fig. 1 Schematic diagram showing the reduction and oxidation steps in selective oxidation of propylene via chemical looping for a catalyst, Cat., supported on an oxygen carrier, here, strontium ferrite. | ||
Studies on chemical looping for selective oxidation of ethylene19,22,23 have used Ag deposited on a non-stoichiometric oxide, strontium ferrite, SrFeO3−δ, with perovskite structure, demonstrating chemical looping epoxidation of ethylene to ethylene oxide (EO). Strontium ferrite was chosen as a suitable oxygen carrier for selective oxidation reactions, as SrFeO3−δ undergoes reversible oxygen release at relatively low temperatures (<300 °C),19,24 and can be produced inexpensively via solid-state methods.25
The ratio of the perovskite phase, SrFeO3, to Ruddlesden–Popper phase, Sr3Fe2O7, was found to strongly influence the rate of release of oxygen from the SrFeO3 support.22 Catalysts for chemical looping epoxidation of ethylene have been found to give stable performance over 33 cycles,23 producing EO at 67% selectivity, with 19% conversion of ethylene. Perovskite supports have also been investigated26,27 for propylene epoxidation and found to improve modestly the selectivity towards PO, by facilitating the dissociation of O2 molecules, but under conventional operating conditions with oxygen in the gas phase, rather than chemical looping.
In this work, Ag, AgCl, and Ag/Au catalysts, supported on SrFeO3 perovskite, were investigated for chemical looping oxidation of propylene. Catalysts containing AgCl and/or Au were found to give low levels of complete combustion, and high selectivity towards propan-1-ol. Interestingly, the AgCl and AgCl/Au catalysts produced a substantially different distribution of products as compared to typical distributions reported for direct epoxidation of propylene over Ag- and Au-based catalysts.5 The results provide an insight into potential future challenges in applying a chemical looping approach to propylene oxidation, and indicate that catalysts composed of AgCl and, or AgAu alloy supported on SrFeO3 may be suitable for production of propan-1-ol from propylene via a chemical looping approach.
2. Materials and methods
2.1. Material preparation
Active particles, composed of a metal catalyst supported on strontium ferrite, were prepared using various deposition methods. Strontium ferrite perovskite (SrFeO3−δ, written as SrFeO3 for brevity) was prepared using a solid-state method.25 Stoichiometric quantities of SrCO3 (Sigma-Aldrich, 98%) and Fe2O3 (Fisher Scientific, 95%) were manually mixed to form a homogeneous powder; then, 50 mL ethanol (Sigma-Aldrich, >99.8%) was added as a binder. The mixture was milled in a planetary ball mill using ceramic balls placed with the mixture in a ceramic jar; ball-milling was carried out for 3 h at 25 Hz, then, the mixture was dried for 24 h at 50 °C. The resulting powder was sieved to 180–355 μm, 50–180 μm, and <50 μm size fractions, and calcined in air for 12 h at 1000 °C. Particles of SrFeO3 were impregnated with metal catalysts using incipient wetness impregnation.A series of mixed AgCl/Au-D catalysts supported on SrFeO3, with target metal loadings x wt% Ag and (10–x) wt% Au (x = 2.5, 5, 7.5), was prepared by varying the volume of Ag and Au solutions added (noting that AgNO3 reacts with AuCl3 in solution to form AgCl). The solutions of Ag and Au precursor were prepared separately, then added sequentially to the support material in appropriate ratios to achieve the desired loading, followed by overnight drying and calcination at 650 °C for 5 h. Catalysts prepared by impregnation with AgNO3 and AuCl3 were designated xAgCl/(10–x)Au-D/SrFeO3.
One batch of this catalyst was also prepared by impregnating SrFeO3 with AgNO3 and AgCl3 solutions, followed by drying at 120 °C and calcination at 700 °C for 5 h. Calcination at 700 °C as opposed to 650 °C induced thermal decomposition of AgCl,29 giving a final loading of 3.4 wt% AgCl and a 5.1 wt% loading of Ag/AgAu. The sample calcined at 700 °C was designated Ag–AgCl/Au-H/SrFeO3.
All samples investigated are summarised in Table 1.
| Sample | Catalyst | Measured amount of catalyst, wt% | Catalyst precursors used | Heat treatment |
|---|---|---|---|---|
| a Letters in the sample codes stand for: D – impregnated with AuCl3, H – impregnated with AuCl3 followed by calcination at 700 °C to partially decompose AgCl, L – impregnated with chloride-free Au–(β-ala). | ||||
| Ag/SrFeO3 | Ag | 11.8 wt% Ag | AgNO3(aq) | 120 °C for 12 h; 650 °C for 5 h |
| Au-D/SrFeO3a | Au | 7.3 wt% Au | Alkaline AuCl3(aq) | 120 °C for 12 h; 650 °C for 5 h |
| AgCl/SrFeO3 | AgCl | 1.1 wt% Ag, 8.8 wt% AgCl | AgNO3(aq), treated with conc. HCl(aq) | 120 °C for 12 h; 650 °C for 5 h |
| xAgCl/(10–x)Au/SrFeO3a (x = 2.5, 5, 7.5) | AgCl/Au | x = 2.5: 3.4 wt% AgCl, 7.9 wt% AgAu | AgNO3(aq), alkaline AuCl3(aq) | 120 °C for 12 h; 650 °C for 5 h |
| x = 5: 5.1 wt% AgCl, 5.1 wt% AgAu | ||||
| x = 7.5: 6.9 wt% AgCl, 5.1 wt% AgAu | ||||
| Ag–AgCl/Au-H/SrFeO3a | Ag/AgCl/Au | 3.4 wt% AgCl, 5.1 wt% (Ag + AgAu) | AgNO3(aq), alkaline AuCl3(aq) | 120 °C for 12 h; 700 °C for 5 h |
| Ag/Au-L/SrFeO3a | Ag/Au | 4.6 wt% Ag, 0.02 wt% Au | AgNO3(aq), Au–(β-ala)(aq) | 120 °C for 12 h; 650 °C for 5 h |
2.2. Material characterisation
Powder X-ray diffraction (XRD) patterns were measured using a Bruker D8 Advance diffractometer, over the angular range 5–80° with step size 0.05° and step time 2 s, using Cu-Kα radiation. Phase composition was determined with Profex software,31 using reference structures from the ICSD database32 (collection codes are given in the ESI,† Table S1). Scanning electron microscopy (SEM) images were taken using a Tescan Mira3 FEG-SEM microscope with an accelerating voltage of 5 kV, and secondary electron (SE) and back-scattered electron (BSE) detectors in parallel. Scanning transmission electron microscopy (STEM) images were taken using a Thermo Scientific (FEI) Talos F200X G2 TEM with a SuperX energy-dispersive X-ray spectroscopy (EDS) detector.X-ray photoelectron spectroscopy (XPS) measurements were taken using an Escalab 250Xi spectrometer, with 20 eV scan pass energy. Scans were calibrated with respect to the Au 4f7/2 peak at 84.00 eV,33 collected on an Au foil (Alfa Aesar, 99.9975+%). Samples of Ag foil (Alfa Aesar, 99.998%) and AgCl powder (Acros Organics, 99+%) were also scanned to determine the binding energy of 3d5/2 for metallic Ag0 and Ag+. Collected results were deconvoluted to assess the electronic states for elements in the analysed samples. The analysis was performed using CasaXPS software.34
2.3. Experiments in a packed bed reactor
The performance of the prepared materials was determined using a small scale packed-bed reactor, operated in the chemical looping mode. A bed of active particles, composed of a metal catalyst impregnated on SrFeO3 (1.50 g, 180–300 μm) was placed in a quartz tube, in between two layers of SiC (Alfa Aesar, 46 grit), with 2.00 g of SiC below and 3.00 g of SiC above the bed of active particles. A schematic of the experimental rig is given in the ESI,† Fig. S1. The bed was assembled in a quartz reactor tube (200 mm length, i.d. 8 mm), with a sintered disk supporting the bed 75 mm from the tube base. The reactor was secured using Swagelok Ultra-Torr vacuum fittings with fluorocarbon FKM O-rings, and wrapped with a heating tape (LewVac, 200 W). A K-type thermocouple, positioned in the centre of the active material, was used to control the setpoint temperature. The reactor was heated under air flow for 2 h prior to experiments, to ensure the setpoint temperature was achieved throughout the bed, and to remove carbonate and hydroxide species from the SrFeO3 surface.35 The gases used in experiments were 5 vol% propylene (balance Ar, BOC, 4.96 or 5.13 vol%) for reduction, N2 (BOC) for purging, and air (BOC) for re-oxidation. Gas flows to the reactor were set to 200 mL min−1 (NTP). Reduction and oxidation cycles were performed by passing gases over the material in the sequence N2–C3H6–N2–air. Generally, in a single cycle, the active bed was reduced in C3H6 for 1.5 min, followed by a 2 min purge step in N2, and then re-oxidised in air for 15 min. The effect of reduction time was also investigated, varying the reduction time from 1.5 min to 60 min, but keeping the 2 min purge in N2 and 15 min reoxidation in air. The active bed was periodically regenerated by heating the packed reactor tube to 650 °C for 5 h ex situ in static air.The gas from the reactor outlet was sampled manually using a gas syringe, withdrawing a 10 mL sample immediately after the reactor tube. Samples were collected 45 s after the start of each reduction step in C3H6/N2. The gas composition was measured with an Agilent 7890A gas-chromatograph (GC), using parallel Agilent PoraBOND-Q and Hayesep-Q/MolSieve 13A columns, connected to FID and TCD detectors, respectively. The FID channel was used to quantify propylene, propylene oxide (PO), propanal, acetone, propan-1-ol, propan-2-ol, and allyl alcohol (AA), and the TCD channel was used to quantify CO2. No CO, or other carbon-containing products, were detected above 5 ppm. The GC measurements were calibrated using two gas mixtures: (1) 1000 ppm propylene/1000 ppm propylene oxide/balance N2; (2) 1000 ppm CO2/balance N2; both BOC. From measurements of calibration gas mixtures, the accuracy of the FID detection system was estimated to be within ±10% of the true value based on variability of results collected by measuring samples of known compositions. The detection threshold was 1–2 ppm for C3 components containing oxygen. The accuracy of the TCD was estimated to be ±4% of the true value, with a detection threshold of 10 ppm CO2. For other organic components, gas mixtures for calibration were generated using an Owlstone V-OVG vapour generator fed with pure liquid components (acetone: VWR, 99%; propan-1-ol: Sigma-Aldrich, 99+%; propan-2-ol, VWR, 99%; propanal: Acros Organics, 99+%; allyl alcohol: Sigma-Aldrich, 99+%). Retention times for each component were consistent to within ±0.05 min for all measurements.
Instantaneous conversion of propylene, X, was estimated from the concentrations measured at the reactor outlet, using (1).
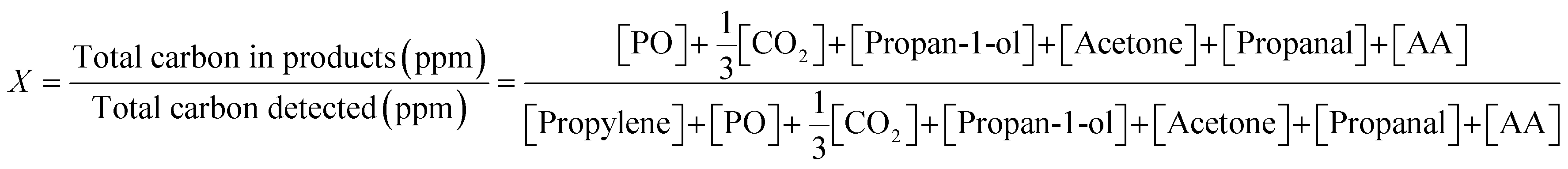 | (1) |
Selectivity towards a given product, SA, (i.e. PO or propan-1-ol) was determined using (2).
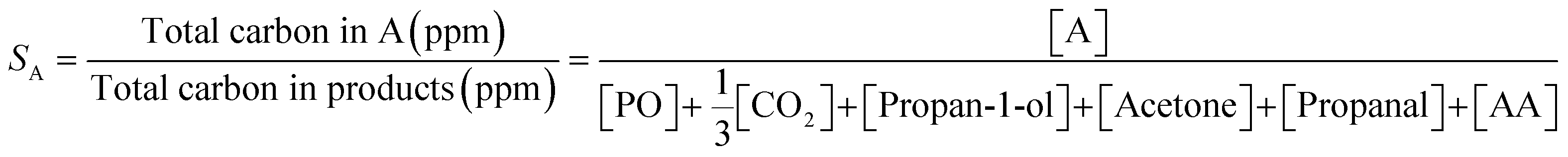 | (2) |
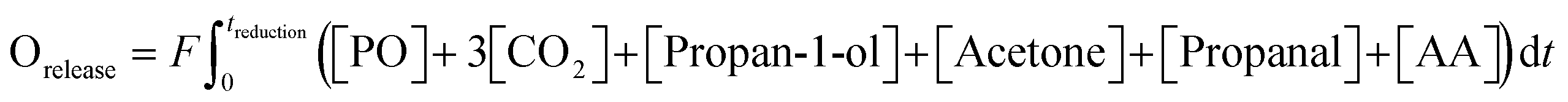 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the two combustion products:
:1 ratio of the two combustion products: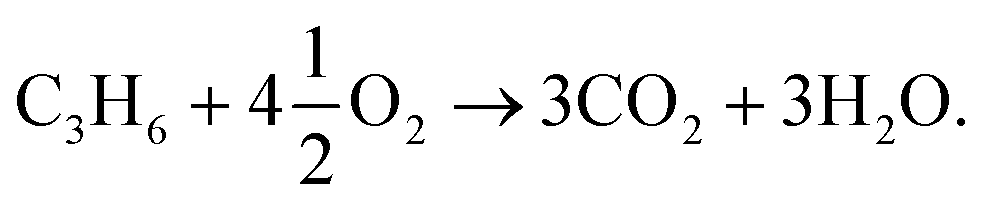 | (R1) |
To confirm the validity of measurements, the carbon balance over each cycle was estimated using (4),
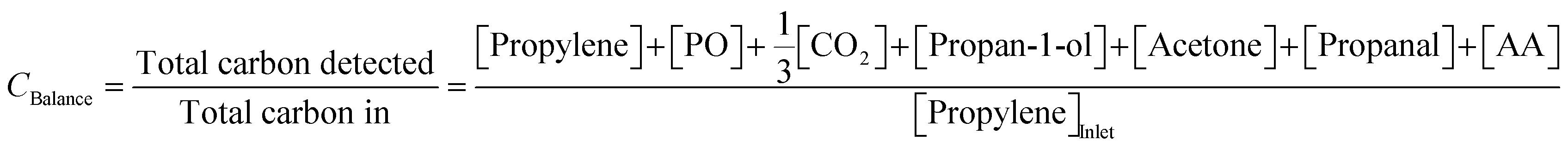 | (4) |
To determine potential side-reactions of oxidation products, chemical looping oxidation experiments were also performed using a propylene feed stream with PO or propanal added. An electrical tube furnace was used to heat the reactor tube as opposed to heating tape, with the active bed placed in the isothermal region of the furnace, using a rig previously described by Gebers et al.23 An inlet gas mixture containing propylene and PO was produced by blending 5.13 vol% propylene (balance Ar) with 1000 ppm propylene/1000 ppm propylene oxide (balance N2), using 100 mL min−1 of each mixture for a nominal composition of 2.56 vol% propylene and 500 ppm propylene oxide. In another experiment, an inlet gas mixture containing propylene and propanal was produced by passing 5.13 vol% propylene through a vapour generator (Owlstone, V-OVG) at 200 mL min−1, loaded with propanal (Acros Organics, 99+%), thus, generating a stream containing 5.13 vol% propylene and ∼1200 ppm propanal. The composition of gas mixtures blended in-house was confirmed with the GC. Similarly, a reaction of PO with hydrogen was investigated by blending 5 vol% H2 (balance N2; Air Liquide) with 1000 ppm propylene/1000 ppm propylene oxide (balance N2), for a nominal composition of 500 ppm PO, 500 ppm propylene and 2.5 vol% H2. The mixture of PO, propylene, and H2 was then passed over the active bed, with the outlet stream composition measured using the GC.
3. Results
3.1. Material characterisation
Prepared catalysts were characterised with XRD and the results are presented in Fig. 2 and S3.† From the XRD patterns for AgCl/SrFeO3, AgCl/Au-D/SrFeO3 and Ag–AgCl/Au-H/SrFeO3, peaks at 2θ = 32.1° and 46.0° were detected, as shown in Fig. 2, corresponding to the AgCl crystalline phase. The characteristic peaks for AgCl were absent in the patterns for Ag/SrFeO3 and Ag/Au-L/SrFeO3. For the AgCl/Au-D/SrFeO3 samples, no metallic Ag was detected, confirming AgCl and AgAu (alloyed Ag and Au) as the main phases containing silver.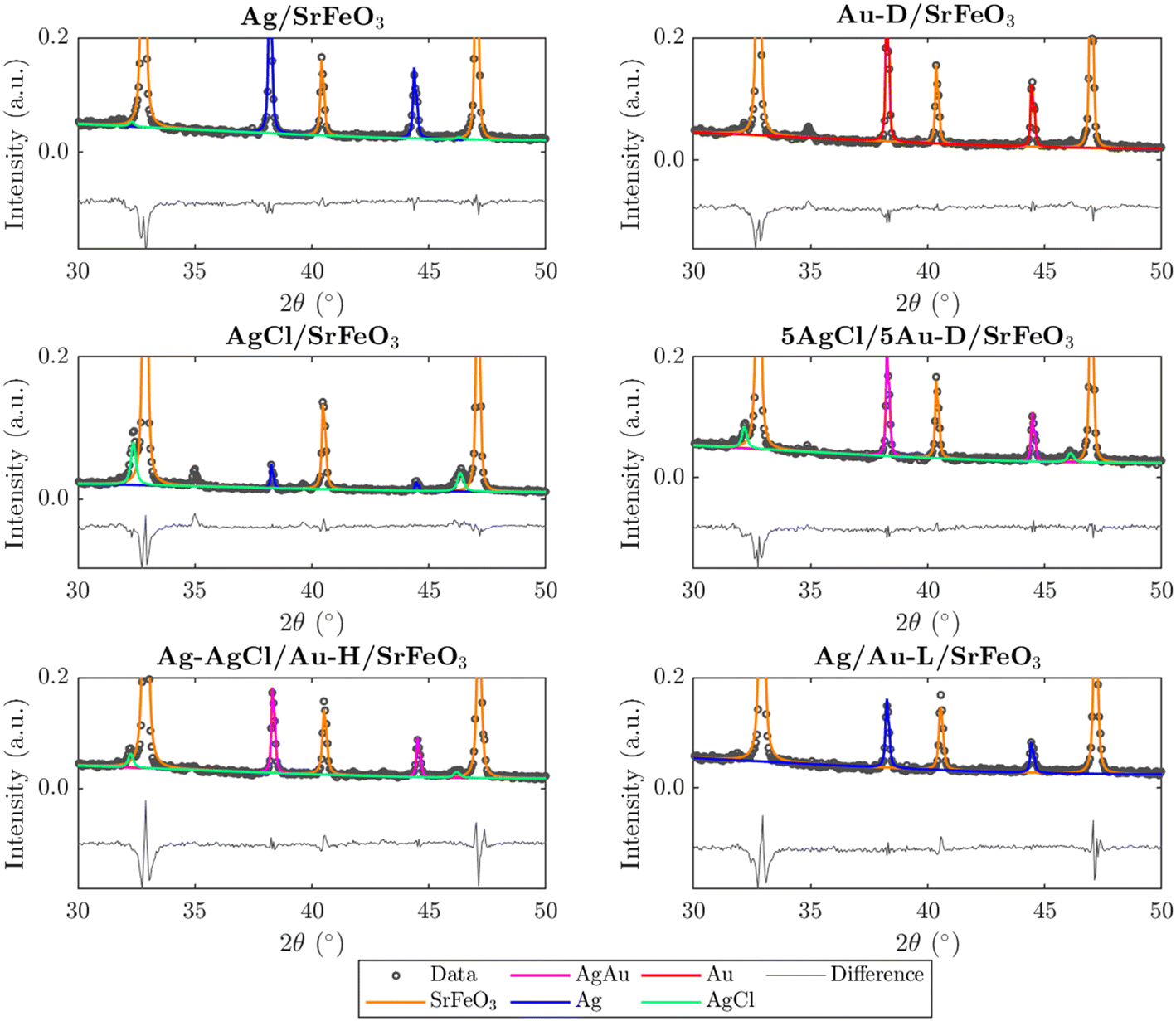 | ||
| Fig. 2 Sections of collected XRD patterns for Ag/SrFeO3, Au-D/SrFeO3, AgCl/SrFeO3, 5AgCl/5Au-D/SrFeO3, Ag–AgCl/Au-H/SrFeO3, and Ag/Au-L/SrFeO3. Circles indicate experimental measurements; lines indicate fitted model using reference patterns. | ||
The estimated compositions calculated for each sample are presented in Table S2.† The oxygen carrier was approximately pure SrFeO3 (>95%). For the Ag–AgCl/Au-H/SrFeO3 sample, calcined at 700 °C, Fig. 2 shows a less prominent AgCl peak than in the Au-D samples calcined at 650 °C, giving an AgCl loading of 3.4 wt% as compared to 5.1 wt%. The smaller AgCl peak confirms that calcination at 700 °C induced thermal decomposition of AgCl, which starts around 650–700 °C.29 No AgCl was detected in the sample prepared using the Au–(β-ala) precursor (Ag/Au-L/SrFeO3), where all Ag and Au were present as metallic nanoparticles, with an Ag loading of 4.6 wt%. Given the close proximity of the Ag, Au, and AgAu XRD peaks, and the low Au loading (∼0.02 wt%) determined from ICP measurements (Table 1), separate AgAu and Au peaks could not be refined for the Ag/Au-L/SrFeO3 sample.
In Fig. S4,† XRD patterns for fresh, spent, and regenerated AgCl/SrFeO3 are shown, with an AgCl fraction of ∼10 wt% calculated for all three samples. The presence of AgCl in the spent sample indicates minimal stripping of chloride due to the reaction between propylene and AgCl, suggesting greater stability of bulk AgCl under reaction conditions as compared to Ag promoted with Cl− ions at the surface.10,11
The presence of AgCl on the surface of the 5Ag/5Au-D/SrFeO3 sample was confirmed via STEM-EDS surface mapping of crushed particles of supported catalysts, shown in Fig. 3 (further STEM-EDS maps are shown in Fig. S5 in the ESI†). Particles composed of AgCl and AgAu were detected on the surface, with AgAu forming larger particles and AgCl smaller (Sauter mean dia. of 266 nm and 111 nm, respectively). Low atomic fractions of Cl were detected in particles of AgAu, and low atomic fractions of Au in particles of AgCl (<5.0 atCl% and <0.6 atAu%, respectively for all analysed particles), demonstrating limited overlap between AgAu and AgCl clusters. Analysis of the AgCl particles gave a molar ratio of Ag to Cl in the range ∼1–2 with an Ag:Cl ratio of 1 corresponding to pure AgCl, and an Ag:Cl ratio of 2 corresponding to 50 at% AgCl and 50 at% Ag. Thus, Ag was present either as particles of AgCl or clusters of metallic Ag with AgCl. For areas of bare SrFeO3 support, no Ag or Au was detected, and the Sr:Fe atomic ratio was close to 1 in all cases.
 | ||
| Fig. 3 STEM-EDS maps of crushed 5Ag/5Au-D/SrFeO3 particles, showing distribution of (a) Ag, (b) Au, (c) Cl, (d) Sr. Area 1 corresponds to bare SrFeO3 support with no Ag or Au detected, Area 2 corresponds to a particle of AgAu, Area 3 corresponds to a particle of AgCl. | ||
The surface of the catalyst particles was further characterised using SEM images (shown in the ESI,† Fig. S6 and S7). Notably, from Fig. S7† for 5Ag/5Au-D/SrFeO3, two distinct surface particle morphologies were observed, with larger (∼425 nm), approximately spherical particles, and smaller (∼135 nm), elongated particles visible (particle size distributions are given in the ESI,† Fig. S8). Additionally, the larger particles appeared brighter in the BSE image, corresponding to greater atomic number,36 which indicates the mixture of AgAu, as compared to the smaller, darker particles, composed of AgCl.
The surface states of the elements in the supported catalysts were determined by XPS, with the full analysis described in the ESI,† section S5. The XPS spectra for the 5Ag/5Au-D/SrFeO3 sample, presented in Fig. 4a and b, demonstrate shifts in binding energy for Ag 3d and Au 4f in AgAu, thus, indicating an alloying behaviour between the two metals, with the extent of the shifts in agreement with values reported in literature.37–40 Only two distinct peaks were detected in the O 1s spectra for 5AgCl/5Au-D/SrFeO3 and AgCl/SrFeO3. The oxygen feature at a lower binding energy of 528.3–530.4 eV (shown in red in Fig. 4c) was assigned to lattice oxygen in SrFeO3,41 while the feature at a higher binding energy of 530.8–533.8 eV (shown in blue in Fig. 4c) was assigned to carbonate or hydroxide species adsorbed at the surface of the SrFeO3.41,42 Additionally, the XPS spectra recorded for the O 1s peak suggest the presence of small amounts of AgOx species at the surface of Ag/SrFeO3 (assigned at 529.5 eV, shown in green in Fig. 4c) that were not detected for any of the other catalyst samples. In Fig. 4d, the AgCl standard and AgCl/SrFeO3 samples show the Cl 2p3/2 peak at 198.8 eV, and 2p3/2–2p1/2 peak separation of c. 1.6 eV, in good agreement with literature.43 A peak shift of −1.5 eV between the 2p3/2 AgCl standard and the AgCl/SrFeO3 sample was observed, which is similar to the shift reported for thin layers (∼5 nm) of AgCl in contact with Ag.43,44
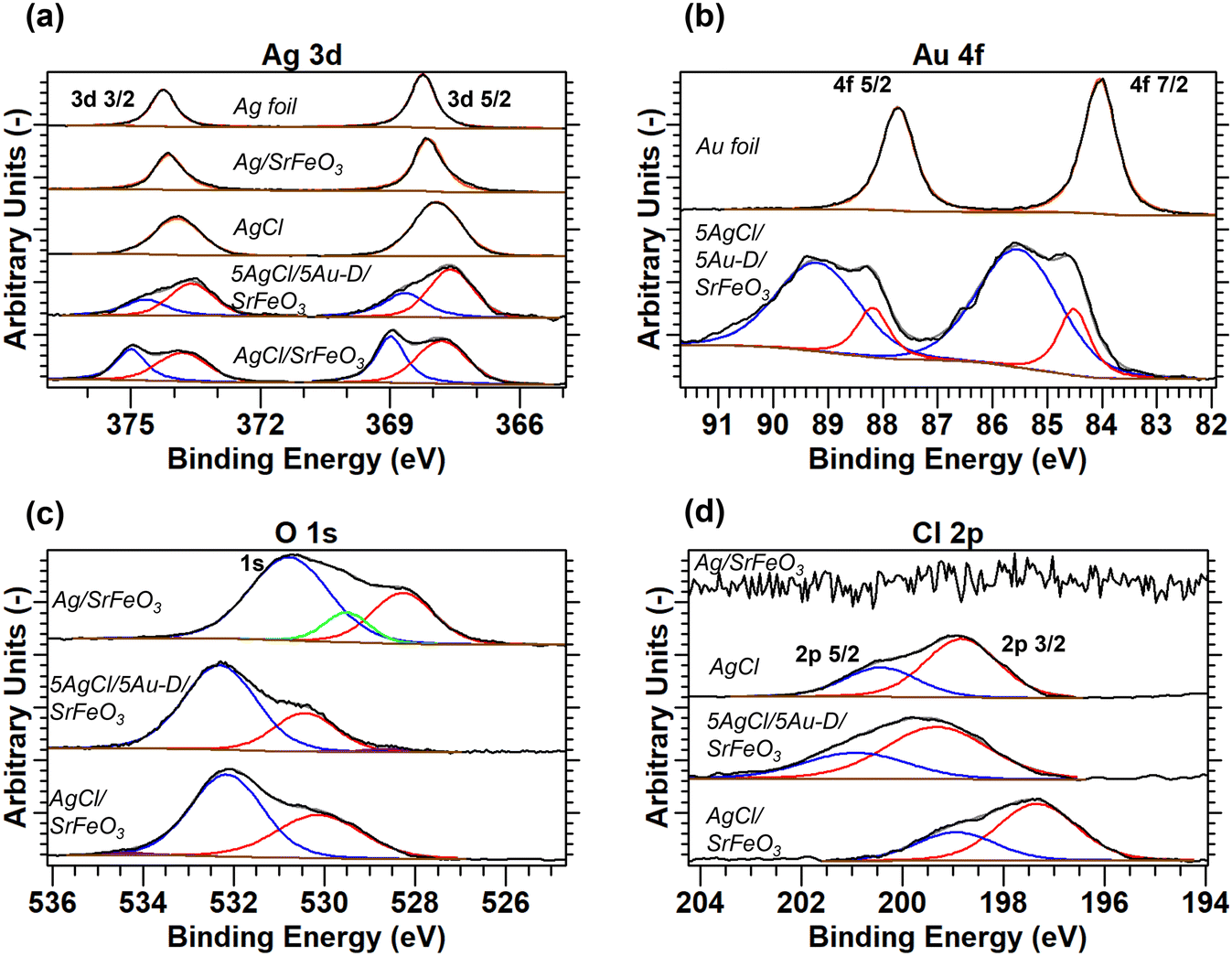 | ||
| Fig. 4 XPS spectra for (a) Ag 3d, (b) Au 4f, (c) Cl 2p, and (d) O 1s scans, with deconvoluted peaks fitted to experimental measurements. Measurements calibrated with respect to Au 4f7/2 peak at BE = 84.0 eV. Full details of peak deconvolution are given in the ESI,† Table S4. | ||
3.2. Performance in chemical looping experiments
Results from experiments carried out in the packed bed for 5AgCl/5Au-D/SrFeO3 catalysts are presented in Fig. 5. The catalysts showed considerable activity towards propan-1-ol formation, with outlet concentrations of ∼100–500 ppm propan-1-ol, a product not detected in previous studies of propylene oxidation over AgCl or Au catalysts8,10,11,13,14 Trace amounts of PO, acetone, propanal, and propan-2-ol (∼5 ppm) were also measured. Over five cycles of reduction and re-oxidation, the outlet concentrations of propan-1-ol and CO2 remained approximately stable, indicating minimal catalyst deactivation with cycling. For the sample of 5Ag/5Au-D/SrFeO3, increasing the reactor temperature from 260 °C to 300 °C resulted in greater overall conversion of propylene, from ∼0.5 to ∼1.5%, but with limited effect on selectivity towards propan-1-ol, which remained at 60 ± 4% over the temperature range. The approximately constant selectivity with temperature is surprising; seemingly, the apparent rates of the formation of propan-1-ol and of complete combustion have similar temperature dependence over the range 260 °C to 300 °C, and hence, the ratio of propan-1-ol to CO2 in the reaction products does not change significantly.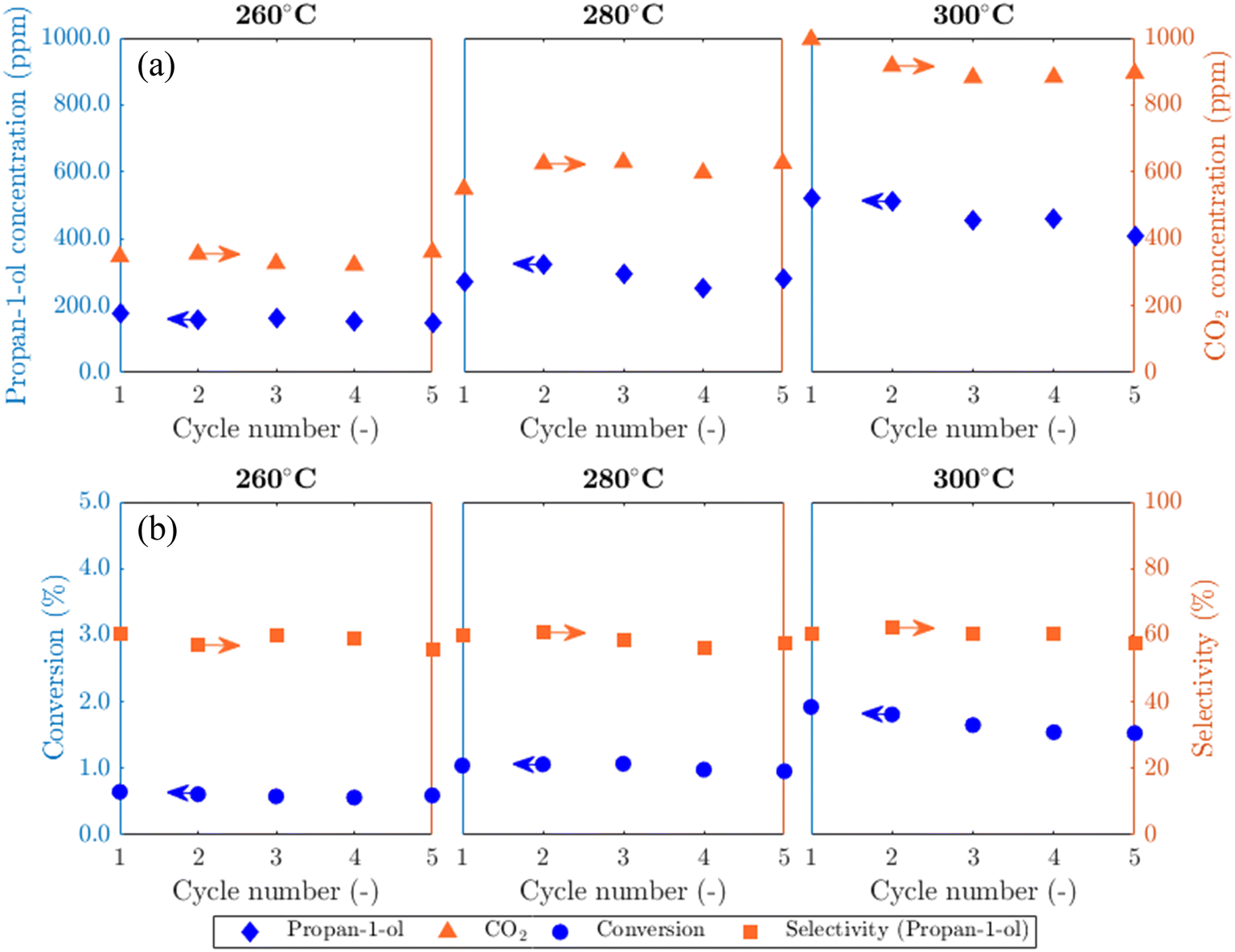 | ||
| Fig. 5 (a) Concentrations of propan-1-ol and CO2 and (b) calculated propylene conversion and selectivity towards propan-1-ol (shown on left and right axes, respectively) over the temperature range 260–300 °C for 5AgCl/5Au-D/SrFeO3. | ||
The effect of altering flowrate of the propylene feed (FC3H6) to the bed of 5Ag/5Au-D/SFO catalyst was also investigated; results are shown in Fig. S13.† The obtained trends are similar to those in Fig. 5 when increasing the temperature, namely, a longer residence time (0.5FC3H6) resulted in the improved conversion of C3H6. For that case, the selectivity to propan-1-ol dropped to ∼42%. When the residence time decreased (2FC3H6), the selectivity remained similar to that at 1FC3H6, namely, ∼60%. This minimal change in selectivity with temperature and residence time of propylene gives evidence that competing parallel and, or consecutive reactions are at play. Possible reactions that lead to the formation of propan-1-ol are further discussed in section 4.2.
Longer reduction steps (120 min) were performed over two AgCl/Au-D/SrFeO3 catalysts (Fig. S10†), showing total oxygen release greater than the oxygen available from Ag2O or from oxygen adsorbed at the catalyst surface (calculations for O are given in the ESI,† section S7, eqn (S2)–(S4)). For the more active sample (7.5AgCl/2.5Au-D/SrFeO3), the rate of oxygen release decreased with time as expected when oxygen for reaction is sourced from a solid oxygen carrier,45 here, SrFeO3. Given that the overall propylene conversion in the standard cycling experiments (Fig. 5a) was low, a reduction time of longer than 1.5 min may be achievable without impacting the catalytic performance, as the amount of oxygen accumulated in the gaseous products after 1.5 min of reduction corresponds to a very small change in SrFeO3−δ stoichiometry (Δδ ≪ 0.01). To determine the feasibility of increasing the length of the reduction step in chemical looping, redox cycling was performed over 7.5AgCl/2.5Au-D/SrFeO3, with the duration of the reduction step increased each cycle, as presented in Fig. 6. Fig. 6b shows that conversion of propylene rapidly decreases with reduction time up to ∼8–9 min reduction, but later declines slowly. The observed behaviour can be modelled as the sum of two exponential decay functions, given in Fig. 6b. This therefore suggests that the rate limiting step may change over the course of extended reduction, potentially from kinetic limitation by the surface reaction, to mass-transfer limitation from oxygen diffusing from bulk SrFeO3 to the catalyst surface. Selectivity to propan-1-ol remains ∼40% if reduction is shorter than 10 min, as shown in Fig. 6c. Decrease in performance with prolonged reduction has been previously observed for chemical looping epoxidation of ethylene19,23 and, to some degree, associated with the decreasing availability of oxygen from SrFeO3.
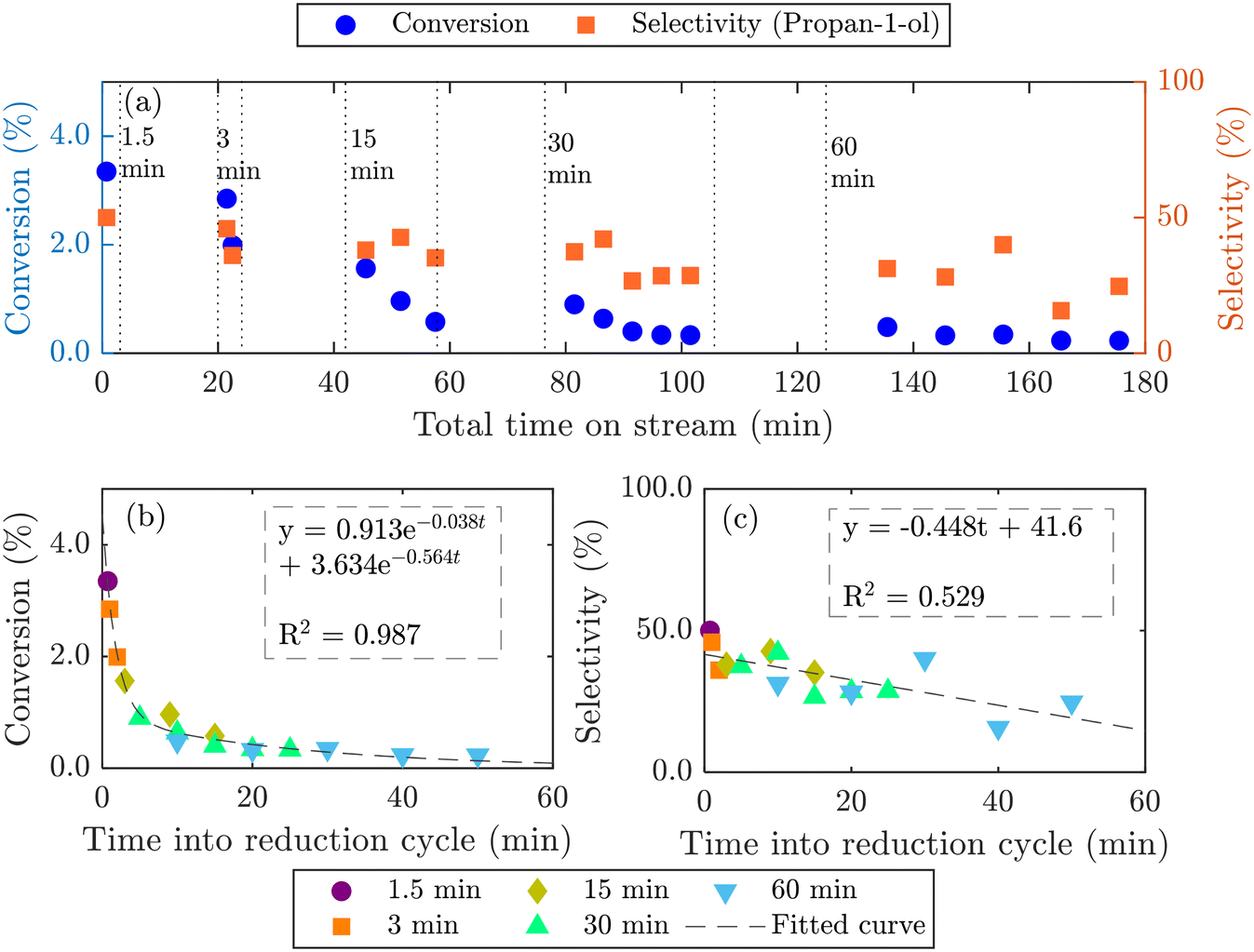 | ||
| Fig. 6 Chemical looping over 7.5AgCl/2.5Au-D/SrFeO3, with reduction time increased from 1.5 to 60 min. Vertical dotted lines in (a) indicate start and end of each reduction step. Change in conversion of propylene (b), and in selectivity towards propan-1-ol (c). | ||
The effect of altering the Ag to Au ratio in xAgCl/(10–x)Au/SrFeO3 catalysts was investigated, with average values of conversion of propylene and selectivity towards propan-1-ol and PO shown in Fig. 7, where error bars indicate the standard deviation taken from 5 redox cycles. Increasing the Ag loading in the Ag–Au mix at the catalyst surface increased the overall conversion of propylene, but the change was primarily driven by complete combustion rather than selective oxidation to desired products (outlet concentrations for each component are given in the ESI,† Fig. S9). Therefore, combustion of propylene likely occurs on Ag sites, as previously reported in studies of propylene oxidation with gaseous oxygen.5 Additionally, for all catalyst compositions, higher temperatures resulted in an increase in total conversion of propylene, but again, with unclear and only little effect on selectivity to propan-1-ol.
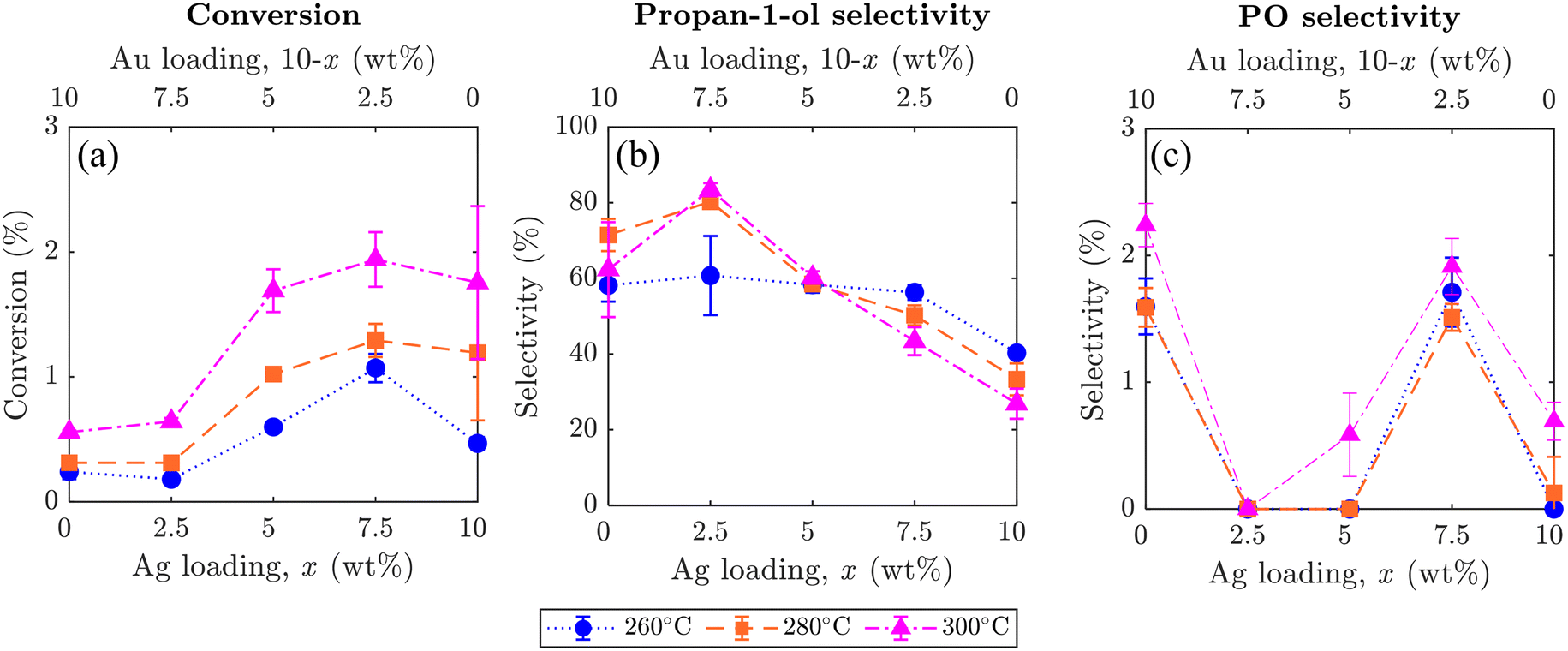 | ||
| Fig. 7 Graphs showing average values of (a) propylene conversion, (b) selectivity towards propan-1-ol, and (c) selectivity towards PO, for xAgCl/(10–x)Au-D/SrFeO3 samples (x = 0, 2.5, 5, 7.5, 10). Error bars show standard deviation over 5 cycles, noting that some error bars are smaller than the datapoint markers. Sample at x = 0 corresponds to Au-D/SrFeO3; sample at x = 10 corresponds to AgCl/SrFeO3. | ||
Selectivity towards propan-1-ol reached a maximum of ∼80% for the sample with a loading of 2.5AgCl/7.5Au-D/SrFeO3, with the selectivity decreasing with higher total Ag loading. The highest propan-1-ol concentration of ∼470 ppm in the outlet stream was observed in experiments with the 7.5AgCl/2.5Au-D/SrFeO3 sample (presented in Fig. S9†) at 300 °C. However, the result was accompanied by a high concentration of CO2, giving a lower overall selectivity for propan-1-ol than the sample of 2.5AgCl/7.5Au-D/SrFeO3.
For PO, the oxygenate product detected with second-highest concentration (Fig. 7), altering the loading of Ag and Au in the AgCl/Au-D/SrFeO3 samples had only limited effect on selectivity. For samples where appreciable concentrations of PO (up to 20 ppm, in Fig. S9†) were detected at the reactor outlet, namely Au-D/SrFeO3 and 7.5AgCl/2.5Au-D/SrFeO3, selectivities towards PO remained low, regardless of the operating temperatures, primarily, because the main products were still CO2 and propan-1-ol.
Results comparing the performance of Ag/SrFeO3 and AgCl/SrFeO3 are shown in Fig. 8. For the Ag/SrFeO3 sample, a notable decrease in catalyst activity was observed after the first chemical looping cycle, with PO concentration decreasing to near-zero, and CO2 concentration more than halving for all cycles. Contrastingly, the AgCl/SrFeO3 sample showed comparatively stable performance over 5 cycles, with substantially greater propan-1-ol concentrations than observed with the Ag/SrFeO3 samples, and lower concentrations of CO2. All cycles over AgCl/SrFeO3 gave low concentrations of PO at the outlet, below 6.0 ppm. The rapid decrease in PO formation over Ag/SrFeO3 after the first cycle suggests that PO was formed via reaction with AgOx surface species, as detected by XPS. As Ag2O and AgO are not thermodynamically stable at 260 °C or above,46 the silver oxides would not be regenerated during the oxidation step of chemical looping.
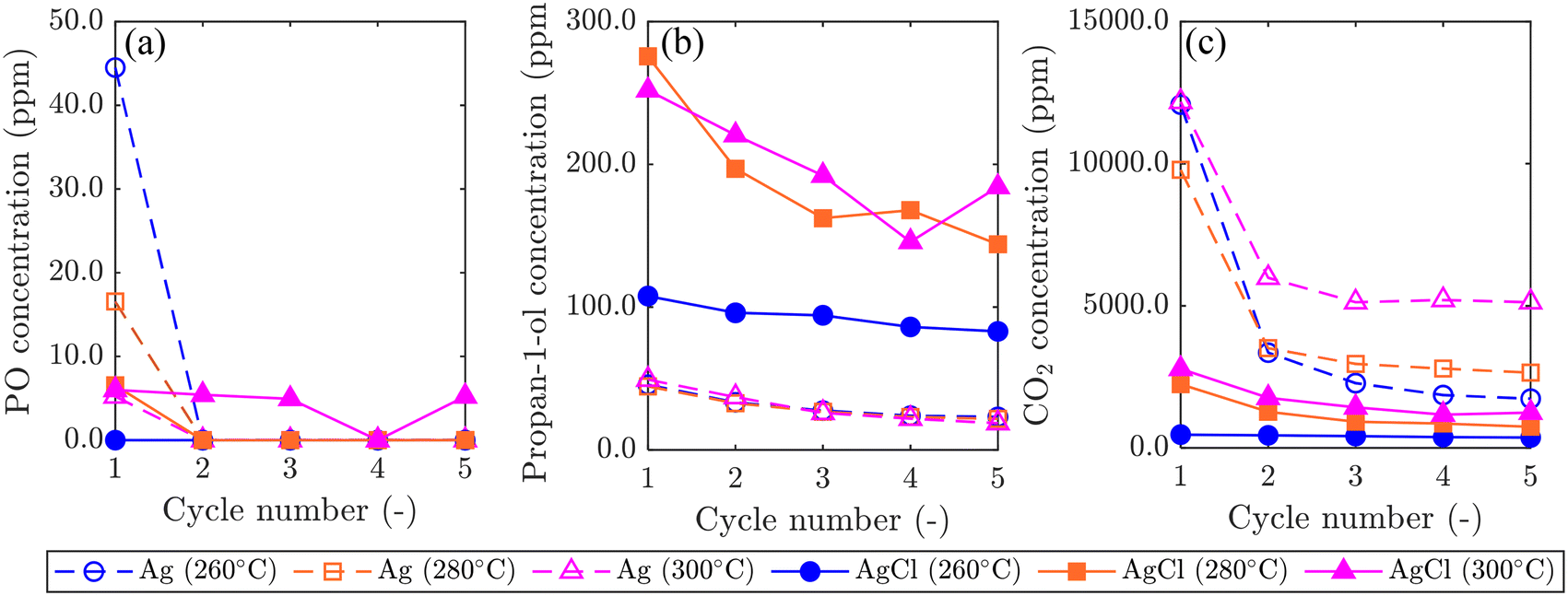 | ||
| Fig. 8 Graphs showing comparison of reaction products: (a) PO, (b) propan-1-ol, and (c) CO2 in experiments over Ag/SrFeO3 and AgCl/SrFeO3, in the temperature range 260–300 °C. Lines between cycles included to help differentiate samples. | ||
No AgOx species were detected at the surface of AgCl/SrFeO3 from XPS measurements, which may explain the lower levels of PO. For both catalysts, raising the temperature from 260 °C to 300 °C resulted in an increase in overall conversion, primarily driven by combustion. Interestingly, however, increasing temperature with Ag/SrFeO3 did not result in a significant change in outlet propan-1-ol concentrations.
The effect of changing the preparation method for Au-containing catalysts was also investigated. In Fig. 9a and b, the product distributions of 5Ag/5Au-D/SrFeO3 (calcined at 650 °C), Ag–AgCl/Au-H/SrFeO3 (calcined at 700 °C to induce thermal decomposition of AgCl), and Ag/Au-L/SrFeO3 (prepared using Au–(β-ala) precursor) are compared.
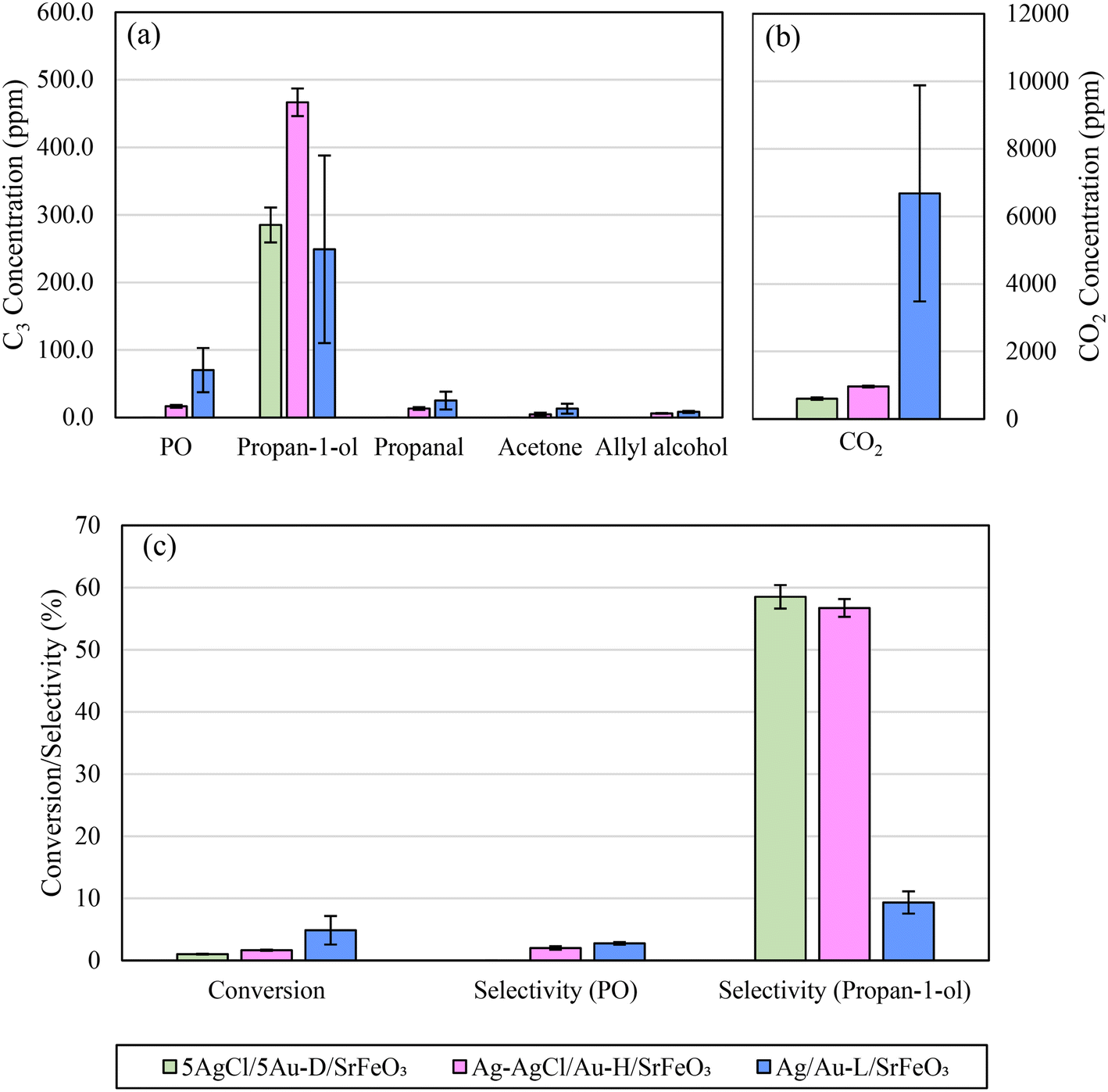 | ||
| Fig. 9 Comparison of (a) concentrations of C3 products, (b) concentrations of CO2, and (c) overall conversion and selectivity, for 5AgCl/5Au-D/SrFeO3, Ag–AgCl/Au-H/SrFeO3, and Ag/Au-L/SrFeO3 at 280 °C. Error bars correspond to standard deviation over 5 cycles. | ||
Under chemical looping conditions at 280 °C, the Ag–AgCl/Au-H/SrFeO3 sample produced a 65% greater concentration of propan-1-ol than the 5AgCl/5Au-D/SrFeO3 sample, with an average outlet concentration of 470 ppm propan-1-ol (Fig. 9 and S11†). However, concentration of CO2 was also ∼60% higher. Propanal, allyl alcohol, and propylene oxide, were detected at low levels (10–20 ppm) for the Ag–AgCl/Au-H/SrFeO3 sample, whereas none were detected for the 5AgCl/5Au-D/SrFeO3.
For the chloride-free Ag/Au-L/SrFeO3 sample containing only small amount of Au (0.02 wt%), a substantially different product distribution was measured as compared to the samples containing AgCl. The highest concentrations of PO (∼70 ppm) were detected in the outlet stream for Ag/Au-L/SrFeO3 as well as a slight decrease in propan-1-ol concentration as compared to 5AgCl/5Au-D/SrFeO3 or Ag–AgCl/Au-H/SrFeO3. However, the selectivity towards C3 oxygenates was markedly lower, due to a substantial increase in complete combustion. Average concentrations of CO2 detected were an order of magnitude greater than for other samples containing gold and were comparable with Ag/SrFeO3. Furthermore, for the Ag/Au-L/SrFeO3 sample, a substantial decrease in activity was measured over the course of 6 cycles (shown in Fig. S12†), in a manner similar to the Ag/SrFeO3 sample. We conclude that the presence of gold, even in minimal quantities (here, 0.02 wt%) was essential for producing PO, but that the overall behaviour of the Ag/Au-L/SrFeO3 catalyst was dictated by the dominant metal, Ag.
4. Discussion
4.1. Effect of Cl and Au on product distribution
In this study, AgCl catalysts were found to favour selective oxidation of propylene, whereas metallic Ag favoured complete combustion, both when comparing Ag/SrFeO3 and AgCl/SrFeO3 (Fig. 8), and when considering 5AgCl/5Au-D/SrFeO3vs. Ag–AgCl/Au-H/SrFeO3 and Ag/Au-L/SrFeO3 catalysts (Fig. 9). In previous studies with gaseous oxygen, unsupported bulk AgCl was found to be essentially inert with respect to oxidation of propylene,8,9 although a three-phase mixture of Ag, AgCl and CuO was demonstrated to be somewhat selective towards formation of propylene oxide.9 Furthermore, Ag catalysts promoted with ∼5 wt% NaCl to form mixed Ag–AgCl catalysts, both without supports7,8 and with non-reactive metal oxide supports,11 have shown appreciable conversion and selectivity towards propylene oxide and acrolein. The lack of activity over AgCl alone is potentially due to strong adsorption of molecular oxygen at the surface of AgCl with limited O2(g) dissociation,47 as compared to favourable dissociation of O2 over Ag (ref. 47) to form reactive Oa–Ag sites.The finding that the addition of Cl inhibits complete combustion, but enhances selective oxidation, is in line with previous investigations into the effect of Cl on propylene oxidation,7,8,10,11,48 however, previous studies focused on small amount of Cl for promoting selective reactions. In studies on direct epoxidation of propylene with O2(g),8 promotion of Ag catalysts NaCl to form a mixed Ag–AgCl catalyst substantially reduced the overall conversion of propylene, accompanied by an approximately proportional increase in selectivity towards propylene oxide. The presence of Cl− was found to suppress total oxidation of propylene by making adsorbed oxygen more electrophilic.8 The fact that, here, the AgCl-containing catalysts were found to be active towards formation of both propan-1-ol and CO2, may be because oxygen is provided from the lattice of the oxygen carrier, SrFeO3, as dissociated O atoms. Additionally, bulk AgCl has been demonstrated to be capable of accommodating and transporting oxygen, for example, Jayaraman and Yang used AgCl as a sorbent for pressure-swing adsorption of oxygen,49 demonstrating its notable adsorption capacity, and relatively rapid oxygen transport through the AgCl structure. Similarly, the results here demonstrate that AgCl on SrFeO3 can provide reactive atomic Oa species also to catalytic reactions. Comparison of oxygen adsorption and dissolution on AgCl, Ag, and Au is provided in the ESI,† section S7.
For the Ag/Au-L/SrFeO3 sample, the gold loading was found to be low (∼0.02 wt%), and as such the Au likely acted as a promoter for the Ag catalyst, rather than forming bulk AgAu alloy, although the exact mechanism remains unclear. The presence of Au strongly affected the process selectivity, with Ag/Au-L/SrFeO3 catalyst producing the highest concentration of PO, and limited propan-1-ol (as shown in Fig. 9). An order of magnitude more CO2 was detected for reaction over the Ag/Au-L/SrFeO3 catalyst as compared to 5AgCl/5Au-D/SrFeO3 and Ag–AgCl/Au-H/SrFeO3, which was expected to take place on the metallic Ag surface present in Ag/Au-L/SrFeO3. The increase in CO2 for the chloride-free sample also supports the hypothesis that Cl species suppress complete combustion.
Given that both AgCl/SrFeO3 and Au-D/SrFeO3 showed appreciable selectivity towards propan-1-ol, both AgCl and Au must be catalytically selective in the chemical looping arrangement towards propan-1-ol. From Fig. 7b, maximum selectivity towards propan-1-ol was observed for the sample of 2.5AgCl/7.5Au/SrFeO3, where both catalysts (AgCl and Au) were present. However, STEM-EDS images (Fig. 3) show little overlap between AgCl and AgAu particles, thus, possible synergy is unclear. Furthermore, from SEM and STEM-EDS (presented in Fig. 3 and S6–S8†), AgCl is present as relatively small particles (∼100 nm), which are likely to be catalytically active, whereas AgAu is present as larger chunks (∼400 nm). For AgAu, the reaction likely occurred at atomic-scale Ag sites at the surface of the large AgAu particles, as demonstrated in previous studies.13,14
The increase in propylene conversion with increasing ratio of Ag to Au on SrFeO3 (as shown in Fig. 7a) suggests a faster rate of reaction at nanoparticles of AgCl catalyst than at nanoparticles of AgAu alloy catalyst. An extended reduction cycle (shown in Fig. S10†) also indicated a markedly faster rate of total release of oxygen in reactions over 7.5AgCl/2.5Au-D/SrFeO3 as compared to 2.5AgCl/7.5Au-D/SrFeO3. For the latter material, the rate of oxygen release was approximately constant over 100 min, suggesting that the process was limited by surface reactions between propylene and Oa species. In contrast, for the particles of 7.5AgCl/2.5Au-D/SrFeO3, rate of oxygen release decreased over 100 min. Reduction times that are substantially longer than 1.5 min may be feasible over the catalysts investigated, however, chemical looping over 7.5AgCl/2.5Au-D/SrFeO3 showed that conversion rapidly decreases for reduction times up to 8–9 min before levelling off (shown in Fig. 7), and that selectivity gradually declines with longer reduction steps. Results presented in Fig. 5, however, confirm that chemical looping with 1.5 min (i.e. where regeneration of the oxygen carrier is more frequent) leads to stable selectivity across at least 5 cycles.
4.2. Mechanisms for propan-1-ol formation
In this work, a different product distribution was detected as compared to any other studies reported in literature5,6 for propylene oxidation over Ag/Au catalysts, with propan-1-ol as a major reaction product. While the main difference is the delivery of oxygen to reactions (here, from SrFeO3, while in other studies from the gas feed), the reason for the selective formation of propan-1-ol rather than any other possible oxygenates is unclear. Thus, we analysed several potential mechanisms for formation of the primary C3 alcohol, starting with direct hydration of propylene by H2O.50 Given that in all experiments complete combustion occurred, water vapour was expected to be present in the reaction mixture. However, a hydration mechanism of propylene would predict formation of both primary and secondary alcohols, with propan-2-ol being the favoured product, because of the stabilisation of the secondary carbocation intermediate via electronic induction from the two methyl groups on either side of the localised positive charge.4,50 Given that <5 ppm of propan-2-ol was detected as compared to up to 500 ppm propan-1-ol under chemical looping conditions, the direct hydration mechanism appears unlikely. Furthermore, addition of 2700 ppm H2O to propylene during reduction did not result in a significant change in outlet propan-1-ol concentration (shown in ESI,† Fig. S18), suggesting that reaction between propylene and water is unlikely.Another possible mechanism considered involves an oxygenated intermediate: PO, which forms and reacts further via the following reactions:
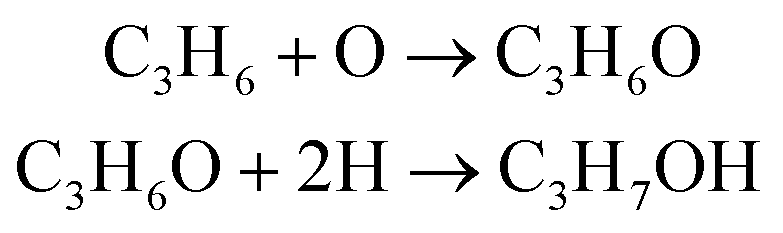 | (R2) |
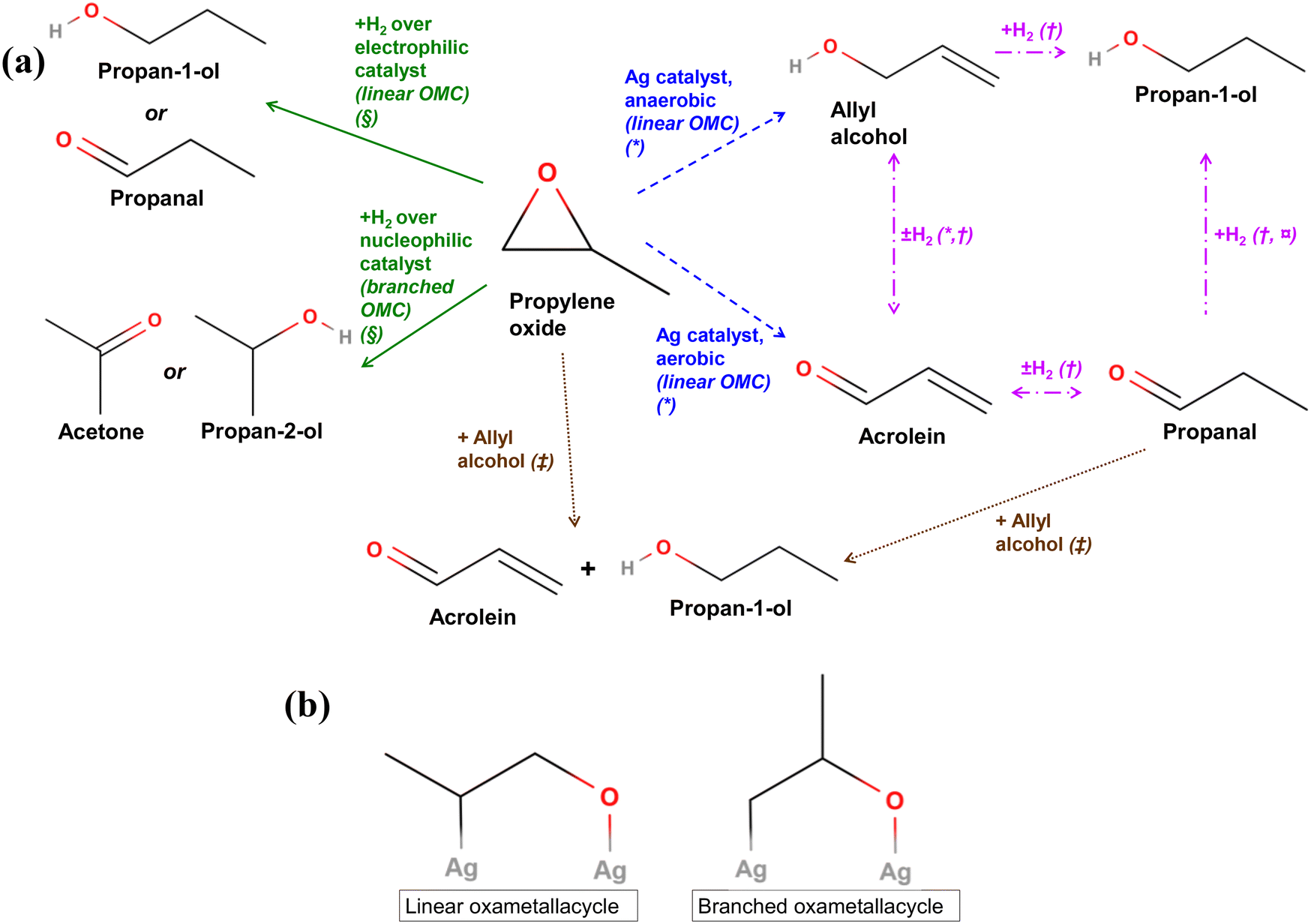 | ||
Fig. 10 (a) Summary of main reactions of PO reported in literature. * = ref. 12, † = ref. 51 and 52, § = ref. 53 and 54,  = ref. 55 and 56, ‡ = ref. 57 and 58. (b) Linear and branched oxametallacycle (OMC) surface species.12,54 = ref. 55 and 56, ‡ = ref. 57 and 58. (b) Linear and branched oxametallacycle (OMC) surface species.12,54 | ||
The mechanisms shown in Fig. 10a involving a C3 oxygenate intermediate reacting with hydrogen to form propan-1-ol clearly require the presence of hydrogen in the reaction stream. A potential source of hydrogen in the chemical looping setup could originate from propylene coking at the catalyst surface. With no oxygen present in the gas-phase during the step when the hydrocarbon is oxidised, any hydrogen formed may be able to go on to react with oxygenate intermediates to form propan-1-ol, or with lattice oxygen to form water. However, in the experiments presented, no hydrogen was detected in the outlet stream. Another possibility is that hydrogen may be supplied from water or surface hydroxide species absorbed on the SrFeO3 surface, which were also possibly detected via XPS (Fig. 4). Moreover, the addition of excess gaseous hydrogen to the propylene feed stream during the reduction step resulted in the preferential reaction of H2 with surface Oa species, or with the SrFeO3 oxygen carrier, to form water, limiting the oxygen available for reaction with C3 species (shown in ESI,† Fig. S19).
Reactions between propylene oxide and H2 have been investigated over various metal catalysts. Bartók and co-workers found that for propylene oxide, reaction over strongly electrophilic Ni or Cu catalysts resulted in the splitting of the more sterically hindered C–O bond in the PO molecule, forming a linear oxametallacycle species (Fig. 10b), which then reacts with H2 and subsequently desorbs to form propan-1-ol or propanal (shown in Fig. 10a, green solid lines).53,54 In contrast, when PO with H2 was passed over less electrophilic catalysts, such as Pt or Pd, splitting of the less sterically hindered C–O bond was favoured, forming a branched oxametallacycle species. The branched surface species, presented in Fig. 10b, could then react with H2, and desorb forming propan-2-ol or acetone. As AgCl is highly electrophilic,8 it may behave similarly to the Ni or Cu surfaces when exposed to PO. Therefore, propan-1-ol may form over xAgCl/(10–x)Au-D/SrFeO3 catalysts by initial formation of PO, followed by the reaction of the PO with H2 at a strongly electrophilic AgCl surface. Gold and gold–silver surfaces might be expected to behave in a similar manner to Pt or Pd, i.e. by forming a non-electrophilic surface. The Au surface would then be expected to favour formation of branched oxametallacycle surface species and, consequently, secondary oxygenate products (acetone and propan-2-ol). However, Au-D/SrFeO3 was primary active towards propan-1-ol oxygenate (shown in Fig. 7), with little to no selectivity towards acetone or propan-2-ol, suggesting electrophilic behaviour and linear oxametallacycle route. The electrophilic behaviour of Au agrees with the finding59 that noble metals impregnated on perovskites transfer electrons to the perovskite oxide on top of which there are deposited, resulting in an electrophilic metal surface.
For reactions over Ag/SrFeO3 (Fig. 8), although Ag surfaces are able to facilitate the formation of OMC species12 resulting in trace concentrations of propan-1-ol, the primary reaction product was CO2, due to catalytic combustion of propylene via allylic hydrogen stripping.5
To determine whether the mechanism of propan-1-ol formation via hydrogenation of PO is plausible, chemical looping experiments were performed with PO added to the feed stream. Fig. 11a presents the measured compositions of the feed gas at the inlet of the packed-bed reactor and after the bed of 5Ag/5Au-D/SrFeO3. For comparison, the gas mixture in one of the experiments contained only propylene (nominal concentration ∼3 vol%), while in the other, contained propylene and PO (∼3 vol% and 400 ppm, respectively).
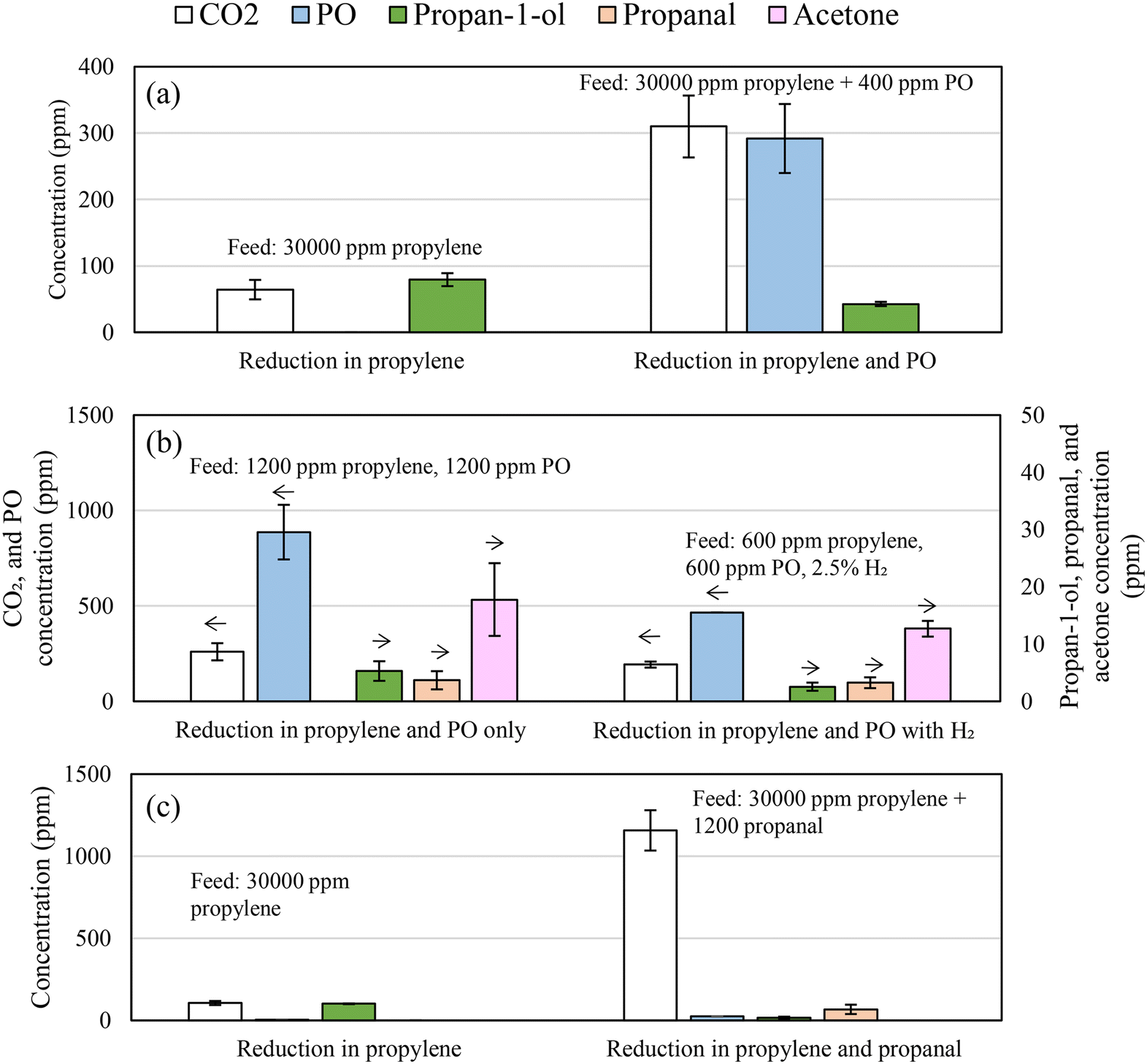 | ||
| Fig. 11 Reaction products for reaction over 5Ag/5Au-D/SrFeO3, for (a) propylene with and without PO added (b) dilute PO/propylene, with and without H2 (c) for propylene with and without propanal added. Error bars show standard deviation from 3 cycles, arrows indicate y-axis for each species. Feed composition given on each figure, with balance gas Ar/N2. | ||
When passing 2.5 vol% propylene over 5AgCl/5Au-D/SrFeO3 with PO in the gas feed, significantly less propan-1-ol was formed, and more CO2. When lower concentrations (500 ppm) of propylene and PO were fed over the bed of 5Ag/5Au-D/SrFeO3 particles, as shown in Fig. 11b, propan-1-ol was not detected in the outlet, with acetone and propanal being the only C3 oxygenate products above 5 ppm. The situation did not change after including H2 in the feed, for an experiment with a ‘PO:propylene:H2’ mixture. However, the additional PO from the feed may compete with propylene for adsorption sites, and once adsorbed, become fully oxidised to CO2, rather than being hydrogenated to form propan-1-ol. Therefore, hydrogenation of PO to form propan-1-ol appears to be feasible only prior to the oxametallacycle desorbing.
Barteau and co-workers12 proposed that for PO adsorbed onto an Ag surface, the gaseous PO, and linear and branched oxametallacycle intermediates are in equilibrium. Therefore, any PO added to the inlet stream would increase the amount of all intermediates that can isomerise to propan-1-ol but also other C3 oxygenates. They also suggested that the linear oxametallacycle is able to undergo a linear 1,2 hydrogen shift, and then desorb as allyl alcohol (shown in Fig. 10a, blue dashed lines). Under aerobic conditions with O2(g), any allyl alcohol formed was found to rapidly become dehydrogenated to form acrolein, which then fully oxidised to CO2. Low levels (6–10 ppm) of allyl alcohol were detected in the outlet stream for reaction of propylene over Ag–AgCl/Au-H/SrFeO3 and Ag/Au-L/SrFeO3 samples (shown in Fig. 9a). Allyl alcohol that is adsorbed on Ag is also able to react with adsorbed hydrogen, Ha, to form propan-1-ol (Fig. 10a, pink dash-dotted lines).51,52 Therefore, allyl alcohol may form by reaction of propylene with Oa without rapidly dehydrogenating to acrolein, and then undergo hydrogenation at the catalyst surface to form propan-1-ol.
Reactions between PO and allyl alcohol, and propanal and allyl alcohol, have been reported by Imanaka and co-workers.57,58 A hydrogen transfer reaction between one molecule of PO or propanal and one molecule of allyl alcohol occurs, to form propan-1-ol and acrolein in a 1:1 molar ratio (Fig. 10a, brown dotted lines). Reaction between PO and allyl alcohol may occur over AgCl/Au/SrFeO3 catalysts; however, the hydrogen transfer reaction would predict acrolein forming in a 1:1 ratio with propan-1-ol. As no acrolein was detected in the chemical looping experiments, the reaction between PO and allyl alcohol is unlikely to contribute significantly to overall propan-1-ol formation reported here. Furthermore, propanal was predicted to also undergo reaction with allyl alcohol to form propan-1-ol and acrolein.58 Given that adding propanal to the experimental feed stream (shown in Fig. 11c) resulted in a moderate decrease in propan-1-ol formation, the reaction between allyl alcohol and propanal is also discounted as a major source of propan-1-ol in the outlet stream.
The reaction of aldehydes and ketones (i.e. propanal and acetone) with hydrogen over gold catalysts to form primary or secondary alcohols, respectively, has been investigated via TPD studies by Pan and co-workers.55,56 The authors found that in the case of hydrogen atoms adsorbed on Au(111), hydrogenation of propanal to propan-1-ol was favoured, and that minimal hydrogenation of acetone to propan-2-ol occurs, as the reaction pathway to form propan-2-ol is not thermodynamically favourable. Hydrogenation of propanal to propan-1-ol would be consistent with the observed experimental results that propan-1-ol is produced in substantially greater quantities over the mixed AgCl/Au catalysts, as opposed to Au alone. To determine if propan-1-ol formation via propanal was feasible, ∼1200 ppm propanal was added to the 5 vol% propylene feed stream over 5AgCl/5Au-D/SrFeO3 catalyst. Fig. 11c shows that the addition of propanal to the inlet feed did not increase the concentration of propan-1-ol measured in the outlet, and instead, showed an increase in complete combustion only. The lack of an increase in propan-1-ol concentration suggests that any propanal formed over AgCl/Au-D/SrFeO3 catalysts undergoes further oxidation to CO2, and so that the reaction mechanism for propan-1-ol formation is unlikely to proceed via propanal.
In summary, the most likely reaction mechanism for propan-1-ol formation is concluded to proceed via initial reaction of propylene with adsorbed surface Oa to form a linear oxametallacycle surface species. The linear oxametallacycle then either reacts directly with hydrogen to form propan-1-ol, or undergoes a hydrogen shift to form allyl alcohol, which is subsequently hydrogenated to propan-1-ol. Further understanding of the surface mechanism may be achieved by performing density-functional theory (DFT) calculations of the hypothesised transition states, or through surface-sensitive experimental measurements (e.g. in situ XPS measurements of Ag and Au surface states over chemical looping cycles).
5. Conclusions
The selective oxidation of propylene via a chemical looping approach was demonstrated with AgCl, AgAu and mixed AgCl/Au catalysts supported on SrFeO3 showing considerable selectivity towards propan-1-ol, with limited formation of other oxygenates. Catalysts containing AgCl were more selective to propan-1-ol as compared to Ag, which promoted complete combustion. Performance of the AgCl/Au catalysts was sensitive to the ratio of Ag and Au, with higher Ag content leading to favoured combustion over any oxygenated products. Selectivity towards propan-1-ol reaching 80% was achieved over 2.5AgCl/7.5Au-D/SrFeO3 catalyst, albeit at <1% propylene conversion. In contrast, Ag/Au-L/SrFeO3, led to PO as a major C3 product. Both results demonstrate that chemical looping can lead to new reaction pathways, strongly depended on the oxygen carrier and the catalyst. The mechanism of the chemical looping formation of propan-1-ol was also discussed, showing that the most likely pathway proceeds by surface reactions of oxametallacycle intermediates, rather than desorption and subsequent reabsorption of oxygenated species.Author contributions
ARPH: material synthesis and characterisation, experiments, formal analysis, methodology, writing—original draft, revision. EJM: supervision, conceptualisation, writing—original draft, revision, funding acquisition. All authors read and approved the final manuscript.Conflicts of interest
The authors have no competing interests to declare that are relevant to the article content.Acknowledgements
ARPH acknowledges financial support from the Cambridge School of Technology Vice-Chancellor's award and Emmanuel College. The research was carried out with funding from EPSRC studentship grant no. 2376181. Thanks are given to Dr H. Greer, Dr C. Truscott, and Dr N. Howard (Yusuf Hamied Department of Chemistry, University of Cambridge) for assistance with electron microscopy imaging and ICP measurements, and to Dr C. Fernandez-Posada (Maxwell Centre, University of Cambridge) for assistance with XPS measurements and analysis. The authors acknowledge use of the Cambridge XPS System, part of the Sir Henry Royce Institute – Cambridge Equipment, EPSRC grant EP/P024947/1. This research was carried out with financial support from UK Research and Innovation, Grant No. EP/V048414/1. Thanks are also given to Professor A. Hayhurst for productive discussions while preparing the manuscript.References
- D. L. Trent, Kirk-Othmer Encycl. Chem. Technol., 2000 DOI:10.1002/0471238961.1618151620180514.a01.
- D. Arntz, A. Fischer, M. Höpp, S. Jacobi, J. Sauer, T. Ohara, T. Sato, N. Shimizu and H. Schwind, Ullmann’s Encycl. Ind. Chem., 2007 DOI:10.1002/14356007.a01_149.pub2.
- M. McCoy, Chem. Eng. News Arch., 2001, 79, 19–20 Search PubMed.
- J. Li, Z. Qin, H. Xu, M. Dong, J. Dong and J. Wang, Ind. Eng. Chem. Res., 2007, 46, 9000–9005 CrossRef CAS.
- S. J. Khatib and S. T. Oyama, Catal. Rev.: Sci. Eng., 2015, 57, 306–344 CrossRef CAS.
- J. Teržan, M. Huš, B. Likozar and P. Djinović, ACS Catal., 2020, 10, 13415–13436 CrossRef.
- G. Lu and X. Zuo, Catal. Lett., 1999, 58, 67–70 CrossRef CAS.
- J. Lu, M. Luo, H. Lei and C. Li, Appl. Catal., A, 2002, 237, 11–19 CrossRef CAS.
- M. Luo, J. Lu and C. Li, Catal. Lett., 2003, 86, 43–49 CrossRef CAS.
- A. Seubsai and S. Senkan, ChemCatChem, 2011, 3, 1751–1754 CrossRef CAS.
- I. D. Charisteidis and K. S. Triantafyllidis, Catal. Today, 2020, 355, 654–664 CrossRef CAS.
- A. Kulkarni, M. Bedolla-Pantoja, S. Singh, R. F. Lobo, M. Mavrikakis and M. A. Barteau, Top. Catal., 2012, 55, 3–12 CrossRef CAS.
- P. Geenen, H. Boss and G. Pott, J. Catal., 1982, 77, 499–510 CrossRef CAS.
- S. Rojluechai, S. Chavadej, J. W. Schwank and V. Meeyoo, Catal. Commun., 2007, 8, 57–64 CrossRef CAS.
- A. Dibenedetto, M. Aresta, C. Fragale, M. Distaso, C. Pastore, A. Venezia, C. Liu and M. Zhang, Catal. Today, 2008, 137, 44–51 CrossRef CAS.
- V. Fattore, Z. Fuhrman, G. Manara and B. Notari, J. Catal., 1975, 37, 215–222 CrossRef CAS.
- V. Fattore, Z. Fuhrman, G. Manara and B. Notari, J. Catal., 1975, 37, 223–231 CrossRef CAS.
- X. Zhu, Q. Imtiaz, F. Donat, C. R. Müller and F. Li, Energy Environ. Sci., 2020, 13, 772–804 RSC.
- M. S. C. Chan, E. Marek, S. A. Scott and J. S. Dennis, J. Catal., 2018, 359, 1–7 CrossRef CAS.
- Y. Gao, L. M. Neal and F. Li, ACS Catal., 2016, 6, 7293–7302 CrossRef CAS.
- T. Sheppard, C. D. Hamill, A. Goguet, D. W. Rooney and J. M. Thompson, Chem. Commun., 2014, 50, 11053–11055 RSC.
- S. Gabra, E. J. Marek, S. Poulston, G. Williams and J. S. Dennis, Appl. Catal., B, 2021, 286, 119821 CrossRef CAS.
- J. C. Gebers, A. R. P. Harrison and E. J. Marek, Discov. Chem. Eng., 2022, 2, 4 CrossRef.
- C. Y. Lau, M. T. Dunstan, W. Hu, C. P. Grey and S. A. Scott, Energy Environ. Sci., 2017, 10, 818–831 RSC.
- E. Marek, W. Hu, M. Gaultois, C. P. Grey and S. A. Scott, Appl. Energy, 2018, 223, 369–382 CrossRef CAS.
- J. Lei, J. Dai, K. B. Tan, J. Huang, G. Zhan and Q. Li, ACS Sustainable Chem. Eng., 2021, 9, 794–808 CrossRef CAS.
- X. Zhang, J. Dai, J. Ding, K. B. Tan, G. Zhan, J. Huang and Q. Li, Catal. Sci. Technol., 2022, 12, 2426–2437 RSC.
- Z. Suo, M. Jin, J. Lu, Z. Wei and C. Li, J. Nat. Gas Chem., 2008, 17, 184–190 CrossRef CAS.
- M. R. H. Siddiqui, S. F. Adil, M. E. Assal, R. Ali and A. A. Al-Warthan, Asian J. Chem., 2013, 25, 3405–3409 CrossRef CAS.
- H. Murayama, T. Hasegawa, Y. Yamamoto, M. Tone, M. Kimura, T. Ishida, T. Honma, M. Okumura, A. Isogai, T. Fujii and M. Tokunaga, J. Catal., 2017, 353, 74–80 CrossRef CAS.
- N. Doebelin and R. Kleeberg, J. Appl. Crystallogr., 2015, 48, 1573–1580 CrossRef CAS PubMed.
- A. Belsky, M. Hellenbrandt, V. L. Karen and P. Luksch, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 364–369 CrossRef PubMed.
- M. P. Seah, I. S. Gilmore and G. Beamson, Surf. Interface Anal., 1998, 26, 642–649 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- E. J. Marek and E. García-Calvo Conde, Chem. Eng. J., 2020, 417, 127981 CrossRef.
- G. E. Lloyd, Mineral. Mag., 1987, 51, 3–19 CrossRef CAS.
- N. N. Kariuki, J. Luo, M. M. Maye, S. A. Hassan, T. Menard, H. R. Naslund, Y. Lin, C. Wang, M. H. Engelhard and C.-J. Zhong, Langmuir, 2004, 20, 11240–11246 CrossRef CAS PubMed.
- S. Malathi, T. Ezhilarasu, T. Abiraman and S. Balasubramanian, Carbohydr. Polym., 2014, 111, 734–743 CrossRef CAS PubMed.
- I. Srnová-Šloufová, B. Vlčková, Z. Bastl and T. L. Hasslett, Langmuir, 2004, 20, 3407–3415 CrossRef PubMed.
- R. J. Chimentão, I. Cota, A. Dafinov, F. Medina, J. E. Sueiras, J. L. G. de la Fuente, J. L. G. Fierro, Y. Cesteros and P. Salagre, J. Mater. Res., 2006, 21, 105–111 CrossRef.
- H. Falcón, J. A. Barbero, J. A. Alonso, M. J. Martínez-Lope and J. L. G. Fierro, Chem. Mater., 2002, 14, 2325–2333 CrossRef.
- A. Abd El-Naser, E. K. Abdel-Khalek, E. Nabhan, D. A. Rayan, M. S. Gaafar and N. S. Abd El-Aal, Philos. Mag., 2020, 101, 710–728 CrossRef.
- V. K. Kaushik, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 273–277 CrossRef CAS.
- Y. Qin, Y. Cui, Z. Tian, Y. Wu and Y. Li, Nanoscale Res. Lett., 2017, 12, 247 CrossRef PubMed.
- B. Bulfin, J. Vieten, S. Richter, J. M. Naik, G. R. Patzke, M. Roeb, C. Sattler and A. Steinfeld, Phys. Chem. Chem. Phys., 2020, 22, 2466–2474 RSC.
- I. Karakaya and W. T. Thompson, J. Phase Equilib., 1992, 13, 137–142 CrossRef CAS.
- S. Kim, S.-C. Lee, C. Lee, M. H. Kim and Y. Lee, Nano Energy, 2018, 48, 134–143 CrossRef CAS.
- J. Lu, J. J. Bravo-Suárez, M. Haruta and S. T. Oyama, Appl. Catal., A, 2006, 302, 283–295 CrossRef CAS.
- A. Jayaraman and R. T. Yang, Chem. Eng. Sci., 2005, 60, 625–634 CrossRef CAS.
- J. E. Logsdon and R. A. Loke, Kirk-Othmer Encycl. Chem. Technol., 2000 DOI:10.1002/0471238961.0919151612150719.a01.
- K. Brandt, M. E. Chiu, D. J. Watson, M. S. Tikhov and R. M. Lambert, J. Am. Chem. Soc., 2009, 131, 17286–17290 CrossRef CAS PubMed.
- M. Bron, D. Teschner, A. Knopgericke, B. Steinhauer, A. Scheybal, M. Havecker, D. Wang, R. Fodisch, D. Honicke and A. Wootsche, J. Catal., 2005, 234, 37–47 CrossRef CAS.
- F. Notheisz, Á. Molnár, Á. Zsigmond and M. Bartók, J. Catal., 1986, 98, 131–137 CrossRef CAS.
- M. Bartók, F. Notheisz and Á. Zsigmond, J. Catal., 1980, 63, 364–371 CrossRef.
- M. Pan, Z. D. Pozun, A. J. Brush, G. Henkelman and C. B. Mullins, ChemCatChem, 2012, 4, 1241–1244 CrossRef CAS.
- M. Pan, A. J. Brush, Z. D. Pozun, H. C. Ham, W.-Y. Yu, G. Henkelman, G. S. Hwang and C. B. Mullins, Chem. Soc. Rev., 2013, 42, 5002 RSC.
- T. Imanaka, Y. Okamoto and S. Teranishi, Bull. Chem. Soc. Jpn., 1972, 45, 1353–1357 CrossRef CAS.
- Y. Okamoto, T. Imanaka and S. Teranishi, Bull. Chem. Soc. Jpn., 1973, 46, 4–8 CrossRef CAS.
- Q. Wang, Y. Gu, W. Zhu, L. Han, F. Pan and C. Song, Adv. Funct. Mater., 2021, 31, 1–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00222a |
| This journal is © The Royal Society of Chemistry 2022 |