
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of a lactose-based biosurfactant by a novel nanodendritic catalyst and evaluating its efficacy as an emulsifier in a food emulsion system†
Maryam Karamiac,
Ali Reza Faraji *bc,
Solmaz Saremnezhadac and
Mostafa Soltaniac
*bc,
Solmaz Saremnezhadac and
Mostafa Soltaniac
aDepartment of Food Science and Technology, Faculty of Pharmacy, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran
bDepartment of Organic Chemistry, Faculty of Pharmaceutical Chemistry, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran. E-mail: alireza_ch57@yahoo.com; a.faraji@iaups.ac.ir
cNutrition and Food Sciences Research Center, Tehran Medical Sciences, Islamic Azad University, Tehran, Iran
First published on 10th November 2022
Abstract
Nonionic lactose fatty acid esters are a class of synthetic biosurfactants with various uses in the food, pharmaceutical, personal care, and cosmetic industries. The objective of this research was the preparation and full characterization of a series of novel metallic encapsulated magnetic core/dendrimer shell composites as catalysts (CoII/MnII G2.0L1/2@SCMBNP) and their use in the chemo- and regioselective synthesis of a biosurfactant for the first time. Surface-active properties (such as contact angle (CA), surface tension (SFT), interfacial tension (IFT), critical micelle concentration (CMC), hydrophilic–lipophilic balance (HLB), foamability (FA) & foam stability (FS), emulsion ability (EmA) & emulsion stability (EmS), surface excess (Γ) and free energy of adsorption (ΔG) were also determined for all synthesized biosurfactants. In comparison to other works, these results suggested that the synthesized lactose fatty acid esters have potential application as synthetic emulsifiers featuring surface properties and are comparable with Ryoto sugar ester L-1695 (sucrose laurate) & Tween-20 (polysorbate 20) as industrial emulsifiers. The optimized conditions for biosurfactant syntheses are 8 days at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of lactose sugar to lauric acid at 50 °C. Lactose ester as a biosurfactant exhibited a decrease of SFT & IFT and was able to stabilize a 20% soybean O/W emulsion. Furthermore, high conversion & yield, excellent chemo- and regioselectivity, and high operational stability over 5 runs were achieved for CoII/MnII-G2.0L1/2@SCMBNP, indicating the suitable efficiency of the catalytic process.
:1 molar ratio of lactose sugar to lauric acid at 50 °C. Lactose ester as a biosurfactant exhibited a decrease of SFT & IFT and was able to stabilize a 20% soybean O/W emulsion. Furthermore, high conversion & yield, excellent chemo- and regioselectivity, and high operational stability over 5 runs were achieved for CoII/MnII-G2.0L1/2@SCMBNP, indicating the suitable efficiency of the catalytic process.
1. Introduction
The surfactant, emulsifier, and wetting agent industries heavily depend on petrochemical feedstocks. With the great interest in both commercial incentives and environmentally friendly processes and also the prediction of scarcities of petroleum-based products, the need for surfactants derived merely from renewable raw materials is an evident target. Therefore, the oil- and sugar biosurfactants can be considered as hydrophobic and hydrophilic renewable sources from oleochemicals (such as palm oil, palm kernel oil, and coconut oil) and carbohydrates (such as glucose from corn starch, sucrose from sugar cane, and lactose from whey solution), respectively.1,2 Thereby, the sugar-based biosurfactants have extensive applications in the food industry (as foaming agents and emulsifiers), cosmetics, special detergents, oral hygiene products, and pharmaceutical industries. Also, carbohydrate-based surfactants exhibit some advantages compared to typical surfactants, such as high water solubility, odorless, non-ionic, tasteless, non-toxic & antimicrobial, anti-fungal, anti-cancer, and insecticidal properties.2–5 Lactose is the principal sugar existing in milk and whey. Whey is a major by-product of cheese manufacturing industry. After the casein curd separates from the milk, following coagulation of the casein proteins through the action of an enzyme (chymosin) or acid, the remaining watery and thin liquid is named whey. Lactose makes up a high proportion (>75%) of the total whey solutions and is considered as one of the most polluting food by-product streams (40–70 g L−1 biochemical oxygen demand (BOD) and 60–80 g L−1 chemical oxygen demand (COD)).6,7 Therefore, utilization and exploitation of whey lead to a reduction of environmental concerns.6Due to the abundance of different hydroxyl groups in lactose, it is necessary to investigate the behavior of catalyst to achieve a “directed catalytic synthesis” for selective lactose esters. Regioselectivity is the preference of chemical breaking/bonding in one direction over all other feasible directions, while chemoselectivity is determining which group reacts. The establishment of regio- and chemoselective conversion is one of the most important assignment in chemical industries because it increases material consumption, superfluous trimming and re-functionalization steps in process.7 Therefore, regio- and chemoselective esterification of sugars is a laborious task related, principally, to their multifunctionality.
In general, carbohydrate-based surfactants are synthesized from sugars and free fatty acids with chemical or biological catalysts. The enzymatic synthesis of sugar esters is typically characterized by regio- and chemoselectivity, excellent conversion, and a high reaction rate compared to chemical catalysts.7–10 However, uneconomical process, inactivation of enzymes in organic solvents (such as DMSO, DMF, and pyridine), and varied activity and reaction conditions in the esterification of different sugars are of the disadvantages of enzymatic synthesis of sugar esters.11 Other catalytic systems, such as Brønsted acids, Lewis acids, metal oxide, resins, clays, and zeolites have been applied instead of costly enzymatic catalysts for the synthesis of sugar esters.7 These catalysts are easily available, cost-effective, and easy to perform.7,12 However, this catalytic synthesis is carried out at a high temperature, has poor chemo-and regioselectivity, has harsh reaction conditions, and leads to the formation of colored by-products. Although zeolites are introduced as efficient catalysts, their application is limited by drawbacks such as deactivation by irreversible adsorption, steric blockage of heavy secondary products, and unsuitable microspores for bulk molecules.13
As an ideal macromolecule, dendritic polymers and their composites have attracted considerable attention because of their unique fractal architecture and physicochemical properties, including their branched three-dimensional framework, nanoscale size, biocompatibility, monodispersity, solubility, high stability, precise molecular weight, tunable terminal functionalities, small inherent viscosity, low toxicity, low cost and other peculiar characteristics. The end terminal functional group on the dendrimer can conjugated with several diverse chemical entities such as ligands, and metals for performing chemical process such as drug/gene delivery, tissue engineering, cancer therapy, biosensing, degradation of pollutant, and catalysis.14,15 Another unique property of dendritic catalysts is that the metallic active sites are very attainable on the periphery.14 These properties are believed to be related, principally, to their globular shapes, tunable nanoscale size, and spatial structure. Therefore, dendrimers are excellent candidates for catalysis as their 3D dimensional structure possess modifiable surfaces with tune-ability, which extended network of peripheral functional group.14–16
One of the catalytic systems not yet used in sugar fatty acid synthesis is dendrimers/dendrons. Therefore, we are the first to report the synthesis of lactose-based surfactant with emulsifying properties by a novel dendritic catalyst. Our strategic planning focuses on combining the prominent features of magnetic beads core/dendrimers shell composite, linker agents, active centers on the periphery, and their applications for the synthesis of a biosurfactant. Highlights of this novel design are to homogenize heterogeneous catalysts, comparative assessment of active centers encapsulated in silica-coated nano magnetic beads (SCMBNP) core/dendrimers shell composite, magnetic separation, prevention from coagulation by the coating of the magnetic core by a silica shell, and fabrication of metallic dendritic catalyst with good monodispersity, abundant surface functional groups, high solubility in reaction media, high capacity for loading metals and also excellent catalytic efficiency in synthesis of a biosurfactant.
The dendronizing method can generate hydrophobic arms on the magnetic nanoparticles core enhancing its compatibility with the organic media and minimize the support for the immobilization of dendritic composite, and the surrounding attached metallic ion can act as a single catalytic site. In fact, the novel synthesized SCMBNP core/dendrimers shell composite has the capacity to encapsulate the insoluble single metallic ion and transport it into the organic media. Additionally, in SCMBNP core/dendrimers shell as an efficient catalyst in the chemo- and regioselective synthesis of lactose fatty ester has been reported for the first time. Pure lactose (PLac) and raw lactose (RLac) obtained directly from whey as alcohol acceptor and dodecanoic acid (LauA, lauric acid) as acyl donor were used as substrates in the magnetic dendrimer-catalyzed esterification.
2. Experimental section
2.1. Equipment for dendrimer characterization and LML detection
The different instruments applied for the detection of synthesized nanodendrimers & surfactants are listed below: CHN (C, H & N elemental analysis, Rapid Heraeus elemental analyzer (Wellesley MA); ICP-AES (Inductively Coupled Plasma Atomic Emission Spectroscopy, PerkinElmer ICP/6500); DLS (Dynamic Light Scattering, Zetasizer Nano-ZS-90 (ZEN 3600,Malvern Instrument)); TEM (Transmission Electron Microscopy, Philips 501 microscope, 80 kV voltage); SEM (Scanning Electron Microscope, Tecnai F30TEM operating at 300 kv); FT-IR (Fourier Transform Infrared Spectroscopy, Shimadzu Varian 4300 Fourier Transform Infrared spectrometer, KBr pellets); TGA (Thermogravimetric Analysis, PerkinElmer TG-DTA 6300, heating rate of 15 °C min−1); XPS (X-ray Electron Spectroscopy, PerkinElmer PHI 5000CESCA system, B. P = 9–10 Torr; VSM (Vibrating-Sample Magnetometer, BHV-55 VSM); XRD (X-ray Diffraction, Bruker D8 Advance diffractometer, CuKα radiation, 40 kv, 20 miliamper); HPLC (High Performance Liquid Chromatography, Agilent 1200 LC System, Detector = Binary SL Pump & Diode Array Detector and a Shodex RI-501 Refractive Index Detector, mobile phase, HPLC grade solvents (MeOH/H2O (85/15, v/v)), flow-rate = 0.2 mL min−1, t = 50 min, T = 40 °C). All dried samples were dissolved in MeOH/H2O (85/15, v/v), filter (13 mm PVDF syringe filter with 0.45 μm pore size); GC/MS (Gas Chromatography Mass Spectroscopy, HP 6890/5973 GC/MS, Shimadzu GC-16A gas chromatograph (GL-16, 5 m−3 mm OV-17 column, 60–220 °C (10 °C min−1), Inj. 230 °C, Det. 240 °C); NMR (Nuclear Magnetic Resonance, Bruker INOVA 500 NMR, Solvent (d6-DMSO)); & Goniometer (ramé-hart goniometer, model OCA 15 plus).2.2. Preparation of nanodendrimers
To prepare bare-nano SCMBNPs, a well-known co-precipitation method was employed.15–17 An aqueous solution of FeCl3·7H2O and FeCl2·4H2O (FeIII/FeII: 2:1) were added to 10 mL HCl (2 M) under a nitrogen atmosphere for 0.5 h. Then, 50 mL of NH3 (37 wt%) solution was added drop-wise which a dark black solid product obtained until the pH value was reached 10. After stirring under nitrogen atmosphere for 0.5 h, the final solution was heated at 70 °C for 0.5 h. The resulted black solid was separated by a magnet and washed with deionized water and dried under vacuum at 80 °C for 13 h. Then, the prepared magnetic nanospheres were dispersed in EtOH/H2O (2:1). The obtained mixture solution was homogenized by ultrasonication for 0.5 h and then 3 mL NH3 (37% w) solution was added drop-wise to the dispersion and stirred vigorously. The silanization of the magnetite core was performed to increase the OH groups on the surface, prevent the aggregation of MBNPs and eliminate the solubility problems in organic solvents when dendrimers were directly grown onto the surface of the MBNPs. The mixture of 0.5 mL tetraethylorthosilicate (TEOS, Sigma Aldrich) in 15 mL EtOH was injected in combination. Then, the process was carried out for 100 min, the magnetic solid was separated by a magnet and washed with EtOH (3 × 10) and H2O (3 × 20) and dried for 10 h. In the end, dark yellow SCMBNPs powder was obtained. In the following steps, the SCMBNP-cored dendrimers grew from the magnetic cores via a step-wise divergent synthesis approach. The G0.0L1@SCMB & G0.0L2@SCMB particles were obtained by refluxing 5.2 g SCMBNPs with 3.5 mL 3-aminopropyl)trimethoxysilane (L1, APTMS, Merck) and 4.0 mL N-3-(trimethoxysilyl) propyl ethylenediamine (L2, 2-AE-3-APTMS, Merck) in 100 mL anhydrous dichloromethane (DCM, Sigma Aldrich) for 24 hours (Fig. 1). The bulk product was filtered off and washed with DCM/MeOH and dried in a vacuum oven at 100 °C for 8 h to prepare G0.0L1@SCMB and G0.0L2@SCMB. 10.0 grams of G0.0L1@SCMB or G0.0L2@SCMB particles were dispersed in 200 mL MeOH and 80 mL methyl acrylate (MA, Merck). The obtained mixture was ultrasonicated and refluxed for 3 h. The solid product (G0.5L1/2@SCMB) was washed with MeOH, and then the mixture was refluxed for 12 h after adding 3.0 g Bis (phthaloyl) diethylenetriamine (BPDEA) as a G1.0L1/2@SCMB or G1.0L2@SCMB. MA and BPDEA were grafted onto the G0.5L1/2@SCMB surface as the precursors of a greatly branched and multi-functionalized G0.5L@SCMB. Then, removal of phthaloyl protective group with N2H4. The resultant G1.0L1@SCMB or G1.0L2@SCMB particles were collected by magnet stick and dried under a vacuum oven. Then, the previous steps are repeated to prepare the second generation dendrimer. In the next step, alkylation with cyanuric chloride ((NCCl)3, Sigma Aldrich) in a solution of N,N-diisopropylethylamine (DIPEA, Merck), and tetrahydrofuran (THF, Sigma Aldrich) for 2 h under Ar atmosphere at <15 °C.15 And then, metformin (Met, Merck), DIPEA in dry dimethylformamide (DMF, Merck), and the G2.0L1@SCMB or G2.0L2@SCMB were dispersed under vigorous stirring at Ar atmosphere at 80 °C for 24 h. The resultant magnetic nano Met-G2.0L1/2@SCMB was collected by an external magnet and washed with DCM/DMF sequentially.15–17 The generated nanocomposite (Met-G2.0L1/2@SCMB particles) were isolated by an external magnet and washed with EtOH/MeOH (50/50, v/v) and toluene (PhMe, Sigma Aldrich) and finally dried at 60 °C for 12 h to afford a brown solid. The functionalized Met-G2.0L@SCMBs that were synthesized with the Met, are identified as G2.0L1@SCMB and G2.0L2@SCMB.Then, Mn/Co-G2.0L2@SCMB were prepared by stirring 60 mg of the G2.0L1@SCMB or G2.0L2@SCMB with 5.4 mmol of Co(OAc)2·4H2O and Mn(OAc)2·4H2O in EtOH (40 mL) under reflux for 24 h. After cooling, the resulting magnetic CoII/MnII-G2.0L1/2@SCMB NPs were isolated by using a magnet bar and washed as like the previous step and finally dried at 50 °C for 12 h to afford a brown solid.
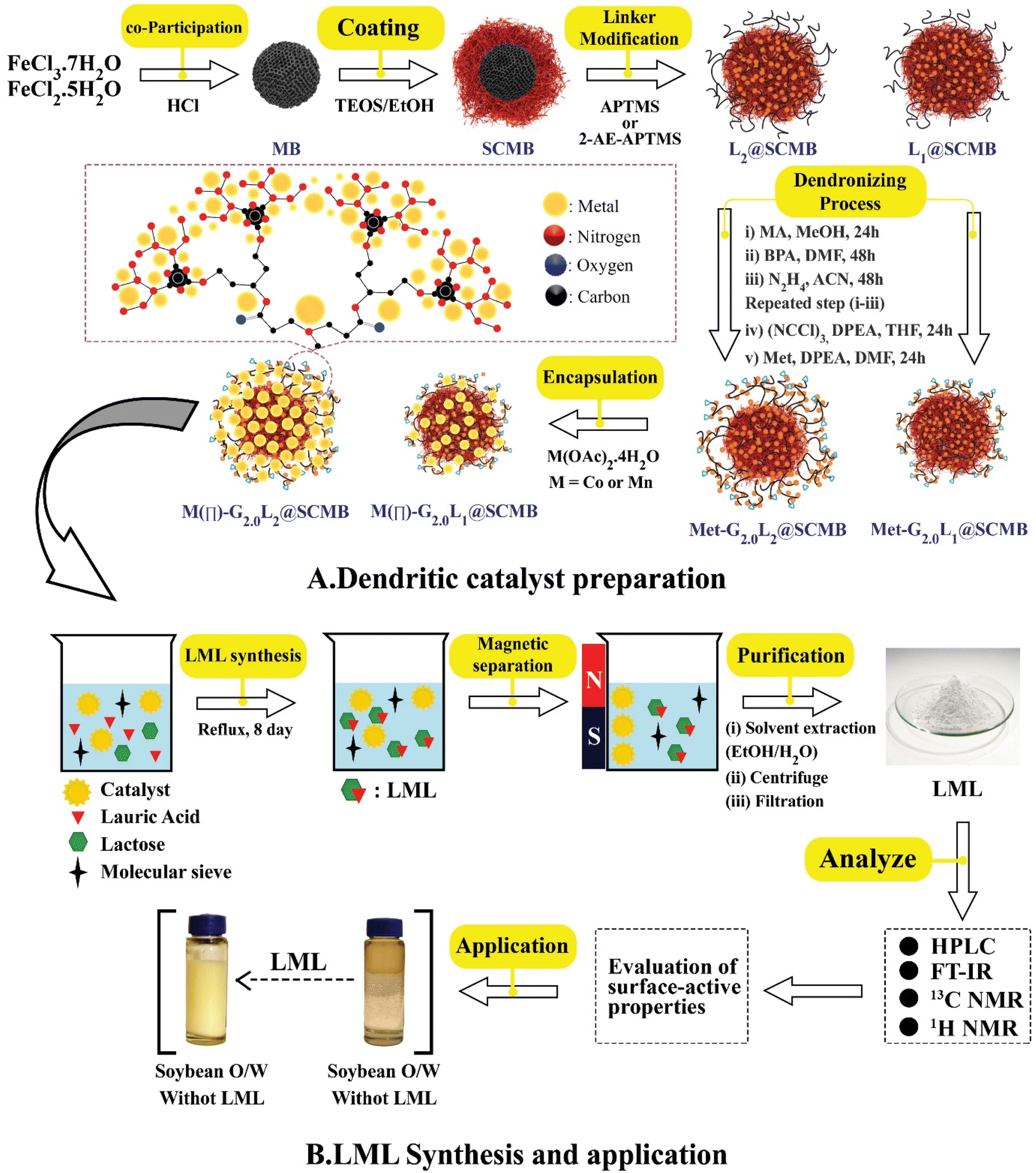 | ||
| Fig. 1 (A) The fabrication flow chart of MII-G2.0L1/2@SCMB (B) the catalytic synthesis of lactose mono lauryl ester (LML) biosurfactant and its application. | ||
2.3. Catalytic production of sugar ester
Synthesis of emulsifier was assembled in a 25 mL glass vial with a plastic cap to prevent solvent evaporation during the long reaction times. 5.0 mL solvent (acetone (ACTN) or acetonitrile (ACN), dried using molecular sieves) was added to 0.25 g lactose monohydrate (Lac, Sigma Aldrich), the desired amount of nano dendritic catalyst (30.0–40.0 mg), activated molecular sieves (MS, 60.0 mg) and 0.280 g lauric acid (LauA, Merck) with the 1:2 molar ratio of Lac:LauA. All reactions were carried out at 50 °C in a shaking water bath. Then, the magnetic nanodendrimer was retrieved using a magnetic stick, washed well with water and ethanol, dried and used in further runs. Aliquots were removed from each vial daily for HPLC analysis.
2.4. Purification of LML
For the purification of LML by solvent extraction method, the applied organic solvent of reaction was evaporated in a vacuum drying oven at 50 °C for 24 h. The extraction of dry LML was carried out in a 50% mixture of EtOH–H2O (40 mL). The mixture was shaken for 4 h in a water bath to extract biosurfactant from the unreacted precursors and byproducts at 50 °C. Then, the obtained homogenized mixture was centrifuged at room temperature for 10 min at 10000 g. The supernatant was filtered and dried in a vacuum drying oven at 50 °C for 48 h to achieve the dried of LML. The obtained sugar ester was identified by thin-layer chromatography (TLC) by silica gel plate using CCl4/n-C6H14 as eluting solvent & iodine tank. In the catalytic process, reaction yields were measured based on the sample's dry weight. The maximum theoretical yield (22.7 mg mL−1) was calculated based on molecular weight (524.2 g mol−1 from GC-MS) for LML as a product and Lac as a limiting substrate. The purity of LML was verified to be greater than 90% by HPLC analysis.
2.5. Preparation of model emulsions
Soybean oil was purchased from a local market (Tehran, Iran) and used for the preparation of emulsion samples. The 20 mL soybean oil was mixed with 80 mL water in a round baker for 10 min to prepare 20% oil-in-water (O/W) samples. The resulting sample (soybean/water 20% v/v) was treated with the synthesized emulsifier at various concentrations. LML (aq) was stirred for 20 min before the addition of 20 mL soybean oil. Immediately, the mixture was vigorously mixed with a high-speed blender (10000 rpm) for 15 min at room temperature and then passed through a microfluidizer. Afterward, emulsion destabilization & emulsification indices were measured (Fig. 1B).
2.6. Surface-active properties
In order to evaluate the emulsifying properties of LML, the hydrophilic–lipophilic balance (HLB) number, critical micelle concentration (CMC), surface tensions at the CMC (γCMC) and minimum surface tensions (γmin), foamability (FA), foaming stability (FS) and surface excess (Γ in mol m−2), area per molecule (A in Å2), Gibbs free energy of adsorption (ΔG in kJ mol−1), emulsifying ability (EmA) and emulsion stability (EmS) of LML were determined. The methods utilized for such purposes are detailed as follows:
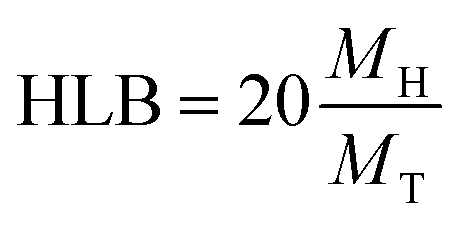 | (1) |
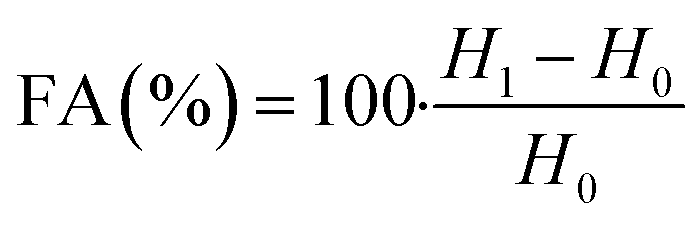 | (2) |
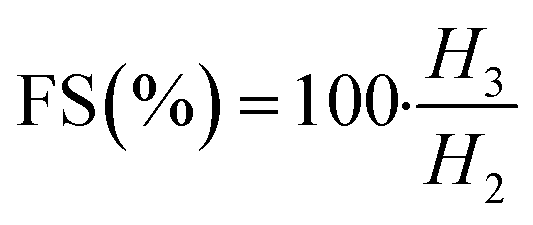 | (3) |
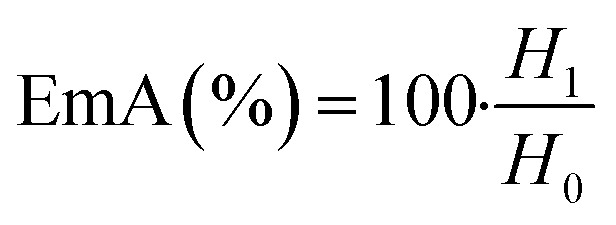 | (4) |
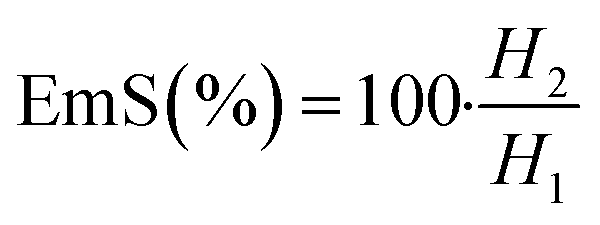 | (5) |
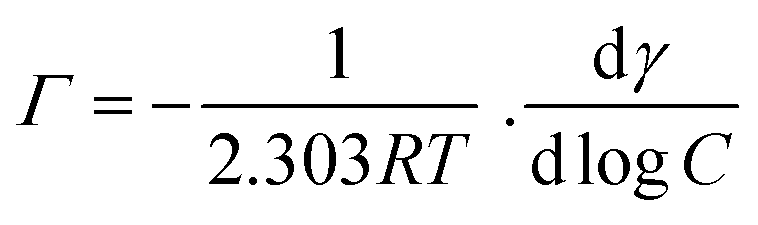 | (6) |
The A depicts the mean area accessible to each molecule forming monolayers. The area of an adsorbed molecule at the surface can be measured using the following equation:
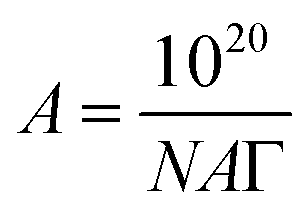 | (7) |
The ΔG of an aqueous solution of LML at the liquid surface can be measured using the following equation:19–21
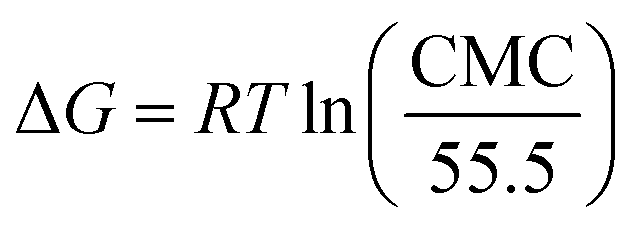 | (8) |
3. Results and discussion
3.1. Structural characterization of nanodendrimers
The physicochemical properties of magnetite beads catalysts, the BET nitrogen adsorption and desorption analyses, and the corresponding SBET (N2 specific surface area), MPD (pore size), and VBJH (pore volume) are listed in Table 1 for all samples. The SBET, MPD, and VBJH of the CoII-G2.0L@SCMBNPs and MnII-G2.0L@SCMBNPs in comparison with magnetic support were decreased (Table 1). These data illustrated that the presence of the organo-modifier on the magnetic pristine decreases the SBET and VBJH by covering the micropore on the magnetic pristine. The low SBET and VBJH value of CoII-G2.0L2@SCMBNPs might be caused by higher metal loading in comparison to other catalysts (Table S1†). The elemental content of the samples was routinely evaluated by AAS (atomic absorption spectroscopy), ICP-OES (induced coupled plasma), and CHN combustion microanalysis. Additionally, throughout the fabrication of the nanodendrimers (G0.5 to G2.0), the identical concentration of L1 and L2 and the excessive amounts of other modifier compounds (MA, BPEA, (NCCl)3 and Met) were used. This excessive amount is related, principally, to decrease the amounts of the unspoiled residual active sites on the SCMBNPs. A change in the color (from yellow to dirty red) of solid products illustrated that the condensation reactions occurred in these stages. In order to determine N-content in the nanodendrimers (G0.5 to G2.0), supplementary assessments were carried out by AAS and BT (back titration) procedure (Table 1). The MII-G2.0L@SCMBNPs were dispersed in HCl (1.0 mM) and stirred for 1 h. The nanodendrimer was collected by a magnet stick and then the supernatant solution was separated. The obtained solution was titrated against a solution of NaOH (1.0 mM) and the equiv. (equivalence point) was determined. From the equiv. point, CHN, and BT values, the N-content in the samples was calculated (see Table S1†).| No. | Catalyst | Lac | CAa (°) | IFTb (mN m−1) | SFTc (mN m−1) | CMCd (mg mL−1) | HLBe |
|---|---|---|---|---|---|---|---|
| a Contact angle of 3.6 mM of aqueous solution of LML on typical microscope slid.b Interficial tension of 3.6 mM of aqueous solution of LML.c Surface tension of 3.6 mM of aqueous solution of LML.d Critical micelle concentration.e Hydrophilic lipophilic balance measured by equiv. HLB = (MH/MT).20, MT = 524.28 g mol−1.f Condition reaction: ([Co-Cat] = 30.0 mg, [Lac] = 250 mg, [LauA] = 278 mg, T = 50 °C, [MS] = 600 mg, ACN = 6 mL).g Condition reaction: ([Mn-Cat] = 40.0 mg, [Lac] = 250 mg, [LauA] = 278 mg, [MS] = 600 mg, T = 50 °C, ACN = 6 mL). | |||||||
| 1 | CoII-G2.0L2@SCMBf | PLac | 77.62 ± 2.9 | 11.07 ± 0.9 | 25.12 ± 0.4 | 0.55 | 14.1 |
| 2 | CoII-G2.0L2@SCMB | RLac | 58.12 ± 3.2 | 10.73 ± 2.5 | 21.73 ± 1.3 | — | 14.1 |
| 3 | MnII-G2.0L2@SCMBg | PLac | 75.38 ± 3.6 | 11.92 ± 0.5 | 26.38 ± 0.7 | 0.63 | 14.1 |
| 4 | MnII-G2.0L2@SCMB | RLac | 55.03 ± 4.2 | 10.42 ± 2.3 | 22.53 ± 2.1 | — | 14.1 |
The content of linked Met on (NCCl)3 was determined by the BT method which is equivalent to a loading of 1.51 and 1.78 mmol of Met per gram of G2.0L1@SCMBNPs and G2.0L2@SCMBNPs, respectively. The dendrimer scaffold has two main sites for metal loading by using various paths: (i) void spaces, and (ii) branching points; and outside surface groups by electrostatic interactions. Therefore, the amount of Co and Mn were estimated, 1.02 ± 0.001 mmol g−1 and 0.97 ± 0.001 mmol g−1 by AAS analyses and, 1.03 and 0.98 by ICP, respectively. Furthermore, the loading rate (the attached average metal ions/surface Met molecules) of MnII-G2.0L2@SCMBNPs and CoII-G2.0L2@SCMBNPs dendrimer are 54 and 57, respectively.
These analyses illustrate that nano metals encapsulated on G2.0L2@SCMBNPs are stabilized by the dendrimer framework. Furthermore, the cobalt and manganese encapsulated on G2.0L1@SCMBNPs by ICP-OES analysis were measured 0.89 ± 0.001 mmol g−1 and 0.83 ± 0.001 mmol g−1, respectively. The results showed that the densities of metal on the surface of G2.0L1@SCMBNPs were not similar. The ICP-OES and AAS analysis of the four CoII/MnII-G2.0L1/2@SCMBNPs demonstrate that magnetic dendrimers containing the L2 as a linker have higher metal loading than two other dendrimers. Obviously, the difference between metal loading leads to a difference in catalyst efficiency, which was confirmed by our experimental results.
In the FT-IR spectra, very important differences among bare-nano SCMBNPs, CoII-G2.0L2@SCMBNPs and MnII-G2.0L2@SCMBNPs were detected (Fig. 2A). In this paper, the (ν), (σ), (s), and (as) represents stretching, bending, symmetric and asymmetric vibration, respectively. The characteristic bands at 545–645, 983, 1095, 1110 and broad band at 3100–3420 cm−1 are attributed to the νs (Fe–O), νas (Si–O–Si), νs (Si–O) and νs (Si–OH), respectively. These peaks confirm the formation of a continuous and very fine layer of inert SiO2 on the surface of the magnetic beads. The primary amine N–H stretches overlapped with Si–OH, Si–Si, and adsorbed water molecules stretches in the FT-IR spectrum of G0.0L1@SCMBNPs and G0.0L2@SCMBNPs. The sp3 H2C–H stretching and NH2 bending vibrations appeared at around 2810 and 1565 cm−1, respectively. Several weak peaks in the area of 1440–1560 cm−1and 2860–2930 cm−1 are attributed to νas (C–C) and νas (C–N) in L1 or L2.15–17 Secondary amine N–H stretches were observed as a very weak peak about 3000–3100 cm−1. Additionally, secondary amine N–H peaks and the N–H deformation peaks appeared at 3000–3100 cm−1 and 1500–1580 cm−1 in the FT-IR spectra of G0.0L1@SCMBNPs and G0.0L2@SCMBNPs, respectively. The appearance of a new peak at 1737 cm−1 demonstrates the attachment of MA to the NH2 groups of the L1 or L2.
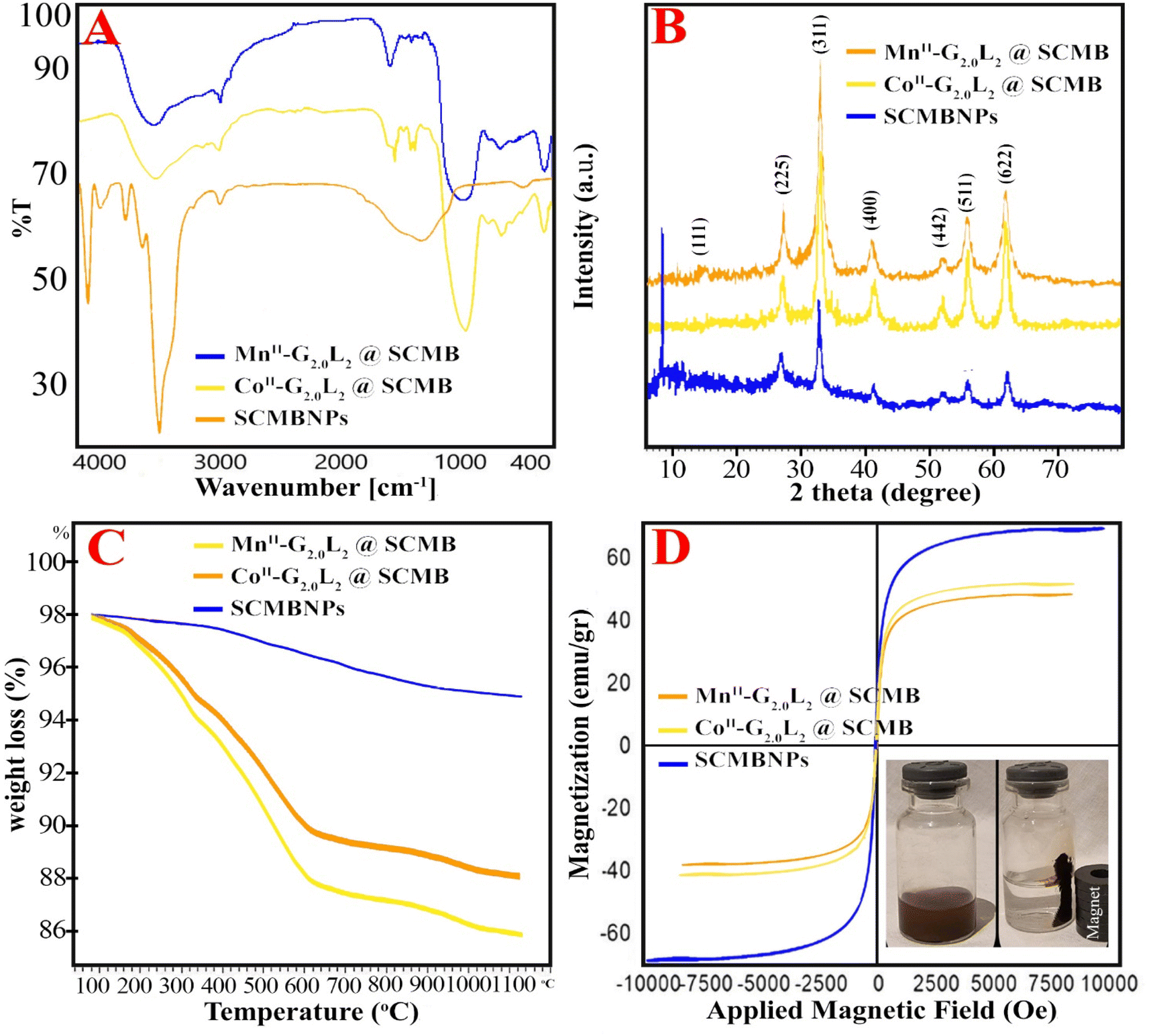 | ||
| Fig. 2 (A) FT-IR, (B) XRD pattern, and (C) TGA profiles, (D) Magnetization curves obtained by VSM of CoII-G2.0L2@SCMB and MnII-G2.0L2@SCMB, (E) XPS analyses of CoII-G2.0L2@SCMB and (F) XPS analyses of MnII-G2.0L2@SCMB. | ||
In the following, nanodendrimer synthesis is performed using a protection/deprotection strategy. Characteristic bands at 1533 and 1710–1778 cm−1 and 1535 cm−1 indicate the phthaloyl group in the mentioned nanodendrimers.15 It can be seen that after 48 h most of the related peaks to the phthaloyl groups were disappeared which confirmed the deprotection of the immobilized amine groups by hydrazine. Subsequently, the appearance of medium peaks at around 1605–1640 cm−1, confirmed the condensation reaction between (NCCl)3 with G1.0/2.0L@SCMBNPs. The strong peaks around 1570–1650 cm−1 and 1250 cm−1 suggest the presence of (NCCl)3 ring.15,16 The weak and new peaks at 417 and 440 cm−1 were assigned to the metal-nitrogen stretching vibration of nanodendrimers after the complex formation of Mn and Co with grafted metformin over modified Met-G2.0L@SCMBNPs, respectively.15–17 The FT-IR spectrum of the nanodendrimers clearly illustrated that the bare-nano SCMBNPs have been successfully modified with organic compounds and heterogenized.
The XRD reflexes of pristine and nano dendrimers are depicted in Fig. 2B. Initially, the diffraction pattern of all samples could be indexed to a fcc (face-centered cubic) structure. Those sharp peaks correspond to the cubic spinal structure of bare-nano SCMBNPs and other samples which attributed to 18.5° (1 1 1), 30.8° (2 2 0), 37° (3 1 1), 44° (4 0 0), 57° (4 2 2), 58.3° (5 1 1) and 64° (4 4 0) plane of the crystal. After the modification process, a little broadened pattern appeared which related to SCMBNPs non-crystalline nature at 15–80°. The weak-broad peaks of cobalt and manganese roughly appeared which had convergence with MII-G2.0L2@SCMB peaks and caused, principally, the increase in the peak intensity at 44–58°.15,17 The data in Fig. 1B lead to the conclusion that the crystal structure and peak intensifying of modification and encapsulation do not remarkably change after the modification process, and no phase separation is observed for all of the samples. The average size of CoII-G2.0L2@SCMB and MnII-G2.0L2@SCMB were determined as about 25 and 28 nm from Debye–Scherrer's equation, respectively.22,23
The thermal stability of all dendritic catalysts and evaluating the composition of samples were done by the thermogravimetric analysis (TGA) under a 5 °C min−1 ramp rate in a nitrogen gas atmosphere. The results showed a little difference in weight loss protocol which corroborate similarity in their structure. The TGA plot starts at 50 °C with a gentle slope and continues to 200 °C and again continues to 800 °C, then stays without changes (Fig. 2C). The first weight losses of physically adsorbed water molecules on the samples were observed at <115 °C. The desorption of water molecules on the dendrimers and decomposition of grafted organic parts are shown at 120–410 °C. Probably, the residual weight is related to the presence of metal oxide and magnetic nanocomposite. The TGA plots of the nanodendrimers indicated that at temperatures below 410 °C, the total weight loss was only 17.5–19.3%. This result illustrated that in the optimum temperature for this reaction (50 °C), the prepared nanodendrimers were completely stable and no weight loss is expected. Also, from TGA plots, the following results are concluded: the metals and organic compounds well immobilized on the pristine, the powerful interaction between the peripheral moiety and surface of pristine, and the little differences in weight loss are related, principally, to the differences in the structure of dendrimers, metal loading and kind of metal (Fig. 2C).
Magnetic properties of the naked SCMBNPs and nanodendrimers were evaluated by the vibrating-sample magnetometer study at room temperature (Fig. 2D). The VSM magnetic hysteresis curves of samples exhibited that all nano dendrimers have magnetic properties; and show diminish compared to naked SCMBNPs. The Ms (magnetization saturation) were found to be 63.4, 47.1, and 43.5 emu g−1 for SCMBNPs, CoII-G2.0L2@SCMB, and MnII-G2.0L2@SCMB, respectively. Against a variable applied magnetic field (−10000 to +10000 Oe), the Ms of SCMBNPs is greater than that of our nanodendrimers. The lower Ms for the nanodendrimers can be related to the incorporation of non-magnetic SiO2 and organic compounds over the iron oxide surface. Still, the latter stands up with adequate magnetism and can be easily attracted by external magnets (inset Fig. 3D). Based on these observations, the following results are concluded: the hysteresis curve is small, the remanence and coercivity are close to zero, the large surface area of nano dendrimers permit the easy spin of the electrons located on the surface of atoms in response to even weak magnetic fields, therefore, the nano dendrimers possess super-paramagnetic behavior and can be effortlessly removed from the reaction medium, and the decrease in Ms value in higher generation dendrimer grafted catalysts is attributed, mainly, to the formation of condensed shell and this, in turn, reflects more non-magnetic behavior.23,24
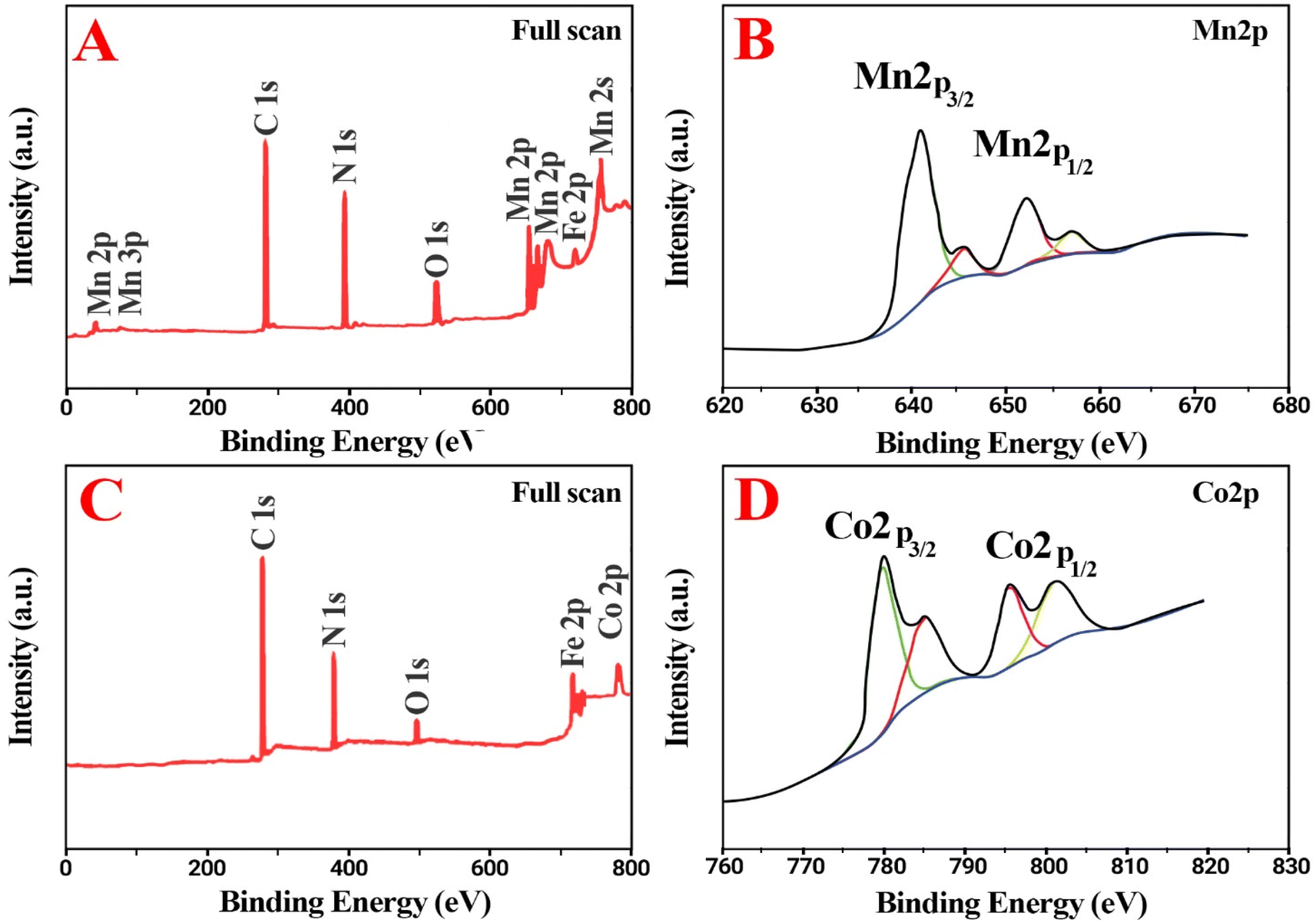 | ||
| Fig. 3 XPS analyses of CoII-G2.0L2@SCMB (A and B) and of MnII-G2.0L2@SCMB (C and D). | ||
The X-ray Photoelectron Spectroscopy (XPS) analyses were done for elucidation of the valence state of the metal nanoparticles in dendrimers (Fig. 3). In dendrimers including manganese and cobalt, the 2p core-level peaks are recorded with high resolution and the characteristic 3p peaks are observed at 34–70 eV BE (Binding Energy).24,25 The XPS spectrum of superior catalyst “CoII-G2.0L2@SCMB and MnII-G2.0L2@SCMB” (Fig. 1E and F) shows the availability of the core-elements such as C (1s), N (1s), O (1s), Fe (2p1/2), Fe (2p3/2), Co (2p1/2), Co (2p3/2) and Mn (2p1/2) and Mn (2p3/2) with the BE 287.4, 401 & 530.78 for C (1s), N (1s) & O (1s), 536–541 for Fe2+, 710–720 for Fe3+, 778–790 eV for Co (2p) and 639–650 for Mn (2p), respectively. As shown in Fig. 2F, this curve is entirely in accordance with asymmetric Mn 2p3/2 peak in Mn at oxidation state +2 which is found at 638.64 ± 0.06 eV with a 2p3/2 to 2p1/2 splitting of 11.10 ± 0.02 eV. The 2p3/2 to 2p1/2 splitting in nanodendrimer is around 11 eV that verifies oxidation sate 2+ for Mn in MnII-G2.0L2@SCMB.24–27 Additionally, The Co(2p3/2) and Co(2p1/2) in Co–N environment generated peaks at 778–790 eV, which is in agreement with +2 oxidation state for cobalt in CoII-G2.0L2@SCMB.26,27
The detailed composition, morphological structure, shape & size of the CoII-G2.0L2@SCMB and MnII-G2.0L2@SCMB were ascertained by FESEM and TEM studies (Fig. 4). From Fig. 4 inferred that surface morphology of nano dendrimers were smooth spherical shape covered by an even distribution of nearly spherical particles, with average diameters near 26–30 nm. Also, the SEM micrographs shown in Fig. 4 indicate that each core–shell nanodendrimer has heterogeneous surface which forms irregular agglomerated globules with diameters from 50–100 nm. This agglomerate formation reveals that the surface energy of the samples is relatively strong. In fact, the surface modification over SCMBNPs by organic molecule and metals can be adjudged from its appearances.28,29 The elemental mapping obviously exhibit that metals dispersed uniformly on the surface of naked SCMBNPs as shown in Fig. 4 https://www.nature.com/articles/s41598-018-19551-3 – Fig. 1. Further, the mapping analysis of individual elements illustrate the presence of the anticipated elements in the chemical composition of the nano dendrimers, namely C, N, O, Si, Fe, Co and Mn. The TEM images of samples shows the most determined particle sizes are in the nanometer ranges and the nano particles are distributed across the naked SCMBNPs surface.
 | ||
| Fig. 4 (A) Microscopic analyses of CoII-G2.0L2@SCMB (A-I) SEM, (A-II) SEM-map, (A-III) TEM image, (B) microscopic analyses of MnII-G2.0L2@SCMB (B-I) SEM, (B-II) SEM-map, (B-III) TEM image, (C) TEM image of recovered MnII-G2.0L2@SCMB (C-I) after 3 run, (C-II) after 5 run, (C-III) after 6 run. | ||
3.2. Identification of LML
The atom numbering, FT-IR, 1H NMR & 13C NMR spectra of the Lac, LauA, and LML are depicted in Fig. 5. FT-IR spectra of Lac, LauA, and LML are shown in Fig. 5A. As it is seen in the spectrum of LML, the structure is verified by the IR bands located at 3300–3400 cm−1 (νas O–H), 1724 cm−1 (νas C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 700–740 & 1472 cm−1 (σs CH2), 1372 cm−1 (σs CH3), and 1050–1070 cm−1 (νas C–O). By comparing the spectrum of LML with LauA, it is clear that the carbonyl bond in the LauA at 1695 cm−1 (νas CO) shifted to 1724 cm−1 (νas CO) in spectrum of LML.3,4,7 These results confirm that the LML is successfully synthesized by the esterification reaction of Lac and LauA in the presence of nanodendrimers and molecular sieves. Analysis of 1H NMR and 13C NMR illustrated that the LML esterified at the C6′ carbon with Lac (Fig. 5B and C). The chemical shifts (δ ppm) in 1H NMR (400 MHz, d6-DMSO) of LML observed at δH = 0.84 (s, 3H, C-12L), 3.9–5.1 (s, 3H,C-12L), (m, 8H, C-2 to C-5 & C-2′ to C-5′), 1.32–4.4 (m, 3H,C-2L to C-11L), 1.32–4.4 (m, 3H,C-2L to C-11L), 6.4 (s, 1H, C-1), 6.7 (s, 1H, C-1′). Also, the characteristic signal in 13C NMR (75 MHz, d6-DMSO) of surfactant appeared at δC = 173(C-1L), 104(C-1′), 92(C-1), 81(C-4), 73(C-5′), 72.7(C-3′), 64 (C-6′), 83(C-5), 89(C-4′), 34(C-2L), 25(C-3L), 28–30(C-4L to C-11L), 14(C-12L). The results illustrate that position of esterification is C-6′. The reasons for this regioselectivity are the following: (1) the order of esterification reaction are alcohol 1° > alcohol 2° > alcohol 3°, based on this order, the C-6′ and C-6 have more sterically available instead of –OH in other position of the lactose, (2) a downfield shift of C-6′ in 13C NMR from 61 in Lac to 64 in LML and the upfield shift of C-5 from 75 in Lac to 73 in LML indicates the occurring of reaction at C-6′. The comparison between spectra obtained for surfactant and raw material (Fig. 5) illustrated that the appeared signals for LML were almost identical to those of literature.11 It should also be stated that the signals of solvent (DMSO) have appeared in 39.5 and 2.50 in 13C NMR and 1H NMR, respectively.7,11 Therefore, it can be concluded that the NMR spectra also confirm the reported data for the catalytic synthesis of LML by nanodendrimers (Fig. 5B and C).
O), 700–740 & 1472 cm−1 (σs CH2), 1372 cm−1 (σs CH3), and 1050–1070 cm−1 (νas C–O). By comparing the spectrum of LML with LauA, it is clear that the carbonyl bond in the LauA at 1695 cm−1 (νas CO) shifted to 1724 cm−1 (νas CO) in spectrum of LML.3,4,7 These results confirm that the LML is successfully synthesized by the esterification reaction of Lac and LauA in the presence of nanodendrimers and molecular sieves. Analysis of 1H NMR and 13C NMR illustrated that the LML esterified at the C6′ carbon with Lac (Fig. 5B and C). The chemical shifts (δ ppm) in 1H NMR (400 MHz, d6-DMSO) of LML observed at δH = 0.84 (s, 3H, C-12L), 3.9–5.1 (s, 3H,C-12L), (m, 8H, C-2 to C-5 & C-2′ to C-5′), 1.32–4.4 (m, 3H,C-2L to C-11L), 1.32–4.4 (m, 3H,C-2L to C-11L), 6.4 (s, 1H, C-1), 6.7 (s, 1H, C-1′). Also, the characteristic signal in 13C NMR (75 MHz, d6-DMSO) of surfactant appeared at δC = 173(C-1L), 104(C-1′), 92(C-1), 81(C-4), 73(C-5′), 72.7(C-3′), 64 (C-6′), 83(C-5), 89(C-4′), 34(C-2L), 25(C-3L), 28–30(C-4L to C-11L), 14(C-12L). The results illustrate that position of esterification is C-6′. The reasons for this regioselectivity are the following: (1) the order of esterification reaction are alcohol 1° > alcohol 2° > alcohol 3°, based on this order, the C-6′ and C-6 have more sterically available instead of –OH in other position of the lactose, (2) a downfield shift of C-6′ in 13C NMR from 61 in Lac to 64 in LML and the upfield shift of C-5 from 75 in Lac to 73 in LML indicates the occurring of reaction at C-6′. The comparison between spectra obtained for surfactant and raw material (Fig. 5) illustrated that the appeared signals for LML were almost identical to those of literature.11 It should also be stated that the signals of solvent (DMSO) have appeared in 39.5 and 2.50 in 13C NMR and 1H NMR, respectively.7,11 Therefore, it can be concluded that the NMR spectra also confirm the reported data for the catalytic synthesis of LML by nanodendrimers (Fig. 5B and C).
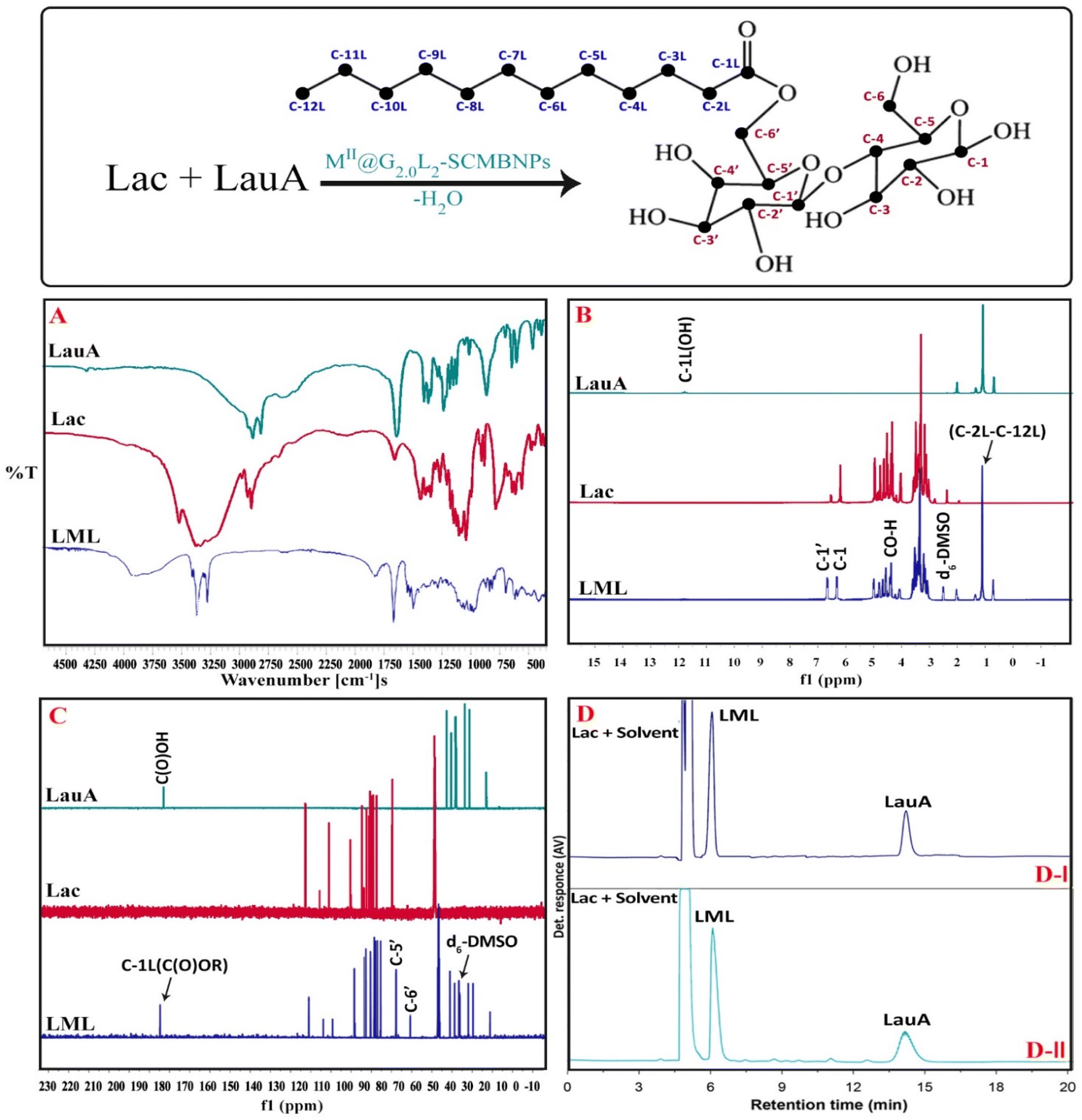 | ||
| Fig. 5 (A) FT-IR spectra of Lac, LauA and LML; (B) 1H NMR (400 MHz, d6-DMSO) of Lac, LauA and LML, (C) 13C NMR (75 MHz, d6-DMSO) of Lac, LauA and LML; (D) HPLC chromatograms of the reaction mixture of Lac and LauA in (DI) 6 day (DII) 8 days, flow rate is 0.2 ml min−1. | ||
Fig. 5D depicts the HPLC chromatograms of the reaction mixture for the catalytic synthesis of LML by CoII-G2.0L2@SCMB. In both HPLC chromatograms, peaks of Lac, LauA, and LML have been identified. With LauA as an excess substrate, LML conversion can be measured by the decrease in LauA amount using a calibration curve. On the other hand, with Lac as a limiting substrate, the yield reaction (%) can be calculated by (actual yield/22.7) × 100 formula. The evolution of the LML peak over time is fast (14–15 min), attaining a conversion (yield) of 92.5% (87%) and 89.4% (83%) in the presence of CoII-G2.0L2@SCMB and MnII-G2.0L2@SCMB at 8 days, respectively. The conversion and yield of the esterification reaction are higher in the beginning, even so, attains a plateau after 6 days, which is presumably related, principally, to the reversibility of the esterification reaction. As depicted in Table S2,† the conversion and yield of the esterification reaction were higher when pure lactose (PLac) was utilized with a Lac-to-LauA ratio of 2:1. The results revealed that the esterification reaction conversion was much slower in the case of raw lactose (RLac) from whey. Whey is a byproduct of the manufacture of cheese or casein that consists of lactose, α-lactalbumin, β-lactoglobulin, immunoglobulins, bovine serum albumin, bovine lactoferrin, and lactoperoxidase and minerals, which can occupy the vacant orbitals of active centers on the nanodendrimers by lone pair electrons of sulfhydryl, hydroxyl and amine groups. This phenomenon caused to inactivation of CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB as catalysts. Fig. 5D displays the evolution of the LML peak in the HPLC chromatograms with time, and Fig. 5 depicts the conversion and yield reaction at different times of the esterification reaction in the presence of CoII-G2.0L2@SCMB and MnII-G2.0L2@SCMB. The appearance of the LML peak is evidently seen as well as the decreasing LauA amount over time. Also, doublet LML peak and multiple LauA esters peaks were not observed in the HPLC chromatograms of mixture reaction.7,11 These results illustrate that the Lac is not able to esterify with multiple LauA in the reactions catalyzed by CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB. It seems that the LML synthesized is too bulky to be able to reach the active center on the CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB to construct a di- or multi-ester lactose, thereby this condition leads to the selective synthesis of LML.
3.3. Effect of pivotal factors in synthesis of LML
The reaction time, temperatures, and type of solvents cause the change in solubility as well as the chemical kinetic reaction mechanism, synthesis rate, catalyst stability, and activity. The LML is the only compound in the reaction mixture which doesn't construction hydrogen bonds, and so it has the weakest intermolecular forces. Large esters such as LML compound compared to small esters tend to construction more slowly. Therefore, in these case, it be necessary to heat the reaction mixture under reflux condition for some time to produce an equilibrium mixture. According to the previous literature,3–8 the reaction temperature profile was evaluated within a temperature range between 40 °C and 55 °C at the below the melting point of the applied solvents. The reaction rate improved upon the increase of temperature from 40 °C to 60 °C, due to the increase in the frequency of efficient collisions between the Lac and LauA molecules.Fig. 6 shows the effect of various solvents on the catalytic synthesis of LML with the investigated nanocatalysts. It was assessed that the different solvents had strong effects on catalytic activity and selectivity. In order to evaluate the influence of the nature of solvents on the syntheses of LML, solvent including ACN (acetonitrile), EtOH (ethanol), THF (tetrahydrofuran), ACTN (acetone), n-HX (hexane), and 2M2B (2-methyl-2-butanol) were used. It illustrates that the change of solvent nature (polar or non-polar) leads to the change in conversion, yield, and selectivity. The lowest conversion was obtained in 2M2B and EtOH since these solvents will participate in competitive adsorption on the catalysts active sites hindering the formation of LML. Selectivity of LML also decreased because, under this condition, the di/multi-ester lactose may be synthesized. ACTN and ACN they had the highest catalytic efficiency for LML synthesis. The reasons could be (1) they are polar and are not coordinating solvents and cannot block the catalyst (2) Lac has good solubility in these solvents (3) manufacture the catalyst-substrates intermediate is stable in these solvents and can be homogeneously dissolved and react in the liquid-phase media.7,11
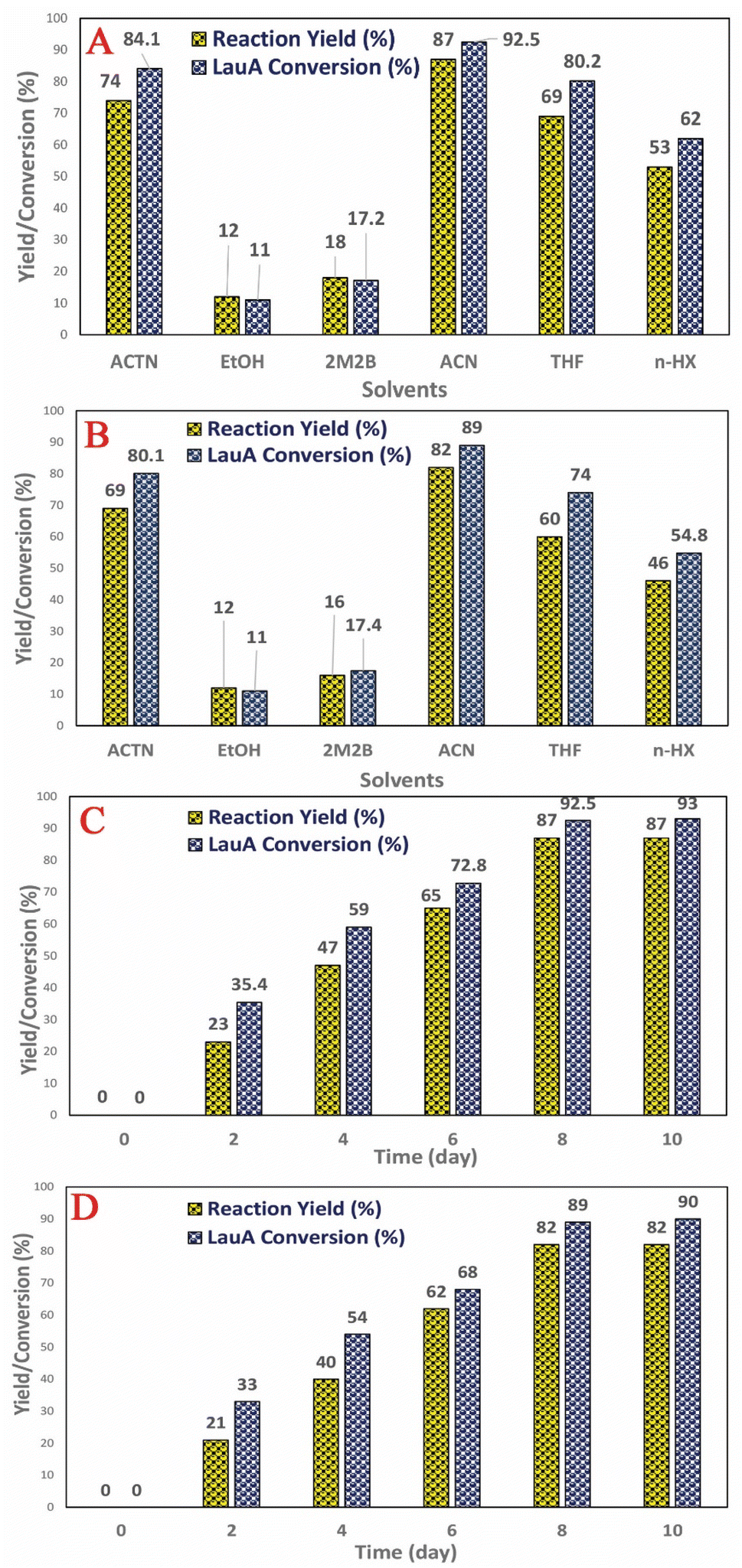 | ||
| Fig. 6 Effect of solvent in the synthesis of bio surfactant by (A) CoII-G2.0L2@SCMB, (B) MnII-G2.0L2@SCMB; effect of time on synthesis of bio surfactant by (C) CoII-G2.0L2@SCMB, (D) MnII-G2.0L2@SCMB. | ||
The CoII-G2.0L2@SCMB displays higher catalytic activity in LML synthesis in comparison to MnII-G2.0L2@SCMB (Table 1). The conversion and yield of LML synthesis by dendritic catalysts increased in the following order: MnII-G2.0L1@SCMB < MnII-G2.0L2@SCMB < MnII-G2.0L2@SCMB < CoII-G2.0L2@SCMB. Higher electronegativity of Co(II) in comparison to Mn(II) in the dendritic framework has been responsible for higher conversion and yield. In regard to yield and conversion (Table 1), several factors such as more electron density and the existence of vacant orbitals in Co, cause faster generation of intermediates, weakening of the metal-compounds bond, and facilitate the release of LML from the surface of the dendritic catalyst and make Co-catalyst more active than Mn-catalyst in our catalytic system.30 In fact, the two more valence electrons in cobalt significantly change the electronic structure independent of the nature of the substrate and linker. Differences in yields appear to be related, principally, to the synergy and reactivity of metal and probably reflect a more appropriate orbital availability arising from occupancy by electrons due to spin states that simplify interaction with the substrates.30–32 The comparison of catalytic performance in the same reaction condition reveals that in addition to the ligand and active center of catalyst, the difference in the nature and length of linker could affect the catalyst activity (MnII-G2.0L1@SCMB < MnII-G2.0L2@SCMB < MnII-G2.0L2@SCMB < CoII-G2.0L2@SCMB). It seems that the long length linker increases flexibility on the surface of pristine through the increase of distances between terminal ligand and surface of SCMBNP. This flexibility causes an increase in loading metal on the periphery of the dendritic catalyst (Table S1†). The long linker promotes surfaces hydrophobicity of the dendritic catalysts which reduce the interaction of the metal with the host pristine and make the catalytic sites more reachable to the substrate [58 and 59]. Moreover, steric effects, the electronic effects of the ligand-metal, electrostatic interactions, and the metal synergy effect, apparently are other functional parameters for the variation in the catalytic efficiency of the synthesized dendritic catalyst.30,32
3.4. Surface-active properties
Non-ionic LML as a biosurfactant is a molecule that combines either hydrophilic head and lipophilic parts (or polar & non-polar groups) and the balance of these groups is introduced as the HLB value. HBL value measured by eqn (1) is a criterion that influences the choice of surfactants for an intended formula, that is W/O or O/W emulsions. Based on HLB values for LML (Table 1), in addition to the facilitation of emulsification, adding LML can also improve the physical stability via adsorption at the O/W interface, decreasing the IFT and impeding the droplets from aggregation.18,19,33 In colloidal and surface chemistry, the CA (°) is the angle formed between the surface and the line tangent to the edge of the drop of the water. Another important characteristic parameter for assessing the efficiency of a biosurfactant is the CMC point, which is defined as the concentration of a biosurfactant above which micelles spontaneously form. Below the CMC point, the SFT decreases with increasing biosurfactant concentration, and above this point, the SFT is extensively independent of the concentration of surfactant. In fact, the addition of a surfactant decreases the interface energy and removes the hydrophobic moiety of surfactant from contact with water. Several parameters such as amphiphile chain length, nature, and structure of the hydrophilic group, temperature, and purity of surfactant have the main influence on the CMC point of surfactant.7,19 One of the illustrious properties of surfactants is diminishing the SFT and IFT of a liquid by decreasing intermolecular forces. Because the force of attraction between the surfactant and water is less than water and water, therefore, physicochemical properties of the synthesized surfactants were measured and then compared to other literature. The SFT curves for the LML synthesized by Co-G2.0L2@SCMB and Mn-G2.0L2@SCMB are shown in Fig. S1.† Table 1 lists the CMC, γCMC, and γmin that were obtained from the SFT diagrams. Due to different conditions of LML synthesis (types of catalysts and solvents), the synthesized LMLs exhibited different purities which caused the difference between CA, IFT, SFT, CMC, γmin and γCMC values (Table 1 and Fig. S1†) in synthesized LMLs.The data of SFT versus the different concentrations of LML were graphed to obtain the CMC, γmin, and γCMC values. As can be seen in Table 1 and Fig. S1,† the LML that was synthesized in ACN by CoII-G2.0L2@SCMB showed the lowest SFT, IFT, and CMC, comparable to the LML synthesis by MnII-G2.0L2@SCMB. As shown in Fig. S1,† the obtained results of CMC, which is 0.55 for LML synthesized by CoII-G2.0L2@SCMB, and 0.63 for LML synthesized by MnII-G2.0L2@SCMB are in good accordance with other reports.3,19 As a result, the aqueous solutions of LML were able to reduce the SFT and IFT, which were lower than those of water (SFTH2O = 67.30 mN m−1 and IFTH2O = 20.80 mN m−1) and comparable with SDS (sodium lauryl sulfate) with SFTSDS = 39.10 mN m−1 and IFTSDS = 4.90 mN m−1.7
The ΔG of surfactant adsorption, is surface excess of Gibbs thermodynamic potential and is widely applied as a basic thermodynamic characterization of surfactant. The Γ (eqn (6)), A (eqn (7)), and ΔG (eqn (8)), of LML for CoII-G2.0L2@SCMB (and MnII-G2.0L2@SCMB) are 1.09 × 10−5 (1.12 × 10−5) mol m−2, 14.7 (14.9) Å2 and −30.54 (−31.76) kJ mol−1, respectively. The surface properties of the synthetized LMLs are comparable with the ones fabricated by other researchers.19 These results illustrate that our catalytic synthesis of LML is a successful approach.
The FA and FS for various concentrations (0.1 to 0.6 g L−1) of the aqueous solution of LML were assessed at room temperature. As shown in Fig. S2† and 6, the FA and FS increased significantly as the concentration of LML enhanced from 0.1 to 0.6 g L−1. At a higher concentration of LML, the viscosity of the solution was ameliorated. As the viscosity increases, bubbles form with difficulty and the speed required for decreasing the thickness of film in the interface of oil and water is reduced. Therefore, this phenomenon principally causes the delay in the rupturing of bubble film.
It is illustrated that the hydrophobic moiety of surfactant was a principal parameter influencing foaming property. Apparently, the surface-adsorbed molecules of surfactant have significant interaction with each other.11,19 LML has a low SFT at equilibrium and exhibits special FA and FS, because it is able to rapidly transfer to the interface to constitute small bubbles with a long half-life time. Additionally, the FS was affected by the concentration of the aqueous solution of LML and standing time. At LML concentrations over than 0.5 g L−1 the FS is reduced about 17% during standing time for 40 min. Thereby, LML showed the good FS at concentrations over 0.5 g L−1, as the foam height descended slowly (Fig. S2† and 7).
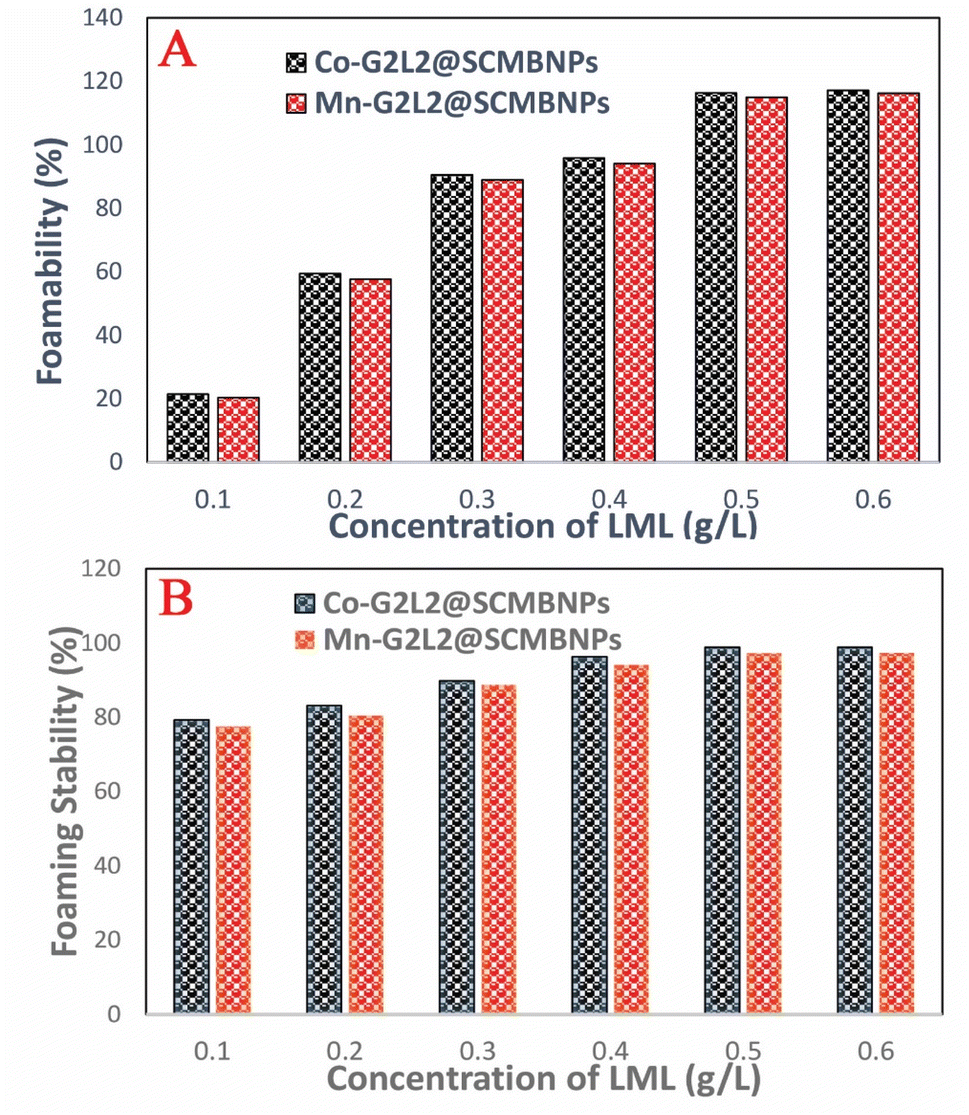 | ||
| Fig. 7 (A) Foamability of various concentration of biosurfactantat T = RT for t = 50 min; (B) foam stability of various concentration of biosurfactant at T = RT for t = 50 min. | ||
Emulsions are composed of two phases and surfactants are of main ingredients of a stable emulsion. The addition of applicable surfactants affects the stability of emulsions by reducing the IFT between oil and water phases.33 EmA and EmS are explained as the system's ability to keep its physicochemical properties unchanged over time. Several phenomena such as coalescence, creaming, flocculation, Ostwald ripening are responsible for the destabilization of emulsions.11,21 In this research, the soybean O/W emulsions (20%) were prepared with a LML content of 0.02% and 0.05% in the aqueous phase, in order to evaluate the influence of the LML on stabilizing of the emulsions. In control samples (emulsion without LML), three layers (oil layer on top, a creaming layer directly underneath, and a clear layer at the bottom) were observed, while, the LML containing emulsions showed no visible oil and clear layers. The EmAs value (eqn (4)) of LML illustrated that LML was efficient for emulsifying the soybean O/W system. The EmAs values (eqn (5)) of synthesized LML by CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB were 54% & 52%, respectively. On the other hand, the results indicated that the EmS by CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB were 72% & 69% after 24 h at 25 °C, respectively. This data confirmed that LML obviously improved the stability of O/W emulsions. The EmS of synthesized LML exceeded 93% and 91% from 0.5 to 2 h for CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB, respectively. Also, the O/W emulsion containing LML had no visible oil or clear layers after 5 days. Indeed, the suitable HLB value (14.1) of LML (0.5 g L−1) allows oil droplets to be stabilized in soybean (20%) emulsion. However, the HLB is not the only factor in the suitability of an emulsifier. On the other hand, LML with moderate hydrophobic moiety has suitable CMC & HLB values and as result is able to be adsorbed at the O/W interface and create consistent surface-interface stability over time.23,36,37 The emulsifying effects of LML and shrinkage of soybean oil droplets were evaluated with TEM & DLS. Decrease of particle size, effective absorption on O/W interface and increase of SFT & IFT occurred when LML was added to the emulsion (20%). Fig. 8 exhibits that a significant number of the droplets are sized up to 10 μm. In case of 20% O/W control emulsion (without LML), the oil droplets were sized between 0.0 and 3000 μm. Furthermore, emulsions containing 0.5 g L−1 LML produced narrow distributions range (day 0), which shifted to wide distribution over time (day 6). Regarding the small size and uniform shape of the oil droplets (Fig. 8 C–E), the probability of coalescence and flocculation of oil droplets are almost low. Additionally, the morphological change of the emulsion (20%) was completely consistent with the DLS results. In the presence of LML, the number of micro-droplets (observed as black dots) has increased while the coalescence and flocculation of soybean oil occurred over time. After 7 days (Fig. 8E), the oil micro-droplets were flocculated and consequently generated the bulk droplets, illustrating the occurrence of emulsion destabilization.
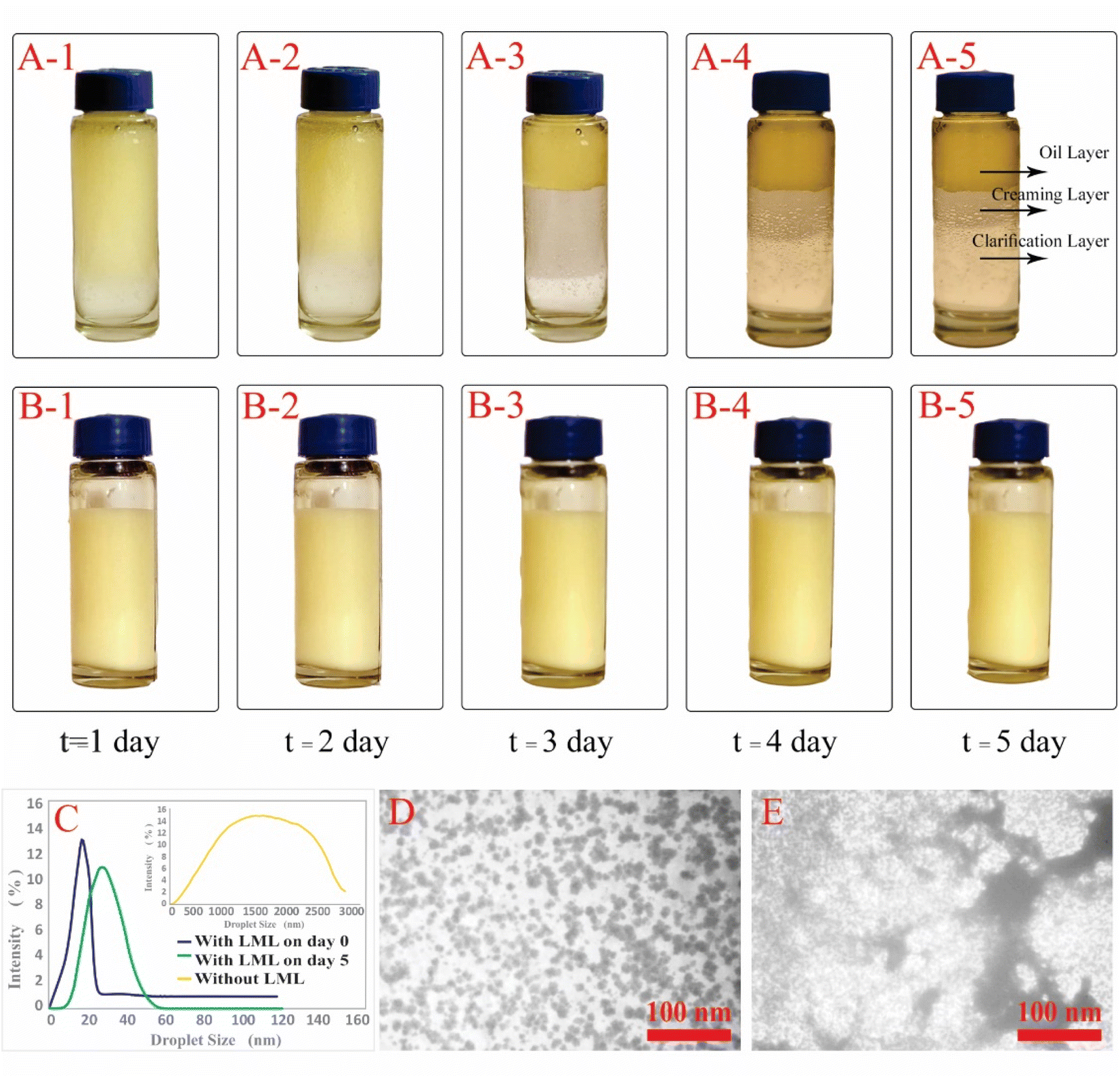 | ||
| Fig. 8 Emulsion stability of soybean O/W emulsion at room temperature after 5 days, (A) without emulsifier, (B) with LML as an emulsifier and (C) particle size distribution of the emulsion at day 0 & 7 and without LML, (D) TEM images of soybean emulsion (20% v/v) at day 0 and (E) TEM images of soybean emulsion (20% v/v) after 7 days. | ||
Generally, these results illustrated that the synthesized LMLs have outstanding emulsion stabilizing properties comparable to Ryoto sugar ester L-1695 (sucrose laurate) & Tween-20 (polysorbate 20) as control industrial emulsifiers.21 Comparison of emulsifier properties of Ryoto sugar ester L-1695 (HLB = 16.0, CMC = 0.44 mM, droplet distribution ranges 0.5–2 μm) & Tween-20 (HLB = 16.7, CMC = 0.42 mM, droplet distribution range 0.8–2 μm) with as-prepared emulsifier confirmed that LML had an excellent performance regarding the decrease of SFT & IFT of the prepared emulsion.
3.5. Proposed pathway for synthesis of biosurfactant
As shown in Scheme 1, coordination of the lactose (via the hydroxyl oxygen) and the lauric acid (via the carbonyl oxygen) requires Brønsted acid sites on the catalyst (step 1). Next, carboxyl oxygen coordinated to metallic site (Mn/Co) to give delocalized carbocation making the carbocation a better electrophile (step 2 and 3). The nucleophile species (the lactose OH) can subsequently attack to the carbonyl group of LauA and produce a reactive tetrahedral intermediate (step 4) and transferred the H+ to the OH groups to form a good leaving group (H2O). The hydroxy group's lactose oxygen atom donates a pair of electrons to a carbon atom which makes a π bond by eliminating H2O.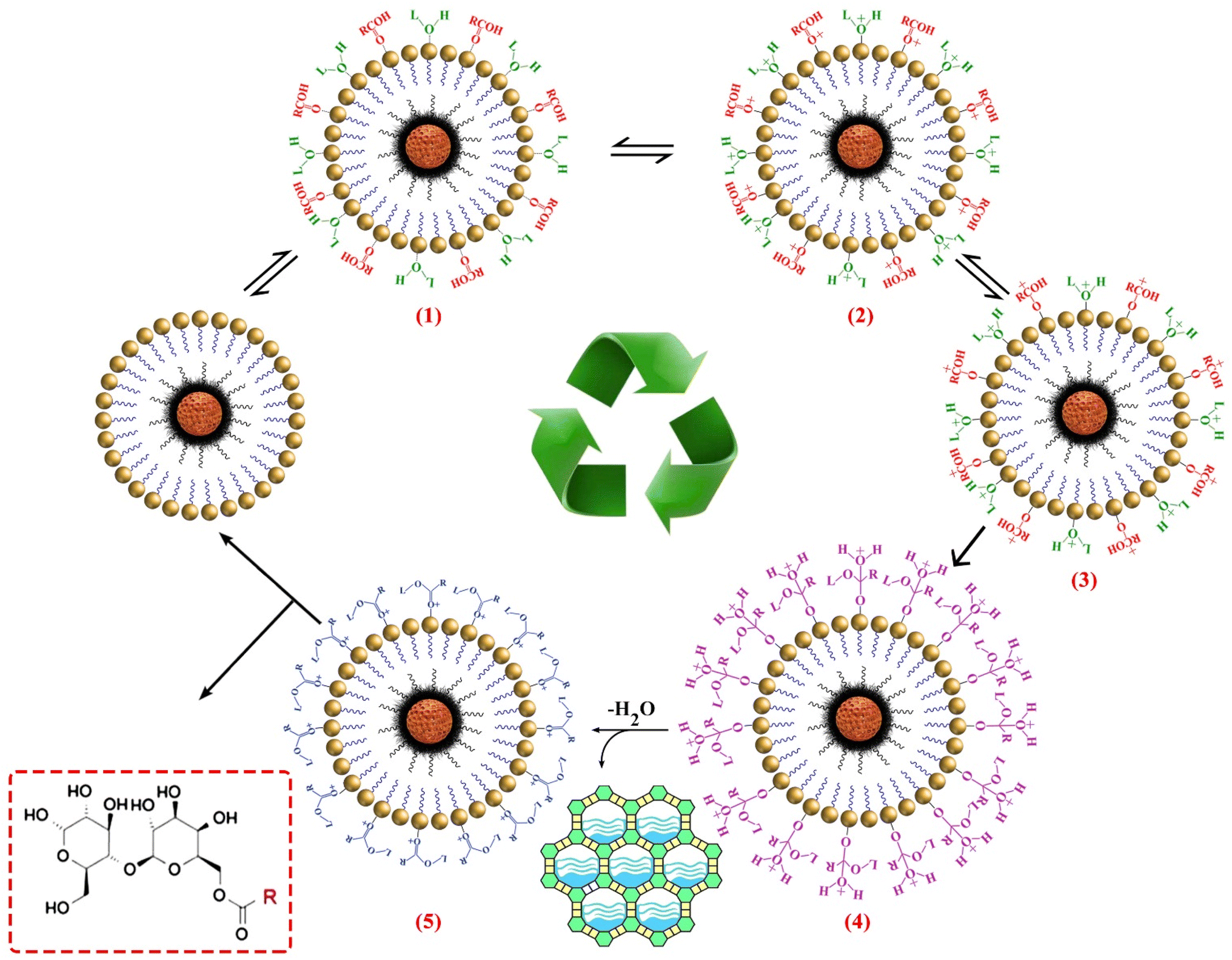 | ||
| Scheme 1 Plausible mechanism of biosurfactant synthesis by the Co/Mn encapsulated on MBNP core/dendrimers shell composite. | ||
Then, H2O eventually adsorbed by molecular sieves (MS) to shift the equilibrium to construction of the LML (steps 4 and 5). In this steps, enhanced amount of H2O in reaction medium might have two negative effects, one deactivation of dendritic catalyst by blocking of active site with H2O, and the second the reaction of reagent with itself. Furthermore, increasing the number of H2O molecules can decrease the yield of the reaction, the MS have a critical role in enhancing of esterification rate.
3.6. Reusability study
We investigated the stability and reusability of CoII-G2.0L2@SCMB and MnII-G2.0L2@SCMB in the synthesis of LML. When the reaction was complete, the magnetic nanodendrimer was separated by an external magnet. The isolated catalyst was washed three times with EtOH and ether, and then vacuum-dried for the next reaction. We performed three reuses and found that the performance of the nano dendrimers was almost unchanged (Fig. S3†). The structure and morphology of the recycled catalysts were studied by ICP-OES, AAS, FT-IR, XRD, and TEM. Additionally, ICP-OES and AAS analysis were performed and the leaching of metal was estimated. The obtained data indicated that the percent of leached CoII and MnII of spent dendritic composites were 8.12 ± 0.13% and 9.35 ± 0.11%, respectively. Also, the XRD analyses and FT-IR of the recovered catalyst showed little change compared to the fresh catalyst. The comparison of XRD analyses of samples showed that some peaks shifted slightly. This shift can be related to distortion of crystalline structure in consecutive runs. TEM images showed that the catalyst was nearly spherical before and after the 5 reaction times. Notably, small aggregates on metal can be observed, which could stem from recycling (Fig. 3C). Since, leaching of the active species from the catalytic system is a serious problem and can challenge the heterogeneous properties of the catalysts, a Sheldon test was carried out to demonstrate the heterogeneity of the system. In this regard, the catalyst removed from the reaction medium after 1 day from the start reaction. Results confirmed that no further progress in surfactant production was done, which illustrate that the catalysts are heterogeneous. CoII or MnII ions grafted on the SCMBNPs and used as catalyst for improving the coordination of metals with metformin. The result showed that the conversion and yield of biosurfactant production are trace which is similar to the conversion and selectivity in the absence of catalyst. The mentioned data illustrated that the LML synthesis occurs due, certainly, to the catalytic nature of the nanostructured organic–inorganic hybrid catalysts.3.7. Uniqueness of our protocol
To illustrate the distinctiveness of our developed protocol, we compared our results with other reports. Enzymes as industrial biocatalysts provide many advantages (such as functioning in mild reaction conditions, high product chemoselectivity, great turnover number and low environmental and physiological toxicity) compared to traditional chemical procedures (such as concentrated mineral acids),34 but being costly, loss in catalytic activity, high sensitivity to denaturation, physical and chemical conditions (e.g., temperature, pH etc.), being unrecoverable & reusable, and mass transfer limitations are of their disadvantages for industrial applications. Although immobilization of the enzymes can partially eliminate disadvantages such as recoverability and improve their stability in broader range of operating conditions, but still their costly, arduous and time-consuming preparation are challenging.34,35 On the other hand, the chemical catalytic systems (e.g., metal oxide, resins, clays, and zeolites etc.) have been utilized for the synthesis of sugar esters. The main advantages of these catalysts over enzymes are their simplicity, availability and their cheaper price.7,12 However, their catalytic procedures are carried out in harsh condition reaction (e.g. high temperature, long reaction time etc.) and produce di- or three esters as by-products. Zeolites become inactive by irreversible adsorption, steric blockage of heavy secondary molecules. They have low reusability and have unsuitable microspores for bulk molecules.35 The comparative study with other catalytic systems depicted that our system has the advantages such as less viscosity than multifunctional organic polymeric catalysts, heterogeneous nature of catalysts,3,14 availability of catalytically active (metal) centers, solubility in organic media due to hydrophobic arms,2–7 simple and low-cost chemical synthesis method, the possibility to use accessible compounds for fabrication on the small size, tremendous surface area-to-volume ratio,14 the stability against leaching, thermal and chemical stability,4 longevity of catalyst, mild condition reaction and less time to esterify,7 excellent conversion and chemoselectivity (specially 6′-O-acyllactose esters). Therefore, using of hybrid nanodendrimers as catalytic systems can be suggested as a novel approach for practical applications.4. Conclusion
CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB are novel synthetic catalysts characterized by their branched structure that have a focal magnetic core and possess a large number of exposed metallic terminals on their surface, which lead to the formation of hydrophobic arms. They are nanometric catalysts that are radially symmetric, globular, mono-dispersed, and heterogeneous. For the first time, the results of the present study introduced that the hybrid SCMBNP core/shell dendrimers containing isolated Lewis acid sites and molecular sieves cooperatively catalyze the regio-chemoselective synthesis of lactose-based biosurfactant. LML is a high-value-added product of lactose and lauric acid reaction and represents a class of biodegradable, non-ionic, low-molecular-weight surfactants (emulsifiers) that has considerable potential in the food, cosmetic, and pharmaceutical industries. This study illustrated that increasing the carbon chain length (L1 & L2) of the linker principally causes more loading of the metal and creates hydrophobic arms on magnetic pristine that is the most important factor influencing the catalytic activity. On the other hand, the difference between the operation of CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB is due, principally, to the difference in the synergistic effect, redox potential, electronegativity, and the electronic structure of active sites. To authenticate the catalytic efficiency, the CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB were applied in the synthesis of biosurfactant by reacting PLac and RLac (derived from whey) with LauA. The low conversion and yield of the RLac are most probably related to the occupation of an active center by coordinating group of β-lactoglobulin, α-lactalbumin, and bovine serum albumin in whey. To elucidate the authenticity of this hypothesis, purity of synthesized LML, structural analysis (FT-IR, 1H NMR, 13C NMR, and HPLC), and surface properties (HLB, CA, IFT, SFT, FA, FS, EmA, EmS, Γ, and ΔG) were estimated and compared to other reports. The results indicated that all properties of the synthesized biosurfactants were in accordance with previous reports. The LML showed a low CA, SFT and IFT compared to the typical surfactant and water. Also, foamability, foaming stability, oil–water emulsifying ability and emulsion stability illustrated that LML is a favorable biosurfactant. Therefore, LML has a high potential for use as a biosurfactant for food and other industries. Additionally, the CoII-G2.0L2@SCMB & MnII-G2.0L2@SCMB could be successfully recovered by a magnetic bar and reused for 5 reaction times with a slight loss of catalytic efficiency.Conflicts of interest
No conflicts of interest exists.Acknowledgements
A. F. acknowledges research council of Tehran Medical Sciences, Islamic Azad University for the research founding of this project.References
- C. J. Drummond and D. Wells, Nonionic Lactose and Lactitol Based Surfactants: Comparison of Some Physico-Chemical Properties, Colloids Surf., A, 1998, 141(1), 131–142 CrossRef CAS.
- M. Ferrer, J. Soliveri, F. J. Plou, N. López-Cortés, D. Reyes-Duarte, M. Christensen, J. L. Copa-Patiño and A. Ballesteros, Synthesis of Sugar Esters in Solvent Mixtures by Lipases from Thermomyces Lanuginosus and Candida Antarctica B, and Their Antimicrobial Properties, Enzyme Microb. Technol., 2005, 36(4), 391–398 CrossRef CAS.
- M. Enayati, Y. Gong, J. M. Goddard and A. Abbaspourrad, Synthesis and Characterization of Lactose Fatty Acid Ester Biosurfactants Using Free and Immobilized Lipases in Organic Solvents, Food Chemistry, 2018, 266, 508–513 CrossRef CAS PubMed.
- M. N. AlFindee, Q. Zhang, Y. P. Subedi, J. P. Shrestha, Y. Kawasaki, M. Grilley, J. Y. Takemoto and C. W. Chang, One-Step Synthesis of Carbohydrate Esters as Antibacterial and Antifungal Agents, Bioorg. Med. Chem., 2018, 26(3), 765–774 CrossRef CAS PubMed.
- B. Pérez, S. Anankanbil and Z. Guo, Synthesis of Sugar Fatty Acid Esters and Their Industrial Utilizations, Fatty Acids, 2017, 329–354 Search PubMed.
- G. W. Smithers, Whey and Whey Proteins—from Gutter-to-Gold, Int. Dairy J., 2008, 18(7), 695–704 CrossRef CAS.
- M. Enayati, Y. Gong and A. Abbaspourrad, Synthesis of Lactose Lauryl Ester in Organic Solvents Using Aluminosilicate Zeolite as a Catalyst, Food Chemistry, 2019, 279, 401–407 CrossRef CAS PubMed.
- N. S. Neta, J. A. Teixeira and L. R. Rodrigues, Sugar Ester Surfactants: Enzymatic Synthesis and Applications in Food Industry, Crit. Rev. Food Sci. Nutr., 2015, 55(5), 595–610 CrossRef CAS PubMed.
- J. F. Kennedy, H. Kumar, P. S. Panesar, S. S. Marwaha, R. Goyal, A. Parmar and S. Kaur, Enzyme-Catalyzed Regioselective Synthesis of Sugar Esters and Related Compounds, J. Chem. Technol. Biotechnol., 2006, 81(6), 866–876 CrossRef CAS.
- N. D. Neta, J. C. dos Santos, S. de Oliveira Sancho, S. Rodrigues, L. R. Gonçalves, L. R. Rodrigues and J. A. Teixeira, Enzymatic Synthesis of Sugar Esters and Their Potential as Surface-Active Stabilizers of Coconut Milk Emulsions, Food Hydrocolloids, 2012, 27(2), 324–331 CrossRef.
- M. K. Walsh, R. A. Bombyk, A. Wagh, A. Bingham and L. M. Berreau, Synthesis of Lactose Monolaurate as Influenced by Various Lipases and Solvents, J. Mol. Catal. B: Enzym., 2009, 60(3–4), 171–177 CrossRef CAS.
- A. Hykkerud and J. M. Marchetti, Esterification ofOleic Acid with Ethanol in the Presence of Amberlyst 15, Biomass Bioenergy, 2016, 95, 340–343 CrossRef CAS.
- L. M. Estiaty, D. Fatimah, D. Suherman and K. Alamsyah, Natural Zeolites as Manure Additive: the Advantages and Disadvantages, Jurnal Zeolit Indonesia, 2005, 4(1), 1–9 Search PubMed.
- W. Sun, S. Mignani, M. Shen and X. Shi, Dendrimer-Based Magnetic Iron Oxide Nanoparticles: Their Synthesis and Biomedical Applications, Drug Discovery Today, 2016, 21(12), 1873–1885 CrossRef CAS PubMed.
- M. B. Wazir, M. Daud, F. Ali and M. A. Al-Harthi, Dendrimer Assisted Dye-Removal: A Critical Review of Adsorption and Catalytic Degradation for Wastewater Treatment, J. Mol. Liq., 2020, 315, 113775 CrossRef CAS.
- M. Arshadi, F. Mousavinia, A. Khalafi-Nezhad, H. Firouzabadi and A. Abbaspourrad, Adsorption of Mercury Ions From Wastewater by a Hyperbranched and Multi-Functionalized Dendrimer Modified Mixed-Oxides nanoparticles, J. Colloid Interface Sci., 2017, 505, 293–306 CrossRef CAS PubMed.
- A. R. Faraji, F. Ashouri, Z. Hekmatian, S. Heydari and S. Mosazadeh, Organosuperbase Dendron Manganese Complex Grafted on Magnetic Nanoparticles; Heterogeneous Catalyst for Green and Selective Oxidation of Ethylbenzene, Cyclohexene and Oximes by Molecular Oxygen, Polyhedron, 2019, 157, 90–106 CrossRef CAS.
- I. K. Hong, S. I. Kim and S. B. Lee, Effects of HLB Value on Oil-in-Water Emulsions: Droplet Size, Rheological Behavior, Zeta-Potential, and Creaming Index, J. Ind. Eng. Chem., 2018, 67, 123–131 CrossRef CAS.
- X. Zhang, F. Song, M. Taxipalati, W. Wei and F. Feng, Comparative Study of Surface-Active Properties and Antimicrobial Activities of Disaccharide Monoesters, PLoS One, 2014, 9(12), e114845 CrossRef PubMed.
- M. Y. Liang, M. G. Banwell, Y. Wang and P. Lan, Effect of Variations in the Fatty Acid Residue of Lactose Monoesters on their Emulsifying Properties and Biological Activities, J. Agric. Food Chem., 2018, 66(47), 12594–12603 CrossRef CAS PubMed.
- S. M. Lee, A. Wagh, G. Sandhu and M. K. Walsh, Emulsification Properties of Lactose Fatty Acid Esters, Food Nutr. Sci., 2018, 9(12), 1341–1357 CAS.
- S. Heydari, D. Habibi, A. R. Faraji and M. Mahmoudabadi, An Overview on the Progress and Development on the Palladium Catalyzed Direct Cyanation, Inorg. Chim. Acta, 2021, 514, 119956 CrossRef CAS.
- X. Huang, C. Xu, J. Ma and F. Chen, Ionothermal Synthesis of Cu-doped Fe3O4 Magnetic Nanoparticles with Enhanced Peroxidase-like Activity for Organic Wastewater Treatment, Adv. Powder Technol., 2018, 29(3), 796–803 CrossRef CAS.
- N. A. Neto, L. E. Nascimento, M. Correa, F. Bohn, M. R. Bomio and F. V. Motta, Characterization and Photocatalytic Application of Ce4+, Co2+, Mn2+ and Ni2+ Doped Fe3O4 Magnetic Nanoparticles Obtained by the Co-Precipitation Method, Mater. Chem. Phys., 2020, 242, 122489 CrossRef.
- R. Foroutan, S. J. Peighambardoust, A. Ahmadi, A. Akbari, S. Farjadfard and B. Ramavandi, Adsorption Mercury, Cobalt, and Nickel with a Reclaimable and Magnetic Composite of Hydroxyapatite/Fe3O4/Polydopamine, J. Environ. Chem. Eng., 2021, 9(4), 105709 CrossRef CAS.
- D. Habibi, S. Heydari, A. Gil, M. Afsharfarnia, A. Faraji, R. Karamian and M. Asadbegy, Application of the Fe3O4@ 1, 10-Phenanthroline-5, 6-Diol@ Mn Nano-Catalyst for the Green Synthesis of Tetrazoles and its Biological Performance, Appl. Organomet. Chem., 2018, 32(2), e4005 CrossRef.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, XPS Determination of Mn Oxidation States in Mn (hydr) Oxides, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- Z. Li, S. Pang, M. Wang, M. Wu, P. Li, J. Bai and X. Yang, Dual-Emission Carbon Dots-Copper Nanoclusters Ratiometric Photoluminescent Nano-Composites for Highly Sensitive and Selective Detection of Hg2+, Ceram. Int., 2021, 47(13), 18238–18245 CrossRef CAS.
- T. R. D. Costa, E. Baldi, A. Figueiró, G. L. Colpani, L. L. Silva, M. Zanetti and M. A. Fiori, Fe3O4@ C Core-Shell Nanoparticles as Adsorbent of Ionic Zinc: Evaluating of the Adsorptive Capacity, Mater. Res., 2019, 22, 1–10 Search PubMed.
- D. Habibi and A. R. Faraji, Preparation, Characterization and Catalytic Activity of a Nano-Co (II)-Catalyst as a High Efficient Heterogeneous Catalyst for the Selective Oxidation of Ethylbenzene, Cyclohexene, and Benzyl Alcohol, C. R. Chim., 2013, 16(10), 888–896 CrossRef CAS.
- S. Bhattacharjee, T. J. Dines and J. A. Anderson, Comparison of Co with Mn and Fe in LDH-Hosted Sulfonato–Salen Catalysts for Olefin Epoxidation, J. Phys. Chem. C, 2008, 112(36), 14124–14130 CrossRef CAS.
- J. G. McAlpin, T. A. Stich, W. H. Casey and R. D. Britt, Comparison of Cobalt and Manganese in the Chemistry of Water Oxidation, Coord. Chem. Rev., 2012, 256(21–22), 2445–2452 CrossRef CAS.
- D. Golodnizky and M. Davidovich-Pinhas, The Effect of the HLB Value of Sucrose Ester on Physiochemical Properties of Bigel Systems, Foods, 2020, 9(12), 1857 CrossRef CAS PubMed.
- N. Bashir, M. Sood and J. D. Bandral, Enzyme Immobilization and its Applications in Food Processing: A Review, Int. J. Chem. Stud., 2020, 8, 254–261 CrossRef CAS.
- J. Chapman, A. E. Ismail and C. Z. Dinu, Industrial Applications of Enzymes: Recent Advances, Techniques, and Outlooks, Catalysts, 2018, 8(6), 238 CrossRef.
- G. Garofalakis, B. S. Murray and D. B. Sarney, Surface Activity and Critical Aggregation Concentration of Pure Sugar Esters with Different Sugar Headgroups, J. Colloid Interface Sci., 2000, 229(2), 391–398 CrossRef CAS PubMed.
- M. Giulietti and A. Bernardo, Crystallization by Antisolvent Addition and Cooling, in Crystallization-science and technology, IntechOpen, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06958j |
| This journal is © The Royal Society of Chemistry 2022 |