
Kinetic and mechanistic insights into Ni-AlKIT-6 catalyzed ethylene oligomerization†
Remi
Beucher
,
Vasile
Hulea
 and
Claudia
Cammarano
*
and
Claudia
Cammarano
*
Institut Charles Gerhardt Montpellier, UMR 5253, ICGM, CNRS, Univ Montpellier, ENSCM, Matériaux Avancés pour la Catalyse et la Santé, 34296 Montpellier, France. E-mail: claudia.cammarano@enscm.fr
First published on 11th October 2021
Abstract
A kinetic study of the ethylene oligomerization on a mesoporous 2 wt% Ni-AlKIT-6 was performed in a fixed bed flow reactor. The temperature ranged from 40 to 120 °C, the inlet ethylene partial pressure from 0.1 to 2.0 MPa and the space velocity (WHSV) was between 37 and 240 h−1. The main products identified at low ethylene conversion were n-butenes (90%) and hexenes (9%). A coordination–insertion mechanism (Cossee–Arlman) was proposed to explain the formation of these primary products. An activation energy of 15.2 kJ mol−1 and a global kinetic order of 1.28 were determined. The experimental data suggest that the kinetically limiting step is the insertion of ethylene into the metal–alkyl bond.
Introduction
Ethylene dimerization/oligomerization is one of the major technologies for the production of higher olefins, which are a key feedstock in the synthesis of plastics (by copolymerization), lubricants (by oligomerization), surfactants (through arylation/sulphonation), alcohols (by hydration) and propylene (by metathesis). The current oligomerization processes use organic solvents and homogeneous catalysts.1–6 In line with sustainable chemistry principles, significant research efforts have been directed to the development of heterogeneous catalysts and processes.4,7,8 Various families of solid catalysts, including complexes on polymers and oxides, metal–organic and covalent organic framework materials, and nickel supported on inorganic porous materials have been examined during the past decades.4,8 Among them, Ni-exchanged zeolites, clays and mesoporous aluminosilicates have shown very promising performances in terms of catalytic activity and selectivity.4,7–20 In addition, they are the only catalysts able to work without solvents and activators/cocatalysts. These materials are bifunctional catalysts, containing both Ni2+ and acid sites. It is generally accepted that the isolated Ni2+ ions are involved in the formation of 1-olefins by ethylene dimerization/oligomerization, while the acid sites catalyze the double bond isomerization and the co-oligomerization of primary olefins.8,12,16In a series of studies, we screened various Ni-based microporous and mesoporous catalysts for the oligomerization of ethylene.7,10,11,13–15,18 The reactions have been conducted in both gas–liquid–solid slurry reactors and gas–solid flow reactors, over a wide range of temperatures (30–150 °C) and pressures (10–40 bar). The best catalysts were the ordered mesoporous materials, namely Ni-MCM-41,9,14,16 Ni-SBA-15,15 and Ni-MCM-48.13 The ethylene transformation over solid catalysts led to a mixture of C4–C6–C8 olefins, with a ratio between the olefins mainly depended on the reaction conditions (T, P, contact time).7–9,14,15,18 In a general manner, at high temperature, contact time, or pressure, the ethylene conversion increased, while the selectivity to butenes decreased, in favor of C6 and C8+ olefins.7–9,14,15 The C4 fraction (butenes) was exclusively linear (with an average composition close to the thermodynamic equilibrium), the C6 oligomers were mainly linear hexenes (∼75%) and the C8+ oligomers were mainly branched hydrocarbons.7–9,15 More recently, we studied the ethylene dimerization over Ni-based catalysts as a stage of a more complex process, designed to convert ethylene to propylene without adding any other reagent. This process consisted of cascade dimerization/isomerization/metathesis reactions, activated by two heterogeneous catalysts: Ni-exchanged mesoporous materials and Mo-, W- or Re-based catalysts.21–25 Working at 60–80 °C, the selectivity to butenes in the dimerization step was higher than 80%.
So far, we have not performed kinetic studies for the dimerization/oligomerization of ethylene. Note also that only few kinetic results on the ethylene dimerization/oligomerization performed in fixed-bed type reactors are available in the literature.16,17,26–29 The studies were mainly performed in the presence of Ni–Y zeolite and Ni–silica alumina catalysts. Riekert found that the reaction over Ni–Y was of the second order with respect to the ethylene concentration and the activation energy was 88 kJ mol−1.26 In contrast, Heveling et al.27 reported a first order over a similar Ni–Y catalyst. First-order kinetics with respect to ethylene has also been reported by Ng and Creaser on Ni–Y.28 The apparent activation energy decreased from 58.6 to 42.1 kJ mol−1 increasing the time on stream. Espinoza et al.29 examined the oligomerization of ethylene over Ni-exchanged silica–alumina. Based on an isothermal plug flow reactor model, they observed a first order reaction. Using experimental observations and single-event microkinetic modeling, Toch et al.30 evaluated the kinetic parameters of intrinsic ethylene oligomerization on a Ni/SiO2–Al2O3 catalyst. The activation energy for insertion and termination step was 76 and 68 kJ mol−1, respectively. More recently, Agirrezabal-Telleria and Iglesia17 examined the kinetic of the ethylene dimerization over Ni-MCM-41 catalyst, at subambient temperatures (243–258 K) and 15 bar. Under these conditions, the reagent (ethylene) condensed within ordered mesopores of the catalyst. A second-order dimerization rate with respect to ethylene pressure was reported. The same reaction order was observed for the ethylene dimerization on N-SSZ-24 zeolite.31
In short, the kinetic studies, performed under different conditions (catalysts, parameters), produced inhomogeneous results. In the present study, we perform an experimental study of the ethylene dimerization and isomerization on a mesoporous 2 wt% Ni-AlKIT-6 in a fixed bed flow reactor, in order to determine the kinetically determined step and the kinetics parameters. Moreover, a mechanism model was proposed.
Experimental
Catalyst preparation
The studied catalyst (Ni-AlKIT-6) was prepared in three successive steps, as shown in the Scheme 1. Initially, a pure siliceous material (sample KIT-6) was prepared by hydrothermal synthesis, using (EO)20(PO)70(EO)20 triblock copolymer (Pluronic P123, Mn = 5800 g mol−1, Aldrich), tetraethyl orthosilicate (TEOS 99%, Aldrich), HCl 37.5% (Aldrich) and n-butanol (99%, Alfa Aesar). The molar ratio of reagents was 1 TEOS/0.017 P123/1.9 HCl/1.3 n-butanol/195 H2O. After stirring for 24 h at 35 °C, the mixture was kept under static conditions at 100 °C for 24 h in an autoclave. The solid was recovered, washed with water, dried at 80 °C and calcined for 8 h in air flow at 550 °C. | ||
| Scheme 1 Main steps involved in the preparation of the Ni-AlKIT-6 catalyst. | ||
Then, the support KIT-6 was treated using sodium aluminate according to a procedure developed by our group,15 to obtain the material Na-AlKIT-6. The solid (2 g of Na-AlKIT-6) was then converted into NH4-AlKIT-6 by ionic exchange (2 h at 30 °C with 100 mL of 0.5 M aqueous solutions of NH4NO3). The sample (2 g of NH4-AlKIT-6) was subjected to a nickel-ion exchange with 100 mL of 0.5 M aqueous solution of Ni(NO3)2 obtained from hexahydrate nickel(II) nitrate (98%, Alfa Aesar), following the same procedure as above. Finally, the exchanged sample was dried and calcined for 5 h in air flow at 550 °C, to obtain the catalyst denoted Ni-AlKIT-6. NH4-AlKIT-6 was calcined for 5 h in air flow at 550 °C to obtain the H-AlKIT-6 sample.
Material characterization
XRD measurements were performed on a Bruker AXS D8 diffractometer. The solid texture was evaluated by nitrogen adsorption/desorption at −196 °C, using a Micrometrics ASAP 2020 analyzer. 27Al MAS NMR spectra of calcinated samples were obtained on a Varian 600 MHz WB Premium Shielded spectrometer, equipped with 3.2 mm o.d. ZrO2 rotors at a rotation speed of 20 kHz. The state of the nickel surface was analyzed by X-ray photoelectron spectroscopy (XPS). The analyses were performed with an ESCALAB 250 (Thermo Electron) equipped with a monochromatic Al Ka source (1486.6 eV). The analyzed zone has a diameter of 400 μm. The acidity of Ni-AlKIT-6 was explored by temperature programmed desorption (TPD) using ammonia as probe molecule (on Micromeritics AUTOCHEM 2910). The sample (around 0.04 g) was pre-treated for 6 h in air flow at 550 °C, and then it was cooled down to 100 °C under helium flow. Adsorption of ammonia on the solid was carried with ammonia diluted at 5% in helium. Desorption of the chemisorbed ammonia is then performed by heating the sample at 20 °C min−1 to 550 °C under a 20 mL min−1 helium flow. Ammonia concentration was measured using a thermal conductivity detector (TCD). Nickel content was determined by atomic absorption spectrometry using a Varian Spectra 220 apparatus and a source emitting monochromatic radiation with a wavelength of 232 nm. The sample (0.04 g) was dissolved in 100 mL of a 2 mol L−1 sodium hydroxide solution and nickel concentrations were measured using an external calibration. The Si/Al ratio were measured by energy-dispersive X-ray spectroscopy. The EDX analyses were performed on an FEI Quanta 200F (15 kV) backscatter instrument.Catalytic tests
The performances of the catalysts were evaluated in a laboratory scale unit equipped with one stainless steel fixed-bed reactor (i.d. of 6 mm, length of 150 mm). The gas flows were monitored by Brooks 5850S HP mass meters. Before each test, the catalysts were activated in situ at 550 °C, for 8 h under a nitrogen flow. The products were analyzed online by GC (Varian CP-3800, FID), using a CP-PoraPLOT Q capillary column (25 m, 0.53 mm, 20 μm) and an auto-sampling valve. Ethylene conversion (X) was calculated from the respective weight concentrations at the inlet (Ci) and outlet (Co) of the reactor (X = 1 − Ci/Co).Product selectivity to the molecule i was Si = y/2 × xi/∑xj, where x stands for molar fraction and y is a coefficient equal to the number of carbon atoms in the molecule.
The reproducibility of the reaction tests was assessed by repeating four tests under the same conditions.
Results and discussion
Characterisation of catalyst
The small-angle XRD patterns of the prepared support KIT-6 showed two diffraction peak d211 (2θ = 0.97°) and d220 (2θ = 1.12°) (Fig. S1†), typical of the well-organized cubic Ia3d symmetry of KIT-6 mesoporous materials.32 Moreover, the XRD pattern is the same for Ni-AlKIT-6 material. This indicate that the KIT-6 structure was preserved despite the various post-synthesis steps. Additionally, the nitrogen adsorption–desorption isotherm of Ni-AlKIT-6 was specific to materials with homogeneous mesopores, with a vertical sharp capillary condensation step (Fig. S2†). The main textural properties obtained from sorption data are summarized in Table 1.The 27Al MAS NMR spectrum of Ni-AlKIT-6 shows a major signal at about 55 ppm, corresponding to the tetrahedrally-coordinated aluminum species (Fig. S3†). This result shows that aluminum is well incorporated into the silica structure. Ni-AlKIT-6 had a Si/Al ratio of 11.0 and a Ni content of 2 ± 0.2 wt% (Table 1), corresponding to a Ni/Al (mol mol−1) ratio of 0.25.
The site occupancies of Na+, NH4+ and Ni2+, determined by EDX measurements, are reported in ESI† (Fig. S4). Na+ amount in Ni-AlKIT-6 is very low, proving ion exchange is efficient. Furthermore, there are almost the same number of sites Ni2+ and NH4+ in the final catalyst.
Knowing that almost all sodium ions are exchanged by NH4+, this result indicates that the exchange rate between NH4+ and Ni2+ is about 50%. During the calcination at 550 °C (see the experimental section), NH4+ ions are decomposed into ammonia and H+, generating the Brønsted acidic sites associated to aluminum atoms. NH3-TPD profile of Ni-AlKIT-6 highlights this acidity (Fig. S5†).
Three signals were detected on the deconvoluted XP spectrum of the Ni-AlKIT-6 catalyst in the Ni 2p region (Fig. S6†). The main signal with a binding energy of 857.7 eV is attributed to isolated Ni(II) species, and more precisely to tetrahedrally coordinated Ni(II).12 This result is confirmed by the presence of its primary satellite peak at 863 eV. The weaker signal at 854.2 eV reveals the presence of NiO, which was not detected by XRD because of its small amount. It is commonly accepted that only the isolated Ni species are precursors of the catalytic dimerization/oligomerization sites.12,21
In summary, a dimerization/isomerization catalyst was prepared and possessed an ordered 3D mesoporosity and a small distribution of the pore size. Ni2+ and H+ sites contained in the material are the catalytic sites for the dimerization and the isomerization respectively.
Catalytic results
External diffusional limitations were evaluated by varying the total flow rate at constant contact time. The results obtained (Fig. S7†), showing that starting from 0.7 L h−1 the ethylene conversion remains constant, indicate that the total flow rate must be greater than 0.7 L h−1 to avoid external diffusional limitations.
Internal diffusional limitations were studied by evaluating the performance of the catalyst as a function of its particles size at constant contact time. The small differences in conversion obtained varying the particles size indicate that there is no internal diffusional limitations under our conditions (Fig. S8†).
As a result of this preliminary study, reactions tests were then performed with a granulometry of 150 μm −250 μm and a total flow rate of 6 L h−1 of a C2H4/N2 mixture (Pethylene = 0.4–2 MPa, Ptotal = 3.0 MPa) thus avoiding internal and external limitations.
According to Fig. 1, butenes are the vast majority of the species formed, representing 89.2% of products. Furthermore, isomers analysis shows that 1-butene accounts for 85% of the butene fraction. Thus, 15% of the butene formed is quickly isomerized into 2-butene. Using the same approach, the extrapolation of hexenes (Fig. 2, left) indicates that they account for about 9.2% of total primary products. Among the hexene isomers, 1-hexene, 2-hexene and 3-methyl-2 pentene were the main species. The proportion between these three isomers as function of conversion is presented in Fig. 2 (right). When the conversion tends to zero, the proportion of 1-hexene remained constant (3.5%), while 2-hexene increases (80%) and 3-methyl-2-pentene decreases (16.5%). Table 2 summarizes the primary products (butenes and hexenes) distribution obtained by extrapolation when the ethylene conversion tends to zero.
 | ||
| Fig. 1 C4 selectivity (left) and 1-C4 isomer ratio (right) vs. ethylene conversion. Reaction conditions: feed = 6 L h−1 of ethylene–nitrogen mixture, catalyst: 0.1 g of Ni-AlKIT-6, T = 40–80 °C, Pethylene = 0.4–2 MPa, Ptotal = 3.0 MPa, contact time = 15–98 s. | ||
 | ||
| Fig. 2 C6 selectivity (left) and C6 isomer proportion (right) vs. ethylene conversion [(◊) 2-hexene (○) 3-methyl-2-pentene (□) 1-hexene]. Reaction conditions: feed = 6 L h−1 of ethylene–nitrogen mixture, catalyst: 0.1 g of Ni-AlKIT-6, T = 40–80 °C, Pethylene = 0.4–2 MPa, Ptotal = 3.0 MPa, contact time = 15–98 s. | ||
| Butene | Hexenes | ||||
|---|---|---|---|---|---|
| 1-C4 | 2-C4 | 1-C6 | 2-C6 | 3-Methyl-2 pentene | |
| Molar selectivity (%) | 75.8 | 13.4 | 0.31 | 7.2 | 1.49 |
 | ||
| Scheme 2 Ethylene dimerization mechanisms in homogeneous catalysis: (a) the Cossee–Arlman mechanism (with a hydride intermediate) and (b) the mechanism with a metallacyclic intermediate. | ||
However, most of the studies on the ethylene oligomerization over Ni-exchanged catalysts seem to converge towards a mechanism analogous to the Cossee–Arlman mechanism.12,30,38,39 Two studies published in the 80s40,41 showed that the coordination of the Ni ions on zeolites and SiO2 surface is similar to that observed in the coordination chemistry. Thus, the dispersed nickel ions can reversibly bind ligands (L) such as C2H4, C3H6, CO, phosphines, or surface oxide ions Os2−, leading to various Ni(L)n+ species. Based on these results, the following mechanism for the formation of 1-butene is proposed (Scheme 3).
 | ||
| Scheme 3 Proposed mechanism for the dimerization of ethylene by Ni-AlKIT-6. (1) Nickel complexation; (2) ethylene complexation; (3) metal–vinyl intermediate; (4) 1-butene formation. | ||
Initially, (1) nickel is bonded by complexation to oxygens on the surface of the support. The first step of the mechanism is the complexation of two ethylene molecules on the nickel (2). This step allows to explain the need of high pressure for the activity of heterogeneous catalysts: high pressures favour the shift of the ethylene complexation equilibrium in the favourable direction. The next step is the formation of a metal–vinyl intermediate (3) by oxidative coupling of an ethylene molecule and a proton transfer. Subsequently, the insertion of coordinated ethylene into the metal–alkyl bond leads to the formation of 1-butene (4). The initial step is regenerated after elimination of the product and coordination of new ethylene molecules. A reaction mechanism able to describe the formation of the identified primary products is proposed in Scheme 4. The starting species (left) is a simplified formula of the structure (3) in Scheme 3. The readsorption of butenes was considered and leads to the formation of the different hexenes observed.
 | ||
| Scheme 4 Proposed mechanism for the formation of the different molecules observed at low conversion on Ni-AlKIT-6. | ||
The conversion results of all the experiments at different temperatures are reported in Fig. S9.† The conversions remain stable from 2 hours of reaction and values were collected at 2.7 h of reaction time in order to ensure that the stationary state is reached. The reaction rate as a function of temperature is shown in Fig. 3 (left).
 | ||
| Fig. 3 Reaction rate vs. temperature (left) Arrhenius plot based on experimental data (right). Reaction conditions: feed = 6 L h−1 of ethylene–nitrogen mixture, 0.1 g of Ni-AlKIT-6, Pethylene = 0.75 MPa, Ptotal = 3.0 MPa. | ||
In order to calculate the activation energy, the Arrhenius plot was applied, plotting the natural logarithm of the reaction rate versus the inverse temperature (Fig. 3 right). We notice that there is no slope break on the Arrhenius plot, signifying that no change in the reaction regime over the studied temperature range occurs. An activation energy of 15.2 kJ mol−1 can be obtained from the slope of the line obtained, comparable to those reported in previous studies. Thus, Seufitelli and Resende42 presented the results of a study on the oligomerization of ethylene at the AlCHE 2018 congress. This team worked with a nickel-based zeolite, which is similar to our catalyst. They determined an activation energy of 11 kJ mol−1. The studies of Zhang and Dalla Lana43 and Volkov et al.44 were carried out with nickel deposited on alumina and resulted in an apparent activation energy of 16.3 kJ mol−1 and between 17 and 22 kJ mol−1 respectively.
In order to study the influence of temperature on selectivities, the values of products selectivities as a function of temperature at isoconversion were compiled in Table 3. We notice that the selectivities of C4, C6 and C8 are strongly close. Thus, there is no influence of temperature on the oligomerization selectivity. On the other hand, the proportion in 1-butene is lower at high temperature than at low temperature. Therefore, the isomerization is strongly improved by temperature.
| T (°C) | Conv.b (%) | t (h) | Product selectivity (%) | C4 selectivity (%) | ||||
|---|---|---|---|---|---|---|---|---|
| C4 | C6 | C8 | 1-C4 | cis-2-C4 | trans-2-C4 | |||
| a Reaction temperature. b Ethylene conversion. c Reaction time. | ||||||||
| 40 | 10.7 | 1.2 | 85.5 | 11.5 | 3.0 | 82 | 9.3 | 8.7 |
| 60 | 10.7 | 1.2 | 85.6 | 10.8 | 3.5 | 78 | 10.8 | 11.2 |
| 80 | 17.4 | 1.8 | 83.1 | 13.2 | 3.6 | 67 | 16.1 | 16.9 |
| 120 | 17.9 | 4.1 | 82.7 | 13.5 | 3.7 | 36 | 31.6 | 32.4 |
 | ||
| Fig. 4 Ethylene conversion vs. partial pressure of the ethylene. Reaction conditions: feed = 6 L h−1 of ethylene–nitrogen mixture, catalyst: 0.1 g of Ni-AlKIT-6, T = 60 °C, Ptotal = 3.0 MPa. | ||
The results of isoconversion selectivities at different partial pressures were reported in Table 4. These results were obtained at different reaction times for the two tests. The selectivities of the different reaction products are similar, which means that there is no significant effect of ethylene pressure on selectivity.
Starting from Fig. 4, the apparent reaction order was calculated as the slope of the log log graph of ethylene consumption rate vs. partial pressure. At low partial pressures (0.1–1.0 MPa), the global order of reaction is 1.28. Other kinetic studies determined the kinetic order of dimerization of ethylene with nickel-based catalysts supported on aluminosilicates. Ng and Creaser45 studied the ethylene dimerization reaction between 50 °C and 70 °C and reported an overall order of 1.31. Zhang and Dalla Lana43 also determined that the reaction is of first order kinetic.
In agreement with other studies, a Langmuir–Hinshelwood mechanism can be used for modelling the dimerization mechanism.17,42 According to this model, the dimerization can be decomposed into different elementary steps described in Scheme 5. Adsorption of two ethylene molecules (a and b), followed by C–C coupling (c) and product desorption (d). The overall reaction rate depending on the slowest step, its expression is related to the limiting step as indicated in the table below (Table 5).
 | ||
| Scheme 5 Elementary steps to decompose the general mechanism of dimerization: (a) adsorption reactant 1; (b) adsorption reactant 2; (c) C–C coupling; (d) product desorption. | ||


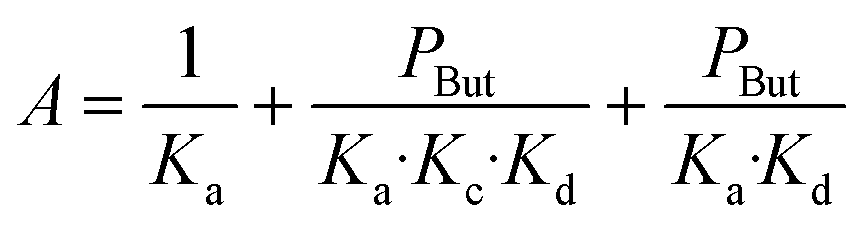





The global order of the reaction previously determined being 1.28, step (a) is not the limiting step. Plotting Peth2/r as a function of ethylene pressure allows us to discern between step (b) and step (c), as it would follow a straight line or a second-degree polynomial plot depending on the limiting step.
This plot reported in Fig. 5 shows that a second order polynomial better fits the experimental points. Thus, it seems more likely that the limiting step is the C–C coupling.
 | ||
| Fig. 5 P eth 2/r vs. ethylene partial pressure (Peth) from experimental points (♦). (A) Comparison of the experimental points to a linear plot (B) comparison of the experimental points to a second-degree polynomial plot. Reaction conditions: feed = 6 L h−1 of ethylene–nitrogen mixture, catalyst: 0.1 g of Ni-AlKIT-6, T = 60 °C, Ptotal = 3.0 MPa. | ||
From Fig. S12,† we can observe that the experimentally observed composition follows a Schultz–Flory distribution, characteristic of linear oligomerisation. This is illustrated by the linear relationship between the logarithms of the mole fractions of the products as a function of their number of ethylene units. A Schultz–Flory distribution suggests that under our conditions there are no undesirable by-reactions such as cracking or co-oligomerisation catalysed by the acid sites.8,46–48 This confirms that a single type of site is active in the material and that the branched oligomers come from species readsorption via a chain-migration mechanism.49
The coefficient α (between 0 and 1) corresponds to the probability that the chain grows by one unit instead of being desorbed from the active site. The lower the number, the more the distribution of oligomers tends towards low molar masses. A coefficient of 0.2 is determined from the slope of log(Xi) = f(i) (Fig. S12†). This relatively low value (<0.5) implies a high proportion of low molar mass oligomers.50 The result obtained is therefore consistent with our experimental data where the products obtained are limited to dimers, trimers and tetramers.
Conclusions
Ethylene dimerization/isomerization catalyzed by Ni-AlKIT-6 was studied under different conditions. Based on the primary products, a mechanism close to Cossee–Arlman was proposed. As shown by Dincă et al.,37 it is possible to effectively discriminate dimerization mechanisms (Arlman–Cossee, metallacycle) by deuterium labeling and mass analysis of the isotopic distribution of the products. This promising technique would provide elements to confirm at a higher level of confidence that dimerization occurs on our Ni-AlKIT-6 catalyst according to a coordination–insertion mechanism. The apparent order of reaction found was 1.28, suggesting that the kinetically determining step is the insertion of ethylene in the metal–alkyl bond at the nickel site neighborhood. In the temperature range studied (40–150 °C) an activation energy of 15.2 kJ mol−1 was found.Author contributions
Authors contribution was here summarized: Remi Beucher: data curation, formal analysis, investigation, methodology, resources, writing. Vasile Hulea: conceptualization, project administration, supervision, validation, visualization, writing. Claudia Cammarano: conceptualization, supervision, validation, visualization, writing.Conflicts of interest
There are no conflicts to declare.References
- J. Skupinska, Chem. Rev., 1991, 91, 613–648 CrossRef CAS.
- A. M. Al-Jarallah, J. A. Anabtawi, M. A. B. Siddiqui, A. M. Aitani and A. W. Al-Sa'doun, Catal. Today, 1992, 14, 1–121 CrossRef.
- C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS.
- H. Olivier-Bourbigou, P. A. R. Breuil, L. Magna, T. Michel, M. F. Espada Pastor and D. Delcroix, Chem. Rev., 2020, 120, 7919–7983 CrossRef CAS.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS.
- H. Olivier-Bourbigou, A. Forestiere, L. Saussine, L. Magna, F. Favre and F. Hugues, Oil, Gas, 2010, 36, 97–102 CAS.
- A. Finiels, F. Fajula and V. Hulea, Catal. Sci. Technol., 2014, 4, 2412–2426 RSC.
- V. Hulea, ACS Catal., 2018, 3263–3279 CrossRef CAS.
- V. Hulea and F. Fajula, J. Catal., 2004, 225, 213–222 CrossRef CAS.
- M. Lallemand, A. Finiels, F. Fajula and V. Hulea, Appl. Catal., A, 2006, 301, 196–201 CrossRef CAS.
- M. Lallemand, O. A. Rusu, E. Dumitriu, A. Finiels, F. Fajula and V. Hulea, Appl. Catal., A, 2008, 338, 37–43 CrossRef CAS.
- S. Moussa, P. Concepción, M. A. Arribas and A. Martínez, ACS Catal., 2018, 3903–3912 CrossRef CAS.
- M. Lallemand, A. Finiels, F. Fajula and V. Hulea, Stud. Surf. Sci. Catal., 2007, 170, 1863–1869 CrossRef.
- M. Lallemand, A. Finiels, F. Fajula and V. Hulea, Chem. Eng. J., 2011, 172, 1078–1082 CrossRef CAS.
- R. D. Andrei, M. I. Popa, F. Fajula and V. Hulea, J. Catal., 2015, 323, 76–84 CrossRef CAS.
- A. Lacarriere, J. Robin, D. Świerczyński, A. Finiels, F. Fajula, F. Luck and V. Hulea, ChemSusChem, 2012, 5, 1787–1792 CrossRef CAS PubMed.
- I. Agirrezabal-Telleria and E. Iglesia, J. Catal., 2017, 352, 505–514 CrossRef CAS.
- R. D. Andrei, E. Borodina, D. Minoux, N. Nesterenko, J.-P. Dath, C. Cammarano and V. Hulea, Ind. Eng. Chem. Res., 2020, 59, 1746–1752 CrossRef CAS.
- I. Elev, B. N. Shelimov and V. B. Kazansky, J. Catal., 1984, 89, 470–477 CrossRef CAS.
- A. Aid, R. D. Andrei, S. Amokrane, C. Cammarano, D. Nibou and V. Hulea, Appl. Clay Sci., 2017, 146, 432–438 CrossRef CAS.
- R. D. Andrei, M. I. Popa, F. Fajula, C. Cammarano, A. A. Khudhair, K. Bouchmella, P. H. Mutin and V. Hulea, ACS Catal., 2015, 5, 2774–2777 CrossRef CAS.
- R. D. Andrei, M. I. Popa, C. Cammarano and V. Hulea, New J. Chem., 2016, 40, 4146–4152 RSC.
- R. Beucher, R. D. Andrei, C. Cammarano, A. Galarneau, F. Fajula and V. Hulea, ACS Catal., 2018, 8, 3636–3640 CrossRef CAS.
- R. Beucher, C. Cammarano, E. Rodríguez-Castellón and V. Hulea, Ind. Eng. Chem. Res., 2020, 59, 7438–7446 CrossRef CAS.
- R. Beucher, C. Cammarano, E. Rodríguez-Castellón and V. Hulea, Catal. Commun., 2020, 144, 106091 CrossRef CAS.
- L. Riekert, J. Catal., 1970, 19, 8–14 CrossRef CAS.
- J. Heveling, C. P. Nicolaides and M. S. Scurrell, J. Chem. Soc., Chem. Commun., 1991, 126–127 RSC.
- F. T. T. Ng and D. C. Creaser, Stud. Surf. Sci. Catal., 1992, 73, 123–131 CrossRef CAS.
- R. L. Espinoza, R. Snel, C. J. Korf and C. P. Nicolaides, Appl. Catal., 1987, 29, 295–303 CrossRef CAS.
- K. Toch, J. W. Thybaut and G. B. Marin, Appl. Catal., A, 2015, 489, 292–304 CrossRef CAS.
- R. Y. Brogaard, M. Kømurcu, M. M. Dyballa, A. Botan, V. Van Speybroeck, U. Olsbye and K. De Wispelaere, ACS Catal., 2019, 9, 5645–5650 CrossRef CAS PubMed.
- F. Kleitz, D. Liu, G. M. Anilkumar, I.-S. Park, L. A. Solovyov, A. N. Shmakov and R. Ryoo, J. Phys. Chem. B, 2003, 107, 14296–14300 CrossRef CAS.
- K. Kimura, A.-I. Hideo and A. Osaki, J. Catal., 1970, 18, 271–280 CrossRef CAS.
- K. Takahashi, H. Tezuka, S. Kitamura, T. Satoh and R. Katoh, Phys. Chem. Chem. Phys., 2010, 12, 1963–1970 RSC.
- T. Matsuda, H. Miura, K. Sugiyama, N. Ohno, S. Keino and A. Kaise, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 1513–1520 RSC.
- D. Roy and R. B. Sunoj, Org. Biomol. Chem., 2010, 8, 1040–1051 RSC.
- E. D. Metzger, R. J. Comito, C. H. Hendon and M. Dincă, J. Am. Chem. Soc., 2017, 139, 757–762 CrossRef CAS PubMed.
- R. Y. Brogaard and U. Olsbye, ACS Catal., 2016, 6, 1205–1214 CrossRef CAS.
- R. Henry, M. Komurcu, Y. Ganjkhanlou, R. Y. Brogaard, L. Lu, K.-J. Jens, G. Berlier and U. Olsbye, Catal. Today, 2018, 299, 154–163 CrossRef CAS.
- L. Bonneviot, D. Olivier and M. Che, J. Mol. Catal., 1983, 21, 415–430 CrossRef CAS.
- F. X. Cai, C. Lepetit, M. Kermarec and D. Olivier, J. Mol. Catal., 1987, 43, 93–116 CrossRef CAS.
- G. Seufitelli and F. Resende, presented in part at the AlCHE2018, October, 2018 Search PubMed.
- Q. Zhang and I. G. Dalla Lana, Chem. Eng. Sci., 1997, 52, 4187–4195 CrossRef CAS.
- A. Volkov, E. Buluchevskii and A. V. Lavrenov, J. Sib. Fed. Univ., Chem., 2013, 6, 352–360 Search PubMed.
- F. T. T. Ng and D. C. Creaser, Appl. Catal., A, 1994, 119, 327–339 CrossRef CAS.
- J. Heveling, A. van der Beek and M. de Pender, Appl. Catal., 1988, 42, 325–336 CrossRef CAS.
- O. Jan and F. L. P. Resende, Fuel Process. Technol., 2018, 179, 269–276 CrossRef CAS.
- R. L. Espinoza, C. P. Nicolaides, C. J. Korf and R. Snel, Appl. Catal., 1987, 31, 259–266 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- G. J. P. Britovsek, R. Malinowski, D. McGuinness, J. Nobbs, A. K. Tomov, A. Wadsley and C. Young, ACS Cent. Sci., 2015, 5, 6922–6925 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00258a |
| This journal is © The Royal Society of Chemistry 2022 |