
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical insights into interfacial stability and ionic transport of Li2OHBr solid electrolyte for all-solid-state batteries†
Bo Liu *ab,
Piguang Liaoa,
Xiaowen Shib,
Yufeng Wena,
Qingdong Goua,
Meidong Yua,
Shenlin Zhoua and
Xinyuan Suna
*ab,
Piguang Liaoa,
Xiaowen Shib,
Yufeng Wena,
Qingdong Goua,
Meidong Yua,
Shenlin Zhoua and
Xinyuan Suna
aCollege of Mathematics and Physics, Jinggangshan University, Ji'an, Jiangxi 343009, China. E-mail: liubo@jgsu.edu.cn
bScience and Technology Innovation Development Center, Ji'an, Jiangxi 343006, China
First published on 2nd December 2022
Abstract
Li-rich antiperovskite materials are promising candidates as inorganic solid electrolytes (ISEs) for all-solid-state Li-ion batteries (ASSLIBs). However, the material faces several pressing issues for its application, concerning the phase stability and electrochemical stability of the synthesized material and the Li-ion transport mechanism in it. Herein, we performed first-principles computational studies on the phase stability, interfacial stability, defect chemistry, and electronic/ionic transport properties of Li2OHBr material. The calculation results show that the Li2OHBr is thermodynamically metastable at 0 K and can be synthesized experimentally. This material exhibits a wider intrinsic electrochemical stability window (0.80–3.15 V) compared with sulfide solid electrolytes. Moreover, the Li2OHBr displays significant chemical stability when in contact with typical cathode materials (LiCoO2, LiMn2O4, LiFePO4) and moisture. The dominant defects of Li2OHBr are predicted to be VLi− and Lii+, corresponding to lower Li-ion migration barriers of 0.38 and 0.49 eV, respectively, while the replacement of some of the OH− by F− is shown to be effective in decreasing migration barriers in Li2OHBr. These findings provide a theoretical framework for further designing high performance ISEs.
1. Introduction
In recent years, inorganic solid electrolytes (ISEs) have received wide attention to replace organic liquid electrolytes currently used in commercial lithium-ion batteries.1 Unlike organic liquid electrolytes, ISEs are nonflammable, nonvolatile, and have no liquid leakage problem, and are also expected to overcome the phenomenon of lithium dendrites, thus they have high safety performance.2 Moreover, ISEs have the potential to improve interfacial stability, which could enable the application of a high-voltage cathode and even lithium metal anode.3 In terms of Li-ion transport properties, several ISEs have been reported, such as the Li10GeP2S12,4 Li7P3S11,5 and lithium-rich anti-perovskites (LRAP),6 with high Li+ conductivities comparable to or even surpassing those of traditional liquid electrolytes.One promising class of ISEs is antiperovskites Li3−nOHnX (n = 0–1, X = Cl, Br) for ASSLIBs.7 For example, Zhao et al. first experimental reported that Li3OCl and Li3O(Cl0.5Br0.5) showed a high ion-conductivity (>10−3 S cm−1 at 300 K) with low activation energies (0.18–0.26 eV).8 Subsequently, a good stability and low Li+ vacancy migration barrier of Li3OX (X = Cl, Br) was verified by first-principles calculations.9 Sugumar et al. reported the successful preparation of Li2OHBr by dry ball-milling of LiOH and LiBr at room temperature, which obtained high ion conductivity of 1.1 × 10−6 S cm−1 with the activation energy of 0.54 eV.10 Recently, Yamamoto et al.11 reported that an ASSLIB composed of Li/Li2OHBr/Fe2(MoO4)3 were fabricated by pressing at room temperature, which exhibited good charge–discharge performance and excellent cycle stability. Zhao and co-workers12 proposed the Li2OHBr as a protective layer for the Li1.5Al0.5Ge1.5(PO4)3 (LAGP) solid electrolyte to prevent the side reaction caused by direct contact between LAGP and Li metal anode. For practical applications, whether Li2OHBr material acts as a solid electrolyte or protective layer, it is crucial that the material shows good thermodynamic stability, electrochemical stability and fast Li-ion diffusion, which are the keys to ameliorating the electrochemical and rate performance of Li2OHBr material. Moreover, Li2OHBr should possess the ability of moisture resistance and oxidation resistance, which will simplify the packaging of ASSLIBs in practice. However, in-depth understanding of these important issues, has been hindered by the complicated synthesis and measurement conditions during the experiments. Therefore, it is critical that we explore the fundamental issues of the phase stability, electrochemical stability, chemical stability and electron/ion transport mechanism of Li2OHBr through reliable theoretical approaches to elucidating the main behind physics mechanism.
In this work, we employ first-principles calculations to assess the phase stability, interfacial stability against electrode material, defect chemistry and electron/ion transport mechanism of anti-perovskites Li2OHBr. We predict a wide electrochemical window and low chemical reactivity for Li2OHBr, ensuring that this material is thermodynamically stable under high-voltage operation and in air. The analysis Li-ion transport mechanism shows the existence of low migration barriers involving charge carriers (VLi−, Lii+) in Li2OHBr. We also study the effect of F− substitution of OH− on the Li+ migration barriers in Li2OHBr. The computational approach in this work can be extended to the design of other ISEs system.
2. Computational methods
All calculations are performed based on density functional theory (DFT) by using the projector augmented wave method, as implemented in the Vienna ab initio Simulation Package (VASP).13 The generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) is applied to treat the electronic exchange–correlation interactions.14 The cutoff energy is set to 520 eV. The atomic force and energy convergence parameters are consistent with Materials Project (MP)15 for all calculations. Based on DFT ground-state energies in the MP database, the phase stability and interfacial stability (including electrochemical and chemical stability) of Li2OHBr are evaluated using the same scheme in the previous work.16The phase stability of Li2OHBr is assessed by computing the energy above convex hull, corresponding the decomposition energy to the thermodynamic phase equilibria. The electrochemical window of Li2OHBr is calculated by using the Li grand potential phase diagram. In this method, the grand potential ϕ of the Li2OHBr is defined as:16
| ϕ[c, μLi] = E[c] − nLi[c]μLi, | (1) |
The chemical stability of Li2OHBr/cathode interfaces are determined by estimating the reaction between the Li2OHBr and cathode with the lowest reaction energy (ΔE),17 namely:
 | (2) |
The defect formation energy Ef(i, q) with a defect i at charge state q is calculated via the following equation:18
| Ef(i, q) = Etot(i, q) − Etot(Li2OHBr, bulk) − nμi + q(εF + EV), | (3) |
3. Results and discussion
3.1 Structural and electronic property
The Li2OHBr orthorhombic structure with space group Cmcm is constructed, as shown in Fig. 1(a). Table 1 lists the optimized lattice parameters of Li2OHBr obtained by DFT calculations, together with the data from Howard et al.19 The lattice parameters are a = 8.015 Å, b = 8.152 Å, and c = 7.944 Å, which are agree with the experiment data (within 2% error). To obtain the electronic properties of Li2OHBr, the electronic band structure of Li2OHBr is evaluated. Since the GGA-PBE functional generally underestimate the band gap, the advanced SCAN meta-GGA functional is used to provide a rigorous result.20 Fig. 1(b) shows that the direct band gap of Li2OHBr is 4.79 eV, and its VBM and CBM are both located at the Γ point of the first Brillouin zone. The large band gap indicates that Li2OHBr is electron insulator, which can effectively block electron leakage and prevent electrode corrosion. The band gap between the valence band maximum (VBM) and the conduction band minimum (CBM) provides an upper limit for the electrochemical window of solid electrolytes. When the chemical potential of the electrode/solid electrolyte is mismatched, i.e., thermodynamically unstable, a chemical reaction between the two materials will occur spontaneously upon contact. | ||
| Fig. 1 (a) Atomic structure and (b) electronic band structure of Li2OHBr. The ball colors green, dark red, red, and gray indicate Li, Br, O, and H sites, respectively. | ||
| System | Calc. (this work) | Exp. (ref. 19) | |||||
|---|---|---|---|---|---|---|---|
| Lattice parameters | a (Å) | b (Å) | c (Å) | a (Å) | b (Å) | c (Å) | |
| 8.015 | 8.152 | 7.944 | 8.010 | 8.030 | 7.880 | ||
| α (°) | β (°) | γ (°) | α (°) | β (°) | γ (°) | ||
| 90 | 90 | 90 | 90 | 90 | 90 | ||
| Atom | Wyckoff | x | y | z | x | y | z |
| O | 8f | 0.000 | 0.744 | 0.514 | 0.000 | 0.741 | 0.513 |
| H | 8f | 0.000 | 0.823 | 0.414 | 0.000 | 0.824 | 0.419 |
| Cl | 8g | 0.749 | 0.489 | 0.250 | 0.747 | 0.487 | 0.250 |
| Li | 8d | 0.250 | 0.250 | 0.000 | 0.250 | 0.250 | 0.000 |
| Li | 4b | 0.000 | 0.500 | 0.000 | 0.000 | 0.500 | 0.000 |
| Li | 4c | 0.000 | 0.195 | 0.250 | 0.000 | 0.197 | 0.250 |
3.2 Phase stability and interfacial stability
The feasibility and complexity level of the experimental synthesis of one given material could be evaluated by its phase stability. In general, for a particular component that does not have a stable phase, the corresponding system will decompose into a stable phase around its component coordinate points. The Li–H–O–Br quaternary phase diagram is constructed at 0 K by minimizing the formation energies of various compositions, as shown in Fig. 2(a). The phase diagram indicates that Li2OHBr is energetically unstable duo to positive formation energy with respect to that form the Li4H3BrO3 and LiBr. However, the energy above hull (Ehull) of Li2OHBr is only 11.6 meV per atom, suggesting that Li2OHBr is likely to be metastable at 0 K against possible decomposition products, and therefore may be stabilized by external conditions (such as pressure, temperature, and entropy).10 For example, the Li2OHBr was synthesized from LiOH and LiBr starting materials by sintering method above 300 °C.21 In fact, successful synthesis of metastable phases has been widely reported for ISEs, such as Li6PS5Cl (21 meV per atom),22 Li10GeP2S12 (25 meV per atom)4 and Li7P3S11 (27 meV per atom).23 | ||
| Fig. 2 (a) Li–H–O–Br quaternary phase diagram. The metastable Li2OHBr is marked in red font. (b) The voltage profiles and phase equilibria of Li2OHBr at different potentials. The light yellow region indicates the electrochemical stability window range. | ||
For the practical application of ASSLIBs, the ISEs should satisfy the conditions of good interfacial stability, including electrochemical stability and chemical stability.24 Using eqn (1), the phase equilibrium of Li2OHBr for a series of lithiation/delithiation reactions is predicted to obtain the electrochemical stability window. The detailed lithiation/delithiation reactions with μLi are listed in Table S1.† As shown in Fig. 2(b), Li2OHBr is oxidized to form Li4H3BrO3 and Br when the oxidation voltage is higher than 3.15 V. Meanwhile, Li2OHBr is reductively decomposed into LiH, LiBr and Li2O starting from 0.80 V. The calculated electrochemical stability window range of Li2OHBr are 0.80–3.15 V vs. Li/Li+. Table 2 shows that Li2OHBr has much wider electrochemical window than that of reported sulfides and oxides solid electrolyte, such as Li10GeP2S12 (1.71–2.14 V), Li7P3S11 (2.28–2.31 V), Li6PS5Cl (1.71–2.01 V), Li2PO2N (0.68–2.63 V) and Li7La3Zr2O12 (0.05–2.91).25 However, it should be pointed out that the dissociation of ISEs depends on kinetic factors, suggesting that the dissociation of the phases may be slowed down or interrupted under certain circumstances, such as slow electron/ion transport in dissociated phases. The above calculations assume complete thermodynamic equilibrium and no kinetic constraints in the reactions. Therefore, ISEs are expected to withstand a wider range of voltages in use than calculated, as measured by cyclic voltammetry (CV) using Li/Li2OHBr/Au cell, the electrochemical potential window of Li2OHBr was 1.7–3.5 V.11
| Solid-state electrolytes | EW vs. Li/Na (V) | Equilibria phase at reduction potential | Equilibria phase at oxidation potential |
|---|---|---|---|
| Li2OHBr (this work) | 0.80–3.15 | LiH, LiBr, Li2O | Li4H3BrO3, Br |
| Li2OHCl (ref. 26) | 0.82–3.15 | LiH, LiCl, Li2O | LiH2ClO5, H2O, LiCl |
| Li3OCl (ref. 9) | 0–2.55 | Li3OCl | Li2O2, LiCl |
| Na3OBr (ref. 27) | 0–1.79 | Na3OBr | Na2O2, NaBr |
| Li10GeP2S12 (ref. 25) | 1.71–2.14 | Li4GeS4, Li2S, P | Li3PS4, GeS2, S |
| Li3PS4 (ref. 25) | 1.71–2.31 | Li2S, P | P2S5, S |
| Na3PS4 (ref. 27) | 1.39–2.45 | Na3P, Na2S | P2S5, S |
| Li7P3S11 (ref. 25) | 2.28–2.31 | Li3PS4, P4S9 | P2S5, S |
| Li6PS5Cl (ref. 25) | 1.71–2.01 | Li2S, LiCl, P | Li3PS4, LiCl, S |
| Li2PO2N (ref. 25) | 0.68–2.63 | Li3P, LiPN2, Li2O | P3N5, Li4P2O7, N2 |
| Li7La3Zr2O12 (ref. 25) | 0.05–2.91 | Zr3O, La2O3, Li2O | Li2O2, La2O3, Li6Zr2O7 |
The chemical stability between the Li2OHBr and various cathodes is calculated by using eqn (2). Three typical cathode materials (e.g., layered LiCoO2, spinel LiMn2O4, olivine LiFePO4) are considered for fully-discharged and half-charged state. Fig. 3 shows the predicted reaction energies between the Li2OHBr and cathode materials, and the corresponding reaction products are listed in Table S2.† The mutual reaction energy ΔEmin of Li2OHBr with both fully-discharged cathodes are predicted to have low reaction energies (0 < |ΔEmin| < 50 meV per atom) in Fig. 3(a). The chemical reactivity sequence for a fully-discharged cathodes with the Li2OHBr is LiFePO4 > LiMn2O4 > LiCoO2, suggesting that LiCoO2 cathode seems to have better interfacial compatibility with the Li2OHBr. An increased chemical reactivity of Li2OHBr with half-charged cathodes are observed from fully-discharged to half-charged cathode states, as shown in Fig. 3(b). It is worth noting that the Li2OHBr has either no reaction or a negligible driving force against LiCoO2 cathode, showing a significant thermodynamic chemical stability. Certainly, for all interfacial reactions ΔEmin < 0, a thermodynamically unstable interface is formed when Li2OHBr is in contact with high-voltage cathodes, which may result in the formation of unwanted interfacial byproducts, thereby reducing the rate capacity and electrochemical performance of ASSLIBs. Therefore, further experiment techniques are awaited to assess potential interfacial products, such as X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS), and electron microscopy (EM).28
 | ||
| Fig. 3 Predicted reaction energies ΔE between Li2OHBr and various cathode materials in both (a) fully-discharged and (b) half-charged states. The reaction energy ΔEmin corresponds to the lowest mutual reaction energy at a ratio of x. | ||
The air stability of ISEs is another issue involved in electrolyte handling and battery assembly in the development of ASSLIBs, where the electrolyte will inevitably be exposed to air and undergo structural changes if it is not chemically stable.29 For example, using the first-principles calculations, Zhang et al.30 revealed the thermodynamic and kinetic mechanism in the reaction of Li10GeP2S12 with H2O in air to produce H2S gas. Here, the stability of Li2OHBr toward air is also studied using reaction energies ΔE calculation for the reaction with moisture, the higher negative value indicates the material is strongly favorable to react with moisture. The estimation of driving force for Li2OHBr when exposed to air via following reaction:
| 3Li2OHBr + 2H2O → 2LiOH2Br + Li4(OH)3Br, | (4) |
| 2Li4(OH)3Br + 3CO2 → 2LiOH2Br + 3Li2CO3 + H2O. | (5) |
The estimated value of ΔE is only −1 meV per atom when Li2OHBr reacts with H2O. While Li4(OH)3Br forms as a hydrolysis intermediate and subsequently reacts favorably with CO2 to produce LiOH2Br, Li2CO3 and H2O (ΔE = −118 meV per atom). Therefore, it is suggested that the Li2OHBr solid electrolyte is stable in dry air. To understand the degradation mechanism of ISEs exposed to air, some experimental techniques, such as in situ scanning/transmission electron microscopy, neutron ray diffraction depth analysis and synchrotron X-ray imaging technologies, have been performed to track local nanoscale chemical evolution and structural information of interfacial phases.31
3.3 Ionic transport mechanism
 , lithium interstitial
, lithium interstitial  , lithium Frenkel defect pair
, lithium Frenkel defect pair  and Li2O Schottky defect pair
and Li2O Schottky defect pair  , as per our previous work.33 Among these defects, lithium Frenkel defect and Li2O Schottky defect pair in Li2OHBr are described as follows:
, as per our previous work.33 Among these defects, lithium Frenkel defect and Li2O Schottky defect pair in Li2OHBr are described as follows:Lithium Frenkel defect:
 | (6) |
Li2O Schottky defect:
 | (7) |
According to the symmetry of Li2OHBr, the possible defect configurations and formation energies are investigated to obtain the lowest energy configuration. The possible defect configurations include two different lithium vacancy defects  , one lithium interstitial defect
, one lithium interstitial defect  , two lithium Frenkel defect pair (Vnear and Vfar), and three Li2O Schottky defect pair (Vadjacent, Vseparated-1, and Vseparated-2) in the ESI of Fig. S1 and S2.† The formation energies of four defect types
, two lithium Frenkel defect pair (Vnear and Vfar), and three Li2O Schottky defect pair (Vadjacent, Vseparated-1, and Vseparated-2) in the ESI of Fig. S1 and S2.† The formation energies of four defect types  are calculated by eqn (3) in the neutral state, as listed in Table 3. By comparing the formation energies, it is found that the dominant defect configuration is Vnear, and the corresponding defect formation energies is 0.34 eV. In contrast, single
are calculated by eqn (3) in the neutral state, as listed in Table 3. By comparing the formation energies, it is found that the dominant defect configuration is Vnear, and the corresponding defect formation energies is 0.34 eV. In contrast, single  and
and  show higher defect formation energies (3.93 eV and 1.29 eV), implying a lower concentration of lithium vacancy and lithium interstitial defect in neutral Li2OHBr. In addition, the defect formation energies of lithium vacancy and interstitial at different charge states q as a function of Fermi level are also calculated in Fig. 4. The results show that the formation energies of Lii+ and VLi− are lower than those of
show higher defect formation energies (3.93 eV and 1.29 eV), implying a lower concentration of lithium vacancy and lithium interstitial defect in neutral Li2OHBr. In addition, the defect formation energies of lithium vacancy and interstitial at different charge states q as a function of Fermi level are also calculated in Fig. 4. The results show that the formation energies of Lii+ and VLi− are lower than those of  ,
,  ,
,  and
and  , suggesting that the Lii+ and VLi− may be the main defect types in Li2OHBr at room temperature. Therefore, charged defects (Lii+ and VLi−) should act as charge carriers in Li2OHBr and will be used to calculate the migration barriers in the next section.
, suggesting that the Lii+ and VLi− may be the main defect types in Li2OHBr at room temperature. Therefore, charged defects (Lii+ and VLi−) should act as charge carriers in Li2OHBr and will be used to calculate the migration barriers in the next section.
| Defect type |
|
|
|
Vnear | Vfar | Vadjacent | Vseparated-1 | Vseparated-2 |
|---|---|---|---|---|---|---|---|---|
| ui (eV) | −1.89 | −1.89 | −1.89 | — | — | −14.35 | −14.35 | −14.35 |
| Ef (eV) | 3.93 | 4.26 | 1.29 | 0.34 | 0.62 | 3.01 | 2.37 | 2.75 |



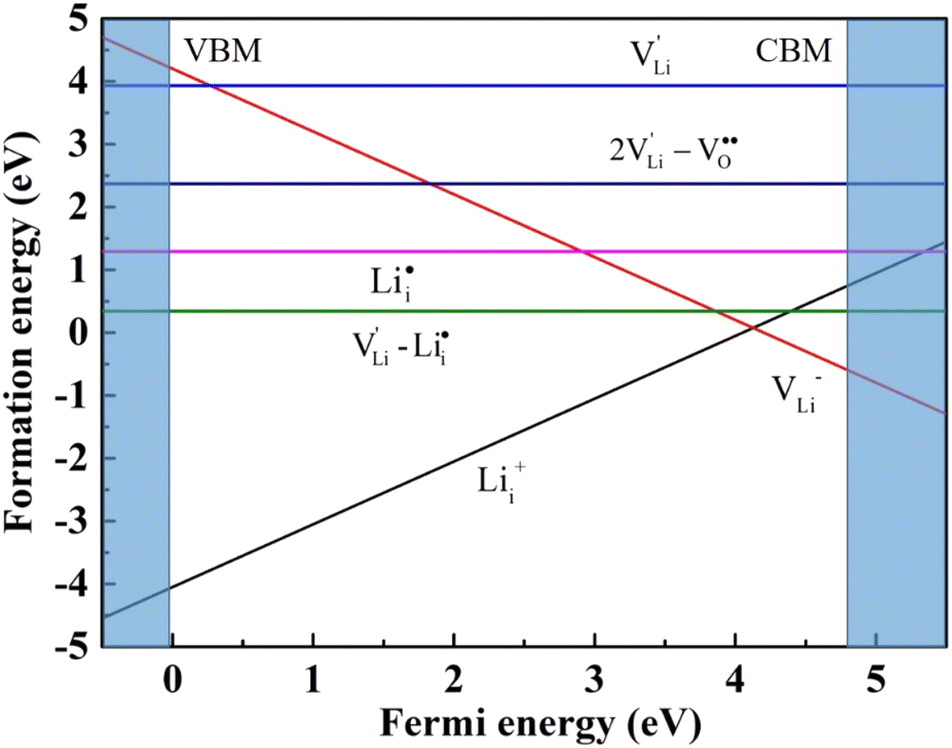 | ||
| Fig. 4 Formation energies of lithium vacancy and interstitial at different charge states. | ||
 | ||
| Fig. 5 Diffusion pathway and migration barriers of VLi− in Li2OHBr (a) along the ab-plane and (b) along the c-axis. | ||
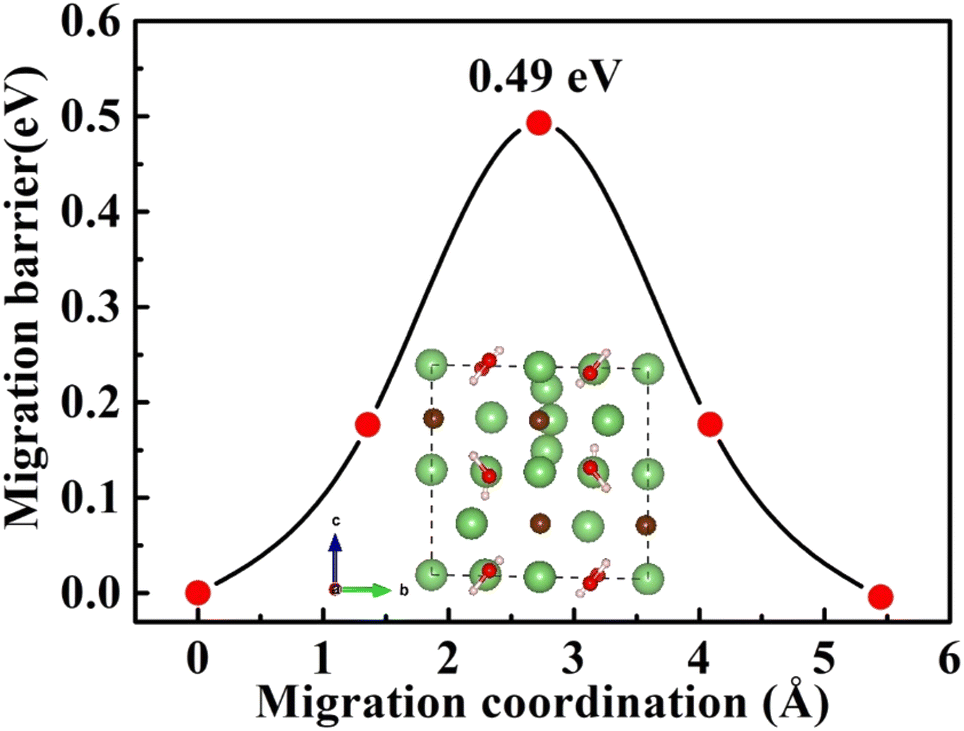 | ||
| Fig. 6 Diffusion pathway and migration barriers of Lii+ in Li2OHBr along the c-axis. | ||
Furthermore, the doping of Li2OHBr with halogen element (F or Cl) may be one of the key factors to enhance the ionic conductivity. In Fig. 7(a) and (b), we present the lithium vacancy migration barriers obtained using CI-NEB calculations for F-doped Li2OHBr, namely Li2(OH)0.875F0.125Br. The results show that the migration barriers of Li2(OH)0.875F0.125Br is 0.37 and 0.33 eV along the ab-plane and c-axis, respectively, which is a lower value than that of pristine Li2OHBr. The main reason is that the substitution of F− for OH− increases the antiperovskite tolerance factor and favors a disordering of the OH− orientation for Li2OHBr, as previous reported by Li et al.21 Therefore, the Li2OHBr can be doped by the substitution of OH− by F− is beneficial to reduce the migration barrier and improve the ionic conductivity.
 | ||
| Fig. 7 Diffusion pathway and migration barriers of VLi− in F-doped Li2OHBr (a) along the ab-plane and (b) along the c-axis. | ||
4. Conclusions
In conclusion, the electronic properties, phase stability, interfacial stability, defect chemistry and Li-ions migration mechanisms of Li2OHBr have been systematically studied by the first-principles calculations. The calculations results indicate that Li2OHBr crystal structure is metastable by thermodynamics analysis. The electronic band structure shows that the Li2OHBr is an insulator with a wide direct band gap. The Li2OHBr has a wide electrochemical stability window that can be matched with the cathode materials. Moreover, the Li2OHBr also exhibits good chemical stability with typical cathode materials and in air. By comparing the defect formation energies in neutral and charged states, it is shown that charged Lii+ and VLi− are the most dominant defect types in Li2OHBr. The Li2OHBr show the low migration barriers by using the CI-NEB calculation, while F-doped Li2OHBr has a lower migration barrier compared with pristine Li2OHBr. This work provides insights into the thermodynamic and kinetic process of Li2OHBr and demonstrates the potential of computational methods in the efficient design of future ISEs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (12004145), the Jiangxi Provincial Natural Science Foundation (20212BAB214032), the Innovation and Entrepreneurship Training Program for College Students in Jiangxi Province (202210419004), the Key Science and Technology Project of Ji'an City (20211015311), the Science and Technology Research Project of Jiangxi Provincial Department of Education (GJJ201030) and the PhD Start-up Fund of Natural Science Foundation of Jinggangshan University (JZB2013). All the calculations were supported by the high-performance computing platform of Shanghai University and Jinggangshan University.References
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- H. Xu, Y. Yu, Z. Wang and G. Shao, Energy Environ. Mater., 2019, 2, 234–250 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- H. Yamane, M. Shibata, Y. Shimane, T. Junke, Y. Seino, S. Adams, K. Minami, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2007, 178, 1163–1167 CrossRef CAS.
- Z. Deng, D. Ni, D. Chen, Y. Bian, S. Li, Z. Wang and Y. Zhao, InfoMat, 2022, 4, e12252 CrossRef CAS.
- J. Zheng, B. Perry and Y. Wu, ACS Mater. Au, 2021, 1, 92–106 CrossRef CAS.
- Y. Zhao and L. L. Daemen, J. Am. Chem. Soc., 2012, 134, 15042–15047 CrossRef CAS PubMed.
- A. Emly, E. Kioupakis and A. Van der Ven, Chem. Mater., 2013, 25, 4663–4670 CrossRef CAS.
- M. K. Sugumar, T. Yamamoto, M. Motoyama and Y. Iriyama, Solid State Ionics, 2020, 349, 115298 CrossRef CAS.
- K. Yoshikawa, T. Yamamoto, M. K. Sugumar, M. Motoyama and Y. Iriyama, Energy Fuels, 2021, 35, 12581–12587 CrossRef CAS.
- L. Gao, R. Zhao, S. Han, S. Li, R. Zou and Y. Zhao, Batteries Supercaps, 2021, 4, 1491–1498 CrossRef CAS.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- B. Liu, D. Wang, M. Avdeev, S. Shi, J. Yang and W. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 948–957 CrossRef CAS.
- B. Liu, J. Liu, J. Yang, D. Wang, C. Ye, D. Wang, M. Avdeev, S. Shi, J. Yang and W. Zhang, J. Power Sources, 2020, 450, 227693 CrossRef CAS.
- S. B. Zhang and J. E. Northrup, Phys. Rev. Lett., 1991, 67, 2339–2342 CrossRef CAS PubMed.
- J. Howard and N. A. W. Holzwarth, Phys. Rev. B, 2019, 99, 014109 CrossRef CAS.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- Y. Li, W. Zhou, S. Xin, S. Li, J. Zhu, X. Lü, Z. Cui, Q. Jia, J. Zhou, Y. Zhao and J. B. Goodenough, Angew. Chem., Int. Ed., 2016, 55, 9965–9968 CrossRef CAS PubMed.
- Z. Deng, Z. Zhu, I.-H. Chu and S. P. Ong, Chem. Mater., 2017, 29, 281–288 CrossRef CAS.
- I.-H. Chu, H. Nguyen, S. Hy, Y.-C. Lin, Z. Wang, Z. Xu, Z. Deng, Y. S. Meng and S. P. Ong, ACS Appl. Mater. Interfaces, 2016, 8, 7843–7853 CrossRef CAS PubMed.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- M. B. Effat, J. Liu, Z. Lu, T. H. Wan, A. Curcio and F. Ciucci, ACS Appl. Mater. Interfaces, 2020, 12, 55011–55022 CrossRef CAS PubMed.
- V. Lacivita, Y. Wang, S.-H. Bo and G. Ceder, J. Mater. Chem. A, 2019, 7, 8144–8155 RSC.
- A. Banerjee, X. Wang, C. Fang, E. A. Wu and Y. S. Meng, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS PubMed.
- W. Xia, Y. Zhao, F. Zhao, K. Adair, R. Zhao, S. Li, R. Zou, Y. Zhao and X. Sun, Chem. Rev., 2022, 122, 3763–3819 CrossRef CAS PubMed.
- J. Zhang, L. Huang and X. Gu, Mater. Adv., 2022, 3, 3143–3150 RSC.
- W. Li, J. Liang, M. Li, K. R. Adair, X. Li, Y. Hu, Q. Xiao, R. Feng, R. Li, L. Zhang, S. Lu, H. Huang, S. Zhao, T.-K. Sham and X. Sun, Chem. Mater., 2020, 32, 7019–7027 CrossRef CAS.
- S. Shi, P. Lu, Z. Liu, Y. Qi, L. G. Hector, H. Li and S. J. Harris, J. Am. Chem. Soc., 2012, 134, 15476–15487 CrossRef CAS PubMed.
- B. Liu, Q. Hu, T. Gao, P. Liao, Y. Wen, Z. Lu, J. Yang, S. Shi and W. Zhang, Journal of Materiomics, 2022, 8, 59–67 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06921k |
| This journal is © The Royal Society of Chemistry 2022 |