
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven proton reduction for semi-hydrogenation of alkynes via organophotoredox/manganese dual catalysis†
Xiao-Yu Wang‡
a,
Yong-Qin He‡b,
Mei Wanga,
Yi Zhoua,
Na Li b,
Xian-Rong Songa,
Zhao-Zhao Zhouc,
Wan-Fa Tian*a and
Qiang Xiao*a
b,
Xian-Rong Songa,
Zhao-Zhao Zhouc,
Wan-Fa Tian*a and
Qiang Xiao*a
aKey Laboratory of Organic Chemistry of Jiangxi Province, Jiangxi Science & Technology Normal University, Nanchang, 330013, P. R. China. E-mail: tianwanfa@yeah.net; xiaoqiang@tsinghua.org.cn
bSchool of Pharmaceutical Science, Nanchang University, Nanchang, 330006, P. R. China
cCollege of Chemistry and Food Science, Nanchang Normal University, Nanchang, P. R. China
First published on 19th December 2022
Abstract
Described here is a unprecedented organophotoredox/manganese dual catalyzed proton reduction and its application for semi-reduction of alkynes. The catalytic active pre-catalyst [Mn-1] can be feasibly be prepared on gram-scale from Mn(acac)2·2H2O in air. This dual catalytic protocol features noble-metal-free catalysts, simple ligand, and mild conditions. Besides, a unique ortho-halogen and -hydroxyl effect was observed to achieve high Z-stereoselectivity.
Catalytic proton reduction is an effective way to generate metal–hydride (M–H) species, which are key intermediates for H2 evolution reactions and hydride transfer reaction.1 A process of reducing a high valent transition metal to its low-valent state via a single electron process, followed by capturing a proton (H+) to form the key M–H species is always involved in these reactions. The non-precious metals, such as Fe,2 Co,3 Ni,4 and Cu4b,5 have exhibited extraordinary activity in this field via electro- or photocatalytic methodologies. Whereas, Mn, as an analogue of the above metals and the third most abundant transition metal in the Earth's crust, has rarely been reported for proton reduction,6 and the existing sporadic examples are limited to electrocatalytic H2 evolution reactions with structurally complex Mn catalysts.
Recently, photocatalytically generated Co–H has demonstrated its efficiency in catalytic hydride transfer reactions.1c,7 We thus wondered if Mn–H species can be formed via photocatalytic methodologies and be used further for reducing unsaturated bonds. We hypothesize that the reaction might proceed as outlined below (Scheme 1A). The excited photocatalyst PS might be reductively quenched by a sacrificial reagent to afford a reductive photocatalyst PS˙−, which can reduce high-valent Mn catalysts to low-valent species. The generated Mn species could react with a proton to form Mn–H, which subsequently undergoes hydrometalation with unsaturated bond followed by protonation to form the hydrogenated product and regenerate high-valent Mn species (Scheme 1A).
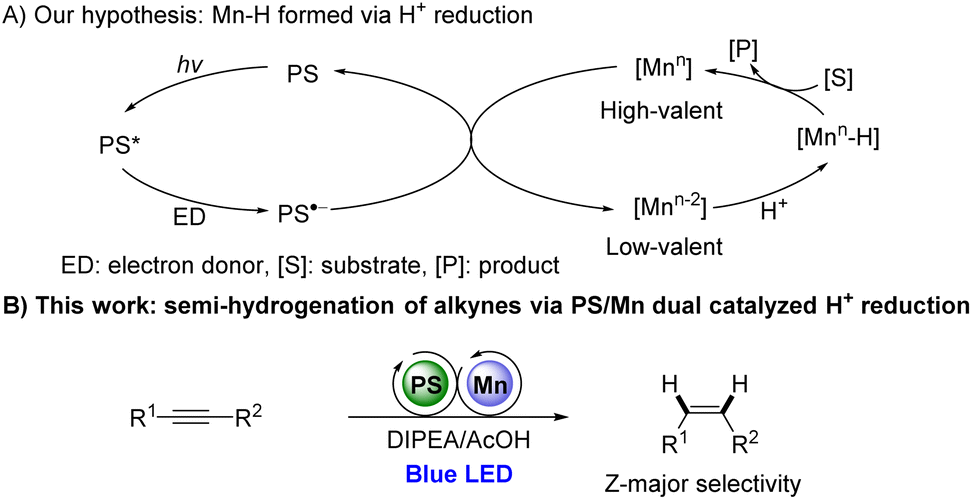 | ||
| Scheme 1 Mn-catalyzed reduction via photocatalytic protons reduction. | ||
Herein, we would like to report the first example of photocatalytically Mn-catalyzed proton reduction for semi-hydrogenation of alkynes by using an [Mn-1]/bipyridines catalytic system with DIPEA/AcOH as hydrogen source (Scheme 1B). To the best of our knowledge, such a Mn and photoredox dual catalytic system for reducing unsaturated bonds has not been reported.
Semi-hydrogenation of alkynes to alkenes is a valuable catalytic process,8 since it leads to configurationally-defined building blocks relevant for the synthesis of pharmaceuticals, agrochemicals, and natural products.9 Applying the earth-abundant transition metal-based catalysts to realize the reduction is the current research direction.10 Despite a few Mn catalytic systems have been established by using H2, NH3BH3, or MeOH as hydrogen source, these methods largely rely on sophisticated but complex Mn-pincer complexes.11 Moreover, these catalytic systems usually require strong base to activate the pre-catalysts accompanied by elevated temperature. Therefore, comparing with the previous work, our protocol is also highlighted by its simple catalyst and mild conditions.
To commend our studies on photocatalytic reduction systems, we selected 1a as a substrate, DIPEA as a sacrificial reagent, AcOH as an additive, and an organic dye 4CzIPN as a photocatalyst, using blue LED irradiation (Table S2†). Mn(acac)2·2H2O was chosen as the precatalyst and various N,N-bidentate ligands L1–L5 were firstly screened (Table S2,† entry 1). Unfortunately, no reaction occurred. Occasionally, we found that the color of Mn(acac)2·2H2O gradually changed from yellowish to black under air. Interestingly, this black Mn compound shows partial catalytic activity with L2 (5,5′-dimethyl-2,2′-bipyridine) as the ligand (not shown). We speculate that Mn(acac)2·2H2O changed in moist air. Realizing that Mn complexes can undergo rapid transformation in air under alkaline conditions,12 we conducted a similar reaction using Mn(acac)2·2H2O as a substrate in the presence of Et3N and H2O. The mixture rapidly turns to black within 10 minutes, and finally afforded [Mn-1] after 2 h stirring (scheme inserted in Fig. 1, see ESI† for detail). A common approach to determine the oxidation of Mn complex is the evaluation of the peak binding energy distance in the multiplet split Mn 3s region in X-ray photoelectron spectra (XPS).13 Since the 3s peak widths of [Mn-1] is 6.2 eV, located at region of MnII, suggesting that [Mn-1] still exists mainly in an oxidation of II (Fig. 1, please see the ESI† for details). HRMS analysis demonstrated the full conversion of Mn(acac)2·2H2O in the reaction that showed in Table 1B (see Fig. S2 and S3†). Furthermore, the UV-vis absorption spectrum of Mn(acac)·2H2O, [Mn-1], and Mn oxides (MnO, Mn2O3, and Mn3O4) were also collected (Fig. S4†). Among them, Mn oxides displayed no absorption at near infrared region (200–400 nm), but Mn(acac)·2H2O and [Mn-1] showed strong absorption at 280 nm and 297 nm respectively, which should be attributed to the π–π* and/or n–π* electron orbital transitions from acetylacetonate. This observation confirmed that the ligand is still anchored to Mn atom in [Mn-1]. Unfortunately, we have not been able to determine the exact structure of [Mn-1] at this stage.
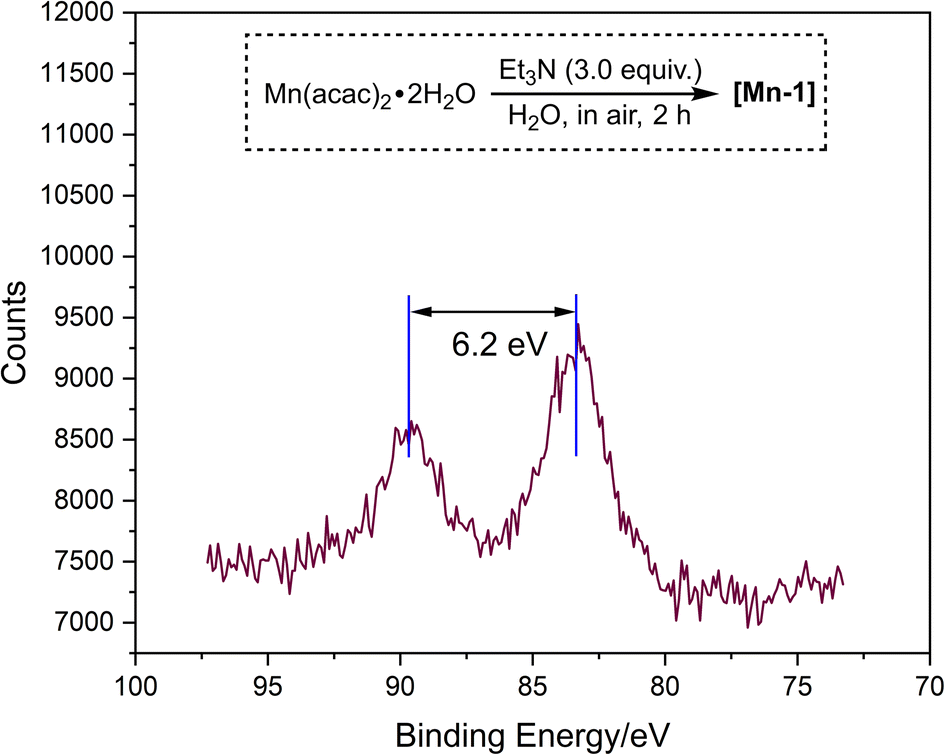 | ||
| Fig. 1 XPS spectra of Mn-3s regions of [Mn-1]. | ||
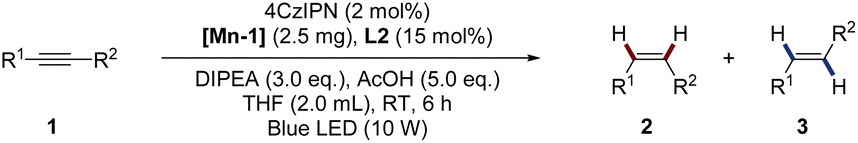
Nonetheless, this Mn complex is catalytically active. Notably, [Mn-1] catalyzes the semi-reduction reaction when employing L2 as the ligand, giving alkene products with a yield of 64% (entry 3). Further ligand's screening revealed that L5 could also catalyze the reaction, but inferior yield was obtained (Table S2,† entries, 2, 4–6). Furthermore, the bidentate phosphine ligand dppp failed to promote the reaction (Table S2,† entry 7). Solvent screening revealed that THF gave the full conversion of 1a to alkenes with a Z/E ratio of 85/15 (Table S2,† entry 8). Other polar solvents such as CH3CN and acetone totally inhibited the semi-hydrogenation (Table S2,† entry 9). However, the MnIII compound Mn(acac)3 failed to catalyze the reaction (Table S2,† entry 10). Similarly, changing the additive of AcOH to H2O totally suppressed the transformation (Table S2,† entry 11). Photocatalyst screening revealed that Ir[ppy]2(dtbbpy)PF6 catalyzed the reaction with low yield (Table S2,† entries 12 and 13). Control experiments confirmed that the photocatalyst, Mn complex, DIPEA, AcOH, and light are all essential to the reaction (Table S2,† entries 14–17). Additionally, Hg poisoning experiments confirmed the homogenous nature of this catalytic system (Table S2,† entry 18). Besides, Mn oxides such as MnO, Mn2O3 and Mn3O4 displayed no activity (Table S2,† entry 19). Of note that none of the over-reduced product 4a was observed under all conditions tested. Meanwhile, we found that the catalytic active pre-catalyst can also be feasible prepared on gram-scale by just evaporating the solvent in the mixture of Mn(acac)2·2H2O and H2O at 80 °C in air (see ESI† for detail).
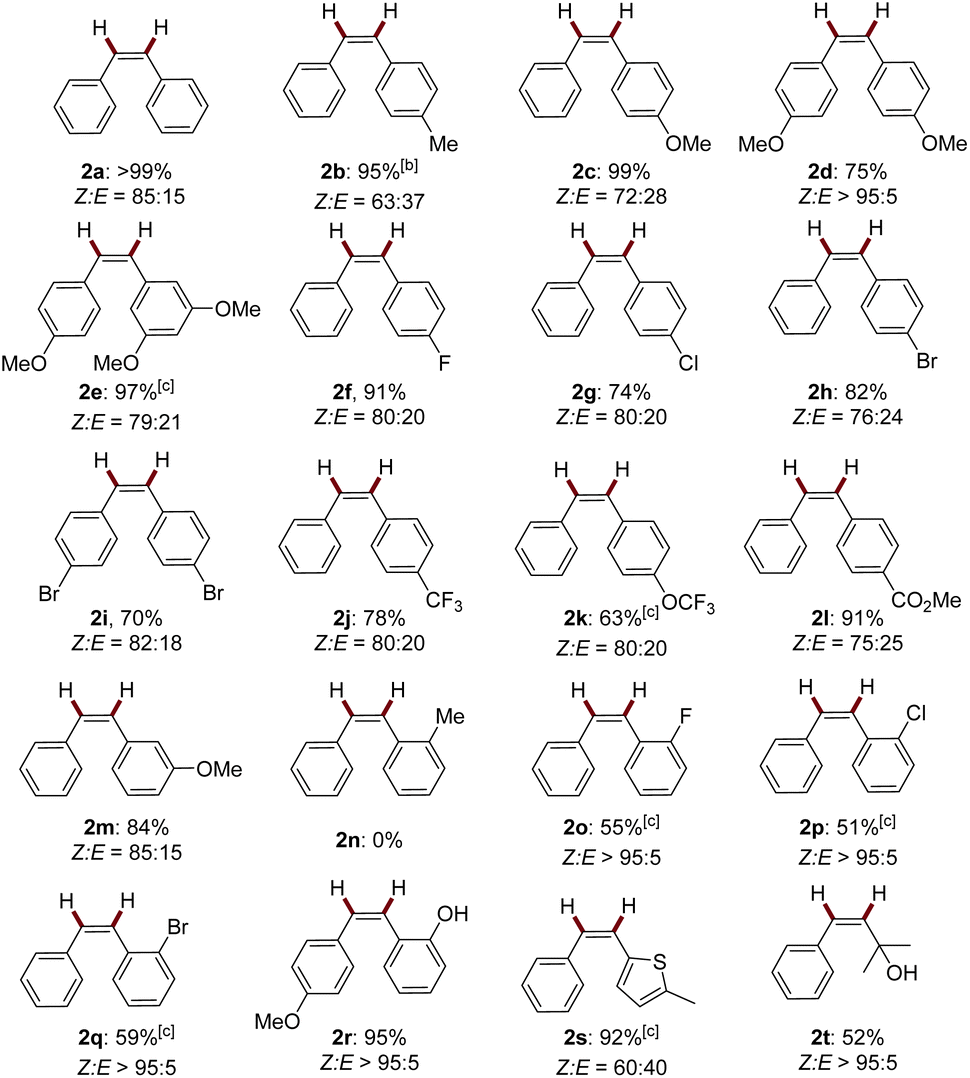
With the optimal conditions established, we evaluated the substrate scope of this dual-photoredox Mn catalytic system (Table 1). A range of electronically- and sterically-differentiated internal alkynes were subjected to the conditions. Notably, para-substituted alkynes were smoothly semi-hydrogenated to alkenes in good to excellent yields with Z-major selectivity (2a–2l, 63–99%, Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E 63:37–>95:5). Reducible groups including chloro, bromo, and ester were tolerated in the reducible conditions. Methoxy at the meta-position did not affect the reaction efficiency, furnishing 2e and 2m in 97% and 84% yields, respectively. Interestingly, ortho-halogen or hydroxyl substituted diphenylalkynes (1o–1r), as well as propargyl alcohol (1t) produced the corresponding alkenes with exclusively Z selectivity with moderate to excellent yields (2o–2r, 2t, 51–95%, Z:E >95:5). By sharp comparison, ortho-methylated diphenylalkyne 1n did not proceed under the standard reaction conditions. The coordination effect may contribute to the exclusive Z-stereoselectivity.14 Specifically, stereochemically well-defined (Z)-ortho-halogen alkenes are important intermediates for diverse functionalization.15 Moreover, the strongly coordinating heterocycle thiophene (1s) was also compatible with the conditions. Terminal alkynes were not compatible under these conditions.
:E 63:37–>95:5). Reducible groups including chloro, bromo, and ester were tolerated in the reducible conditions. Methoxy at the meta-position did not affect the reaction efficiency, furnishing 2e and 2m in 97% and 84% yields, respectively. Interestingly, ortho-halogen or hydroxyl substituted diphenylalkynes (1o–1r), as well as propargyl alcohol (1t) produced the corresponding alkenes with exclusively Z selectivity with moderate to excellent yields (2o–2r, 2t, 51–95%, Z:E >95:5). By sharp comparison, ortho-methylated diphenylalkyne 1n did not proceed under the standard reaction conditions. The coordination effect may contribute to the exclusive Z-stereoselectivity.14 Specifically, stereochemically well-defined (Z)-ortho-halogen alkenes are important intermediates for diverse functionalization.15 Moreover, the strongly coordinating heterocycle thiophene (1s) was also compatible with the conditions. Terminal alkynes were not compatible under these conditions.
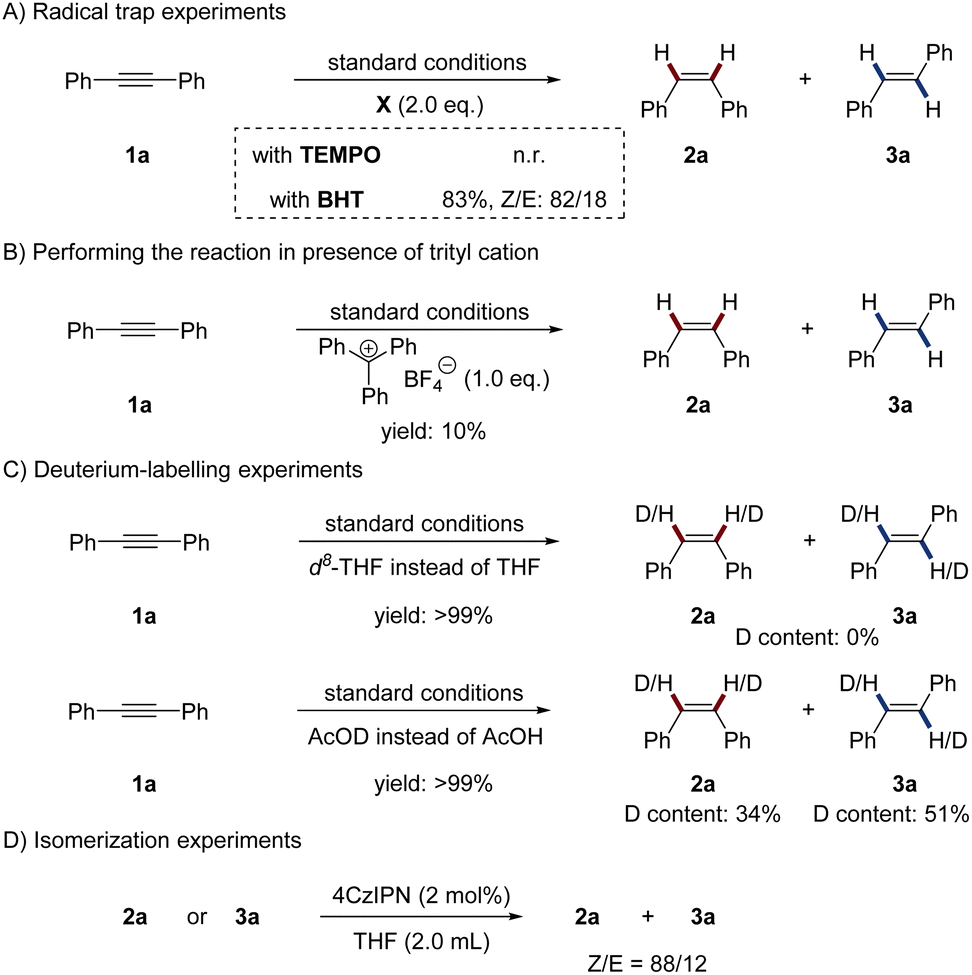
To gain insight into the mechanism of the novel photoredox/manganese dual catalysis, several control experiments were conducted. To probe a potential radical mechanism, the addition of radical traps to the reaction was investigated (Scheme 2A). When 2 equivalents of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) were added to the standard reaction, the reaction was completely inhibited. However, 2,6-di-tert-butyl-4-methylphenol (BHT) only slightly reduced the reaction yield. Realizing that radical traps might react with the metal-hydride to inhibit the reaction, we thus deduced that radical species might not involve in the catalytic cycle.16 We surmised that a Mn–H species might be generated in the reaction and be an essential component for product formation. To test this hypothesis, we performed the reaction in the presence of the trityl cation, which is known to be a hydride abstractor for organometallic compounds.17 As anticipated, the reaction was almost completely inhibited by adding stoichiometric amounts of triphenylcarbenium tetrafluoroborate (Scheme 2B). Deuterium labelling experiments were performed to investigate the H2 source of the reaction (Scheme 2C). When the reaction was conducted in d8-THF, no deuterium was incorporated into the alkenes. Using AcOD instead of AcOH lead to 34% of D-incorporation in 2a and 51% of D-incorporation in 3a. Because DIPEA always serves as both an electron donor and a proton donor in reductive photoredox catalysis,1b,8 we thus deduced that both DIPEA and AcOH work as the H2 source. Next, isomerization experiments were conducted by employing 2a or 3a as substrates without the Mn complex, AcOH and DIPEA (Scheme 2D). The same Z/E selectivity as the standard reaction was observed, indicating that photo-induced energy transfer process is the decisive factor for the stereoselectivity.18
 | ||
| Scheme 2 Control experiments. | ||
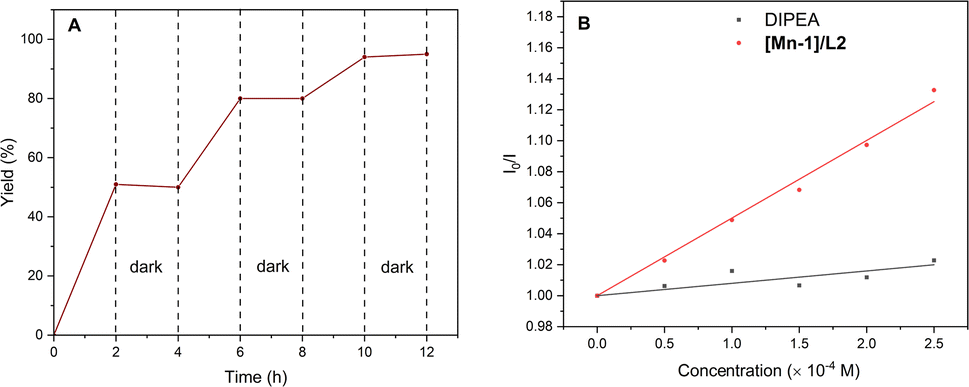
Additional experiments were performed to probe the reaction pathway. First, the light on-off experiments confirmed the intersection of photoredox with Mn catalysis (Fig. 2A). Second, Stern–Volmer luminescence quenching studies showed that only Mn complex can quench the excited photocatalyst 4CzIPN at a high rate, indicating that efficient electron transfer occurs between excited 4CzIPN and the Mn complex (Fig. 2B). Cyclic voltammetry measurements of the [Mn-1]/L2 complex showed two successive reducible peaks positioned at −1.13 V vs. SCE and −1.43 V vs. SCE (see ESI) corresponding to the respective reductive potential of MnII/I and MnI/0. Since these reductive potentials are less negative than the oxidative potential of the excited photocatalyst 4CzIPN* (E1/2 red[PC˙+/PC*] = −1.25 V vs. SCE)19 or within its roughly 200 mV range,20 we thus believe that single electron reduction of Mn complex by the excited photocatalyst is possible.
 | ||
| Fig. 2 (A) Light on-off experiments and (B) Stern–Volmer luminescence quenching plot. | ||
Based on above experiments, we supposed that the reaction should involve a photoreduction of high-valent Mn to low-valent state in present of DIPEA, and Mn–H species formation via capturing of H+ by low-valent Mn, although we haven't been able to determine the exact structure of [Mn-1] yet (Scheme S3†).
In conclusion, we report a novel photocatalytic transfer semi-reduction of alkynes through dual Mn and photoredox catalysis. This protocol enables the semi-reduction of a broad range of alkynes with Z-major or -exclusive selectivity at room temperature using a simple bipyridine ligand. Although the definitive catalyst structure has not yet been determined, we believe this work will stimulate enthusiasm in the area of Mn-photoredox dual catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of China (No. 21861016, 22001101), the Science Foundation of Jiangxi Province (No. 20212BAB203009, 20212BAB213014, and 20212BAB213027), and Jiangxi Science & Technology Normal University (No. 2018BSQD024, Doctor Startup Fund) for financial support.Notes and references
- (a) W. T. Eckenhoff, Coord. Chem. Rev., 2018, 373, 295 CrossRef CAS; (b) A. Mazzeo, S. Santalla, C. Gaviglio, F. Doctorovich and J. Pellegrino, Inorg. Chim. Acta, 2021, 517, 119950 CrossRef CAS; (c) M. Kojima and S. Matsunaga, Trends. Chem., 2020, 2, 410 CrossRef CAS.
- (a) F. Gärtner, B. Sundararaju, A.-E. Surkus, A. Boddien, B. Loges, H. Junge, P. H. Dixneuf and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9962 CrossRef PubMed; (b) A. C. Cavell, C. L. Hartley, D. Liu, C. S. Tribble and W. R. McNamara, Inorg. Chem., 2015, 54, 3325 CrossRef CAS PubMed.
- (a) J. L. Dempsey, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Acc. Chem. Res., 2009, 42, 1995 CrossRef CAS PubMed; (b) J. Willkomm and E. Reisner, Bulletin of Japan Society of Coordination Chemistry, 2018, 71, 18 CrossRef.
- (a) S. Wiese, U. J. Kilgore, D. L. DuBois and R. M. Bullock, ACS Catal., 2012, 2, 720 CrossRef CAS; (b) X.-S. Hong, D. Huo, W.-J. Jiang, W.-J. Long, J.-D. Leng, L. Tong and Z.-Q. Liu, Chemelectrochem, 2020, 7, 4956 CrossRef CAS.
- A. M. Abudayyeh, O. Schott, H. L. C. Feltham, G. S. Hanan and S. Brooker, Inorg. Chem. Front., 2021, 8, 1015 RSC.
- V. Kaim and S. Kaur-Ghumaan, Eur. J. Inorg. Chem., 2019, 2019, 5041 CrossRef CAS.
- (a) P. Rai, K. Maji and B. Maji, Org. Lett., 2019, 21, 3755 CrossRef CAS PubMed; (b) W. F. Tian, Y. Q. He, X. R. Song, H. X. Ding, J. Ye, W. J. Guo and Q. Xiao, Adv. Synth. Catal., 2020, 362, 1032 CrossRef CAS; (c) Y.-L. Li, S.-Q. Zhang, J. Chen and J.-B. Xia, J. Am. Chem. Soc., 2021, 143, 7306 CrossRef CAS PubMed.
- (a) A. M. Kluwer and C. J. Elsevier, in The Handbook of Homogeneous Hydrogenation, ed. J. G. d. Vries. and C. J. Elsevier, 2006, p. 374 Search PubMed; (b) C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313 CrossRef CAS PubMed.
- J. M. J. Williams, in Preparation of alkenes: a practical approach, Oxford, U. K., 1996 Search PubMed.
- D. Baidilov, D. Hayrapetyan and A. Y. Khalimon, Tetrahedron, 2021, 98, 132435 CrossRef CAS.
- (a) A. Brzozowska, L. M. Azofra, V. Zubar, I. Atodiresei, L. Cavallo, M. Rueping and O. El-Sepelgy, ACS Catal., 2018, 8, 4103 CrossRef CAS; (b) M. Garbe, S. Budweg, V. Papa, Z. Wei, H. Hornke, S. Bachmann, M. Scalone, A. Spannenberg, H. Jiao, K. Junge and M. Beller, Catal. Sci. Technol., 2020, 10, 3994 RSC; (c) J. Sklyaruk, V. Zubar, J. C. Borghs and M. Rueping, Org. Lett., 2020, 22, 6067 CrossRef CAS PubMed; (d) V. Zubar, J. Sklyaruk, A. Brzozowska and M. Rueping, Org. Lett., 2020, 22, 5423 CrossRef CAS PubMed; (e) R. A. Farrar-Tobar, S. Weber, Z. Csendes, A. Ammaturo, S. Fleissner, H. Hoffmann, L. F. Veiros and K. Kirchner, ACS Catal., 2022, 12, 2253 CrossRef CAS PubMed; (f) A. Torres-Calis and J. J. Garcia, Catal. Sci. Technol., 2022, 12, 3004 RSC.
- (a) J. J. Morgan, Geochim. Cosmochim. Acta, 2005, 69, 35 CrossRef CAS; (b) M. Sankaralingam, Y.-M. Lee, S. H. Jeon, M. S. Seo, K.-B. Cho and W. Nam, Chem. Commun., 2018, 54, 1209 RSC.
- (a) J. W. Murray, J. G. Dillard, R. Giovanoli, H. Moers and W. Stumm, Geochim. Cosmochim. Acta, 1985, 49, 463 CrossRef CAS; (b) J. L. Junta and M. F. Hochella, Geochim. Cosmochim. Acta, 1994, 58, 4985 CrossRef CAS.
- J. Chen, C. Chen, C. Ji and Z. Lu, Org. Lett., 2016, 18, 1594 CrossRef CAS PubMed.
- (a) Z. Li and R. J. Twieg, Chem.–Eur. J., 2015, 21, 15534 CrossRef CAS PubMed; (b) T. Matsushima, S. Kobayashi and S. Watanabe, J. Org. Chem., 2016, 81, 7799 CrossRef CAS PubMed.
- (a) A. C. Albéniz, P. Espinet, R. López-Fernández and A. Sen, J. Am. Chem. Soc., 2002, 124, 11278 CrossRef PubMed; (b) J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620 CrossRef CAS PubMed.
- (a) D. A. Straus, C. Zhang and T. D. Tilley, J. Organomet. Chem., 1989, 369, C13 CrossRef CAS; (b) S. R. Bahr and P. Boudjouk, J. Org. Chem., 1992, 57, 5545 CrossRef CAS.
- (a) K. Singh, S. J. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275 CrossRef CAS PubMed; (b) W. Cai, H. Fan, D. Ding, Y. Zhang and W. Wang, Chem. Commun., 2017, 53, 12918 RSC; (c) J. B. Metternich, D. G. Artiukhin, M. C. Holland, M. von Bremen-Kühne, J. Neugebauer and R. Gilmour, J. Org. Chem., 2017, 82, 9955 CrossRef CAS PubMed; (d) J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719 CrossRef CAS PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182 CrossRef CAS PubMed.
- S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07920h |
| ‡ X.-Y. Wang and Y.-Q. He contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |