
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioactive 2-pyridone-containing heterocycle syntheses using multicomponent reactions
Diana Hurtado-Rodríguez
,
Angélica Salinas-Torres
,
Hugo Rojas
,
Diana Becerra
* and
Juan-Carlos Castillo
 *
*
Grupo de Catálisis de la UPTC, Escuela de Ciencias Química, Universidad Pedagógica y Tecnológica de Colombia, Avenida Central del Norte 39-115, Tunja, Colombia. E-mail: diana.becerra08@uptc.edu.co; juan.castillo06@uptc.edu.co
First published on 7th December 2022
Abstract
2-Pyridone-containing heterocycles are considered privileged scaffolds in drug discovery due to their behavior as hydrogen bond donors and/or acceptors and nonpeptidic mimics, and remarkable physicochemical properties such as metabolic stability, solubility in water, and lipophilicity. This review provides a comprehensive overview of multicomponent reactions (MCRs) for the synthesis of 2-pyridone-containing heterocycles. In particular, it covers the articles published from 1999 to date related to anticancer, antibacterial, antifungal, anti-inflammatory, α-glucosidase inhibitor, and cardiotonic activities of 2-pyridone-containing heterocycles obtained exclusively by an MCR. The discussion focuses on bioactivity data, synthetic approaches, plausible reaction mechanisms, and molecular docking simulations to facilitate comparison and underscore the applications of the 2-pyridone motif in drug discovery and medicinal chemistry. We also present our conclusions and outlook for the future.
 Diana Hurtado-Rodríguez | Diana Hurtado-Rodríguez was born in Manizales (Colombia) and received her BSc degree at the Universidad Pedagógica y Tecnológica de Colombia (Tunja) in 2023. Currently, she is working on her MSc degree in Chemistry under the supervision of Dr Juan Castillo, Dr Diana Becerra and Prof. Hugo Rojas at the Universidad Pedagógica y Tecnológica de Colombia (Tunja). Her research focuses on the synthesis of 2-pyridone derivatives with potential anticancer activity. |
 Angélica Salinas-Torres | Angélica Salinas-Torres was born in Tunja (Colombia) and received her BSc degree at the Universidad Pedagógica y Tecnológica de Colombia (Tunja) in 2021. Currently, she is working on her MSc degree in Chemistry under the supervision of Dr Juan Castillo, Dr Diana Becerra and Prof. Hugo Rojas at the Universidad Pedagógica y Tecnológica de Colombia (Tunja). Her research focuses on the synthesis of amides with potential anticancer activity. |
 Hugo Rojas | Hugo Rojas was born in Tunja (Colombia). He is a Full Professor at the School of Chemical Sciences at the Universidad Pedagógica y Tecnológica de Colombia (Tunja), leading the Grupo de Catálisis de la UPTC since 1996. He completed his PhD in Chemical Sciences under the supervision of Prof. Patricio Reyes at the Universidad de Concepción (Chile) in 2003. His current research interests involve the areas of heterogeneous catalysis, biocatalysis, photocatalysis, catalysis in organic chemistry, and materials chemistry. |
 Diana Becerra | Diana Becerra was born in Florida (Colombia). After studying Chemistry at the Universidad del Valle (Cali, Colombia), she received her PhD in Chemical Sciences in 2014 under the supervision of Prof. Braulio Insuasty. Then, she became a postdoctoral fellow at the Institut des Sciences Moléculaires de Marseille (France) under the supervision of Prof. Jean Rodriguez and Prof. Damien Bonne. In 2017, she began her academic career at the Universidad Pedagógica y Tecnológica de Colombia (Tunja). Her research interest focuses on the synthesis of biologically active heterocyclic compounds, sustainable chemistry, catalysis in organic chemistry, and supramolecular chemistry. |
 Juan-Carlos Castillo | Juan Castillo was born in Cali (Colombia). After studying Chemistry at the Universidad del Valle (Cali, Colombia), he received his PhD in Chemical Sciences in 2013 under the supervision of Prof. Rodrigo Abonia. He became a postdoctoral fellow at the Institut des Sciences Moléculaires de Marseille (France) under the supervision of Prof. Jean Rodriguez and Prof. Yoann Coquerel. Later, he was a postdoctorate researcher at the Universidad de los Andes (Bogotá, Colombia) under the supervision of Prof. Jaime Portilla. In 2018, he began his academic career at the Universidad Pedagógica y Tecnológica de Colombia (Tunja). His current research interests involve the synthesis of biologically active heterocyclic compounds, sustainable chemistry, catalysis in organic chemistry, and supramolecular chemistry. |
1 Introduction
Multicomponent reactions (MCRs) are convergent reactions that lead to a single product from three or more reactants, where all or most of these atoms are incorporated in the product structure through a cascade of elementary reactions.1–3 The popularity of MCRs lies in the operational simplicity, savings in time and energy (step efficiency), proceeding with high convergence (process efficiency) and bond-forming index (BFI), and compatibility with a range of unprotected orthogonal functional groups.1–3 Thereby, MCRs are considered a powerful alternative to prepare structurally complex and diverse organic molecules with a wide range of applications in polymer chemistry,4 combinational chemistry,5 organic chemistry,6,7 medicinal chemistry,3,8 and chiefly drug discovery programs.9 Many of the classical MCRs are named reactions and all have allowed the incorporation of bioactive scaffold shapes, including Strecker (1850),10 Hantzsch (1881),11 Biginelli (1891),12 Mannich (1912),13 Passerini (1921),14 Kabachnik-Fields (1952),15 Asinger (1956),16 Ugi (1959),17 Gewald (1966),18 Van Leusen (1977),19 and Groebke–Blackburn–Bienaymé (1998).20 Over the past few decades, MCRs have been employed as a powerful tool to create chemical diversity, high throughput generation of bioactive scaffolds, and the large-scale production of drug candidates.21,22 It is estimated that roughly 5% of the marketed drugs have been synthesized through an MCR strategy, including Nifedipine (Hantzsch), Telaprevir (Passerini), and Crixivan (Ugi).22Among all the synthetic and naturally occurring N-heterocycles, 2(1H)-pyridones are six-membered heterocycles possessing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NH groups, which have been employed as privileged scaffolds in medicinal chemistry and drug discovery programs due to their interesting structural features and a broad range of biological activities.23–25 The most important structural characteristic is the tautomeric equilibrium between 2-hydroxypyridine (lactim) and 2(1H)-pyridone (lactam), which predominates in both solid and solution phases.26–28 Additionally, 2(1H)-pyridones are important in drug design due to their capability to: (a) act as bioisosteres for amides, phenyls, pyridines, pyridine N-oxides, and phenols; (b) serve as both hydrogen bond donors and/or acceptors; and (c) achieve better drug-like properties such as low lipophilicity, solubility in water, and metabolic stability under physiological conditions.29,30 For instance, 2-pyridone is widely found in bioactive natural products, including Huperzine A used in China as a treatment for Alzheimer's disease and is available in the U.S. as a nutraceutical (not regulated by the FDA),31 fredericamycin A employed as a novel lead compound for the chemotherapy of human cancers,32 and Camptothecin displayed anticancer activity as an inhibitor of DNA topoisomerase I (Fig. 1).33 Over the past two decades, 2-pyridone derivatives have become a hot point in drug discovery programs with an increasing number of FDA-approved drugs as kinase inhibitors, including the most recent approvals of Ripretinib (2020), Tazemetostat (2020), Doravirine (2018), Duvelisib (2018), and Palbociclib (2015) (Fig. 2).29 In particular, tazemetostat is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2. Thereby, EZH2 inhibitors represent a notable clinical promise for cancer treatment because some anticancer drugs have acquired resistance and suffer from poor bio-distribution and limited brain penetration.34
O and NH groups, which have been employed as privileged scaffolds in medicinal chemistry and drug discovery programs due to their interesting structural features and a broad range of biological activities.23–25 The most important structural characteristic is the tautomeric equilibrium between 2-hydroxypyridine (lactim) and 2(1H)-pyridone (lactam), which predominates in both solid and solution phases.26–28 Additionally, 2(1H)-pyridones are important in drug design due to their capability to: (a) act as bioisosteres for amides, phenyls, pyridines, pyridine N-oxides, and phenols; (b) serve as both hydrogen bond donors and/or acceptors; and (c) achieve better drug-like properties such as low lipophilicity, solubility in water, and metabolic stability under physiological conditions.29,30 For instance, 2-pyridone is widely found in bioactive natural products, including Huperzine A used in China as a treatment for Alzheimer's disease and is available in the U.S. as a nutraceutical (not regulated by the FDA),31 fredericamycin A employed as a novel lead compound for the chemotherapy of human cancers,32 and Camptothecin displayed anticancer activity as an inhibitor of DNA topoisomerase I (Fig. 1).33 Over the past two decades, 2-pyridone derivatives have become a hot point in drug discovery programs with an increasing number of FDA-approved drugs as kinase inhibitors, including the most recent approvals of Ripretinib (2020), Tazemetostat (2020), Doravirine (2018), Duvelisib (2018), and Palbociclib (2015) (Fig. 2).29 In particular, tazemetostat is a potent, selective, and orally bioavailable small-molecule inhibitor of EZH2. Thereby, EZH2 inhibitors represent a notable clinical promise for cancer treatment because some anticancer drugs have acquired resistance and suffer from poor bio-distribution and limited brain penetration.34
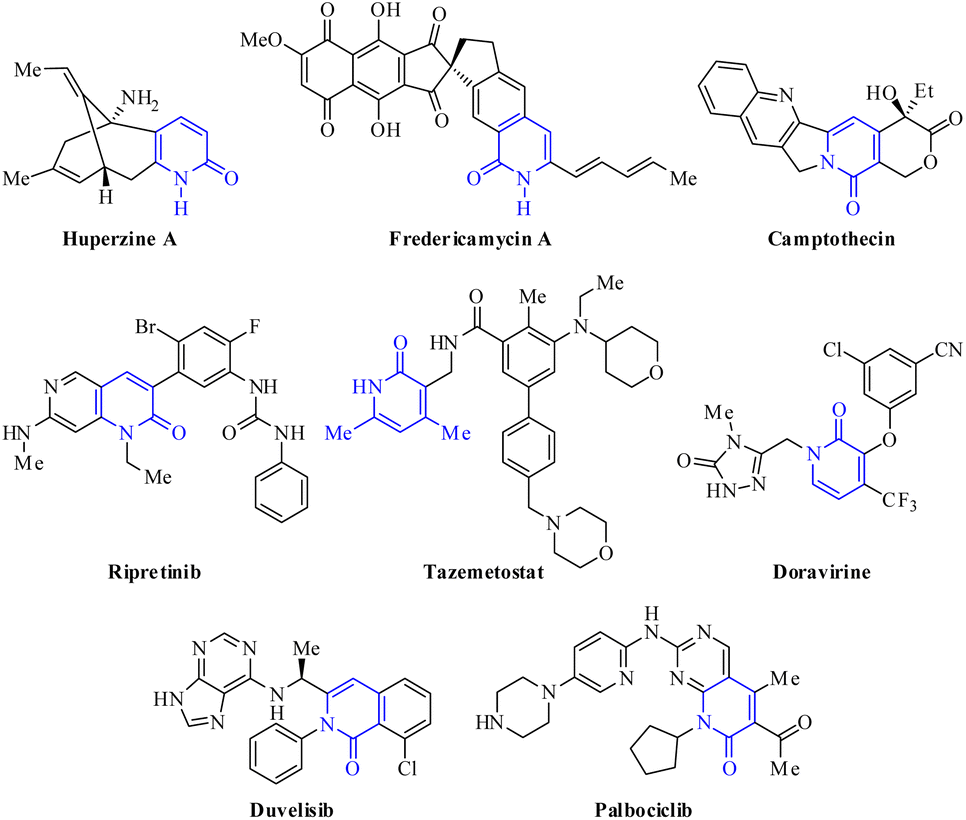 | ||
| Fig. 1 Bioactive natural products and selected examples of FDA-approved drugs containing 2-pyridone core. | ||
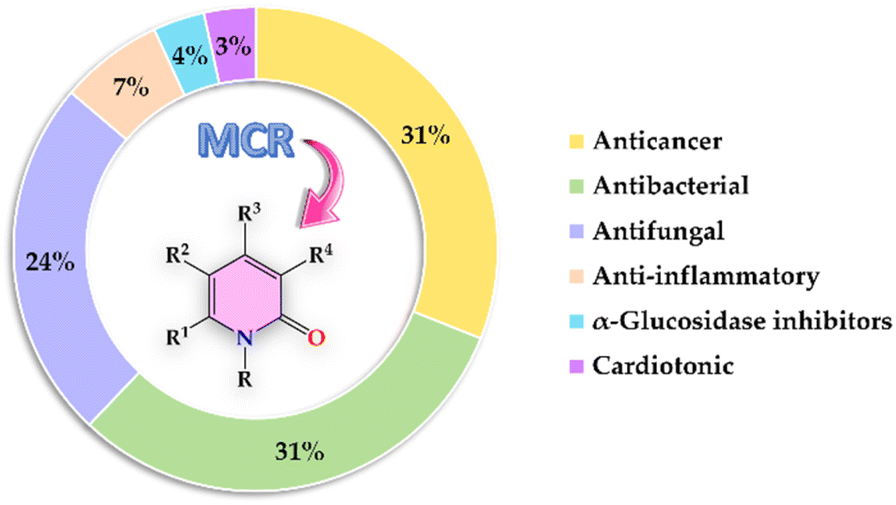 | ||
| Fig. 2 Bibliometric graphic depicting the percentage of articles associated with each biological activity screened from 1999 to date [data were collected searching in Scopus for the keywords: “2-pyridones”, “biological activity”, and “multicomponent reactions”]. | ||
The present review covers articles published from 1999 to date related to anticancer, antibacterial, antifungal, anti-inflammatory, α-glucosidase inhibitor, and cardiotonic activities of 2-pyridones derivatives obtained exclusively by an MCR (Fig. 2). It should be noted that 86% of articles found suitable for this review corresponded to 2-pyridone derivatives with anticancer (31%), antibacterial (31%), and antifungal (24%) activities. For a better comprehension, the present review has been organized and discussed in terms of the biological activities exhibited by 2-pyridone derivatives.
2 Multicomponent synthesis of bioactive 2-pyridone derivatives
2.1. Anticancer activity
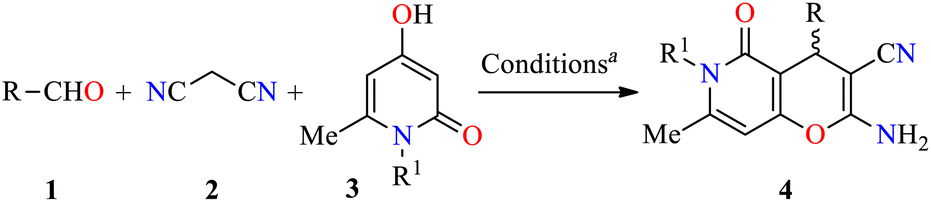
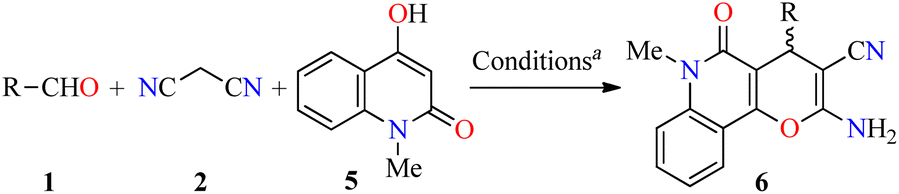
Analysis of the database of U.S. FDA-approved drugs reveals that nearly 60% of unique small-molecule drugs contain an N-heterocycle.35 In particular, N-heterocyclic skeletons are used as building blocks for the production of oncology-active pharmaceutical ingredients (APIs) and many biologically active molecules.36 As a result, a plethora of methodologies for preparing diversely functionalized N-heterocycles with important anticancer activity have been described during the last decade.37–40 In this regarding, Magedov et al. described the synthesis of pyrano[3,2-c]pyridones 4 through a three-component reaction of diverse aromatic aldehydes 1, malononitrile 2, and 4-hydroxy-1,6-dimethylpyridin-2(1H)-one 3 mediated by triethylamine (45 mol%) in refluxing ethanol for 50 min (Table 1).41 The reaction mixture was cooled at room temperature, and the precipitate was filtered and washed with ethanol to afford products 4a–l in 75–98% yields. This protocol was distinguished by its short reaction times, high yields, the use of a green solvent, and broad substrate scope. Moreover, compounds 4a–l were screened for their anticancer activity against HeLa (cervical cancer) cell line. The cells were treated with respective compounds for 48 h and cell viability was assessed through measurements of mitochondrial dehydrogenase activity using the MTT method (Table 1). Data shown are average ± SD of three independent experiments. These compounds showed good to excellent anticancer activity with IC50 values ranging from 0.33 to >100 μM. Remarkably, compounds 4a (R = 3-Br-4-NMe2C6H3, R1 = Me) and 4b (R = 3-Br-4,5-(MeO)2C6H2, R1 = Me) displayed the best activity with IC50 values of 0.33 and 0.58 μM, respectively. Since many clinically used anticancer agents induce apoptosis in cancer cells, pyrano[3,2-c]pyridones 4a–i and 4l were tested for their ability to induce apoptosis in the Jurkat cells using a flow cytometric annexin-V/propidium iodide assay. In summary, the compounds 4a–g were found to be strong inducers of apoptosis (50–60% after 36 h treatment) in Jurkat cells at 5 μM concentration, which is comparable to the Colchicine used at the same concentration. Lastly, the flow cytometric cell cycle analysis was performed with compounds 4a and 4b against the Jurkat cell line, leading to a pronounced cell cycle arrest in the G2/M phase (Table 2). This result is characteristic of antimitotic agents disrupting microtubule assembly, which is also observed with 2-amino-4-aryl-4H-chromene-3-carbonitriles that bind to or near the colchicine binding site on β-tubulin.42
|
||||
|---|---|---|---|---|
| Compound | R | R1 | Yield 4 (%) | IC50b (μM) |
| HeLa | ||||
| a Reaction conditions: aldehyde 1 (0.8 mmol), malononitrile 2 (0.8 mmol), 4-hydroxy-1,6-dimethylpyridin-2(1H)-one 3 (0.8 mmol), triethylamine (45 mol%), EtOH (3 mL), reflux, 50 min.b Compound concentration required to reduce HeLa cell viability by 50% after 48 h treatment relative to 100% DMSO control as assessed with the MTT assay. Data shown are average ± SD of three independent experiments. | ||||
| 4a | 3-Br-4-NMe2C6H3 | Me | 83 | 0.33 ± 0.06 |
| 4b | 3-Br-4,5-(MeO)2C6H2 | Me | 87 | 0.58 ± 0.14 |
| 4c | 3-Br-4-EtO-5-MeOC6H2 | Me | 88 | 1.08 ± 0.8 |
| 4d | 3-Br-4-OH-5-MeOC6H2 | Me | 75 | 2.67 ± 1.1 |
| 4e | 3-Br-4-AcO-5-MeOC6H2 | Me | 83 | 3.50 ± 1.3 |
| 4f | 3-Br-4-F-C6H3 | Me | 84 | 6.33 ± 1.1 |
| 4g | 3-BrC6H4 | Me | 97 | 6.50 ± 1.3 |
| 4h | 3,4-(Cl)2C6H3 | Me | 98 | 18.3 ± 2.9 |
| 4i | 3,4,5-(MeO)3C6H2 | Me | 97 | 43.3 ± 5.1 |
| 4j | 4-iPrC6H4 | Me | 81 | >100 |
| 4k | 3-NO2C6H4 | Me | 97 | 35.0 ± 21.8 |
| 4l | 3,4,5-(MeO)3C6H2 | 3,4-Dimethoxyphenethyl | 80 | 22.7 ± 6.4 |
| Compound | % Relative DNA contenta | ||
|---|---|---|---|
| G0/G1 | S | G2/M | |
| a % Relative DNA content ± SD after 15 h treatment of Jurkat cells with indicated compounds from two independent experiments. | |||
| DMSO | 49.4 ± 2.7 | 26.6 ± 0.1 | 23.9 ± 2.4 |
| 4a (5 μM) | 14.4 ± 1.7 | 18.1 ± 0.6 | 67.5 ± 2.3 |
| 4b (5 μM) | 12.0 ± 3.5 | 19.0 ± 2.0 | 68.5 ± 4.7 |
| Colchicine (25 μM) | 19.0 ± 0.2 | 23.1 ± 0.1 | 57.9 ± 0.1 |
In a similar way to previously described,41 Magedov et al. reported the synthesis of pyrano[3,2-c]quinolones 6a–y in 64–97% yields via a three-component approach utilizing commercially available 4-hydroxy-1-methylquinolin-2(1H)-one 5 under the same reaction conditions (Table 3).43 The anticancer activity of all synthesized compounds was evaluated against HeLa (cervical cancer) and MCF-7 (breast cancer) cell lines. The cells were treated with respective compounds for 48 h, and cell viability was assessed through measurements of mitochondrial dehydrogenase activity using the MTT method (Table 3). Data shown are average ± SD of two independent experiments. Overall, compounds 6a–y showed excellent activity against HeLa and MCF-7 cancer cell lines with GI50 values ranging from 0.013 to >50 μM and 0.003 to >50 μM, respectively. Because many clinically used anticancer agents induce apoptosis in cancer cells, the compounds 6t (R = 3-Br-4-OH-5-MeOC6H2) and 6u (R = 3-Br-4,5-(MeO)2C6H2) were tested for their ability to induce apoptosis in Jurkat (model for human T-cell leukemia) cells using a flow cytometric annexin-V/propidium iodide assay. Although compound 6t is a strong apoptosis inducer at 100 nM, its potency drops as the concentration is reduced to 10 nM. In contrast, compound 6u retains its potency at this low concentration. Moreover, the flow cytometric cell cycle analysis was performed with compounds 6t and 6u against the Jurkat cell line, showing a pronounced cell cycle arrest in the G2/M phase (Table 4). Lastly, the effect of compounds 6t and 6u on in vitro tubulin polymerization was performed to support the proposed antitubulin mechanism of action. In this assay, the microtubule formation is monitored by the increase in fluorescence intensity of the reaction mixture. Paclitaxel exhibited a potent enhancement of the microtubule formation relative to the effect of DMSO control. In contrast, compounds 6t and 6u displayed a potent microtubule destabilizing effect like the known tubulin polymerization inhibitor Podophyllotoxin.
|
||||
|---|---|---|---|---|
| Compound | R | Yield 6 (%) | GI50b (μM) | |
| HeLa | MCF-7 | |||
| a Reaction conditions: aldehyde 1 (0.8 mmol), malononitrile 2 (0.8 mmol), 4-hydroxy-1-methylquinolin-2(1H)-one 5 (0.8 mmol), triethylamine (45 mol%), EtOH (3 mL), reflux, 50 min.b Compound concentration required to reduce HeLa and MCF-7 cells viability by 50% after 48 h treatment relative to 100% DMSO control as assessed with the MTT assay. Data shown are average ± SD of two independent experiments. | ||||
| 6a | C6H5 | 78 | 0.39 ± 0.01 | 3.38 ± 0.53 |
| 6b | 3,4,5-(MeO)3C6H2 | 86 | 0.24 ± 0.02 | 1.0 ± 0.3 |
| 6c | 3-OH-4-MeOC6H3 | 82 | >50 | >50 |
| 6d | 3-MeO-4-OHC6H3 | 84 | 0.63 ± 0.05 | 0.5 ± 0.14 |
| 6e | 3-MeO-4-OH-5-NO2C6H2 | 89 | 0.63 ± 0.02 | 0.71 ± 0.12 |
| 6f | 3-NO2C6H4 | 77 | 0.32 ± 0.02 | 1.1 ± 0.1 |
| 6g | 2-NO2C6H4 | 73 | 5.0 ± 1.4 | 3.5 ± 0.3 |
| 6h | 4-Pyridinyl | 79 | >50 | 32 ± 2 |
| 6i | 3-Pyridinyl | 81 | 2.0 ± 0.1 | >50 |
| 6j | 2-Furanyl | 75 | 22 ± 1 | 35 ± 2 |
| 6k | 5-Methyl-2-furanyl | 78 | 3.9 ± 0.2 | 5.1 ± 0.1 |
| 6l | 2,3-(Cl)2C6H3 | 95 | 1.8 ± 0.2 | >50 |
| 6m | 2,6-(Cl)2C6H3 | 88 | >50 | 36 ± 2 |
| 6n | 3,4-(Cl)2C6H3 | 97 | 0.3 ± 0.03 | 2.5 ± 0.3 |
| 6o | 3-ClC6H4 | 91 | 0.15 ± 0.04 | 2.2 ± 0.2 |
| 6p | 3-FC6H4 | 90 | 0.30 ± 0.01 | 1.0 ± 0.3 |
| 6q | 3-BrC6H4 | 93 | 0.74 ± 0.03 | 0.003 ± 0.001 |
| 6r | 3-Br-4-FC6H3 | 95 | 0.27 ± 0.03 | 0.81 ± 0.08 |
| 6s | 3,5-(Br)2-4-OHC6H2 | 92 | 0.27 ± 0.02 | 0.43 ± 0.01 |
| 6t | 3-Br-4-OH-5-MeOC6H2 | 94 | 0.047 ± 0.01 | 0.39 ± 0.16 |
| 6u | 3-Br-4,5-(MeO)2C6H2 | 95 | 0.014 ± 0.003 | 0.38 ± 0.03 |
| 6v | 3,5-(Br)2C6H3 | 82 | 0.077 ± 0.006 | 0.075 ± 0.007 |
| 6w | 3-Br-4-MeOC6H3 | 64 | 0.41 ± 0.04 | 0.5 ± 0.1 |
| 6x | 5-Bromopyridin-3-yl | 85 | 0.013 ± 0.003 | 0.015 ± 0.008 |
| 6y | 3-Br-4-AcO-5-MeOC6H2 | 84 | 0.18 ± 0.02 | 0.025 ± 0.06 |
| Compound | % Relative DNA contenta | ||
|---|---|---|---|
| G0/G1 | S | G2/M | |
| a % Relative DNA content ± SD after 24 h treatment of Jurkat cells with indicated compounds from two independent experiments. Compounds 6t and 6u are used at 1 μM employing the flow cytometric Vybrant Orange staining assay. | |||
| DMSO | 56 ± 2 | 21 ± 3 | 20 ± 2 |
| 6t | 27 ± 3 | 22 ± 2 | 47 ± 3 |
| 6u | 20 ± 2 | 28 ± 2 | 49 ± 2 |
Importantly, a series of 4,6-diaryl-2-oxo-1,2-dihydropyridine-3-carbonitriles 9a–o were synthesized through a four-component reaction of various (hetero)aromatic aldehydes 1, substituted acetophenones 7, ethyl cyanoacetate 8, and an excess of ammonium acetate in refluxing ethanol for 10 to 12 h (Scheme 1).44 The precipitate obtained was subjected either to re-crystallization from a mixture of DMF/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v), or to column chromatography on silica gel to afford products 9a–o in 62–92% yields. All compounds were evaluated for their in vitro anticancer activity against the human colon adenocarcinoma cell line HT29 using the MTT method. However, only the compounds 9b (R = 3-ClC6H4, R1 = C6H5) and 9d (R = 2-EtOC6H4, R1 = 3-thiophenyl) showed a significant anticancer activity with IC50 values of 2.1 and 1.2 μM, respectively.
:10, v/v), or to column chromatography on silica gel to afford products 9a–o in 62–92% yields. All compounds were evaluated for their in vitro anticancer activity against the human colon adenocarcinoma cell line HT29 using the MTT method. However, only the compounds 9b (R = 3-ClC6H4, R1 = C6H5) and 9d (R = 2-EtOC6H4, R1 = 3-thiophenyl) showed a significant anticancer activity with IC50 values of 2.1 and 1.2 μM, respectively.
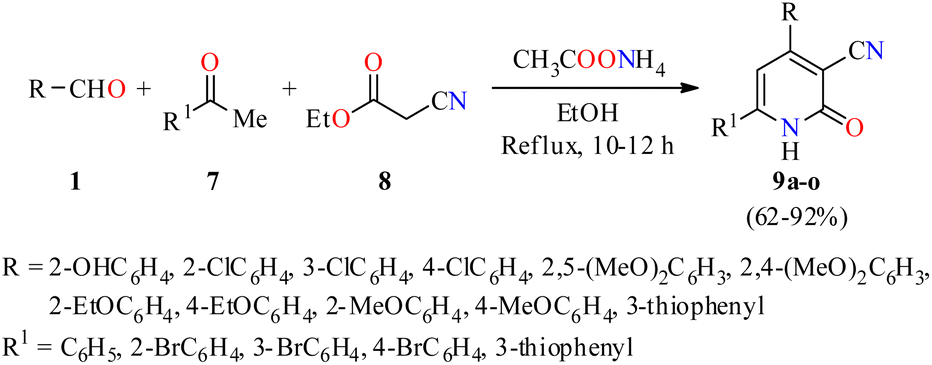 | ||
| Scheme 1 Four-component synthesis and anticancer evaluation of 4,6-diaryl-2-oxo-1,2-dihydropyridine-3-carbonitriles 9. | ||
Later, a molecular docking simulation was performed to investigate the interactions of compound 9d with Pim-1 kinase (PDB ID code: 2OBJ) (Fig. 3). It was found that the oxygen atom of the carbonyl group and NH moiety form two hydrogen bonds with Lys67. Moreover, the nitrogen atom of the cyano group forms a hydrogen bond with Ser189, while the oxygen atom of the ethoxy group is involved in extra hydrogen bonding through a water molecule with Asp167, Asp186, and Ser189 residues.
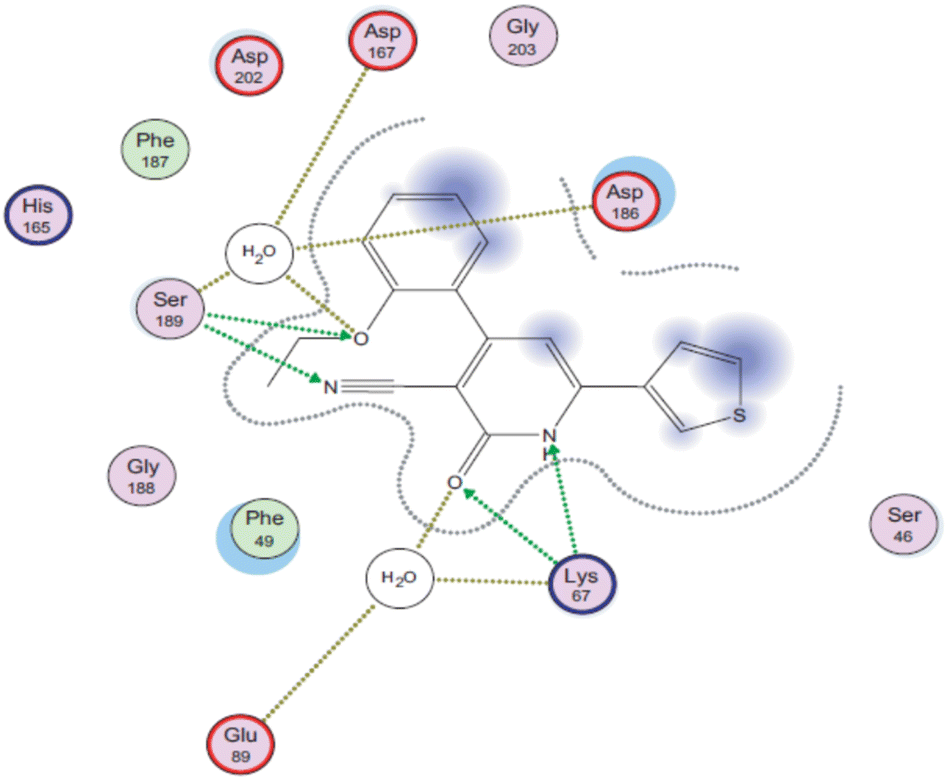 | ||
| Fig. 3 2D interactions of compound 9d with Pim-1 kinase (PDB ID code: 2OBJ). Reproduced with permission from ref. 44. Copyright Elsevier Inc., 2022. | ||
To date, few reports are available in the literature regarding the eco-friendly synthesis of chromene-containing 2(1H)-pyridones. In this regard, the solventless synthesis of 4-aryl-6-[benzo[f]coumarin-3-yl]-3-cyano-2(1H)-pyridones 11a–d was developed through a four-component reaction of aromatic aldehydes 1, malononitrile 2, and acetyl-3H-benzo[f]chromen-3-one 10 in the presence of sodium hydroxide at 75 °C for 45 min (Table 5).45 Later, the reaction mixture was poured into water, and then washed with water, filtered, dried, and recrystallized from EtOH to furnish products 11a–d in 71–87% yields. Some highlights of this protocol were associated with the absence of solvent, shorter reaction time, and a simple workup procedure. Lastly, 2(1H)-pyridone derivatives 11a–c were evaluated for their in vitro anticancer activity against liver HepG2 and breast MCF-7 cell lines using the MTT method in the presence of 5-fluorouracil as a reference drug. In summary, compounds 11a–c displayed a moderate cytotoxic effect with IC50 values in the range of 53.6–77.6 μM and 56.3–78.3 μM for HepG2 and MCF-7 cell lines, respectively, when compared to 5-fluorouracil (IC50 = 9.30 and 13.1 μM, respectively).
|
||||
|---|---|---|---|---|
| Compound | R | Yield 11 (%) | IC50c (μM) | |
| HePG2 | MCF-7 | |||
| a Reaction conditions: aldehyde 1 (1 mmol), malononitrile 2 (1.5 mmol), 2-acetyl-3H-benzo[f]chromen-3-one 10 (1 mmol), NaOH (1.5 mmol), 75 °C, 45 min.b Standard drug for the study.c Compound concentration required to reduce HePG2 and MCF-7 cells viability by 50% after 48 h treatment relative to 100% DMSO control as assessed with the MTT assay. | ||||
| 11a | C6H5 | 87 | 77.6 | 78.3 |
| 11b | 4-OHC6H4 | 85 | 58.1 | 59.9 |
| 11c | 4-MeOC6H4 | 79 | 53.6 | 56.3 |
| 11d | 4-Me2NC6H4 | 71 | — | — |
| 5-Fluorouracilb | — | — | 9.30 | 13.1 |
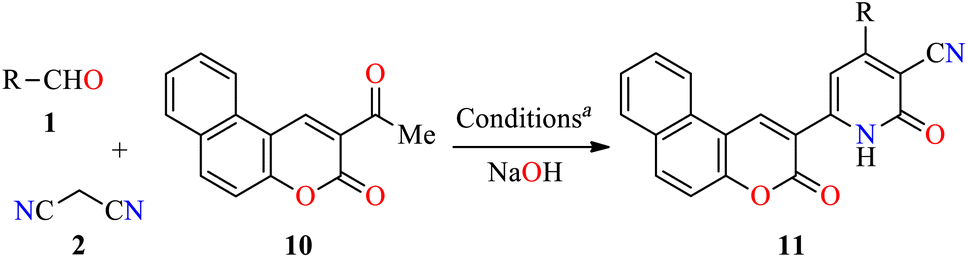
Importantly, the presence of electron donating groups such as hydroxy or methoxy substituents attached to the aromatic ring play an important role in the molecular interactions and have a profound effect on antiproliferative activity. In this regard, 3,4,6-triaryl-2(1H)-pyridones 13a–p were synthesized in 58–82% yields through a one-pot three-component reaction of aromatic aldehydes 1, substituted acetophenones 7, and phenyl acetamides 12 in the presence of sodium hydride in DMSO at 130 °C (Scheme 2).46 All synthesized compounds 13a–p were evaluated for their in vitro anticancer activity against MCF-7 and MDA-MB-231 breast cancer cell lines using the MTT method in the presence of Nolvadex (tamoxifen citrate) as a standard drug. Data shown are average ± SD of two independent experiments. Remarkably, compound 13p (R1 = 4-(2-(piperidin-1-yl)ethoxy, (R2 = R3 = MeO, R4 = R5 = R6 = R7 = H) showed the most significant cytotoxic activity against MCF-7 and MDA-MB-231 cell lines with IC50 values of 35.7 ± 5.8 μM and 16.8 ± 2.3 μM, respectively, when compared to Nolvadex (IC50 = 8.8 ± 1.2 μM and 9.0 ± 1.3 μM, respectively). Cell cycle analysis showed that compound 13p induced arrest of cells in the G1-phase and reduction in the S-phase cells in a dose-dependent manner. Moreover, compound 13p decreased ROS generation in a dose-dependent manner and induced ROS-independent mitochondrial-mediated apoptosis in MDA-MB-231 cells. Lastly, the apoptotic activity of compound 13p was also confirmed by DNA fragmentation and by expression of pro-apoptotic genes such as BAD, BAK, and BimL.
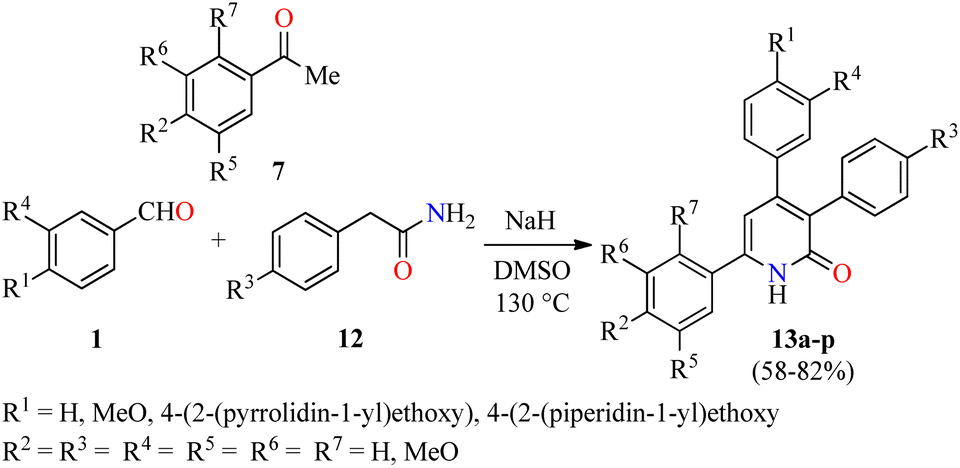 | ||
| Scheme 2 One-pot three-component synthesis and anticancer evaluation of 3,4,6-triaryl-2(1H)-pyridones 13. | ||
1,3-Dipolar cycloaddition reaction is one of the efficient ways for the regio- and stereoselective construction of five-membered heterocycles.47 In this sense, regioselective three-component synthesis of 4-hydroxyquinolin-2(1H)-one grafted spiropyrrolidines 17 has been accomplished through a 1,3-dipolar cycloaddition reaction of various azomethine ylides derived from isatin derivatives 15 and sarcosine 16 with 4-hydroxyquinolin-2(1H)-ones 14 as dipolarophile using a mixture of MeOH/H2O (2:1, v/v) at reflux for 2.5–3.0 h (Table 6).48 The reaction mixture was cooled at room temperature, and the products were filtered and recrystallized in a mixture of MeOH/DMF (7:3, v/v) to afford products 17a–g in 46–92% yields. Although the detailed mechanism of the reaction is not fully established, the regioselective formation of spiropyrrolidines 17 could involve a decarboxylative condensation of isatins 15 with sarcosine 16 to give azomethine ylides I, which undergo a 1,3-dipolar cycloaddition reaction with dipolarophiles 14. The regioselectivity in the product formation could be explained by considering the secondary orbital interaction (SOI) between the orbital of the carbonyl group of dipolarophiles 14 and those of azomethine ylides I. Moreover, the regio- and stereochemical outcome of the 1,3-dipolar cycloaddition reaction is ascertained by X-ray crystallography and spectroscopic techniques. The compounds 17 were screened for their in vitro anticancer activity against HCT-116 (colon cancer) and HeLa (cervical cancer) cell lines using the MTT method in the presence of Doxorubicin as a standard drug. Data shown are average ± SD of two independent experiments. Overall, compounds 17d (R = R1 = H, R2 = NO2) and 17c (R = R2 = H, R1 = Me) showed the best anticancer activity against the HCT-116 cell line with IC50 values of 9.3 and 9.6 μM, respectively, when compared to doxorubicin (IC50 = 2.4 μM). Furthermore, compound 17b (R = Cl, R1 = R2 = H) showed a good anticancer activity against the HeLa cell line with an IC50 value of 10.7 μM, in comparison to Doxorubicin (IC50 = 1.8 μM).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | R2 | Yield 17 (%) | IC50c (μM) | |
| HCT-116 | HeLa | |||||
| a Reaction conditions: 4-hydroxyquinoline 14 (1.5 mmol), isatine 15 (1.5 mmol), sarcosine 16 (1.5 mmol), MeOH/H2O (2:1, v/v), reflux, 2.5–3.0 h.b Standard drug for the study.c Compound concentration required to reduce HCT-116 and HeLa cells viability by 50% after 48 h treatment relative to 100% DMSO control as assessed with the MTT assay. Data shown are average ± SD of two independent experiments. |
||||||
| 17a | H | H | H | 92 | 83.2 ± 0.42 | 15.1 ± 0.18 |
| 17b | Cl | H | H | 88 | — | 10.7 ± 0.15 |
| 17c | H | Me | H | 62 | 9.6 ± 0.14 | 28.4 ± 0.30 |
| 17d | H | H | NO2 | 54 | 9.3 ± 0.12 | 16.7 ± 0.14 |
| 17e | H | Me | NO2 | 46 | 10.9 ± 0.18 | 25.5 ± 0.28 |
| 17f | H | Cl | H | 87 | 12.0 ± 0.20 | 34.6 ± 0.17 |
| 17g | H | Br | H | 82 | — | — |
| Doxorubicinb | — | — | — | — | 2.4 ± 0.10 | 1.8 ± 0.05 |
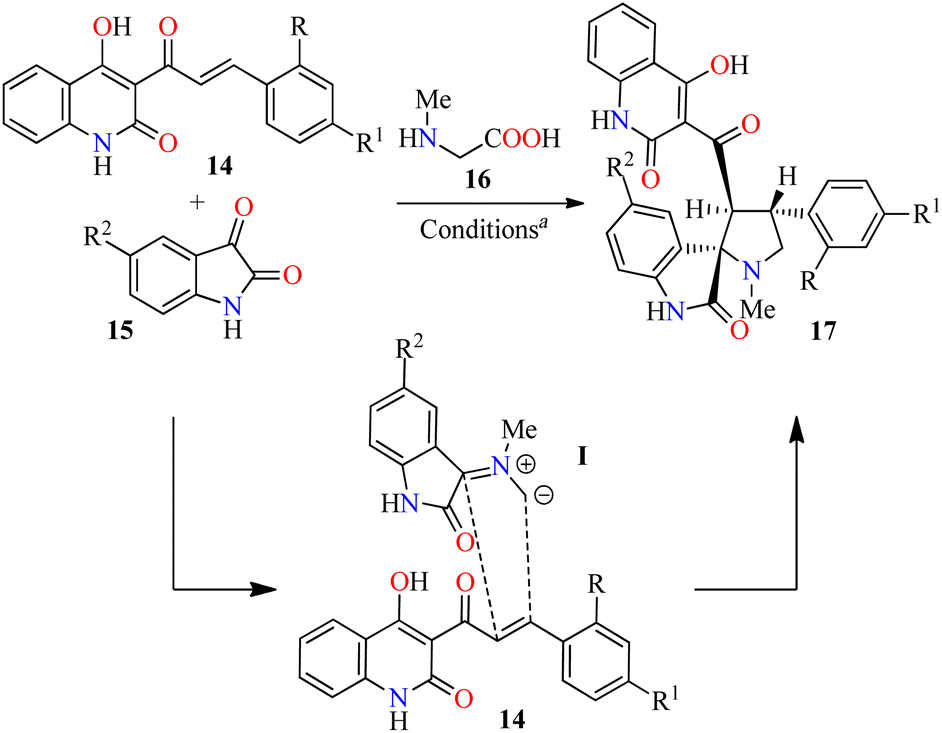
Alternatively, Tangella and co-workers described the regio- and diastereoselective synthesis of trans-1,2-dihydrobenzofuro[3,2-c]quinolinones 20 in 77–90% yields via a three-component reaction of (hetero)aromatic aldehydes 1, α-bromoacetophenones 18, and 4-hydroxybenzo[h]quinolin-2(1H)-ones 19 in pyridine at 100 °C for 6 h (Scheme 3A).49 The yields were found to be higher in case of substrates with electron-donating groups (i.e. R = 3,4,5-(MeO)3C6H2, R1 = 4-MeOC6H4, 90%) compared to substrates with electron-withdrawing groups (i.e. R = 4-NO2C6H4, R1 = C6H5, 77%). Moreover, heteroaryl aldehydes (R = 3-pyridyl and 2-thiophenyl) were found to be well tolerated and the corresponding products 20h–j were obtained in 81–87% yields. The plausible reaction mechanism is briefly outlined in Scheme 3B.49 The first step involves the formation of arylidinebenzo[h]quinoline-2,4-diones II through a pyridine-catalyzed Knoevenagel condensation of aldehydes 1 with 4-hydroxycoumarin derivatives 19. The second step proceeds via a Michael addition of pyridinium ylides III with Knoevenagel adducts II to afford keto tautomers IV, which are in tautomeric equilibrium with enol tautomers V. Lastly, intermediates V undergo a diastereoselective intramolecular cyclization to give desired products 20. Importantly, this protocol selectively generated trans-2,3-dihydrofuran derivatives due to the steric hindrance of the large substituents at C-2 (aryl) and C-3 (aryl or hetaryl), which are opposing one another in the transition state and the carbanion intermediate. In addition, pyridine plays different roles such as base, solvent, and a source of pyridinium ylides.
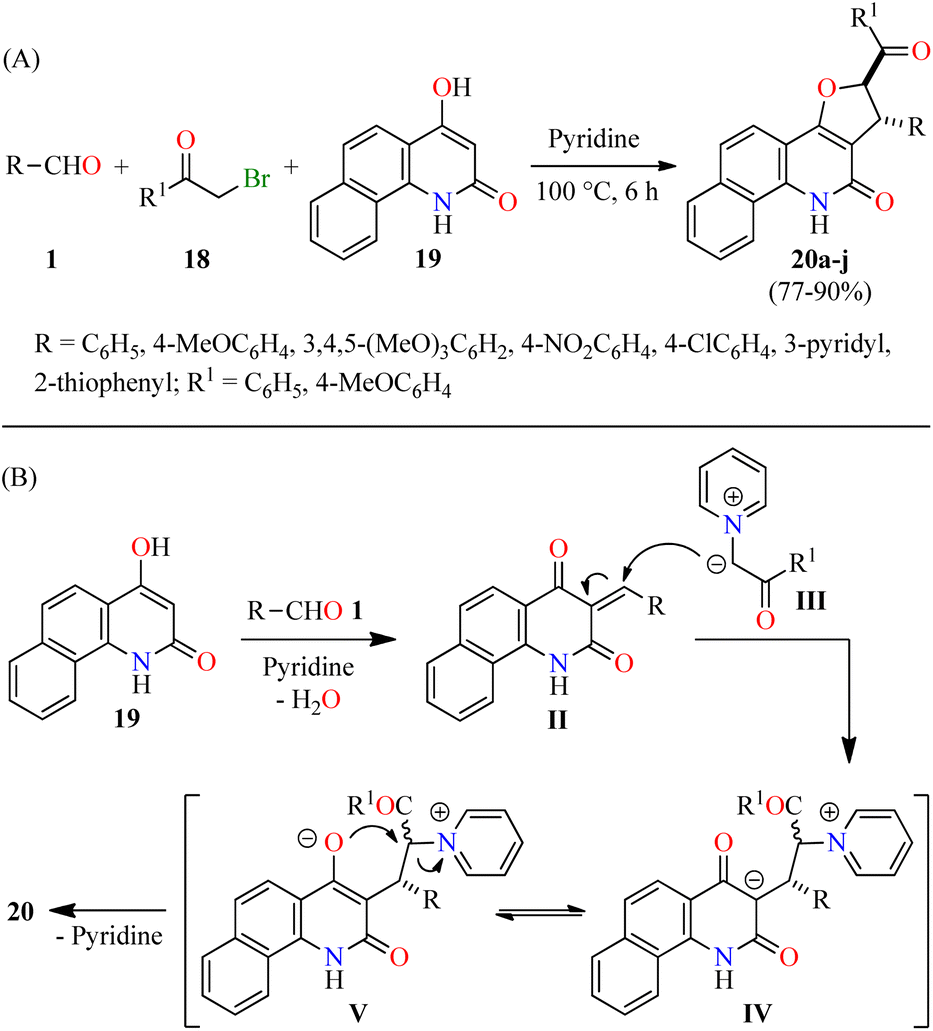 | ||
| Scheme 3 (A) Three-component synthesis and anticancer evaluation of trans-2,3-dihydrofuran derivatives 20. (B) Plausible reaction mechanism for the formation of 20. | ||
All compounds 20a–j were evaluated for their in vitro cytotoxic activity against A549 (lung cancer), DU-145 (prostate cancer), HeLa (cervical cancer), MCF-7 (breast cancer), and normal mouse fibroblast (NIH 3T3) cell lines using the MTT colorimetric assay in the presence of Doxorubicin as a standard drug.49 The screening results revealed that all synthesized compounds showed selective cytotoxicity to cancer cells (IC50 values ranging from 2.14 to 17.94 μM) with a very weak effect on normal cells (IC50 values ranging from 70.43 to >100 μM). Most of the compounds showed prominent activity against the MCF-7 cell line compared to other tested cancer cell lines. In particular, compounds 20c (R = R1 = 4-MeOC6H4) and 20i (R = 2-thiophenyl, R1 = 4-MeOC6H4) displayed the best anticancer activity against the MCF-7 cell line with IC50 values of 2.56 and 2.14 μM, respectively, when compared to Doxorubicin (IC50 = 1.43 μM). Also, compound 20c displayed high activity against A549 and DU-145 cell lines with IC50 values of 6.05 and 3.28 μM, respectively, in comparison to Doxorubicin (IC50 = 1.21 and 2.14 μM, respectively). In addition, compounds 20d (R = 3,4,5-(MeO)3C6H2, R1 = C6H5) and 20f (R = 4-NO2C6H4, R1 = C6H5) exerted significant activity against the HeLa cell line with IC50 values of 4.89 and 4.35 μM, respectively (IC50 = 1.63 μM for Doxorubicin).
A literature survey revealed that 4,6-diaryl-3-cyano-2-pyridones displayed a potent anticancer activity by inhibition of the Pim-1 kinase.50–52 In this regard, a novel series of substituted N-(2-ethylphenyl)-2-oxo-pyridine-3-carbonitriles 22 were synthesized in 25–55% yields through a piperidine-catalyzed three-component reaction of aromatic aldehydes 1, substituted acetophenones 7, and 2-cyano-N-(2-ethylphenyl)acetamide 21a in refluxing ethanol for 5–9 h (Scheme 4).53 This multicomponent protocol was extended to active methylene compounds such as diethyl malonate 23 and malononitrile 2 to afford fully substituted 2-pyridone derivatives 24 and 25, respectively, in moderate yields. The anticancer activity of synthesized compounds was evaluated by the National Cancer Institute (NCI), which selected 14 compounds for one-dose screening. Among them, compound 25g (R = 3,4-(MeO)2C6H3) was selected for five-dose screening, which displayed a potent anticancer activity with GI50 values ranging from 3.15 to 4.09 μM against leukemia, CNS, colon, and ovarian cancer cell panels, and moderate cytotoxicity against the other cancer cell lines using the MTT method. Interestingly, cell cycle analysis against the OVCAR-3 (ovarian cancer) cell line showed that compound 25g induced apoptosis and arrested the cell cycle at the G2/M phase, which was accompanied by a reduction in the S-phase cells (Table 7).
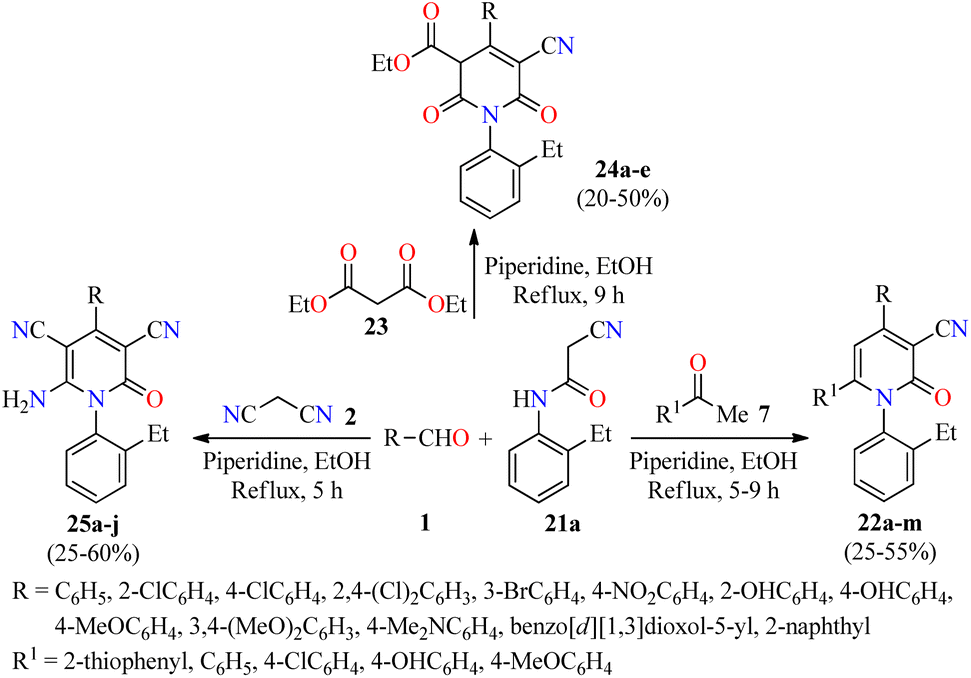 | ||
| Scheme 4 Three-component synthesis and anticancer evaluation of highly substituted 2-pyridone derivatives 22, 24, and 25. | ||
On the other hand, the provirus integration in Maloney (Pim) kinases are attractive drug targets because they are involved in cancer-specific pathways and upregulated in different hematological and solid cancers.54 Among them, Pim-1 kinase has a significant role in the regulation of the cell cycle through phosphorylation of Cdc25 phosphatases and cell cycle inhibitors.54 As a result, compound 25g was screened against Pim-1, Pim-2, and Pim-3 kinases using quercetin as a reference drug.53 The results revealed more selectivity towards Pim-1 kinase (IC50 = 1.89 μM) with a percentage inhibition of 64.6%, when compared to Quercetin (IC50 = 1.22 μM). Lastly, molecular docking studies were performed to investigate the interactions of compound 25g with Pim-1 kinase (PDB ID code: 2OBJ) (Fig. 4). The nitrogen atom of cyano groups in positions 3 and 5 forms two hydrogen bonds with Asn172 and Lys67, respectively. Also, the oxygen atom of the methoxy group in position 3 forms a hydrogen bond with Asp186. Besides, 4-phenyl and 2-ethyl phenyl rings interact with lle185 and Gly48 through an arene-H bonding, respectively.
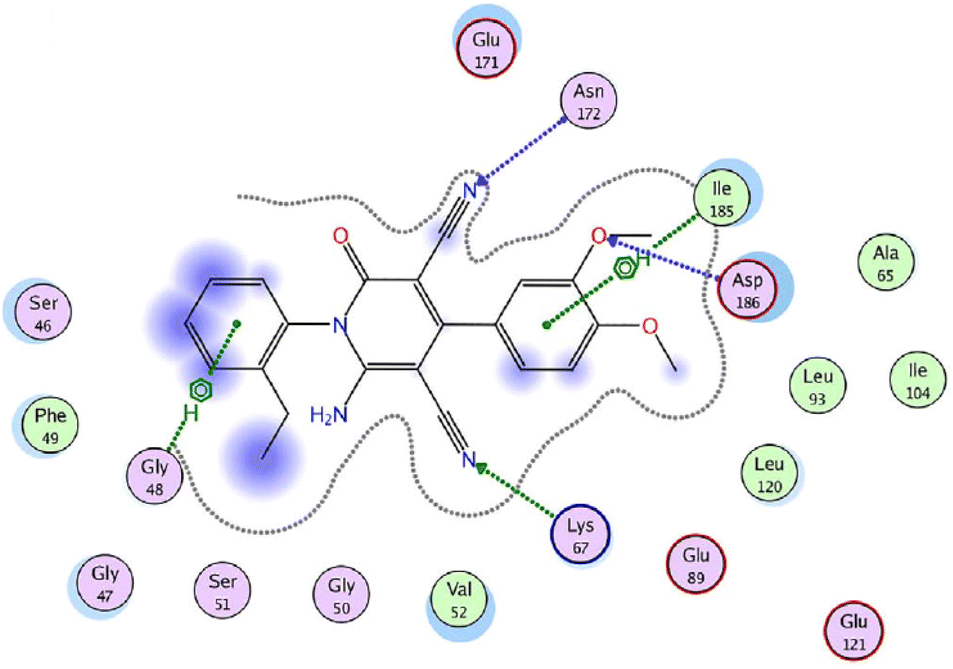 | ||
| Fig. 4 2D interactions of compound 25g with Pim-1 kinase (PDB ID code: 2OBJ). Reproduced with permission from ref. 53. Copyright John Wiley & Sons Inc., 2022. | ||
Recently, a series of 4,6-diaryl-3-cyano-2(1H)-pyridones 26a–n were synthesized through a four-component reaction of various (hetero)aromatic aldehydes 1, substituted acetophenones 7, ethyl cyanoacetate 8, and an excess of ammonium acetate in refluxing n-butanol for 1–3 h (Scheme 5).55 The precipitate was filtered, dried, and recrystallized from the appropriate solvent such as EtOH, AcOH, DMF, EtOH/AcOH (1:1, v/v), and DMF/AcOH (1:1, v/v) to afford products 26a–n in 35–70% yields. Selected compounds were screened for their in vitro anticancer activity against HepG2 (liver cancer) and THLE2 (normal liver cells) using the MTT assay in the presence of 5-fluorouracil as a reference drug. Data are expressed as mean ± SD of three independent experiments. Interestingly, the compound 26n (R = 4-MeOC6H4, R1 = 3-H2NC6H4) showed good activity against HepG2 and THLE2 cells with IC50 values of 19.2 ± 1.01 μM and 44.11 ± 0.89 μM, respectively, in comparison to 5-fluorouracil (IC50 = 6.98 μM for HepG2 cancer cell line). Moreover, the compound 26l (R = 3-ClC6H4, R1 = 4-ClC6H4) displayed a moderate activity against HepG2 and THLE2 cells with IC50 values of 43.7 ± 1.23 μM and 48.2 ± 0.98 μM, respectively. Overall, tested compounds were safe against normal liver cells with IC50 values ranging from 44.1 to ≥ 50 μM.
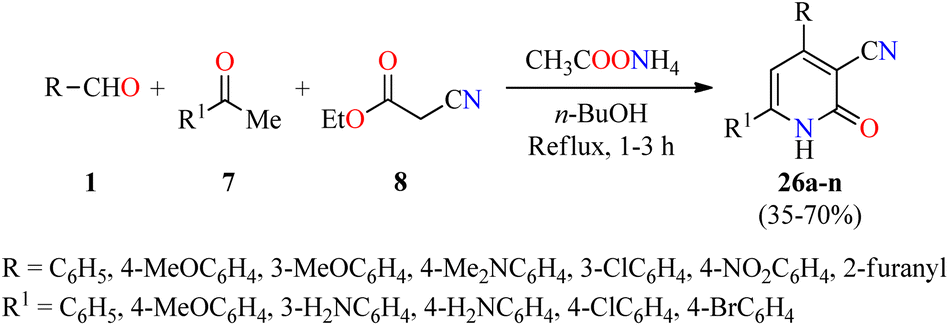 | ||
| Scheme 5 Four-component synthesis and anticancer evaluation of 2(1H)-pyridone derivatives 26. | ||
2.2. Antibacterial activity
Over the past few decades antibacterial drugs (i.e. Amphotericin B, Fluconazole, Penicillin, Chloramphenicol, among others) become less effective or even ineffective due to their emerging antimicrobial resistance. In regards to this case, innovative synthetic strategies will be required to develop new molecules with potential antibacterial properties against a wide range of bacteria.56,57 In this regard, an efficient three-component reaction of 2-oxo-1,2-dihydroquinoline-3-carbaldehydes 27, cyclohexane-1,3-dione derivatives 28, and (thio)urea 29a (R2 = H) catalyzed by La(OTf)3 (10 mol%) in ethanol at room temperature for 1.5–2.0 h afforded N-allylquinolone derivatives 30a–p (Scheme 6).58 This multicomponent approach was successfully extended to N-phenylthiourea 29b (R2 = C6H5) under similar reaction conditions for 3.0–3.5 h to give N-allylquinolone derivatives 30q–x. After the completion of the reaction, the solid was filtered, washed with ethanol, and purified by leaching in an equal volume ratio of chloroform and methanol to obtain products 30 in good yields. Subsequently, compounds 30a–x were screened against Gram-negative (Escherichia coli and Pseudomonas aeruginosa) and Gram-positive (Staphylococcus aureus and Streptococcus pyogenes) bacterial strains by using the agar well diffusion method in the presence of ciprofloxacin as a standard drug. Interestingly, the compound 30h (R = R1 = Me, R2 = H, X = S) showed the highest activity against Pseudomonas aeruginosa with a MIC value of 20 μg mL−1, while compounds 30r (R = H, R1 = Me, R2 = C6H5, X = S) and 30x (R = Cl, R1 = Me, R2 = C6H5, X = S) were the most active against Escherichia coli with a MIC value of 50 μg mL−1, in comparison to Ciprofloxacin (MIC = 25 μg mL−1 for both strains). Moreover, compounds 30f (R = R1 = Me, R2 = H, X = O) and 30w (R = Cl, R1 = H, R2 = C6H5, X = S) displayed a good activity against Staphylococcus aureus with a MIC value of 62.5 μg mL−1, when compared to ciprofloxacin (MIC = 50 μg mL−1). Lastly, compounds 30e (R = Me, R1 = R2 = H, X = O), 30g (R = Me, R1 = R2 = H, X = S), 30m (R = Cl, R1 = R2 = H, X = O), and 30p (R = Cl, R1 = Me, R2 = H, X = S) exerted a moderate MIC value of 100 μg mL−1 against Streptococcus pyogenes, in comparison to Ciprofloxacin (MIC = 50 μg mL−1).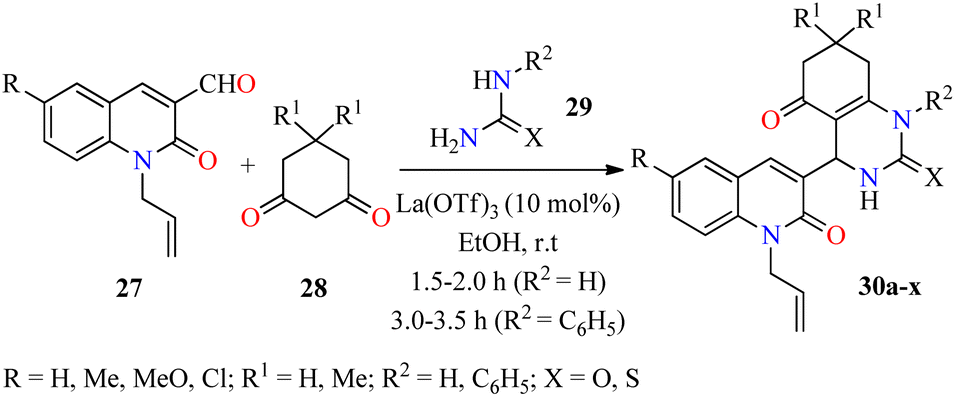 | ||
| Scheme 6 La(OTf)3-catalyzed three-component synthesis and antibacterial evaluation of N-allylquinolone derivatives 30. | ||
In a similar way to previously mentioned,58 the same authors reported the solventless synthesis of N-allylquinolone derivatives 32 in 80–87% yields through a solventless three-component reaction of N-allyl quinolone-3-carbaldehydes 27, cyclohexane-1,3-dione derivatives 28, and 2-hydroxy-1,4-naphthoquinone 31 catalyzed by ceric ammonium nitrate (5 mol%) in a microwave oven at 420 W for 6 min (Scheme 7A).59 This multicomponent approach was successfully extended to malononitrile/iso-propylcyanoacetate 2/33 under similar reaction conditions at 420 W for 4 min to give N-allylquinolone derivatives 34 in 81–88% yields. This methodology was distinguished by its high yields, short reaction times, and solvent-free conditions. The plausible mechanistic pathways for products 32 and 34 are illustrated in Scheme 7B and C, respectively. It was supposed that the reaction occurred via the ortho-quinone methides VI, which are formed by the nucleophilic addition of 2-hydroxy-1,4-naphthoquinone 31 to quinolone-3-carbaldehydes 27 catalyzed with CAN. Subsequent attack of cyclic 1,3-dicarbonyl compounds 28 (enol form) to the ortho-quinone methides VI gives intermediates VII, which undergo 6-exo-trig intramolecular cyclization/elimination sequence to afford compounds 32. Alternatively, the Michael addition of malononitrile/iso-propylcyanoacetate 2/33 to the ortho-quinone methides VI gives intermediates VIII, which undergo 6-exo-dig intramolecular cyclization/tautomerization sequence to furnish compounds 34. The rapid formation of compounds 34 may be due to the nucleophilic attack of the hydroxy group on the highly electrophilic carbon atom of the nitrile group (VIII) compared to the carbon atom of the carbonyl group (VII).
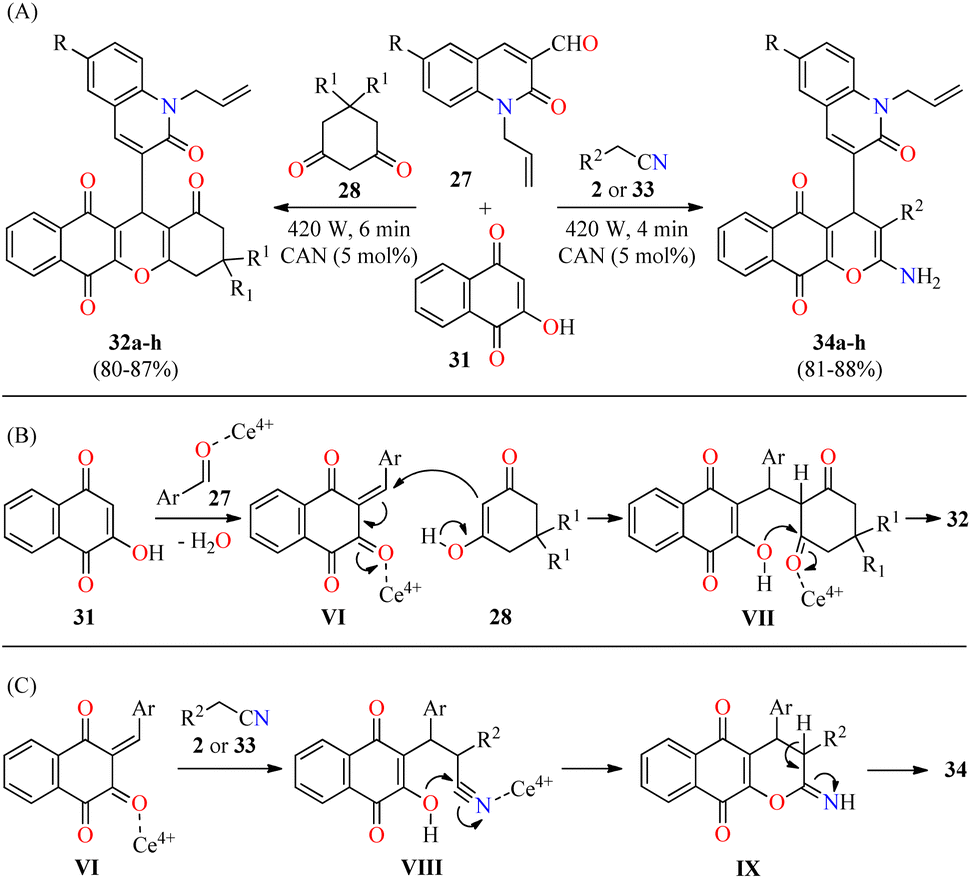 | ||
| Scheme 7 (A) CAN-catalyzed three-component synthesis of N-allylquinolone derivatives 32 and 34 and their in vitro antibacterial activity against four bacterial strains. (B) Plausible reaction mechanism for the formation of 32. (C) Plausible reaction mechanism for the formation of 34. | ||
The compounds 32a–h and 34a–h were screened for their antibacterial activity against two Gram-positive (Staphylococcus aureus and Streptococcus pyogenes) and two Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacterial strains using Ciprofloxacin as a standard drug. The compounds 32 and 34 showed moderate to excellent antibacterial activity with MIC values in the range of 20 to 500 μg mL−1, in comparison to Ciprofloxacin (MIC = 25–50 μg mL−1). In particular, compounds 34a (R = H, R2 = CN) and 32d (R = Cl, R1 = H) showed the highest activity against Staphylococcus aureus and Streptococcus pyogenes with MIC values of 62.5 and 50 μg mL−1, respectively. Remarkably, compounds 34f (R = Me, R2 = COO-i-Pr) and 32c (R = MeO, R1 = H) displayed an excellent potency against Escherichia coli and Pseudomonas aeruginosa with MIC values of 25 and 20 μg mL−1, respectively, which are equipotent to Ciprofloxacin (MIC = 25 μg mL−1 for both strains).
The 4-aryl-6-[benzo[f]coumarin-3-yl]-3-cyano-2(1H)-pyridones 11a–d obtained by a four-component process and discussed in Section 2.1. Anticancer activity (Table 5),45 were also screened for their antibacterial activity against two Gram-positive (Bacillus subtilis and Enterococcus faecalis) and one Gram-negative (Salmonella typhimurium) bacterial strains by using the disk diffusion method in the presence of Ampicillin and Chloramphenicol as standard drugs (Table 8). The compounds showed a diameter of growth of inhibition zone in the range of 10–13 mm, 9–11 mm, and 8–12 mm against Bacillus subtilis, Enterococcus faecalis, and Salmonella typhimurium, respectively, in comparison to Ampicillin (13–25 mm) and Chloramphenicol (18–24 mm). Overall, the Gram-negative bacterium was more susceptible to the synthesized compounds than Gram-positive bacteria. Remarkably, compounds 11d (R = 4-Me2NC6H4) and 11c (R = 4-MeOC6H4) showed the highest activity against Salmonella typhimurium with a diameter of growth of inhibition zone of 11 and 12 mm, respectively, when compared to Ampicillin (13 mm) and Chloramphenicol (18 mm).
Interestingly, a series of coumarin–pyridone hybrids 37 were obtained in 79–93% yields through a one-pot three-component reaction of (E)-3-(3-arylacryloyl)-2H-chromen-2-ones 35, ethyl 2-nitroacetate 36, and an excess of ammonium acetate in refluxing n-BuOH for 3–10 h (Scheme 8).60 The reaction mixture was cooled to room temperature and the precipitate was filtered, washed, and recrystallized from EtOH to obtain the product 37. This approach shows a broad substrate scope and excellent functional-group tolerance with diverse electron-rich and electron-deficient substituents. A plausible mechanism proposed by the authors is shown in Scheme 9. Initially, α,β-unsaturated ketones 35 reacted with ethyl 2-nitroacetate 36 through a Michael addition/condensation sequence mediated by ammonium acetate to give enamine-type intermediates 39, which subsequently underwent an intramolecular cyclization to generate 3,4-dihydropyridin-2(1H)-ones 40. Lastly, the elimination of HNO2 afforded coumarin–pyridone hybrids 37. The compounds 37a–n were screened for their antibacterial activity against three Gram-positive (Bacillus subtilis, Klebsiella pneumoniae, and Staphylococcus aureus) and two Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacterial strains using Ciprofloxacin as a standard drug. It was found that compounds 37d (R = 4-MeOC6H4), 37i (R = 3-NO2C6H4), 37k (R = 3-Br-4-MeOC6H3), and 37o (R = 2-pyridyl) exhibited a moderate activity with MIC values in the range of 60–80 μg mL−1, in comparison to Ciprofloxacin (MIC = 0.03–8.0 μg mL−1).
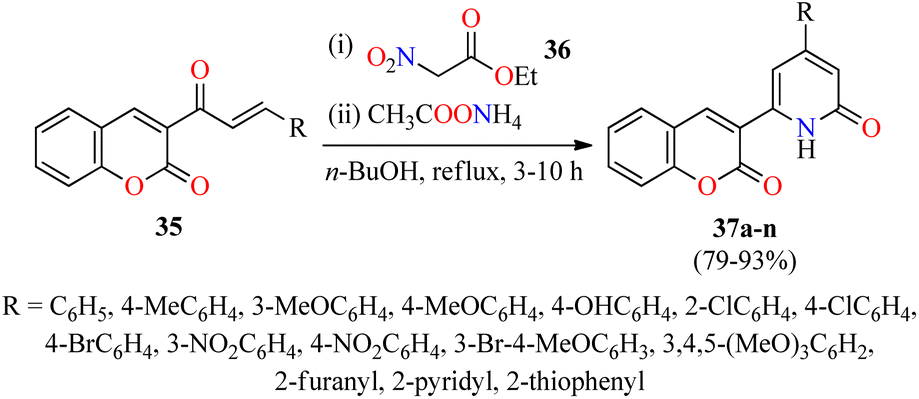 | ||
| Scheme 8 One-pot three-component synthesis of coumarin-pyridone hybrids 37 for evaluation of their antibacterial activity. | ||
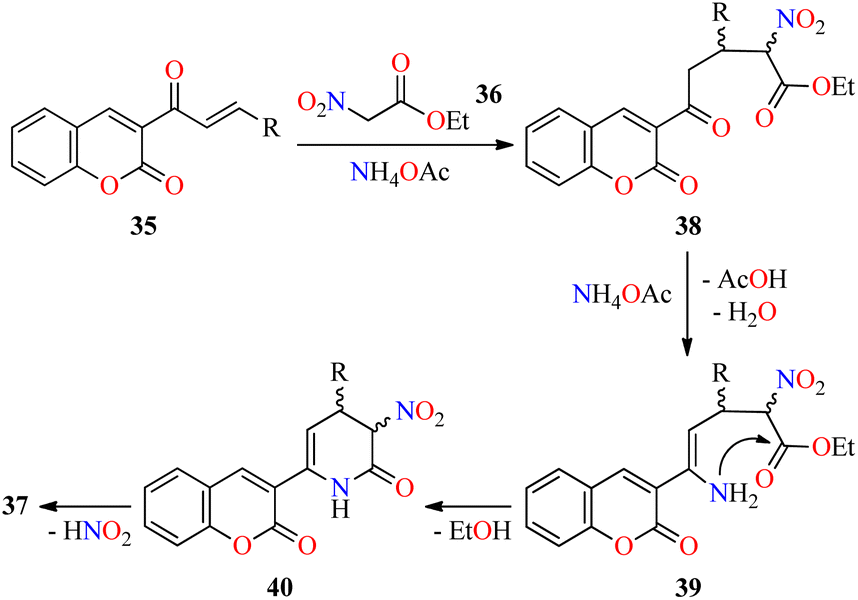 | ||
| Scheme 9 Plausible mechanism for the synthesis of coumarin–pyridone hybrids 37. | ||
Alternatively, Pandit and co-workers described the piperidine-catalyzed synthesis of fully substituted 2(1H)-pyridone derivatives 43 via a three-component reaction of (hetero)aromatic aldehydes 1, acetoacetanilide 41, and cyanoacetamide 42 in refluxing ethanol for 7–8 h (Scheme 10).61 The reaction mixture was cooled in a refrigerator overnight and the precipitate was filtered, washed with EtOH, and dried to obtain the products 43 in 89–93% yields. Selected compounds were screened for their antibacterial activity against two Gram-positive (Bacillus subtilis and Staphylococcus aureus) and two Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacterial strains. Overall, the compounds 43c (R = 2-BrC6H4) and 43q (R = 2-furanyl) showed a diameter of growth of inhibition zone in the range of 3.50–4.25 mm and 1.50–3.75 mm for Gram-positive and Gram-negative bacterial strains, respectively.
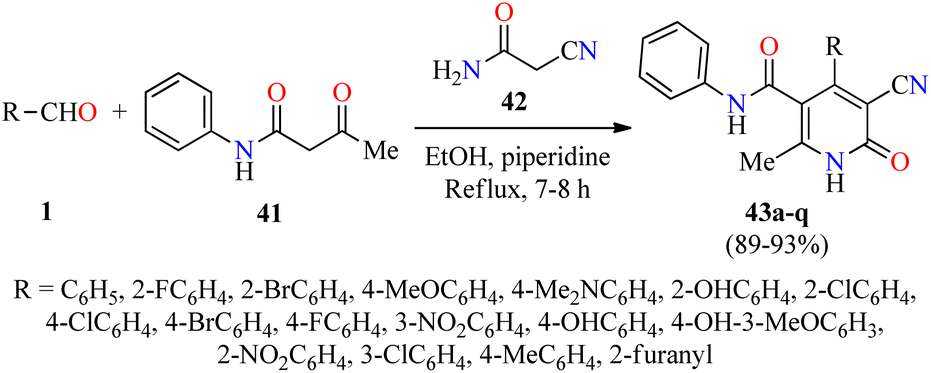 | ||
| Scheme 10 Piperidine-catalyzed three-component synthesis of 2(1H)-pyridone derivatives 43 for evaluation of their antibacterial activity. | ||
Quinolone analogs have a broad spectrum of antimicrobial properties. For instance, Krishna and colleagues described the synthesis of quinolone–pyridine hybrids 45 in 63–85% yields through a time-efficient four-component reaction of (hetero)aromatic aldehydes 1, malononitrile 2, 3-acetyl-4-hydroxy-2(1H)-quinolone 44, and an excess of ammonium acetate catalyzed by NbCl5 (15 mol%) in DMF at 75 °C for 30 min (Scheme 11).62 The compounds 45a–p were screened for their antibacterial activity against two Gram-positive (Staphylococcus aureus and Bacillus cereus) and three Gram-negative (Klebsiella planticola, Escherichia coli, and Pseudomonas aeruginosa) bacterial strains using Ciprofloxacin hydrochloride as a standard drug. It was found that compound 45e (R = 4-MeOC6H4) exhibited an excellent activity against Staphylococcus aureus and Bacillus cereus with a MIC value of 2.34 μg mL−1, in comparison to Ciprofloxacin hydrochloride (MIC = 4.68 and 2.34 μg mL−1, respectively). In case of Klebsiella planticola, compounds 45c (R = 4-ClC6H4), 45h (R = 2-ClC6H4), and 45p (R = 2-furanyl) showed the same activity than standard drug with a MIC value of 4.68 μg mL−1. Lastly, compounds 45e (R = 4-MeOC6H4), 45f (R = 4-i-BuC6H4), and 45p (R = 2-furanyl) exhibited higher antibacterial activity than standard drug against Escherichia coli with a MIC value of 2.34 μg mL−1, while the compound 45i (R = 2-BrC6H4) displayed a good potency against Pseudomonas aeruginosa with a MIC value of 4.68 μg mL−1, in comparison to Ciprofloxacin hydrochloride (MIC = 4.68 and 2.34 μg mL−1, respectively).
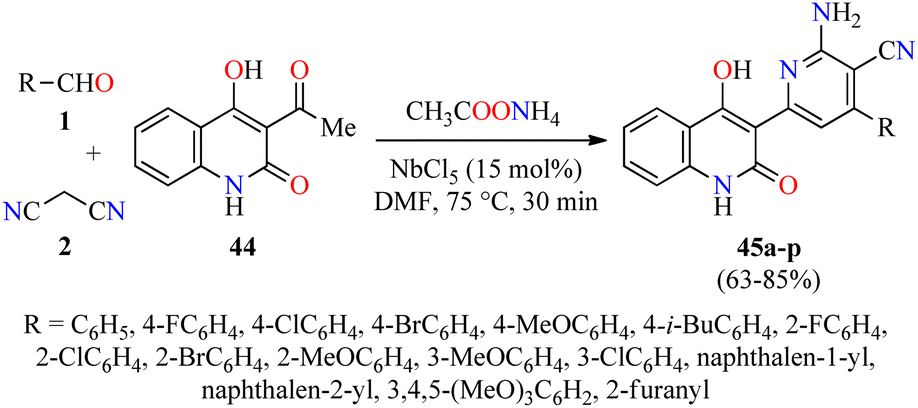 | ||
| Scheme 11 NbCl5-catalyzed four-component synthesis of quinolone–pyridine hybrids 45 for evaluation of their antibacterial activity. | ||
A year later, the same authors employed a similar strategy for the synthesis of quinolone–pyrimidine hybrids 47 by a three-component reaction of (hetero)aromatic aldehydes 1,3-acetyl-4-hydroxy-2(1H)-quinolone 44, and an excess of guanidine hydrochloride 46 in an aqueous solution of KOH under refluxing ethanol for 6 h (Scheme 12).63 Purification of products by column chromatography was unsuccessful due to their poor solubility. For that reason, the crude product was diluted with 100 mL of water and the pH was adjusted to 2.0 by using a hydrochloric acid solution (6.0 M). The resulting solid was filtered, washed with water, dried, and recrystallized in ethanol to give the products 47 in 78–93% yields. All synthesized compounds 47a–t were screened for their antibacterial activity against two Gram-positive (Staphylococcus aureus and Bacillus cereus) and three Gram-negative (Klebsiella planticola, Escherichia coli, and Pseudomonas aeruginosa) bacterial strains using Neomycin as a standard drug. It was found that compound 47s (R = 3-MeO-4-BnOC6H3) exhibited the highest activity against Staphylococcus aureus, while compounds 47k (R = 3,4,5-(MeO)3C6H2) and 47m (R = 3-OHC6H4) showed the best activity against Bacillus cereus with a MIC value of 0.58 μg mL−1 in both cases, when compared to Neomycin (MIC = 18.75 μg mL−1 for both strains). In addition, compounds 47m and 47r (R = 2,5-(MeO)2C6H3) resulted to be more potent than Neomycin against Klebsiella planticola and Pseudomonas aeruginosa with a MIC value of 0.58 μg mL−1, respectively. The compounds 47c (R = 4-ClC6H4), 47f (R = 4-i-BuC6H4), 47k, and 47n (R = 4-BnOC6H4) showed an excellent antibacterial activity against Escherichia coli with a MIC value of 1.17 μg mL−1, in comparison to Neomycin (MIC = 18.75 μg mL−1).
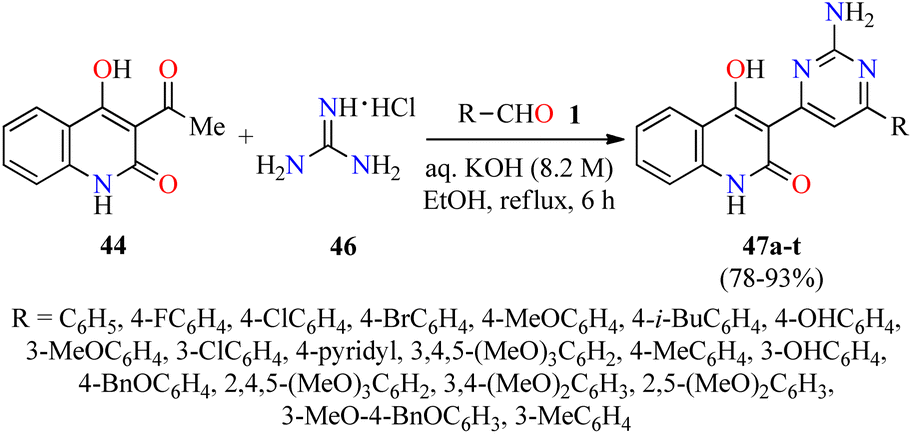 | ||
| Scheme 12 Three-component synthesis of quinolone–pyrimidine hybrids 47 for evaluation of their antibacterial activity. | ||
Recently, El-Hashasha and co-workers described the time-efficient synthesis of 4,6-diaryl-3-cyano-2(1H)-pyridone 48 by a four-component reaction of 4-methoxybenzaldehyde 1, 4-bromoacetophenone 7, ethyl cyanoacetate 8, and an excess of ammonium acetate in refluxing ethanol for 20 min (Scheme 13A).64 The precipitate was filtered, dried, and recrystallized from methanol to afford product 48 in 89% yield. The plausible mechanism reaction proceeds via a Knoevenagel condensation of 4-methoxybenzaldehyde 1 with ethyl cyanoacetate 8 to afford cyanoacrylate X, which undergoes a Michael addition with 4-bromoacetophenone 7 to give intermediate XI (Scheme 13B). The condensation reaction of 1,5-dicarbonyl compound XI with ammonia gives enamine intermediate XII, which undergoes an intramolecular cyclization/dehydrogenation sequence to deliver the desired product 48. Interestingly, the functionalization of compound 48 afforded pyridine and triazole derivatives in high yields. Lastly, compound 48 was screened for its antibacterial activity against Staphylococcus aureus and Escherichia coli as Gram-positive and Gram-negative bacterial strains, respectively, using Cefoxitin as a standard drug. It was found that compound 48 displayed a diameter of growth of the inhibition zone of 13 mm and 12 mm against Staphylococcus aureus and Escherichia coli, respectively, in comparison to Cefoxitin (25 mm for both strains).
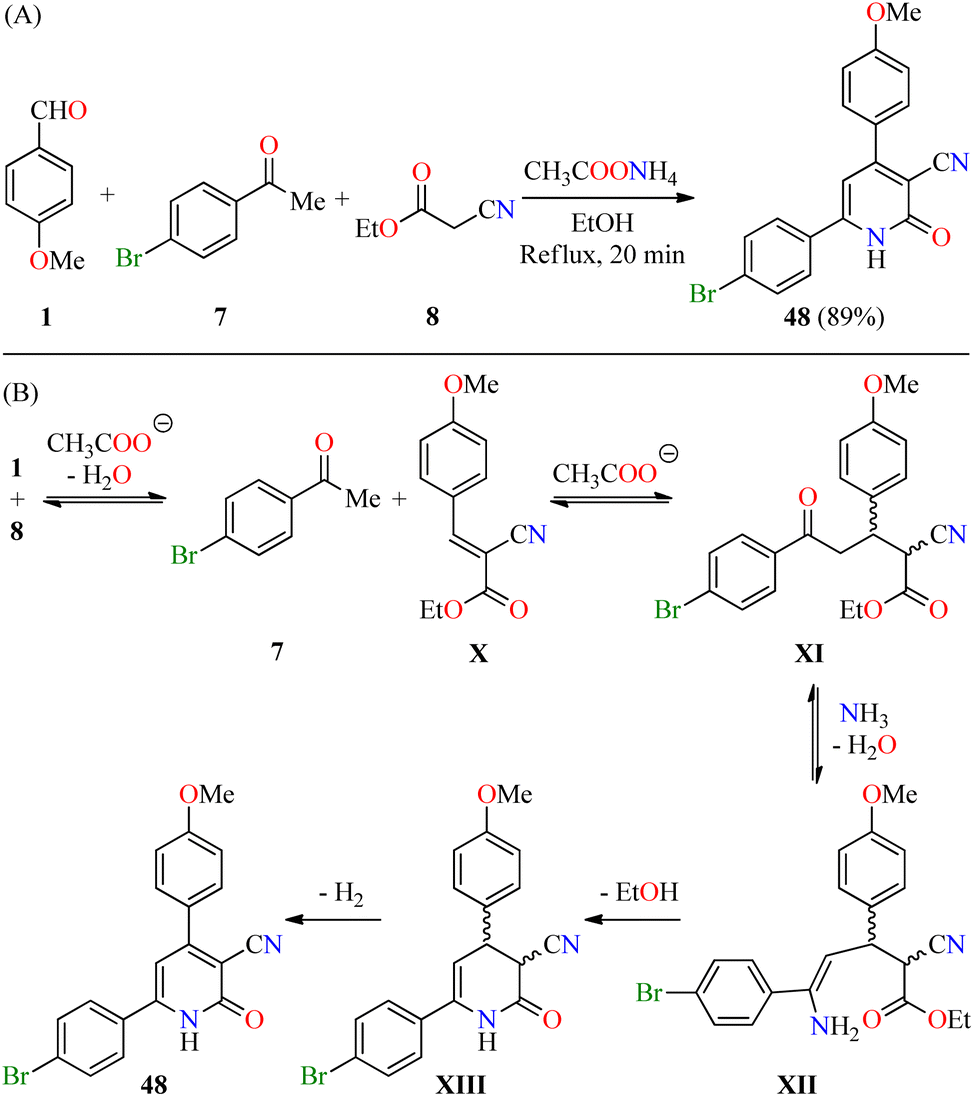 | ||
| Scheme 13 (A) Four-component synthesis of the 4,6-diaryl-3-cyano-2(1H)-pyridone 48 and its in vitro antibacterial activity against two bacterial strains. (B) Plausible reaction mechanism for the formation of 48. | ||
Interestingly, 2-pyridone-3-carboxylic acids 52 were synthesized in 52–86% yields through a three-component reaction of 3-formylchromones 49, Meldrum's acid 50, and primary amines 51 in the presence of diammonium hydrogen phosphate (DAHP, 20 mol%) as a basic catalyst in water at 70 °C for 3 h (Scheme 14).65 This protocol was distinguished by its operational simplicity, good yields, short reaction times, and structural diversity of products by the use of different aliphatic and aromatic amines. The compounds 52a–p were screened for their antibacterial activity against two Gram-positive (Staphylococcus aureus and Methicillin-resistant staphylococcus aureus) and two Gram-negative (Escherichia coli and Acinetobacter baumannii) bacterial strains using Cefixime and Ciprofloxacin as standard drugs. It was found that compounds 52p (R = 4-MeO, R1 = cyclohexyl) and 52b (R = H, R1 = allyl) exhibited the highest activity against Staphylococcus aureus with MIC values of 2.96 and 4.67 μg mL−1, respectively, in comparison to Cefixime (MIC = 1 μg mL−1) and Ciprofloxacin (MIC = 0.25 μg mL−1). In addition, the compound 52o (R = 4-MeO, R1 = allyl) showed the best activity against Methicillin-resistant Staphylococcus aureus with a MIC value of 82.33 μg mL−1, when compared to Cefixime (MIC = 32 μg mL−1) and Ciprofloxacin (MIC = 64 μg mL−1). In case of Escherichia coli, the compound 52h (R = H, R1 = 3,4-dimethoxyphenethyl) showed a moderate activity with a MIC value of 12.79 μg mL−1, while all synthesized compounds 52a–p displayed a low antibacterial activity against Acinetobacter baumannii with MIC values ranging from 329.32 to >500 μg mL−1, when compared to Cefixime (MIC = 4 and 32 μg mL−1, respectively) and Ciprofloxacin (MIC = 0.5 and 64 μg mL−1, respectively).
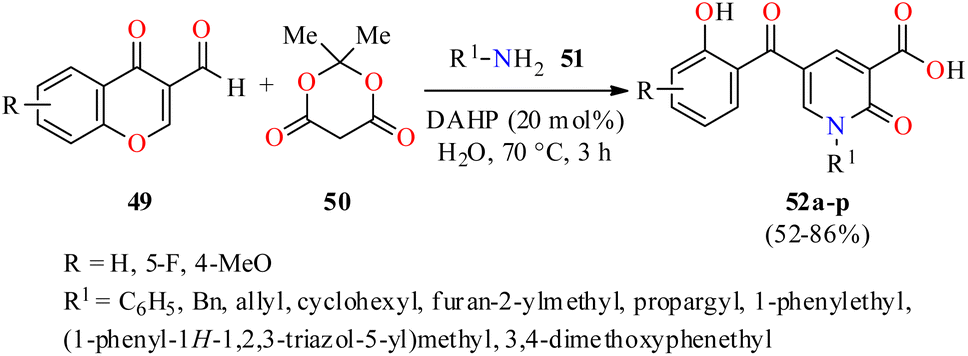 | ||
| Scheme 14 DAHP-catalyzed three-component synthesis of 2-pyridone-3-carboxylic acids 52 and their in vitro antibacterial activity against four bacterial strains. | ||
Lastly, molecular docking studies were performed to investigate the interactions of compound 52p (R = 4-MeO, R1 = cyclohexyl) with the DNA gyrase active site (PDB ID code: 5CDQ) (Fig. 5).65 2-Hydroxy-4-methoxybenzoyl interacted with Arg458 and Asp437 through π-cation and H-bonding, respectively. Also, N-cyclohexane and carbonyl groups occupied the proximity of Gly81 and formed H-bonding interaction with Ser84, respectively. Interestingly, the electron-donating property of the methoxy substitution at the benzoyl ring increased the tendency of the compound 52p to make π-cation interaction with Arg458.
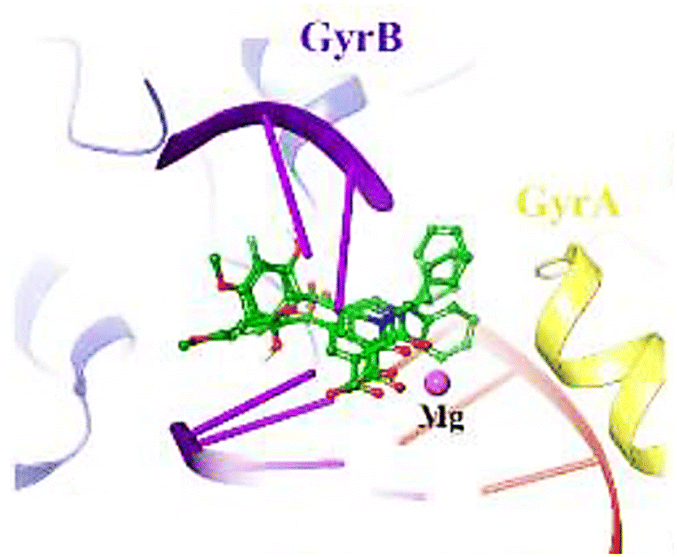 | ||
| Fig. 5 3D Interactions of compound 52p with Staphylococcus aureus DNA gyrase (PDB ID code: 5CDQ). GyrA and GyrB subunit of DNA gyrase are in yellow and blue, respectively. | ||
2.3. Antifungal activity
For decades, fungal infections have been difficult health conditions to treat due to their high toxicity, low efficacy rates, long duration of treatment, and resistance to clinically antifungal drugs.66 These reasons have prompted the development of new antifungal agents with distinct action or multitargeted combination therapy.66–68 In this way, N-allylquinolone derivatives 30 were obtained through a La(OTf)3-catalyzed three-component reaction and discussed in Section 2.2. Antibacterial activity (Scheme 6).58 The antifungal activity of these compounds was evaluated against Candida albicans, Aspergillus niger, and Aspergillus clavatus using Nystatin as a standard drug. Overall, N-allylquinolone derivatives 30 showed MIC values in the range of 100 to >1000 μg mL−1 for three fungal strains, in comparison to Nystatin (MIC = 100 μg mL−1 for all strains). Interestingly, the compound 30r (R = H, R1 = Me, R2 = C6H5, X = S) showed a significant activity against Candida albicans with a MIC value of 200 μg mL−1. Also, compounds 30w (R = Cl, R1 = H, R2 = C6H5, X = S) and 30x (R = Cl, R1 = Me, R2 = C6H5, X = S) displayed the same activity that Nystatin against Aspergillus niger and Aspergillus clavatus, respectively, with a MIC value of 100 μg mL−1.On the other hand, N-allylquinolone derivatives 32 and 34 obtained by a solvent-free microwave-assisted multicomponent process and discussed in Section 2.2. Antibacterial activity (Scheme 7),59 were also screened for their antifungal activity against Candida albicans, Aspergillus niger, and Aspergillus clavatus using Nystatin as a standard drug. The compounds 32a (R = R1 = H) and 34a (R = H, R2 = CN) showed a moderate activity against Candida albicans with a MIC value of 250 μg mL−1, in comparison to Nystatin (MIC = 100 μg mL−1). Importantly, compounds 32c (R = MeO, R1 = H) and 34h (R = Cl, R2 = COO-i-Pr) exerted an excellent activity against Aspergillus niger, while the compounds 32d (R = Cl, R1 = H), 32h (R = Cl, R1 = Me), and 34d (R = Cl, R2 = CN) displayed the highest potency against Aspergillus clavatus with a MIC value of 100 μg mL−1 in all cases, when compared to Nystatin (MIC = 100 μg mL−1 for both strains).
Importantly, coumarin–pyridone hybrids 37 obtained through a one-pot three-component approach and discussed in Section 2.2. Antibacterial activity (Scheme 8),60 were also screened for their antifungal activity against Candida albicans, Aspergillus fumigatus, and Aspergillus niger using Amphotericin B as a standard drug. It was found that compounds 37d (R = 4-MeOC6H4) and 37m (R = 2-furanyl) exhibited a moderate activity with MIC values in the range of 60–75 μg mL−1 against three fungal strains, in comparison to Amphotericin B (MIC = 0.5–1.0 μg mL−1, respectively).
Alternatively, fully substituted 2(1H)-pyridone derivatives 43 obtained by a piperidine-catalyzed three-component approach and discussed in Section 2.2. Antibacterial activity (Scheme 10),61 were also screened for their antifungal activity against Aspergillus niger. It was found that compounds 43a (R = C6H5), 43b (R = 2-FC6H4), 43c (R = 2-BrC6H4), 43d (R = 4-MeOC6H4), 43e (R = 4-Me2NC6H4), and 43q (R = 2-furanyl) showed a diameter of growth of inhibition zone in the range of 3.00–3.75 mm against Aspergillus niger. Unfortunately, the authors did not use a standard drug for this study.
Alternatively, Krishna and colleagues described the synthesis of quinolone–pyridine hybrids 45 via an NbCl5-catalyzed four-component reaction and discussed in Section 2.2. Antibacterial activity (Scheme 11),62 were also screened for their antifungal activity against Candida albicans, Candida parapsilosis, Candida glabrata, Candida aaseri, Aspergillus niger, and Issatchenkia hanoiensis using Miconazole as a standard drug. Particularly, the compound 45n (R = naphthalene-2-yl) showed the highest activity against tested Candida and fungal pathogens with MIC values ranging from 1.67 to 9.37 μg mL−1, in comparison to Miconazole (MIC = 4.68–9.37 μg mL−1). Moreover, compounds 45b (R = 4-FC6H4), 45d (R = 4-BrC6H4), 45f (R = 4-i-BuC6H4), 45n, and 45p (R = 2-furanyl) showed an excellent antifungal activity against Candida strains with MIC values in the range of 2.34–9.37 μg mL−1, in comparison to Miconazole (MIC = 4.68–9.37 μg mL−1). In cases of Aspergillus niger and Issatchenkia hanoiensis, compounds 45a (R = C6H5), 45d, and 45n displayed a high potency with MIC values in the range of 1.67–9.37 μg mL−1, respectively, in comparison to Miconazole (MIC = 9.37 μg mL−1).
A year later, the same authors used a similar strategy for the synthesis of quinolone–pyrimidine hybrids 47 by a three-component protocol as discussed in Section 2.2. Antibacterial activity (Scheme 12),63 were also tested for their antifungal activity against Candida albicans, Candida parapsilosis, Candida glabrata, Candida aaseri, Aspergillus niger, and Issatchenkia hanoiensis using Miconazole as a standard drug. Overall, compounds 47h (R = 3-MeOC6H4), 47r (R = 2,5-(MeO)2C6H3), and 47t (R = 3-MeC6H4) showed the highest antifungal activity against Candida strains with MIC values ranging from 0.58 to 1.17 μg mL−1, in comparison to Miconazole (MIC = 4.68–9.37 μg mL−1). In cases of Aspergillus niger and Issatchenkia hanoiensis, compounds 47r and 47n (R = 4-BnOC6H4) exerted an excellent activity with a MIC value of 0.58 μg mL−1, respectively, in comparison to Miconazole (MIC = 9.37 μg mL−1).
Lastly, 2-pyridone-3-carboxylic acids 52 obtained through a DAHP-catalyzed three-component approach and discussed in Section 2.2. Antibacterial activity (Scheme 14),65 were also screened for their antifungal activity against Candida albicans using Nystatin as a standard drug. Unfortunately, all compounds 52a–p exhibited a low antifungal activity with a MIC value of >500 μg mL−1, in comparison to Nystatin (MIC = 64 μg mL−1).
2.4. Anti-inflammatory activity
Inflammation is a self-protective procedure to eliminate the injurious stimuli and to start the healing process. Importantly, inflammation is a critical component of tumor progression.69,70 Thus, the anti-inflammatory therapy is efficacious towards early neoplastic progression and malignant conversion.69,70 Most of the commercially available anti-inflammatory drugs are found to be highly unsafe for long-term use due to their adverse effects.71 As a result, new heterocyclic molecules are being developed and a number of them are in the advanced stages of clinical trials.71 Recently, the utilization of 2-cyano-N-(4-hydroxyphenyl)acetamide has been explored in the construction of fully substituted 2-pyridone derivatives with biomedical features.72 In 2021, Fayed and colleagues reported the piperidine-catalyzed three-component reaction of aromatic aldehydes 1, 2-cyano-N-(4-hydroxyphenyl)acetamide 21b, and cyanoacetamide 42 in refluxing ethanol for 6 h to afford 2(1H)-pyridone derivatives 53a–d in 58–69% yields (Scheme 15).72 This multicomponent approach was successfully extended to ethyl acetoacetate 54 under similar reaction conditions for 2 h to give 2-pyridone derivatives 55a and 55b in 81% and 76% yields, respectively. Some compounds were screened for their in vivo anti-inflammatory activity employing the carrageenan-stimulated rat hind paw edema method.73 The anti-inflammatory behavior of compounds 53b (R = 4-MeOC6H4), 53c (R = 4-BrC6H4), 55a (R = 4-MeOC6H4), and 55b (R = 4-ClC6H4) in a 100 mg kg−1 ratio with a rat's body weight were compared with the standard Phenylbutazone (100 mg kg−1). In particular, compound 53b displayed the highest anti-inflammatory activity with a percentage of inhibition of 9.2%, 24.6%, 42.2%, and 40.6% at 1, 2, 3, and 4 hours, respectively, which was only superior to the anti-inflammatory effect of Phenylbutazone at 1 hour (0.72%, 29.9%, 41.2%, and 43.3%, respectively). Lastly, compounds 53b, 53c, 55a, and 55b were screened for their in vitro anti-inflammatory activity using the interleukin-6 kits and Phenylbutazone as a standard drug.74 As expected, compound 53b displayed the highest anti-inflammatory activity with a percentage of IL-6 inhibition of 65.1%, when compared to Phenylbutazone (85.5%). It should be noted that compounds 53c, 55a, and 55b showed a percentage of IL-6 inhibition of 49.5%, 31.4%, and 41.2%, respectively.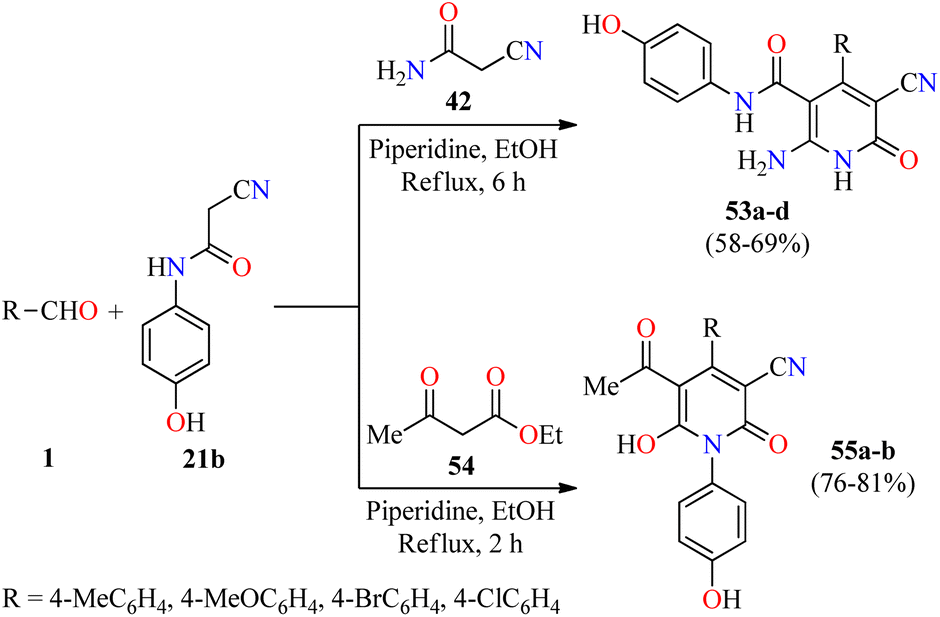 | ||
| Scheme 15 Piperidine-catalyzed three-component synthesis of fully substituted 2-pyridone derivatives 53 and 55 with anti-inflammatory activity. | ||
In 2022, the same authors described the piperidine-catalyzed three-component reaction of aromatic aldehydes 1, 2-cyano-N-(4-hydroxyphenyl)acetamide 21b, and ethyl acetoacetate 54 or acetylacetone 56 in refluxing ethanol for 2 h to afford fully substituted 2-pyridone derivatives 55a–c and 57a–c in 76–87% and 62–89% yields, respectively (Scheme 16).75 This multicomponent approach was successfully extended to malononitrile 2 or ethyl 2-cyanoacetate 8 under similar reaction conditions for 3–5 h to give 2-pyridone derivatives 58a–f and 59a–b in 68–82% and 67–77% yields, respectively. Selected compounds were screened for their in vivo anti-inflammatory activity employing the carrageenan-stimulated rat hind paw edema method using Phenylbutazone as a reference drug. In summary, compounds exerted a percentage of inhibition in the range of 10.3–52.6%, 18.4–57.6%, and 27.2–45.6% at 2, 3, and 4 hours, respectively, in comparison to Phenylbutazone (29.9%, 41.2%, and 43.3%, respectively). Most of the tested compounds showed a higher anti-inflammatory effect than Phenylbutazone. Particularly, compounds 58b (R = 4-ClC6H4) and 58f (R = 4-MeOC6H4) displayed an excellent percentage of inhibition of (52.6% and 57.6%) and (44.2% and 50.2%) at 2 and 3 hours, respectively. In addition, compounds 58a (R = C6H5), 58c (R = 4-FC6H4), and 58f showed a percentage of inhibition of 44.2%, 44.2%, and 45.6%, respectively, at 4 hour. Lastly, selected compounds were screened for their in vitro anti-inflammatory activity using the interleukin-6 kits and Phenylbutazone as a standard drug.75 As expected, compound 58f showed almost the same activity than Phenylbutazone (85.5%) with a percentage of IL-6 inhibition of 85.0%. In addition, compounds 58a and 58c showed a good percentage of IL-6 inhibition corresponding to 79.2% and 67.1%, respectively.
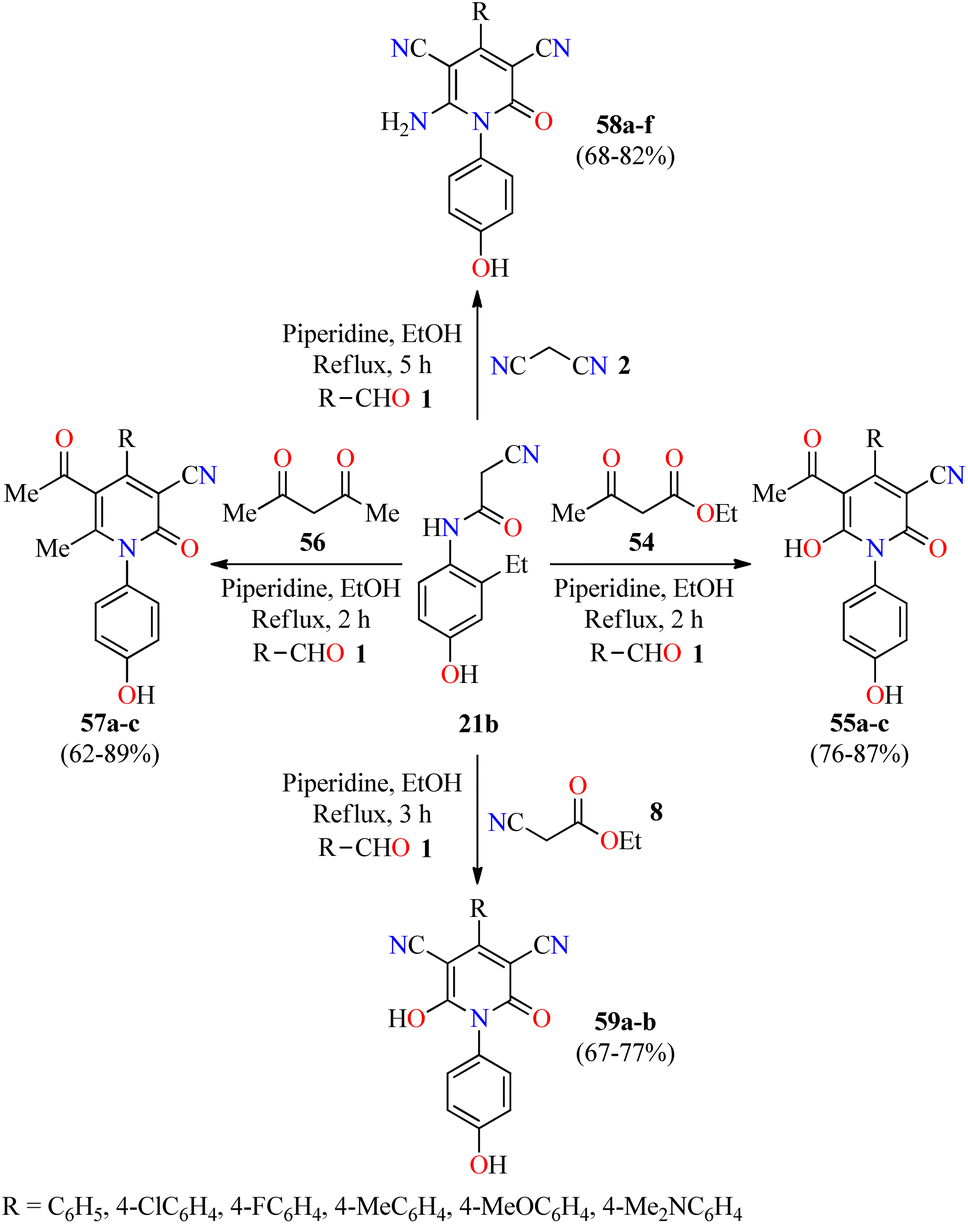 | ||
| Scheme 16 Three-component synthesis and anti-inflammatory evaluation of 2-pyridone derivatives 55, 57, 58, and 59. | ||
Molecular docking studies were performed to investigate the interactions of compound 58f (R = 4-MeOC6H4) with the active site of IL-6 (PDB ID code: 1N26) (Fig. 6).75 The docking research of Phenylbutazone (standard drug) showed that arene–H interaction with Ile194 increased fixation within the active region of IL-6. For compound 58f, the oxygen atom of the hydroxyl group and nitrogen atom of the amino group formed two H-bonding interactions with Thr188 and Cys102, respectively.
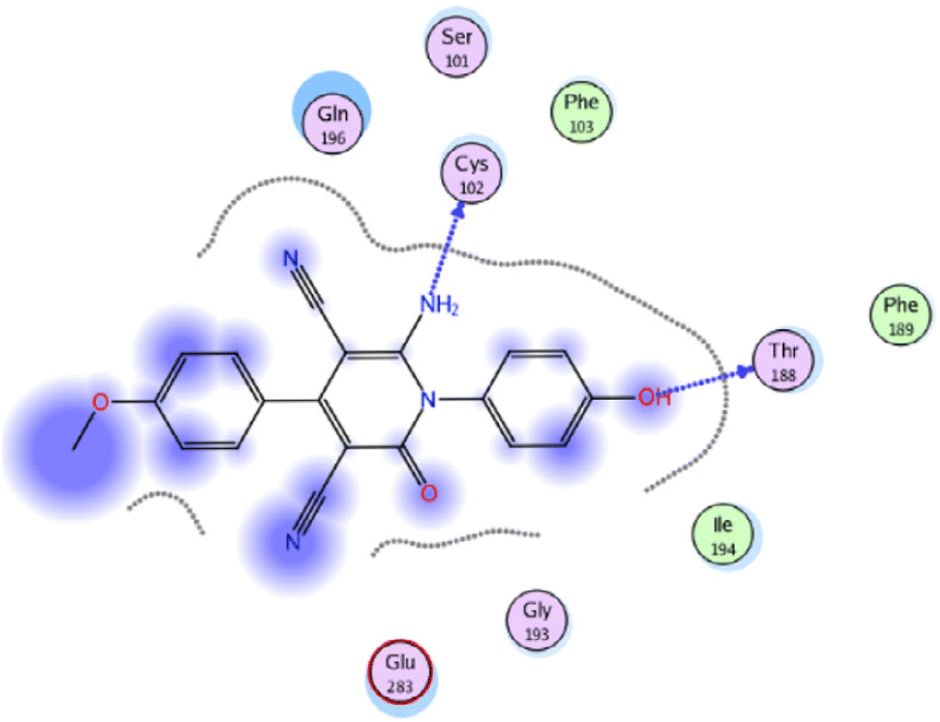 | ||
| Fig. 6 2D Interactions of compound 58f with the active site of IL-6 (PDB ID code: 1N26). Reproduced with permission from ref. 75. Copyright Elsevier Inc., 2022. | ||
2.5. α-Glucosidase inhibitory activity
Type 2 diabetes is the most common form of diabetes, which is a challenging metabolic disease characterized by insulin resistance, leading to hyperglycemia or abnormal blood glucose levels and damage to various physiological processes and organs.76,77 The tremendous increase in the number of patients affected by this disease and the side effects of antidiabetic drugs have promoted the search for new heterocyclic compounds with better safety and antidiabetic activity.76,77 In particular, the incorporation of azoles such as pyrazole, imidazole, and triazole, among others, is required in the design of new antihyperglycemic agents with higher activity than acarbose.78 Recently, Duan and colleagues reported the cascade synthesis of functionalized 3-bis(indol-3-yl)methylquinoline-2(1H)-ones 62 in 40–66% yields through a pseudo-three-component between indoles 60 and 2-acyl-N-acrylaniline derivatives 61 catalyzed by Pd(OAc)2 (10 mol%) in the presence of an excess of TFA as an additive and TBPB as an oxidant in a mixture of DMF/DMSO (9:1, 3.0 mL) at 90 °C for 16–36 h (Scheme 17).79 Notably, three C–C bonds and one ring are formed in one step by Pd-catalyzed C-3 alkenylation of indoles with 2-acyl-N-acrylaniline derivatives. Lastly, selected quinolone-bis(indolyl)methane hybrids 62 were screened against α-glucosidase using Acarbose as a reference drug. Most of the compounds showed a higher α-glycosidase inhibition than Acarbose. Remarkably, compounds 62c (R = R1 = R2 = R4 = H, R3 = C6H5), 62e (R = R1 = R3 = R4 = H, R2 = Bn), and 62l (R = 5-Me, R1 = R3 = R4 = H, R2 = PMB) showed the highest inhibitory activity with IC50 values of 27.5, 21.8, and 15.6 μM, respectively, when compared to Acarbose (IC50 = 154.7 μM).
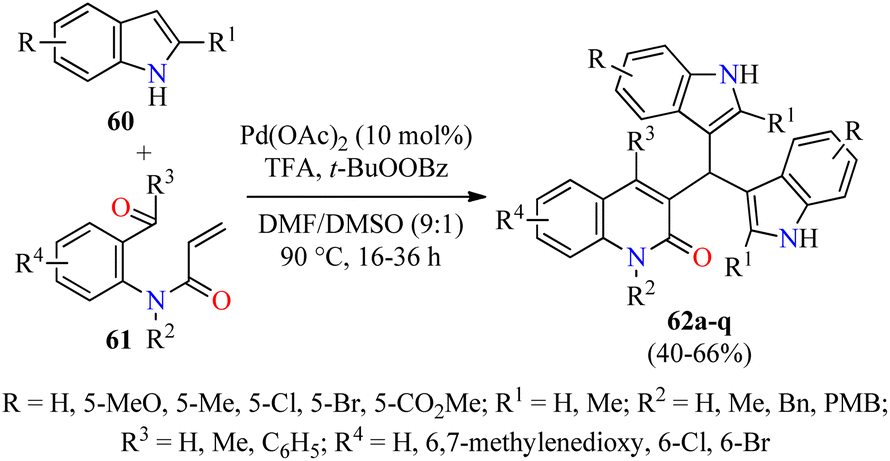 | ||
| Scheme 17 Pd(OAc)2-Catalyzed pseudo-three-component synthesis of 3-bis(indol-3-yl)methylquinoline-2(1H)-ones 62 as α-glucosidase inhibitors. | ||
As illustrated in Scheme 18, the cascade reaction is triggered by Pd-catalyzed C-3 alkenylation of indoles 60 with 2-acyl-N-acrylanilines 61 to give α,β-unsaturated amides 63, which underwent an intramolecular cyclization to generate intermediates 64. The benzylic alcohol moiety of 64 is easily eliminated to form allylic-type carbocations 65. Lastly, the cascade process is completed by the nucleophilic attack of indoles to the stabilized intermediates 65 to deliver products 62.
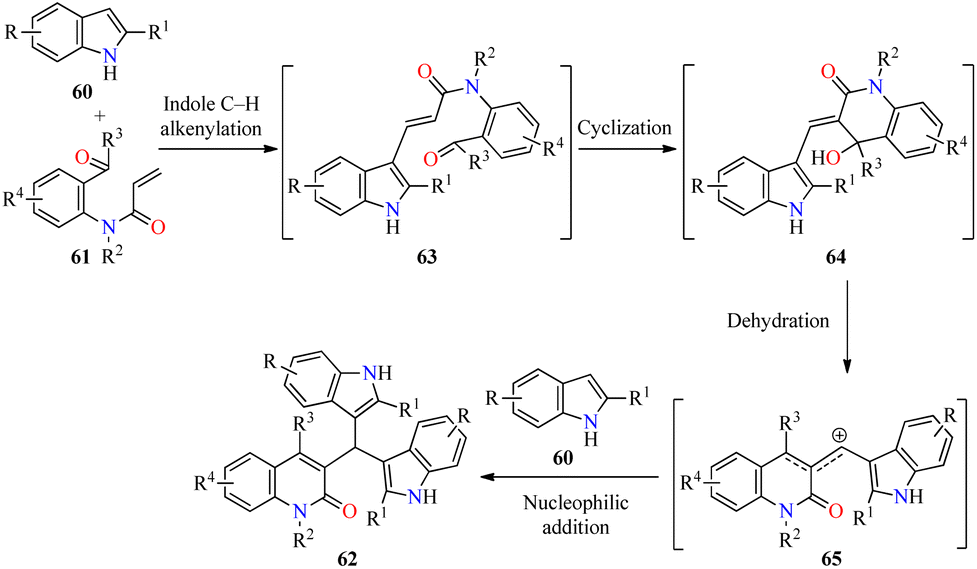 | ||
| Scheme 18 Plausible mechanism for the synthesis of 3-bis(indol-3-yl)methylquinoline-2(1H)-ones 62. | ||
2.6. Cardiotonic activity
Cardiotonic drugs are used for treating cardiac insufficiency because they increase the contractile power of the myocardium improving its capability and efficacy.80 In particular, 2-pyridone-based drugs such as Amrinone, Milrinone, and Olprinone have been employed as potent cardiotonic agents during the last decades.81–84 In this way, a series of 3-cyano-2(1H)-pyridone derivatives 66 were synthesized through a four-component reaction of aromatic aldehydes 1, substituted acetophenones 7, ethyl cyanoacetate 8, and an excess of ammonium acetate in refluxing ethanol for 6 h (Scheme 19).85 The reaction mixture was cooled and the precipitate was filtered, washed successively with water, dried, and recrystallized to afford products 66a–o in 31–40% yields. These compounds were evaluated for their cardiotonic activity using the spontaneously beating atria model from Reserpine-treated guinea pigs.86 Isoproterenol sulfate at a concentration of 5 × 10−4 M was used as a reference standard to be able to follow the changes in both the atria contractility and frequency rate. The best pharmacological profile was obtained with compound 66i (R = 4-OHC6H4, R1 = 3,4-(MeO)2C6H3), which displayed selectivity for increasing the force of contraction (108.7% change over control) rather than the frequency rate (40.8% change over control) at 5 × 10−4 M concentration, in comparison to isoproterenol sulfate (83.7 and 76.4%, respectively).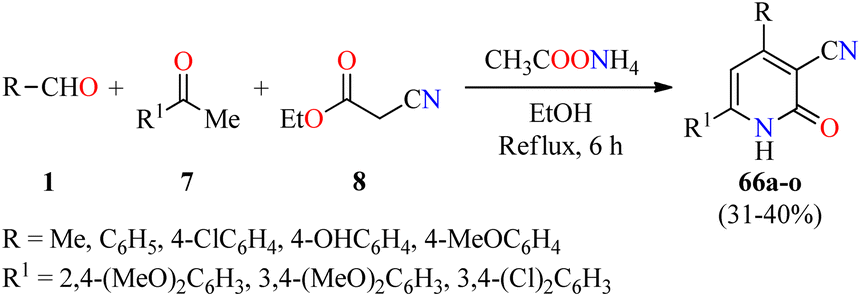 | ||
| Scheme 19 Four-component synthesis of 2(1H)-pyridone derivatives 66 as nonsteroidal cardiotonic agents. | ||
3 Conclusions and perspectives
2-Pyridone-containing heterocycles play a significant role in drug discovery and medicinal chemistry due to their interesting structural features and a broad range of biological activities. In particular, 2-pyridones can act as bioisosteres for amides; serve as both hydrogen bond donors and/or acceptors; and achieve better drug-like properties such as low lipophilicity, solubility in water, and metabolic stability under physiological conditions. As a result, an increasing number of FDA-approved drugs containing the 2-pyridone motif have been marketed as kinase inhibitors. Most of the preparative routes toward commercial drugs and biologically active compounds involve batch processes and multistep sequences, respectively. To meet the global demand for large-scale manufacturing of higher-value products at a reduced cost, synthetic organic chemists have shifted their attention to continuous flow reactors due to their several advantages over classical batch reactors. In this sense, MCRs can be positively affected by the use of flow processes in terms of yield, selectivity, reaction time, and real-time monitoring. Although there are several 2-pyridone-based drugs and drug candidates, we find very few reports on the multicomponent synthesis of bioactive 2-pyridone derivatives. This review aims to illustrate a variety of MCR-based approaches applied to the synthesis of bioactive 2-pyridone-containing heterocycles, demonstrating the value of this strategy in drug discovery and medicinal chemistry. Although many works have been published on the multistep synthesis of bioactive 2-pyridones, we hope that the reader will gain an appreciation that multicomponent reactions (MCRs) can lead to an overall process with high atom-economy and step-efficiency.Author contributions
The five individuals listed as authors have contributed substantially to the development of this review, and no other person was involved with its development. The contribution of authors is as follows: Miss Diana Hurtado-Rodríguez. Papers analysis with anticancer activity and original draw composition. Miss Angélica Salinas-Torres. Papers analysis with anticancer activity and original draw composition. Prof. Dr Hugo Rojas. Manuscript review and editing. Dr Diana Becerra. Papers analysis with anti-inflammatory, α-glucosidase inhibitor, and cardiotonic activities, literature investigation, writing, manuscript review and editing, and conceptualization. Dr Juan-Carlos Castillo. Papers analysis with antibacterial and antifungal activities, literature investigation, writing, manuscript review and editing, conceptualization, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from Universidad Pedagógica y Tecnológica de Colombia. The authors thank to the Dirección de Investigaciones at the Universidad Pedagógica y Tecnológica de Colombia for financial support (project numbers SGI-3073 and SGI-3312).Notes and references
- A. Dömling, W. Wang and K. Wang, Chemistry and biology of multicomponent reactions, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- C. G. Neochoritis, T. Zhao and A. Dömling, Tetrazoles via multicomponent reactions, Chem. Rev., 2019, 119, 1970–2042 CrossRef CAS PubMed.
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Synthesis of biologically active molecules through multicomponent reactions, Molecules, 2020, 25, 505 CrossRef PubMed.
- R. Kakuchi, Multicomponent reactions in polymer synthesis, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS PubMed.
- R. Abonia, J. Castillo, B. Insuasty, J. Quiroga, M. Nogueras and J. Cobo, Efficient catalyst-free four-component synthesis of novel γ-aminoethers mediated by a Mannich type reaction, ACS Comb. Sci., 2013, 15, 2–9 CrossRef CAS PubMed.
- J. D. Sunderhaus and S. F. Martin, Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds, Chem. - Eur. J., 2009, 15, 1300–1308 CrossRef CAS.
- S. E. John, S. Gulati and N. Shankaraiah, Recent advances in multi-component reactions and their mechanistic insights: a triennium review, Org. Chem. Front., 2021, 8, 4237–4287 RSC.
- D. Becerra, R. Abonia and J.-C. Castillo, Recent applications of the multicomponent synthesis for bioactive pyrazole derivatives, Molecules, 2022, 27, 4723 CrossRef CAS PubMed.
- H. A. Younus, M. Al-Rashida, A. Hameed, M. Uroos, U. Salar, S. Rana and K. M. Khan, Multicomponent reactions (MCR) in medicinal chemistry: a patent review (2010–2020), Expert Opin. Ther. Pat., 2021, 31, 267–289 CrossRef CAS PubMed.
- A. Strecker, Ueber die künstliche Bildung der Milchsäure und einen neuen, dem glycocoll homologen körper, Ann. Chem. Pharm., 1850, 75, 27–45 CrossRef.
- A. Hantzsch, Condensationprodukte aus aldehydammoniak und ketoniartigen verbindungen, Chem. Ber., 1881, 14, 1637–1638 CrossRef.
- P. Biginelli, Ueber aldehyduramide des acetessigäthers, Chem. Ber., 1891, 24, 1317 CrossRef.
- C. Mannich and W. Krösche, Ueber ein kondensationsprodukt aus formaldehyd, ammoniak und antipyrin, Arch. Pharm., 1912, 250, 647–667 CrossRef CAS.
- M. Passerini, Sopra gli isonitrili (I). Composto del p-isonitrilazobenzolo con acetone ed acido acetico, Gazz. Chim. Ital., 1921, 51, 126–129 CAS.
- E. K. Fields, The synthesis of esters of substituted amino phosphonic acids, J. Am. Chem. Soc., 1952, 74, 1528–1531 CrossRef CAS.
- F. Asinger, Über die gemeinsame einwirkung von schwefel und ammoniak auf ketone, Angew. Chem., 1956, 68, 413 CrossRef CAS.
- I. Ugi, R. Meyr, U. Fetzer and C. Steinbrückner, Versuche mit isonitrilen, Angew. Chem., 1959, 71, 373–388 Search PubMed.
- K. Gewald, E. Schinke and H. Böttcher, Heterocyclen aus CH-aciden nitrilen, VIII. 2-Amino-thiophene aus methylenaktiven nitrilen, carbonylverbindungen und schwefel, Chem. Ber., 1966, 99, 94–100 CrossRef CAS.
- O. H. Oldenziel, D. Van Leusen and A. M. Van Leusen, Chemistry of sulfonylmethyl isocyanides. 13. A general one-step synthesis of nitriles from ketones using tosylmethyl isocyanide. Introduction of a one-carbon unit, J. Org. Chem., 1977, 42, 3114–3118 CrossRef CAS.
- H. Bienaymé and K. A. Bouzid, New heterocyclic multicomponent reaction for the combinatorial synthesis of fused 3-aminoimidazoles, Angew. Chem., Int. Ed., 1998, 37, 2234–2237 CrossRef.
- R. Abonia, J. Castillo, B. Insuasty, J. Quiroga, M. Nogueras and J. Cobo, An efficient synthesis of 7-(arylmethyl)-3-tert-butyl-1-phenyl-6,7-dihydro-1H,4H-pyrazolo[3,4-d][1,3]oxazines, Eur. J. Org. Chem., 2010, 2010, 6454–6463 CrossRef.
- A. Boltjes and A. Dömling, The Groebke-Blackburn-Bienaymé reaction, Eur. J. Chem., 2019, 2019, 7007–7049 CrossRef CAS PubMed.
- M. M. K. Amer, M. A. Aziz, W. S. Shehab, M. H. Abdellattif and S. M. Mouneir, Recent advances in chemistry and pharmacological aspects of 2-pyridone scaffolds, J. Saudi Chem. Soc., 2021, 25, 101259 CrossRef CAS.
- S. Sangwan, N. Yadav, R. Kumar, S. Chauhan, V. Dhanda, P. Walia and A. Duhan, A score years' update in the synthesis and biological evaluation of medicinally important 2-pyridones, Eur. J. Med. Chem., 2022, 232, 114199 CrossRef CAS PubMed.
- S. Lin, C. Liu, X. Zhao, X. Han, X. Li, Y. Ye and Z. Li, Recent advances of pyridinone in medicinal chemistry, Front. Chem., 2022, 10, 869860 CrossRef CAS PubMed.
- O. Bensaude, M. Chevrier and J. E. Dubois, Lactim-lactam tautomeric equilibriums of 2-hydroxypyridines. 1. Cation binding, dimerization, and interconversion mechanism in aprotic solvents. A spectroscopic and temperature-jump kinetic study, J. Am. Chem. Soc., 1978, 100, 7055–7060 CrossRef CAS.
- O. Bensaude, M. Chevrier and J. E. Dubois, Lactim/lactam tautomeric interconversion mechanism in water/polar aprotic solvent water systems. 2. Hydration of 2-hydroxypyridines. Evidence for a bifunctional water-catalyzed proton transfer, J. Am. Chem. Soc., 1979, 101, 2423–2429 CrossRef CAS.
- A. Loppinet-Serani, F. Charbonnier, C. Rolando and I. Huc, Role of lactam vs. lactim tautomers in 2(1H)-pyridone catalysis of aromatic nucleophilic substitution, J. Chem. Soc., Perkin Trans. 2, 1998, 2, 937–942 RSC.
- Y. Zhang and A. Pike, Pyridones in drug discovery: recent advances, Bioorg. Med. Chem. Lett., 2021, 38, 127849 CrossRef CAS PubMed.
- C. Laurence, K. A. Brameld, J. Graton, J.-Y. Le Questel and E. Renault, The pK(BHX) database: toward a better understanding of hydrogen-bond basicity for medicinal chemists, J. Med. Chem., 2009, 52, 4073–4086 CrossRef CAS PubMed.
- G. Yang, Y. Wang, J. Tian and J.-P. Liu, Huperzine A for Alzheimer's disease: a systematic review and meta-analysis of randomized clinical trials, PLoS One, 2013, 8, e74916 CrossRef CAS PubMed.
- R. C. Pandey, M. W. Toussaint, R. M. Stroshane, C. C. Kalita, A. A. Aszalas, A. L. Gawetson, T. T. Wei, K. M. Byrne, R. F. Geoghegan and R. J. White, Fredericamycin A, a new antitumor antibiotic I. Production, isolation and physicochemical properties, J. Antibiot., 1981, 34, 1389–1401 CrossRef CAS PubMed.
- V. J. Venditto and E. E. Simanek, Cancer therapies utilizing the camptothecins: a review of the in vivo literature, Mol. Pharmaceutics, 2010, 7, 307–349 CrossRef CAS PubMed.
- P. Mellini, B. Marrocco, D. Borovika, L. Polletta, I. Carnevale, S. Saladini, G. Stazi, C. Zwergel, P. Trapencieris, E. Ferretti, M. Tafani, S. Valente and A. Mai, Pyrazole-based inhibitors of enhancer of zeste homologue 2 induce apoptosis and autophagy in cancer cells, Philos. Trans. R. Soc., B, 2018, 373, 20170150 CrossRef PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US. FDA approved pharmaceuticals, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A review on recent advances in nitrogen-containing molecules and their biological applications, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- R. Abonia, D. Insuasty, J. Castillo, B. Insuasty, J. Quiroga, M. Nogueras and J. Cobo, Synthesis of novel quinoline-2-one based chalcones of potential anti-tumor activity, Eur. J. Med. Chem., 2012, 57, 29–40 CrossRef CAS PubMed.
- B. Insuasty, D. Becerra, J. Quiroga, R. Abonia, M. Nogueras and J. Cobo, Microwave-assisted synthesis of pyrimido[4,5-b][1,6]naphthyridin-4(3H)-ones with potential antitumor activity, Eur. J. Med. Chem., 2013, 60, 1–9 CrossRef CAS PubMed.
- A. Salinas-Torres, J. Portilla, H. Rojas, D. Becerra and J.-C. Castillo, Synthesis, spectroscopic analysis, and in vitro anticancer evaluation of 2-(phenylsulfonyl)-2H-1,2,3-triazole, Molbank, 2022, 2022, M1387 CrossRef CAS.
- A. Salinas-Torres, E. Jiménez, D. Becerra, J. J. Martínez, H. Rojas, J.-C. Castillo and M. A. Macías, Synthesis, anticancer evaluation, thermal and X-ray crystallographic analysis of 2-oxo-2H-chromen-7-yl 4-chlorobenzoate using a conductively heated sealed-vessel reactor, J. Mol. Struct., 2023, 1274, 134414 CrossRef CAS.
- I. V. Magedov, M. Manpadi, N. M. Evdokimov, E. M. Elias, E. Rozhkova, M. A. Ogasawara, J. D. Bettale, N. M. Przheval’skii, S. Rogelj and A. Kornienko, Antiproliferative and apoptosis inducing properties of pyrano[3,2-c]pyridones accessible by a one-step multicomponent synthesis, Bioorg. Med. Chem. Lett., 2007, 17, 3872–3876 CrossRef CAS PubMed.
- W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, Y. Wang, J. Zhao, S. Jia, J. Herich, D. Labreque, R. Storer, K. Meerovitch, D. Bouffard, R. Rej, R. Denis, C. Blais, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng, S. Kasibhatla and S. X. Cai, Discovery of 4-aryl-4H-chromenes as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. 1. Structure−activity relationships of the 4-aryl group, J. Med. Chem., 2004, 47, 6299–6310 CrossRef CAS PubMed.
- I. V. Magedov, M. Manpadi, M. A. Ogasawara, A. S. Dhawan, S. Rogelj, S. V. slambrouck, W. F. A. Steelant, N. M. Evdokimov, P. Y. Uglinskii, E. M. Elias, E. J. Knee, P. Tongwa, M. Y. Antipin and A. Kornienko, Structural simplification of bioactive natural products with multicomponent synthesis. 2. Antiproliferative and antitubulin activities of pyrano[3,2-c]pyridones and pyrano[3,2-c]quinolones, J. Med. Chem., 2008, 51, 2561–2570 CrossRef CAS PubMed.
- A. H. Abadi, D. A. Abouel-Ella, J. Lehmann, H. N. Tinsley, B. D. Gary, G. A. Piazza and M. A. O. Abdel-Fattah, Discovery of colon tumor cell growth inhibitory agents through a combinatorial approach, Eur. J. Med. Chem., 2010, 45, 90–97 CrossRef CAS PubMed.
- A. A. Fadda, K. S. Mohamed, H. M. Refat and E. E. El-Bialy, Synthesis, characterization and biological activity of some novel coumarin derivatives containing pyridine moiety, Heterocycles, 2015, 91, 134–148 CrossRef CAS.
- M. I. Ansari, A. Arun, M. K. Hussain, R. Konwar and K. Hajela, Discovery of 3,4,6-triaryl-2-pyridones as potential anticancer agents that promote ROS-independent mitochondrial-mediated apoptosis in human breast carcinoma cells, ChemistrySelect, 2016, 1, 4255–4264 CrossRef CAS.
- J.-C. Castillo, N.-F. Bravo, L.-V. Tamayo, P.-D. Mestizo, J. Hurtado, M. Macías and J. Portilla, Water-compatible synthesis of 1,2,3-triazoles under ultrasonic conditions by a Cu(I) complex-mediated click reaction, ACS Omega, 2020, 5, 30148–30159 CrossRef CAS PubMed.
- M. Sankaran, C. Uvarani, K. Chandraprakash, S. U. Lekshmi, S. Suparna, J. Platts and P. S. Mohan, A regioselective multicomponent protocol for the synthesis of novel bioactive 4-hydroxyquinolin-2(1H)-one grafted monospiropyrrolidine and thiapyrrolizidine hybrids, Mol. Diversity, 2014, 18, 269–283 CrossRef CAS PubMed.
- Y. Tangella, K. L. Manasa, V. L. Nayak, M. Sathish, B. Sridhar, A. Alarifi, N. Nagesh and A. Kamal, An efficient one-pot approach for the regio- and diastereoselective synthesis of trans-dihydrofuran derivatives: cytotoxicity and DNA-binding studies, Org. Biomol. Chem., 2017, 15, 6837–6853 RSC.
- E. A. Marwa, M. M. E. Mostafa, M. F. Salwa, A. M. Mona and A. H. Aly, Design, synthesis and docking study of pyridine and thieno[2,3-b]pyridine derivatives as anticancer PIM-1 kinase inhibitors, Bioorg. Chem., 2018, 80, 674–692 CrossRef PubMed.
- I. W. Cheney, S. Yan, T. Appleby, H. Walker, T. Vo, N. Yao, R. Hamatake, Z. Hong and J. Z. Wu, Identification and structure–activity relationships of substituted pyridones as inhibitors of Pim-1 kinase, Bioorg. Med. Chem. Lett., 2007, 17, 1679–1683 CrossRef CAS PubMed.
- A. C. Pierce, M. Jacobs and C. Stuver-Moody, Docking study yields four novel inhibitors of the protooncogene Pim-1 kinase, J. Med. Chem., 2008, 51, 1972–1975 CrossRef CAS PubMed.
- M. M. F. Ismail, A. M. Farrag and D. Abou-El-Ela, Synthesis, anticancer screening, and in silico ADME prediction of novel 2-pyridones as Pim inhibitors, J. Heterocycl. Chem., 2020, 57, 3442–3460 CAS.
- T. Mochizuki, C. Kitanaka, K. Noguchi, T. Muramatsu, A. Asai and Y. Kuchino, Physical and functional interactions between Pim-1 kinase and Cdc25A phosphatase. Implications for the Pim-1-mediated activation of the c-Myc signaling pathway, J. Biol. Chem., 1999, 274, 18659–18666 CrossRef CAS PubMed.
- A. T. A. Boraei, E. H. Eltamany, I. A. I. Ali, S. M. Gebriel and M. S. Nafie, Synthesis of new substituted pyridine derivatives as potent anti-liver cancer agents through apoptosis induction: in vitro, in vivo, and in silico integrated approaches, Bioorg. Chem., 2021, 111, 104877 CrossRef CAS PubMed.
- C. Walsh, Molecular mechanisms that confer antibacterial drug resistance, Nature, 2000, 406, 775–781 CrossRef CAS PubMed.
- B. Khameneh, M. Iranshahy, V. Soheili and B. S. F. Bazzaz, Review on plant antimicrobials: a mechanistic viewpoint, Antimicrob. Resist. Infect. Control., 2019, 8, 118 CrossRef PubMed.
- H. H. Jardosh and M. P. Patel, Lanthanum triflate-triggered synthesis of tetrahydroquinazolinone derivatives of N-allylquinolone and their biological assessment, J. Serb. Chem. Soc., 2012, 77, 1561–1570 CrossRef CAS.
- H. H. Jardosh and M. P. Patel, Microwave-induced CAN promoted atom-economic synthesis of 1H-benzo[b]xanthene and 4H-benzo[g]chromene derivatives of N-allyl quinolone and their antimicrobial activity, Med. Chem. Res., 2013, 22, 2954–2963 CrossRef CAS.
- R. Khajuria, S. Mahajan, Ambica and K. K. Kapoor, Expeditious synthesis of coumarin-pyridone conjugates molecules and their anti-microbial evaluation, J. Chem. Sci., 2017, 129, 1549–1557 CrossRef CAS.
- A. B. Pandit, M. M. Savant and K. D. Ladva, An efficient one-pot synthesis of highly substituted pyridone derivatives and their antimicrobial and antifungal activity, J. Heterocycl. Chem., 2018, 55, 983–987 CrossRef CAS.
- A. Krishna and S. Sarveswari, One-pot synthesis of 2-amino-6-(1,2-dihydro-4-hydroxy-2-oxoquinolin-3-yl)-4-arylpyridine-3-carbonitriles catalysed by NbCl5 and their in vitro antimicrobial studies, ChemistrySelect, 2019, 4, 9987–9992 CrossRef CAS.
- A. Krishna, V. Vijayakumar and S. Sarveswari, Synthesis of new 3-(2-amino-6-arylpyrimidin-4-yl)-4-hydroxyquinolin-2(1H)-ones and their in vitro antimicrobial and “DPPH” scavenging activity evaluation, ChemistrySelect, 2020, 5, 7967–7972 CrossRef CAS.
- M. A. El-Hashash, S. S. Shaban and R. S. Ali, Synthesis of 3-cyano-2-pyridone derivative and its utility in the synthesis of some heterocyclic compounds with expecting antimicrobial activity, J. Heterocycl. Chem., 2021, 58, 329–339 CrossRef CAS.
- E. M. Ahadi, H. Azizian, V. F. Vavsari, A. Aliahmadi, Z. Shahsavari, H. R. Bijanzadeh and S. Balalaie, Synthesis and decarboxylation of functionalized 2-pyridone-3-carboxylic acids and evaluation of their antimicrobial activity and molecular docking, Iran. J. Pharm. Res., 2021, 20, 456–475 CAS.
- A. Geronikaki, M. Fesatidou, V. Kartsev and F. Macaev, Synthesis and biological evaluation of potent antifungal agents, Curr. Top. Med. Chem., 2013, 13, 2684–2733 CrossRef CAS PubMed.
- G. Bouz and M. Doležal, Advances in antifungal drug development: an up-to-date mini review, Pharmaceuticals, 2021, 14, 1312 CrossRef CAS PubMed.
- N.-R. Elejalde, M. Macías, J.-C. Castillo, M. Sortino, L. Svetaz, S. Zacchino and J. Portilla, Synthesis and in vitro antifungal evaluation of novel N-substituted 4-aryl-2-methylimidazoles, ChemistrySelect, 2018, 3, 5220–5227 CrossRef CAS.
- L. Coussens and Z. Werb, Inflammation and cancer, Nature, 2002, 420, 860–867 CrossRef CAS PubMed.
- K. L. Rock and H. Kono, The inflammatory response to cell death, Annu. Rev. Pathol.: Mech. Dis., 2008, 3, 99–126 CrossRef CAS PubMed.
- S. Wongrakpanich, A. Wongrakpanich, K. Melhado and J. Rangaswami, A comprehensive review of non-steroidal anti-inflammatory drug use in the elderly, Aging Dis., 2018, 9, 143–150 CrossRef PubMed.
- E. A. Fayed, A. H. Bayoumi, A. S. Saleh, E. M. E. Al-Arab and Y. A. Ammar, In vivo and in vitro anti-inflammatory, antipyretic and ulcerogenic activities of pyridone and chromenopyridone derivatives, physicochemical and pharmacokinetic studies, Bioorg. Chem., 2021, 109, 104742 CrossRef CAS PubMed.
- C. A. Wintere, D. A. Risley and G. W. Nuss, Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs, Proc. Soc. Exp. Biol. Med., 1962, 111, 544–547 CrossRef PubMed.
- M. A. Croce, T. C. Fabian, J. H. Patton, S. M. Melton, M. Moore and L. L. Trenthem, Partial liquid ventilation decreases the inflammatory response in the alveolar environment of trauma patients, J. Trauma, 1998, 45, 273–282 CrossRef CAS PubMed.
- E. A. Fayed, E. M. E. Al-Arab, A. S. Saleh, A. H. Bayoumi and Y. A. Ammar, Design, synthesis, in silico studies, in vivo and in vitro assessment of pyridones and thiazolidinones as anti-inflammatory, antipyretic and ulcerogenic hits, J. Mol. Struct., 2022, 1260, 132839 CrossRef CAS.
- L. Suresh, P. Onkara, P. S. V. Kumar, Y. Pydisetty and G. V. P. Chandramouli, Ionic liquid-promoted multicomponent synthesis of fused tetrazolo[1,5-a]pyrimidines as α-glucosidase inhibitors, Bioorg, Med. Chem. Lett., 2016, 26, 4007–4014 CrossRef CAS PubMed.
- A. Chaudhury, C. Duvoor, V. S. R. Dendi, S. Kraleti, A. Chada, R. Ravilla, A. Marco, N. S. Shekhawat, M. T. Montales, K. Kuriakose, A. Sasapu, A. Beebe, N. Patil, C. K. Musham, G. P. Lohani and W. Mirza, Clinical review of antidiabetic drugs: implications for type 2 diabetes mellitus management, Front. Endocrinol, 2017, 8, 6 Search PubMed.
- M. Özil, M. Emirik, A. Beldüz and S. Ülker, Molecular docking studies and synthesis of novel bisbenzimidazole derivatives as inhibitors of α-glucosidase, Bioorg. Med. Chem., 2016, 24, 5103–5114 CrossRef PubMed.
- L. Duan, M.-X. Zuo, K.-Q. Xie, Y.-C. Liu, W.-Z. Qiu, L.-P. Wang and S. Liu, Palladium-catalyzed cascade synthesis of novel quinolone-bis(indolyl)methane hybrids as promising α-glucosidase inhibitors, Synthesis, 2020, 52, 1680–1686 CrossRef CAS.
- A. Y. Bagrov, J. I. Shapiro and O. V. Fedorova, Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets, Pharmacol. Rev., 2009, 61, 9–38 CrossRef CAS PubMed.
- N. A. Klein, S. J. Siskind, W. H. Frishman, E. H. Sonnelblick and T. H. LeJemtel, Hemodynamic comparison of intravenous amrinone and dobutamine in patients with chronic congestive heart failure, Am. J. Cardiol., 1981, 48, 170–175 CrossRef CAS PubMed.
- R. A. Young and A. Ward, Milrinone: a preliminary review of its pharmacological properties and therapeutic use, Drugs, 1988, 36, 158–192 CrossRef CAS PubMed.
- R. Murakami, K. Sano, Y. Murakami, T. Shimda and S. Morioka, Effects of intracoronary infusion of an inotropic agent, E-1020 (loprinone hydrochloride), on cardiac function: evaluation of left ventricular contractile performance using the end-systolic pressure-volume relationship, Int. J. Cardiol., 1995, 51, 57–63 CrossRef CAS PubMed.
- A. Abadi, O. Al-Deeb, A. Al-Afify and H. El-Kashef, Synthesis of 4-alkyl(aryl)-6-aryl-3-cyano-2(1H)-pyridinones and their 2-imino isosteres as nonsteroidal cardiotonic agents, Farmaco, 1999, 54, 195–201 CrossRef CAS PubMed.
- P. Dorigo, D. Fraccarollo, G. Santostasi, R. M. Gaion, I. Maragno, M. Floreani and F. Carpenedo, Pharmacological characterization of a new milrinone analogue, Farmaco, 1994, 49, 19–23 CAS.
- M. K. Robinson, T. L. Nusair, E. R. Fletcher and H. L. Ritz, A review of the Buehler guinea pig skin sensitization test and its use in a risk assessment process for human skin sensitization, Toxicology, 1990, 61, 91–107 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |