
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Data-rich process development of immobilized biocatalysts in flow
Jacob H.
Forstater
 * and
Shane T.
Grosser
* and
Shane T.
Grosser
Process Research and Development, Merck & Co., Inc., Rahway, NJ 07065, USA. E-mail: Jacob.forstater@merck.com
First published on 7th January 2022
Abstract
Immobilization has been widely applied to improve the productivity and lifetime of select enzyme classes used in commodity chemical synthesis. Advances in enzyme engineering capabilities and high throughput experimentation have dramatically increased the use of novel enzyme classes and their applications to pharmaceutical synthesis, where immobilization is used to increase enzyme loading and stability and improve protein-product isolation. Conventional approaches to immobilization development are time and resource-intensive and cannot meet pharmaceutical development's demanding timelines. We developed a data-rich methodology that uses a medium pressure chromatography system to automate the screening and development of affinity-based enzyme immobilizations. We integrated inline PAT to characterize the immobilization process in real-time and facilitate rapid decision making. Most critically, this approach served to significantly reduce the amounts of enzyme and resin required for meaningful process development, transforming immobilization process development and optimization from a manual 6–8 week process to an automated overnight process. This paper demonstrates the development and application of data-rich experimental methods to rapidly identify, develop, and optimize robust, scalable immobilization processes, provide improved fundamental understanding, and describes how this methodology has enabled multiple successful commercial-scale immobilizations.
Introduction
Enzyme immobilization is an established practice in commodity chemical synthesis that enables enzyme reuse and improves the batch productivity of common enzymes.1–3 Recently, significant advances in enzyme engineering have dramatically improved the speed of enzyme development and expanded the scope and utility of enzyme-catalyzed transformations in pharmaceutical synthesis.4–6 Immobilization is increasingly used to solve challenges across the biocatalytic process development and commercialization lifecycle. For example, immobilization facilitates utilization of high loadings of low activity early enzyme variants, improves protein-product isolation, and enables the deployment of multienzyme cascades.7,8Many commodity immobilized enzymes are immobilized by covalent linkage to a functionalized support or adsorption to a polymeric resin;9 both approaches require significant development efforts that are specific to the enzyme and reaction chemistry and often result in considerable enzyme activity losses. Affinity chromatography is a commonly used protein purification technique that employs specific interactions between the protein and a ligand to capture and purify a target protein from a crude feedstock.10 After immobilizing the target protein to the resin, contaminants are washed from the resin. Subsequently, the immobilized-protein is eluted by competitive displacement or changing the buffer composition to conditions that disfavor the affinity interaction. This approach is attractive for pharmaceutical applications and allows for the selective immobilization of the target enzyme using resins and methods that are well established for pharmaceutical synthesis and have regulatory precedent to enable resin reuse. One such affinity technique is immobilized metal affinity chromatography (IMAC), which exploits the coordination of histidines' aromatic nitrogen with a divalent transition metal cation to capture proteins using a metal-functionalized resin. A short polyhistidine sequence is appended to the target protein's N or C terminus to enable the requisite interactions for immobilization.11–13 This approach results in selective immobilization of the protein with binding strengths greater than most antibody–antigen interactions.14
Recently, we,7 along with other groups15,16 have utilized IMAC as a platform for immobilization. To date, no systematic methodology has been presented to enable rapid development and optimization of these immobilizations for specific needs such as improved purity, high enzyme loading, or limiting downstream protein burden in pharmaceutical synthesis. Manual methods for screening and developing these immobilizations are labor and material intensive, relying on iterative resin and condition screening and batch reaction execution.1 These approaches cannot meet pharmaceutical process development needs and cannot compete with the pace of enzyme evolution or accommodate ongoing changes to the synthetic route. As a result of these inefficiencies, immobilization is generally the option of last resort, used to solve engineering challenges in previously established processes rather than as a tool to enable biocatalytic process development and manufacture.
We developed a semi-automated, data-rich immobilization development platform that enables rapid discovery, process development, and optimization of metal affinity immobilizations for immobilized biocatalysis to meet these challenges. The system automates screening of protein immobilization conditions such as buffer, pH, mobile phase additives, and protein concentration. Inline pH, UV, and conductivity measurements enable real-time monitoring of protein and buffer composition to provide immediate feedback to process sensitivities, while automated fraction collection enables offline analysis using other analytical techniques.
Using this platform, we significantly improved the efficiency of our development process. Before this effort, a standard immobilization required multiple months of labor and grams of protein to develop. With this platform, a typical immobilization development can be completed in a matter of days, is highly automated, and requires only milligrams quantities of unpurified protein. We have successfully utilized this combined approach to develop robust immobilized enzymes, which have enabled multiple biocatalytic processes, including a multienzyme immobilized biocatalytic cascade used to synthesize islatravir.7
This paper identifies the conditions necessary to obtain quantitative measurements of immobilized yield and relative affinity with minimal length screens. Furthermore, we utilize this system to elucidate the relationship the chelating ligand and metal have on the immobilized affinity and purity and demonstrate that imidazole, a histidine analog, can be used to both vary the purity and yield of the immobilization. Finally, we detail how these measurements can be used to target specific application requirements across the development and commercialization lifecycle and enable design of both flow and batch-based immobilizations.
Methods
Reagents
Recombinantly produced galactose oxidase was obtained as a lyophilized crude cell-free extract, as detailed previously.7 Protein solutions were made by the dissolution of lyophilized cell-free-extract into the desired buffer. Buffers were made from pH-adjusted 1 M sodium phosphate buffer (Boston Bioproducts), pH adjusted 2 M imidazole (Teknova, pH 8.0), 5 M NaCl (Sigma Aldrich), and UHPLC-grade water (Thermo Scientific). Except where stated otherwise, all other reagents were obtained from Sigma Aldrich and used without further purification.Agarose IDA and NTA resins (Bio Works) were obtained as 1 mL columns (28 mm bed height × 7 mm ID), pre-charged with the desired metal; the mean particle diameter was specified by the vendor as 45 μm. All columns were cleaned and re-charged with metal according to their manufacturer's specifications.
Ni-NTA Superflow (Qiagen), Sepharose 6FF IMAC (Cytiva), Nuvia IMAC Ni (Bio-rad), were obtained as prepacked 0.5 mL columns (25 mm bed height × 5 mm ID, Repligen). For experiments employing alternative metals, the resins were stripped and charged according to the manufacturer's specification.
High-pressure size exclusion chromatography (HP-SEC)
Size exclusion chromatography was performed on an Agilent 1100 HPLC using an isocratic method (0.25 ml min−1 flowrate, 12 minutes run time) with a Waters XBridge BEH200 Size Exclusion Column (4.6 mm × 150 mm, 2.5 μm), which was heated to 35 °C. The mobile phase consisted of 100 mM sodium phosphate buffer (pH 7) + 50 mM KCl in UHPLC-grade water. A diode array detector (DAD) was used to detect the absorbance at 220 and 280 nm. SEC calibration was performed with a commercial molecular weight standard (Waters BEH200 Standard), purified galactose oxidase was used to assess detector response linearity for quantitation.FPLC
An AKTA Avant 25 FPLC (Cytiva, Upsala, Sweden) was utilized for experiments detailed in the manuscript. The system was configured with an additional fixed wavelength UV monitor (U9-L, 2 mm pathlength) between the sample delivery pump and the column to enable monitoring of the column inlet stream UV absorbance. The binary pump outlet streams were joined by a y-shaped mixer and dynamically mixed using a 1.4 mL dynamic mixer containing a magnetic stir bar, which was agitated perpendicular to the flowing stream (Cytiva). A 1 M NaCl solution was used to calibrate the gradient delay and response. The system delay volumes were calibrated by monitoring the UV and conductance changes associated with injecting non-interacting tracers (1% acetone and 10 g L−1 dextran blue in 1 M NaCl were both used) at different positions with the system.17 Chromatograms were analyzed in Unicorn Evaluation Classic (Cytiva) and integrated using a morphological baseline.The system was also configured with a loop selection valve (V9-L, Cytiva) and an autosampler (Alias Bio, Spark Holland) to facilitate automated sample introduction or calibrated sample delivery. The loop volume on the sampler or V9-L valve was configured for the experiment.
Lyophilized cell-free lysate was dissolved in the desired mobile phase. A dedicated pump (P9, Cytiva) was used to fill a sample loop or directly inject the protein solution into the column. During the process, the effluent was monitored by UV, conductivity, and pH.
For 1 mL scale columns, 5.0 mL of a 7.5 g L−1 protein solution in the desired mobile phase was injected, subsequently low-affinity or non-specifically bound proteins were eluted with a 5 CV (column volume) isocratic step containing 15 mM imidazole dissolved in mobile phase A (B = 3%). Next, a linear gradient between 15 mM (B = 3%) and 500 mM (B = 100%) imidazole eluted the bound protein. The final imidazole concentration was maintained an additional 2 CV to compensate for the gradient delay associated with the binary mixer volume (1.4 mL) and system dead volume. Finally, the inlet composition was adjusted back to mobile phase A and the column re-equilibrated.
A modified protocol was used for 0.5 mL scale experiments (25 mm bed height × 5 mm ID columns). The inline mixer size was reduced to 0.6 mL to minimize the gradient delay and 2.0 mL of a 7.1 g L−1 protein solution dissolved in mobile phase A was employed. Following injection, the columns were washed with 20 CV mobile phase A, followed by a 3 CV isocratic step containing 15 mM imidazole dissolved in mobile phase A (B = 3%). Subsequently a modified linear gradient between 15 mM (B = 3%) and 400 mM (B = 80%), with a duration of 10 CV was used to elute bound protein. This was followed by a 5 CV isocratic step at 500 mM imidazole to verify complete removal of the target protein.
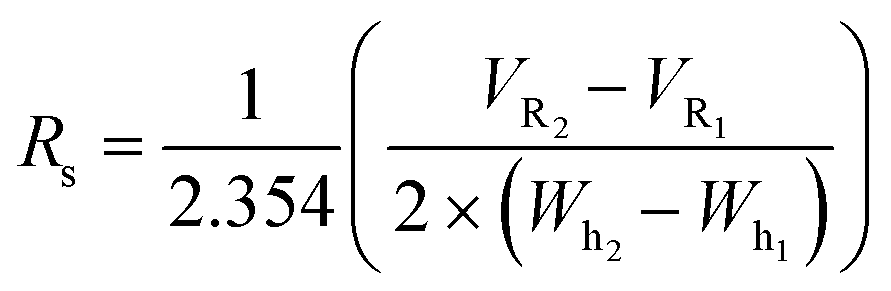
Normalized gradient slope
Normalized gradient slope was calculated, as detailed in Carta,18 according to the following equation: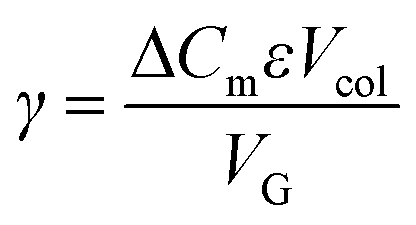 , where ΔCm is net change in mobile phase modifier concentration (imidazole) accomplished over the linear gradient, Vcol is the column volume, VG is the total gradient volume, and ε = 0.34 is column extraparticle porosity as determined experimentally by monitoring the elution volume of a pore-impermeant, non-interacting tracer (blue dextran, 2 MDa MW, Sigma Aldrich).
, where ΔCm is net change in mobile phase modifier concentration (imidazole) accomplished over the linear gradient, Vcol is the column volume, VG is the total gradient volume, and ε = 0.34 is column extraparticle porosity as determined experimentally by monitoring the elution volume of a pore-impermeant, non-interacting tracer (blue dextran, 2 MDa MW, Sigma Aldrich).
Results and discussion
The conventional drivers for traditional immobilization modalities – enzyme reuse and batch productivity – are less applicable for the high potency, low volume small-molecules that dominate pharmaceutical company pipelines. Instead, immobilization is critical for pharmaceutical manufacture, enabling early process development and route scouting activities during enzyme evolution prior to identifying optimized enzymes or reaction conditions. As a result, we focused our efforts on developing an immobilization platform based on a single immobilization modality and establishing a methodology to enable rapid development and optimization of the immobilization.Initially, we evaluated conventional immobilization modalities such as adsorption, covalent linkage, or encapsulation. However, these approaches introduced several challenges into reaction process development, where early enzyme hits may have low activity, stability, or expression levels, and substrate availability is limited. Additionally, these methods typically required significant immobilization process development for each enzyme.
To eliminate laborious method development for each enzyme, we pursued an immobilization strategy based on immobilized metal affinity chromatography (IMAC) as our first-line immobilization platform. IMAC is well established as a protein purification platform. It exploits the strong non-covalent interaction between an undercoordinated divalent metal (connected to a chelating resin) and a series of consecutive histidine residues (6–12) that are genetically engineered to one of the termini. By tailoring the specific immobilization conditions, the enzyme is selectively immobilized to the resin, while residual host cell proteins are rejected by washing the resin. In a typical protein purification, the resin-bound, highly-purified protein would be eluted for use. However, to use the immobilized enzyme as our catalyst, we left the enzyme on the resin and subsequently equilibrated it in the desired reaction buffer. This process prepares a highly pure immobilized enzyme with the potential for resin reuse, should it be desired.
Resin selectivity and binding affinity
We first sought to establish a screening methodology capable of assessing immobilization under process relevant conditions. Immobilizations were performed under conditions that promote histidine complexation with immobilized divalent metals. Crude protein was dissolved in a high strength alkaline buffer operating above the histidine pKa (50 mM sodium phosphate, pH 8). This approach ensured histidine was deprotonated, a requirement for complexation with the metal. 500 mM NaCl was included in the buffer to suppress electrostatic repulsion between proteins and minimize electrostatic attraction of contaminant proteins with the resin.10,13Screening was performed with small chromatography columns packed with the desired resin. FPLC was used to automate fluidic handling, including mobile phase and protein delivery, and integrate inline process-relevant monitoring (pH, conductivity, UV) of the column effluent used to characterize the immobilization process. Following the protein sample injection, the column was washed to remove low-affinity (non-specific) immobilized proteins.
For immobilization development, we sought to identify the impact of process-related parameters such as the immobilization buffer, protein, or resin chemistry on the enzyme's binding affinity to the resin and the immobilization selectivity for our desired enzyme. The histidine analog, imidazole, was used to competitively displace immobilized enzyme, and the elution was monitored. To resolve differences in protein binding affinity, the bound protein was eluted by applying a linear imidazole gradient—this approach served to separate proteins based on the strength of their interactions with the resin.19,20
To establish conditions that resolve differences in affinity or capacity, we first investigated the impact of gradient elution steepness on peak separation, resolution, and product recovery for identical injections, as seen in Fig. 1A. The column was equilibrated in mobile phase A, followed by a 5 mL injection of 7.5 mg mL−1 protein solution in the same mobile phase. The column was then washed with 5 CV of mobile phase A, followed by an elution under a linear gradient of imidazole from 0 to 500 mM, dissolved in mobile phase A. The gradient slope, γ, was varied from 8.5 to 34.
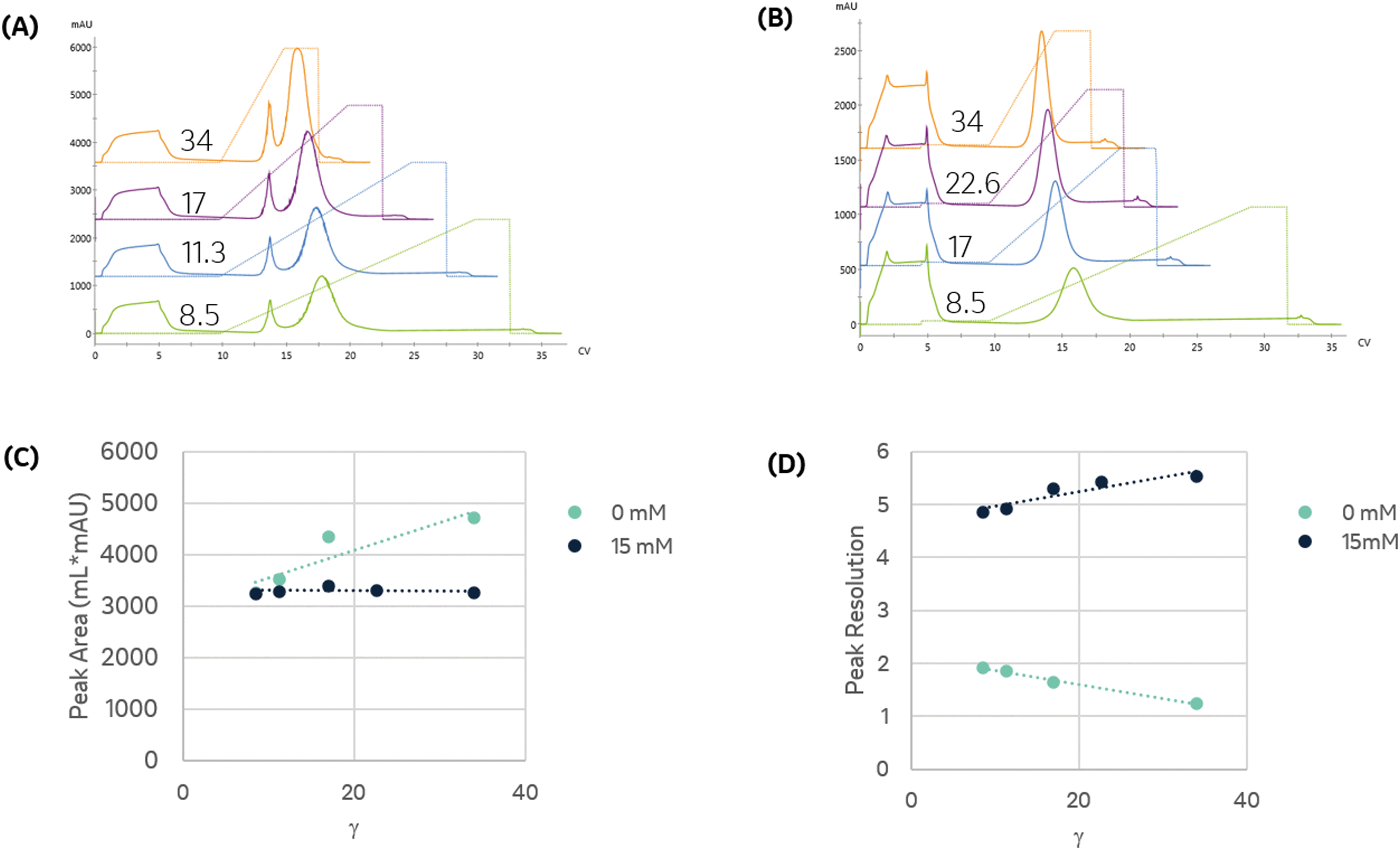 | ||
| Fig. 1 The binding and elution of protein from the column were monitored at 280 nm to assess the impact of linear gradient slope and imidazole. (A) Binding and elution chromatograms produced for identical injections of protein dissolved in mobile phase A and subject to a linear gradient of imidazole of a varying slope, γ, as indicated in the figure. A pre-elution wash with mobile phase A was used to remove unbound protein prior to elution. (B) Impact of including 15 mM imidazole in the protein solution and pre-elution wash on the linear gradient elution from 15 to 500 mM imidazole as a function of γ. (C) Comparison of the primary eluting peak area, corresponding to the immobilized enzyme, for trials shown in (A) and (B). (D) Resolution of the primary eluting peak for trials shown in (A) and (B). | ||
We found that the elution profile was highly dependent on the gradient slope γ. The first peak in each chromatogram corresponds to the column effluent during the injection of the protein solution. It contains unbound protein contents that flow through during the protein injection and the subsequent wash with the same buffer. For the shallowest gradient, γ = 8.5, two unique peaks elute. The first peak corresponds to weak binding or non-specifically bound proteins that interact with the metal through residues such as tryptophan, cysteine, or carboxylic acids residues.11 Due to their low affinity, they are competitively displaced by lower imidazole concentrations. The later peak only elutes at significantly higher imidazole concentrations. This observation is consistent with the disruption of the high affinity (kD ∼ 10 nM) interaction between the metal and the polyhistidine tag placed on the enzyme.14 HP-SEC analysis of these two peaks confirmed the low-affinity peak consisted primarily of E. coli host cell proteins, whereas the high-affinity peak was exceptionally pure (96.1% target enzyme). These findings demonstrate that IMAC immobilization retains the desired enzyme and successfully purifies it from a crude cellular lysate. The high imidazole concentrations necessary to elute the enzyme are consistent with the high-affinity interaction between histidine residues and nickel and demonstrate the significant retention of the desired enzyme on the resin. These findings suggest IMAC immobilization is an effective method to both purify and immobilize an enzyme for biocatalytic applications.
Shorter gradients, characterized by a larger γ, resulted in substantial overlap or co-elution of both species. As a result, the later eluting peak area grows monotonically with γ, as seen in Fig. 1C, and we cannot calculate the peak concentration or resin specificity. For symmetric Gaussian peaks, a peak resolution, Rs of 1.5, would correspond to less than 0.15% peak overlap. Due to the skewed Gaussian peak shape, we required a minimum R of 2.0 to quantify the peak area. Even at the most extended gradients, γ = 8.5, Rs does not exceed this value, as shown in Fig. 1D.
Given the similar elution times observed for both peaks in the gradient elution length study, we sought to improve the immobilized enzyme peak resolution by removing the contaminating low-affinity proteins prior to the gradient elution. We first investigated if we could reduce non-specific adsorption by including low levels of imidazole (15 mM) in the sample and mobile phase. As seen in Fig. 1B, the high-affinity peak is well resolved for even the largest γ values, suggesting the content in the first eluting peak seen in Fig. 1A was removed in the column wash or injection cycle. The resolved peak purity was 95.7% and did not vary with γ.
Notably, for samples containing 15 mM imidazole, the measured area of the eluted peak did not exhibit any dependence on γ, as seen in Fig. 1C. As γ decreases, the total peak area of the samples without imidazole converge with peak area of the imidazole-containing samples, suggesting that the addition of imidazole before the gradient improved resolution but did not impact the quantity of immobilized enzyme. These findings demonstrate that the addition of imidazole before the elution is an effective strategy to enable fast quantitative screening of immobilized enzymes with very short gradients.
Steeper gradients (high γ) produce a larger axial imidazole gradient in the column, resulting in peak compression due to the higher characteristic velocity for peak contents at the end of the peak.18 For the trials in Fig. 1B, this approach leads to improved resolution with γ, as seen in Fig. 1D. Increasing peak compression concentrates the protein into a narrower peak, which can be more easily detected, enabling small total protein utilization in screens.
These results establish critical parameters for assessing immobilization specificity and for rapidly quantifying the amount of immobilized enzyme. We subsequently applied this methodology to systematically identify the impact of resin metal and chelator on the immobilization selectivity and purity.
Role of metal
For successful implementation in pharmaceutically relevant biocatalytic transformations, the divalent metal must be appropriately selected based on process compatibility and regulatory considerations. For example, many enzymes are metal-dependent or metal-sensitive. Nickel and cobalt are highly selective for the polyhistidine tag and are widely employed in IMAC purifications. However, both metals also have low permissible limits in pharmaceutical products compared to iron or copper and must be carefully controlled.21 To accommodate the range of potential process requirements, we systematically evaluated the impact of metal and chelating ligand on immobilization.The choice of metal impacts binding strength and specificity. We investigated three different divalent metals (nickel, cobalt, and copper). We did not employ iron due to the low affinity of Fe2+ with amino acid ligands and propensity for Fe2+ to oxidize to Fe3+, which has significant interactions with other residues, leading to off-target immobilization.22,23
We compared the binding and elution of protein with NTA-agarose resins charged with either nickel, cobalt, or copper to identify metal-related differences in binding specificity. To assess the specificity of each metal, imidazole was omitted from the protein and pre-elution wash solutions. As seen in Fig. 2A, the first peak retention was similar for all metals; however, the separation from the second peak differed significantly. The resolution of all three peaks did not meet our requirement of Rs > 2. To qualitatively compare the area, the relative area of the second peak is reported. Compared to nickel, the cobalt and copper second peak areas were 91.9% and 90.9%, respectively.
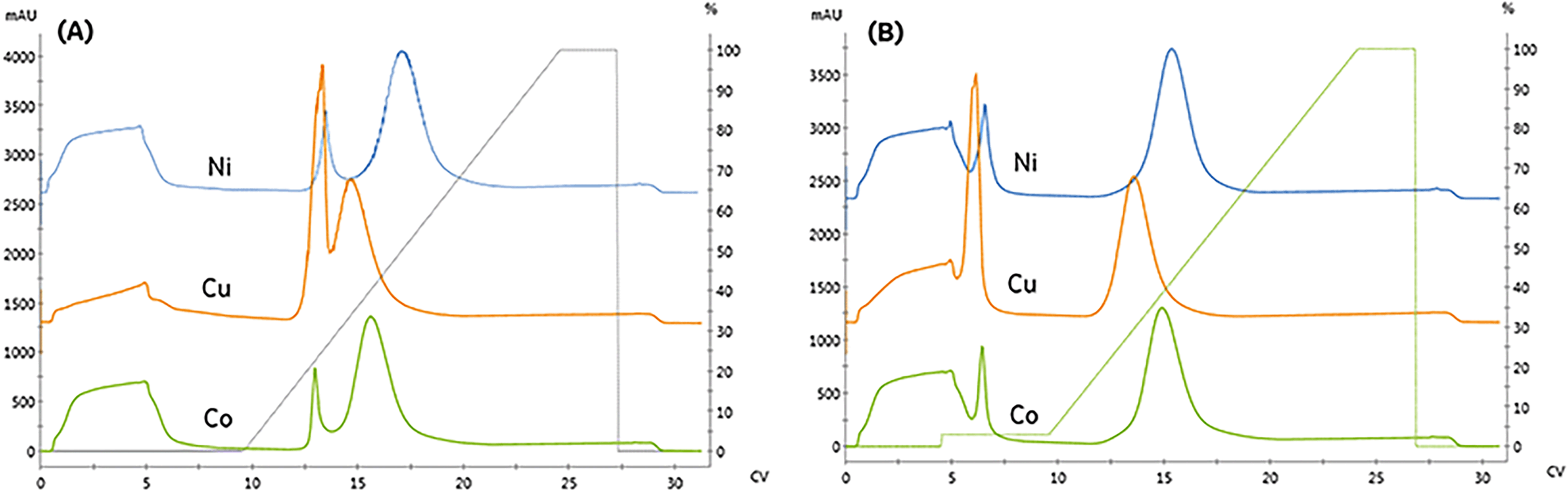 | ||
| Fig. 2 Comparing the binding and elution profiles when the resin is charged with different divalent metals (nickel, copper, and cobalt). A fixed linear-gradient, γ = 11.3, was employed for trials where: (A) no imidazole is included in the sample or pre-elution wash (B) 15 mM imidazole is included in the pre-elution wash. | ||
Affinity immobilization simultaneously purifies and immobilizes an enzyme, producing a high density immobilized biocatalyst devoid of interfering host cell protein. To identify potential contaminants, HP-SEC was used to characterize the immobilized fraction's purity profile as a function of the metal ion. The area percentage of the molecular weight range matching purified enzyme was used to quantify the purity. To compare differences in host cell protein selectivity, the impurities were classified based on whether their molecular weight was higher than enzyme or lower, as tabulated in Table 1. In all cases, the high-affinity peak was purer than the crude lysate.
| Metal | Imidazole (mM) | FPLC | SEC | Impurity % (SEC) | ||||
|---|---|---|---|---|---|---|---|---|
| Sample | Wash | Normalized relative area % | R s | Specificity | Purity (%) | HMW | LMW | |
| Co | 0 | 0 | 91.9 | 1.5 | 83 | 97.3 | 0.0 | 2.7 |
| Co | 0 | 15 | 90.0 | 4.61 | 100 | 96.0 | 0.0 | 4.0 |
| Co | 15 | 15 | 98.3 | 2.58 | 100 | 96.5 | 0.0 | 3.5 |
| Cu | 0 | 0 | 90.9 | 0.63 | 60 | 84.7 | 8.9 | 6.4 |
| Cu | 0 | 15 | 81.7 | 4.04 | 100 | 96.9 | 0.0 | 3.1 |
| Cu | 15 | 15 | 80.9 | 4.2 | 100 | 95.3 | 1.0 | 3.7 |
| Ni | 0 | 0 | 100.0 | 1.86 | 85 | 96.1 | 0.0 | 3.9 |
| Ni | 0 | 15 | 90.5 | 4.58 | 100 | 97.3 | 0.0 | 2.7 |
| Ni | 15 | 15 | 91.4 | 5.06 | 100 | 95.7 | 0.9 | 3.4 |
| Crude | — | — | — | — | — | 67.9 | 15.7 | 16.4 |
The copper resin exhibited significant non-specific adsorption in the high-affinity peak. These results are consistent with previous studies demonstrating that copper can immobilize proteins with a single histidine residue, whereas nickel and cobalt both require two histidine residues to immobilize.11 As seen in Fig. 2B, the addition of imidazole as a pre-elution wash eliminated the low-affinity peak from all metals. The imidazole wash effectively displaced the large population of contaminant proteins bound to the copper resin. As a result, the immobilized content purity was upgraded from 84.7% to 96.9% and resulted in the rejection of high molecular weight contaminants, as detailed in Table 1.
The higher binding capacity and lower selectivity of copper has important implications for design of immobilized processes. Copper retains a significant quantity of non-specific protein during the immobilization. These species and the desired enzyme elute under similar imidazole concentrations. Therefore, the use of copper-NTA based resins are not appropriate for applications requiring high capacity and high purity.
There are multiple chelate properties that impact the immobilization selectivity, including coordination number and geometry.24 With this improved understanding of metal-dependent differences in immobilized affinity and purity for NTA, we next investigated the impact of utilizing a lower denticity chelating ligand that is more widely commercially available than NTA.
Role of ligand
For pharmaceutical synthesis, the supply chain and process economics for obtaining the resin are critical. The tetradentate chelator, NTA, is often used in biopurifications due to the high metal stability. However, the resin is not widely used outside of protein purification or at large scales due to the high cost of the derivatized-NTA ligand necessary for manufacture.25 In contrast, many process-scale chelating resins use the tridentate chelator, iminodiacetic acid (IDA), which can be manufactured by direct conjugation of IDA to the resin. These resins are more widely available and less expensive.We next investigated the impact of chelating ligand denticity on protein affinity, specificity, immobilized purity, and binding capacity using an IDA-agarose resin. To facilitate direct comparison with NTA, the IDA and NTA resins used in this study were derived from the same base bead. Similar linear gradient elution experiments were used to examine the impact of chelating ligand denticity and metal and determine appropriate screening conditions to facilitate quantitative comparisons.
As seen in Fig. 3A and Table 2, the high-affinity peak area and imidazole content necessary for elution varied with metal, similarly to the NTA resin. However, unlike the NTA resin, the imidazole concentrations required to elute from low affinity peak varied across metals. Again, the high-affinity elution peak resolution was below our Rs = 2.0 threshold set for quantitation, although qualitatively, cobalt exhibited the highest specificity.
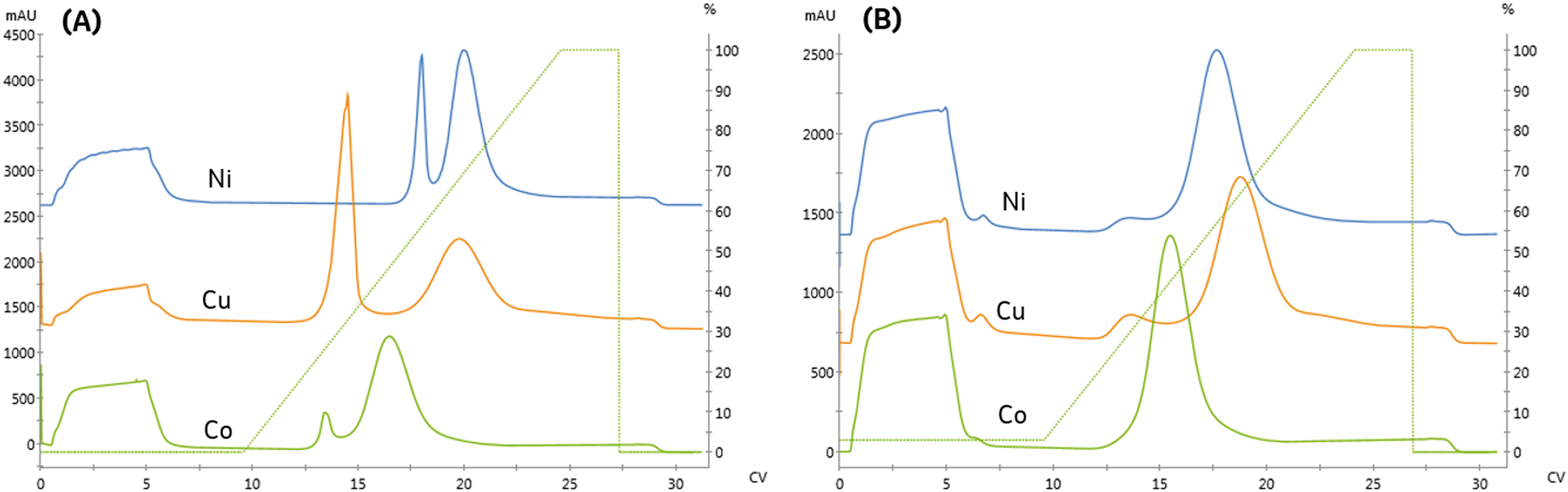 | ||
| Fig. 3 Comparing the binding and elution profiles when the IDA resin is charged with different divalent metals (nickel, copper, and cobalt). A fixed linear gradient, γ = 11.3 was employed for trials where: (A) no imidazole is included in the sample or pre-elution wash (B) 15 mM imidazole is included in the sample and pre-elution wash. | ||
| Metal | Imidazole (mM) | FPLC | SEC | Impurity % (SEC) | ||||
|---|---|---|---|---|---|---|---|---|
| Sample | Wash | Normalized relative area % | R s | Specificity | Purity (%) | HMW | LMW | |
| Co | 0 | 0 | 90.1 | 1.37 | 91 | 92.6 | 1.2 | 6.8 |
| Co | 15 | 15 | 111.6 | 1.94 | 100 | 94.0 | 0.0 | 6.0 |
| Cu | 0 | 0 | 81.9 | 1.86 | 55 | 91.5 | 0.2 | 8.1 |
| Cu | 15 | 15 | 85.1 | 1.36 | 93 | 92.9 | 0.0 | 7.1 |
| Ni | 0 | 0 | 100.0 | 1.18 | 79 | 94.7 | 0.2 | 5.1 |
| Ni | 15 | 15 | 85.6 | 1.15 | 96 | 99.0 | 0.0 | 1.0 |
IDA is expected to provide a higher binding capacity and lower specificity than NTA due to the additional metal chelate site available for complexation. Consistent with this prediction, the high-affinity peak area on the IDA resin was the same or greater than observed on NTA. These differences are likely less significant due to differences in the grafting density of the ligands to the resin and the use of a spacer arm to conjugate NTA to the resin, which reduces steric hindrance limitations on protein binding.
Previous studies have noted metal-specific differences in the number and arrangement of histidine residues necessary for immobilization.11 For instance, copper-IDA is capable of immobilizing proteins containing only a single histidine. In contrast, nickel requires two proximal histidine residues, and cobalt requires they reside on an alpha-helical segment of the protein.24 Consistent with these observations, significant non-specific binding on the copper and nickel IDA resins was observed, as seen in Fig. 3A. In contrast to NTA, residual low-affinity species are not entirely removed from either of these metals when 15 mM imidazole is included in the immobilization and wash solutions, as indicated in Fig. 3B by the presence of a low-affinity elution peak in the copper and nickel chromatograms.
These findings demonstrate that immobilized affinity, capacity, and selectivity can be modulated by both the metal and ligand for design of an immobilized process. While IDA resins can provide increased capacity and lower cost, the decreased specificity, and the high concentrations of imidazole, which will be necessary to off-compete undesired species, will be unsuitable for many applications. For applications that necessitate rejection of interfering host cell proteins, the process would require use of high imidazole concentrations, which would also co-elute a significant portion of the enzyme.
Implications for immobilization process development
This methodology can be readily adapted to screening other process parameters or execute designed experiments. For example, we have utilized this methodology to assess the impact of resin pore size, chemistry, or mechanical properties. Similarly, we applied it to assess immobilized enzyme stability under reaction conditions or in the presence of interfering reagents by applying the suspect reagents as a gradient and to identify the concentration which results in enzyme removal.This methodology can also be utilized to streamline the optimization of process parameters. For example, for process scale immobilizations, either in batch or column, removal of non-specifically bound proteins is accomplished by immobilizing the enzyme with a basal concentration of imidazole and then extensively washing the immobilized enzyme with an imidazole containing solution until no further protein is observed in the effluent. While immobilization is critical to the elimination of the undesired protein, excess imidazole will result in the removal of the desired enzyme.
The appropriate imidazole concentration will depend on the immobilization buffer conditions, target and contaminant proteins, and the resin. To identify the minimal imidazole content necessary to remove the non-specific binding, the imidazole concentration was varied during the post-immobilization wash and the amount of protein removed was quantified. The immobilized protein was then eluted with an imidazole gradient to monitor the elimination of the low-affinity elution peak. As seen in Fig. 4A and B, the amount of protein removed during the wash step increased with imidazole concentration. When no imidazole was included in the wash step, a small low-affinity peak in the gradient elution and a shoulder intercepting the high-affinity peak are observed.
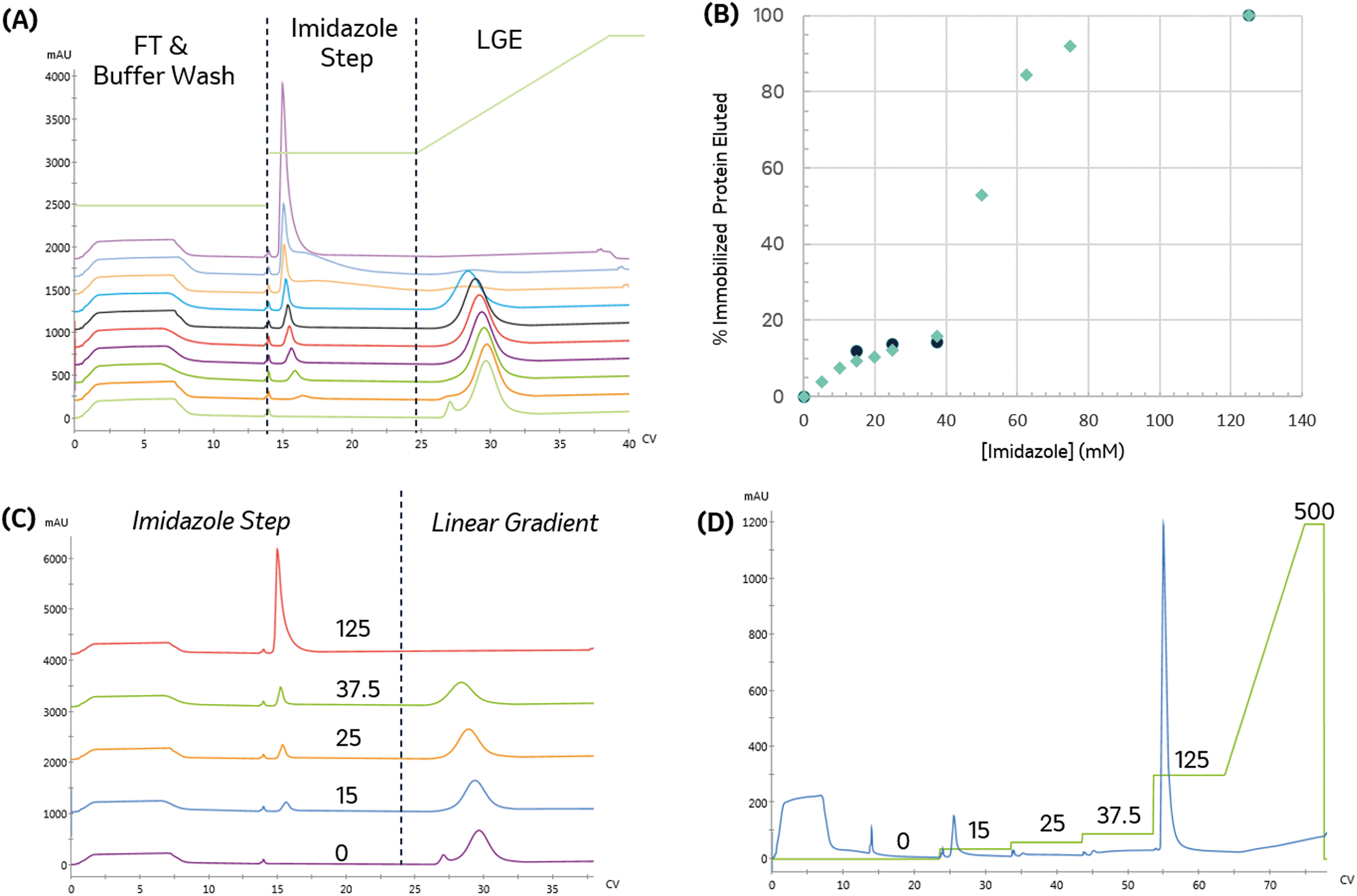 | ||
| Fig. 4 Impact of increasing imidazole content in the pre-elution wash. (A) Elution profile with increasing imidazole content in the pre-elution step. From bottom-up: 0, 5, 10, 15, 20, 25, 37.5, 62.5, 125, and 500 mM. (B) Percentage of bound protein removed as a function of imidazole pre-elution wash concentration. Green diamonds indicate percentage of bound protein removed by individual imidazole steps; black squares show cumulative bound protein removed for a sequence of increasing imidazole steps, (C) plot showing select election profiles in more detail, for different imidazole step concentrations (indicated on the plot), followed by a linear gradient elution (D) bound protein can also be quantified and eluted through a series of increasing imidazole step washes. The imidazole concentration profile is shown in green with the concentrations indicated above. Increasing the imidazole concentration used in the pre-elution wash decreases bound content removed in the gradient elution. | ||
To identify a range of imidazole concentrations for further investigation, we compared the amount of protein (by area percent) removed in the wash step as a function of imidazole concentration, as shown in Fig. 4B. The amount of protein removed in the wash increases linearly until reaching a plateau around 37.5 mM imidazole. Above this value, a large increase in protein content removed is observed, consistent with achieving the imidazole concentration needed to start eluting the immobilized enzyme, which makes up approximately 68% of the total protein content in the crude lysate.
For biocatalytic applications where endogenous host cell proteins negatively impact the reaction (e.g. off-target activity to product or substrate or consumption of a necessary co-factor), the primary immobilization objective is to reject and clear non-specific binders efficiently, and this particular example demonstrates that 37.5 mM imidazole effectively removes low-affinity species without significantly removing target enzyme, resulting in 15.8% reduction in bound protein. It also suggests that, in cases where such purity levels are not necessary, concentrations lower than this target could be effectively used to off-compete non-specific immobilizing species. Additional experiments with washing can then be performed to assess the contents of the effluent stream.
With appropriate initial range-finding experiments, a more efficient screen using a single run with a series of wash steps can be established using increasing imidazole concentrations. Fig. 4C and D compare the elution profile obtained from the runs employing varying imidazole concentrations in the wash steps, as shown in Fig. 4A, and the chromatogram produced by concatenating those wash steps into a single run. As expected, both chromatograms demonstrate that a 125 mM imidazole is necessary to remove all protein from the resin.
To directly compare the two different methods for executing this experiment, the cumulative protein content removed during the series of sequential imidazole washes is calculated. As seen in Fig. 4B, the cumulative protein content is in good agreement with the previous experiment and allowed us to obtain similar data in a shorter amount of time with 90% less protein.
In this example, the column washing process parameters are directly transferable to a batch displacement wash process, and the efficient protein utilization highlights the benefits of this data-rich, automated approach. Further improvements in efficiency were obtained by implementing conditional logic, which instructs the system to proceed to the next step when the UV trace reaches specific stability conditions (data not shown).
Early resin screening for immobilization of galactose oxidase for use in islatravir
For the development of islatravir,7 initial resin screening focused on maximizing the immobilized loading capacity and selectivity for early galactose oxidase enzyme variants. Galactose oxidase is a copper-dependent enzyme that was used to perform an oxidation in the islatravir synthetic pathway. In addition to oxygen, the reaction also requires two non-immobilized auxiliary enzymes that maintain the correct oxidation state. We utilized large pore size resins to maximize oxygen mass transfer and access of non-immobilized auxiliary enzymes.26Due to the extremely limited sample quantity of galactose oxidase and low expression levels, we developed a modified screening method to minimize sample consumption and guide further experimentation. The screen utilized a 0.5 mL column instead of a 1.0 mL column and consumed approximately 1/3 of the sample compared to the trials shown previously in this paper. Following injection of the protein sample, dissolved in mobile phase A, an extended wash of the column with mobile phase A was performed to ensure complete removal of non-immobilized proteins. Subsequently, the low-affinity proteins were eluted with an isocratic wash containing 15 mM Imidazole in mobile phase A. This was followed by truncated linear gradient elution from 15 mM to 400 mM, followed by an isocratic wash with 500 mM to verify complete removal of any immobilized protein.
Three Ni-NTA IMAC resins were first examined. As seen in Fig. 5 and detailed in Table 3, both high flow agarose resins had similar performance. In contrast, the UNOsphere (acrylamido) resin, Bio-rad Nuvia IMAC, was capable of immobilizing nearly twice as much enzyme, based on the area of the high-affinity peak, with substantially improved selectivity. This resin also exhibited slightly improved retention, suggesting improved affinity.
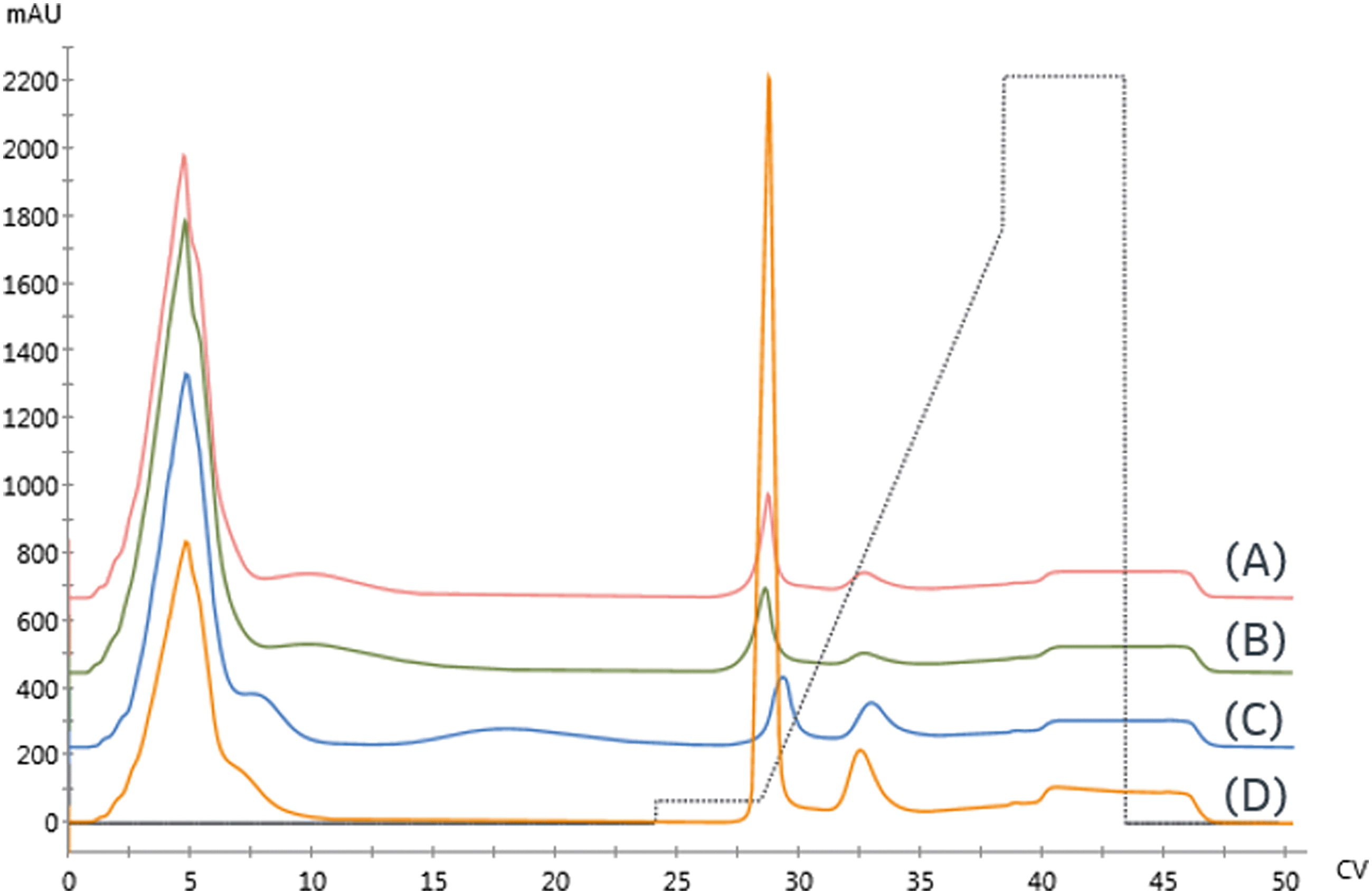 | ||
| Fig. 5 Initial resin screen for galactose oxidase using 0.5 mL columns with the following resins: (A) GE Ni Sepharose 6FF (B) Ni-NTA Superflow (C) Nuvia IMAC – Ni (D) Nuvia IMAC – Cu. Gradient profile is shown by dashed lines. | ||
| Resin | Metal | Base bead | Normalized relative area % | R s | Specificity | Normalized retention |
|---|---|---|---|---|---|---|
| GE Sepharose 6FF | Ni | Agarose | 42% | 2.53 | 24 | 0.993 |
| Qiagen Ni-NTA Superflow | Ni | Agarose | 51% | 2.01 | 24 | 0.992 |
| Bio-rad Nuvia IMAC | Ni | UNOsphere | 100% | 1.92 | 45 | 1.00 |
| Bio-rad Nuvia IMAC | Cu | UNOsphere | 143% | 2.44 | 18 | 0.987 |
A Cu-charged version of Nuvia IMAC was also examined due to concerns that the nickel might displace the enzyme's copper center, resulting in loss of activity. As seen in Fig. 5, there is a significant increase in non-specific binding, as demonstrated by the low-affinity peak area. These findings are consistent with copper's lower specificity. The increased area of the high-affinity peak and reduced retention are consistent with the increase in non-specific binding.
Based on these findings and subsequent reaction performance, Nuvia IMAC was chosen for use in immobilized route development, as we have previously detailed.7
Conclusion
In summary, we have established a data-rich automated methodology that enables rapid screening and development of IMAC-based enzyme immobilizations to enable pharmaceutically relevant biocatalytic synthesis. We applied this method to systematically elucidate the impact of metal and chelating ligand on immobilization affinity and selectivity. We demonstrated that imidazole can be efficiently used to tune the selectivity of the immobilization and utilized automation to optimize the imidazole concentration to meet specific purity goals. Finally, we demonstrated how this method was used to enable resin selection for the synthesis of islatravir. Implementing automated execution of immobilization process development significantly reduces the development time and material consumption while improving fundamental understanding of the immobilization process itself and the methodology can be readily adapted for development of other immobilization modalities and online reaction execution.Conflicts of interest
There authors declare no competing financial interests.Acknowledgements
The authors would like to thank Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, for providing funding for this work. The authors also would like to thank Drs. Rebecca Ruck, Aaron Cote, and John Welsh for their helpful feedback, All are members of Process Research and Development, Merck & Co., Inc., Kenilworth, NJ, USA.References
- L. Cao, Carrier-bound Immobilized Enzymes: Principles, Application and Design, Wiley-VCH, 2005 Search PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- J. Boudrant, J. M. Woodley and R. Fernandez-Lafuente, Process Biochem., 2020, 90, 66–80 CrossRef CAS.
- J. M. Woodley, in Modern Biocatalysis: Advances Towards Synthetic Biological Systems, 2018, pp. 516–538 Search PubMed.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G. D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CAS.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- J. P. Smith, M. L. D. Liu, M. L. Lauro, M. Balasubramanian, J. H. Forstater, S. T. Grosser, Z. E. X. Dance, T. A. Rhodes, X. D. Bu and K. S. Booksh, Analyst, 2020, 145, 7571–7581 RSC.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 35–54 Search PubMed.
- J. Porath, J. Carlsson, I. Olsson and G. Belfrage, Nature, 1975, 258, 598–599 CrossRef CAS PubMed.
- E. Sulkowski, BioEssays, 1989, 10, 170–175 CrossRef CAS PubMed.
- Y. J. Kim and S. M. Cramer, J. Chromatogr., 1991, 549, 89–99 CrossRef CAS.
- V. Gaberc-Porekar and V. Menart, J. Biochem. Biophys. Methods, 2001, 49, 335–360 CrossRef CAS PubMed.
- S. Knecht, D. Ricklin, A. N. Eberle and B. Ernst, J. Mol. Recognit., 2009, 22, 270–279 CrossRef CAS PubMed.
- M. P. Thompson, S. R. Derrington, R. S. Heath, J. L. Porter, J. Mangas-Sanchez, P. N. Devine, M. D. Truppo and N. J. Turner, Tetrahedron, 2019, 75, 327–334 CrossRef CAS.
- W. Bohmer, A. Volkov, K. Engelmark Cassimjee and F. G. Mutti, Adv. Synth. Catal., 2020, 362, 1858–1867 CrossRef PubMed.
- T. Hahn, T. Huuk, V. Heuveline and J. Hubbuch, J. Chem. Educ., 2015, 92, 1497–1502 CrossRef CAS.
- G. Carta and A. Jungbauer, Protein Chromatography, Wiley-VCH, 2010 Search PubMed.
- S. Vunnum, S. R. Gallant, Y. J. Kim and S. M. Cramer, Chem. Eng. Sci., 1995, 50, 1785–1803 CrossRef CAS.
- R. D. Johnson, R. J. Todd and F. H. Arnold, J. Chromatogr. A, 1996, 725, 225–235 CrossRef CAS PubMed.
- Q3D(R1) Elemental Impurities: Guidance for Industry, Docket FDA–2013–D–1156, United State Food and Drug Administration, Silver Spring, MD, 2020 Search PubMed.
- M. Zachariou and M. T. Hearn, J. Protein Chem., 1995, 14, 419–430 CrossRef CAS PubMed.
- N. Ramadan and J. Porath, J. Chromatogr., 1985, 321, 93–104 CrossRef CAS PubMed.
- E. K. M. Ueda, P. W. Gout and L. Morganti, J. Chromatogr. A, 2003, 988, 1–23 CrossRef CAS PubMed.
- E. Hochuli, H. Döbeli and A. Schacher, J. Chromatogr. A, 1987, 411, 177–184 CrossRef CAS PubMed.
- R. M. Lindeque and J. M. Woodley, Org. Process Res. Dev., 2020, 24, 2055–2063 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |