
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–organic frameworks incorporating azobenzene-based ligands as a heterogeneous Lewis-acid catalyst for cyanosilylation of imines†
Aasim Saeeda,
Xiao-Yu Zhanga,
Zi-Qing Huanga,
Xin-Yang Zhaoa,
Lei Xub,
Yue Zhao *a,
Wei-Yin Sun*a and
Jing Zhao*a
*a,
Wei-Yin Sun*a and
Jing Zhao*a
aCoordination Chemistry Institute, State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: Jingzhao@nju.edu.cn; zhaoyue@nju.edu.cn; sunwy@nju.edu.cn; Tel: +86 25 89681957
bJiangsu Key Lab of Data Engineering and Knowledge Service, School of Information Management, Nanjing University, Nanjing 210023, China
First published on 12th December 2022
Abstract
In this work, two novel metal–organic frameworks (MOFs) were synthesized by the reaction of azobenzene-based ligands and Zn(NO3)2/CdCO3 under solvothermal conditions with the formula of {[Zn2(abtc)(azpy)(H2O)2]·4H2O}n (1) and {[Cd(abtc)0.5(azpy)0.5(H2O)]·3H2O}n (2) (H4abtc = 3,3′,5,5′-azobenzene tetracarboxylic acid, azpy = 4,4′-azobipyridine). According to the single-crystal X-ray diffraction (SC-XRD) analysis, complexes 1 and 2 possessed quite similar structures except for the coordination modes of the central metal nodes attributed to the difference between the cationic radius of Zn(II) and Cd(II). The Zn(II) cations in 1 adopted a distorted seesaw coordination geometry and the coordination between Zn(II) and organic linkers gave two-dimensional (2D) coordination networks, while the Cd(II) cations in 2 could also bind with the carboxylate groups from neighboring coordination networks to form a three-dimensional (3D) coordination framework. Furthermore, complexes 1 and 2 showed high catalytic activity as heterogeneous Lewis-acid catalysts towards the cyanosilylation of imines with satisfactory reusability under mild conditions and the similar catalytic performance of 1 and 2 could be attributed to the similarity in their structures. A prudent mechanism has been proposed as well to elucidate the role of complexes 1 and 2 in the catalytic process.
Introduction
As a class of important intermediates, cyanohydrins have been widely used in the preparation of various high-value chemicals in chemistry and biology, such as α-amino acids, β-amino alcohols, α-hydroxy esters, and so on due to the versatile reactivity of the cyano group.1–5 Among different synthetic approaches, the catalytic addition to unsaturated bonds (mainly C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, and CO bonds) by employing trimethylsilyl cyanide (TMSCN) as the cyanating reagents to give cyanohydrin trimethylsilyl ethers has been considered as one of the most efficient procedures because of the advantages of this methodology such as safety, simple operation, atom economy, no side reactions, high isolated-yields, low dissociation energy of Si–C bond and so on.6–9 Hence, the development of catalysts for the transformation from TMSCN to cyanohydrins has drawn intensive attention and been frequently at the forefront of synthetic chemistry. In the past several decades, varieties of catalysts have been demonstrated to be efficient in this methodology but most of them are homogeneous, which leads to great difficulties in the separation and reusability of the catalysts.10–17 In this regard, heterogeneous catalysts have caught researchers' attention because their inherent stability and ease of recovery make it simple but efficient to be separated and reused.18–24 Therefore, it is of great significance to develop well-performed heterogeneous catalysts for the synthesis of cyanohydrins.
C, CN, and CO bonds) by employing trimethylsilyl cyanide (TMSCN) as the cyanating reagents to give cyanohydrin trimethylsilyl ethers has been considered as one of the most efficient procedures because of the advantages of this methodology such as safety, simple operation, atom economy, no side reactions, high isolated-yields, low dissociation energy of Si–C bond and so on.6–9 Hence, the development of catalysts for the transformation from TMSCN to cyanohydrins has drawn intensive attention and been frequently at the forefront of synthetic chemistry. In the past several decades, varieties of catalysts have been demonstrated to be efficient in this methodology but most of them are homogeneous, which leads to great difficulties in the separation and reusability of the catalysts.10–17 In this regard, heterogeneous catalysts have caught researchers' attention because their inherent stability and ease of recovery make it simple but efficient to be separated and reused.18–24 Therefore, it is of great significance to develop well-performed heterogeneous catalysts for the synthesis of cyanohydrins.
Metal–organic frameworks (MOFs) are a class of burgeoning inorganic–organic porous materials constructed from the coordination of multidentate organic linkers and metal cations or clusters.25–27 Due to the ultrahigh porosity, large surface area, structural diversity and tunability, and easy modification, MOFs have stimulated great scientific interest in the past several decades and found diverse applications in various fields including gas adsorption/storage/separation, chemical sensing, drug delivery, nonlinear optics, energy storage/conversion, and catalysis, among which heterogeneous catalysis was one of the earliest proposed and demonstrated applications.28–42 Compared with other heterogeneous catalysts, MOFs exhibited many superior characteristics derived from their structural nature. For example, the ordered architecture enables the uniform dispersion and the high density of catalytic sites to improve the catalytic utility; the large enough pore could encapsulate reactants in a confined environment, which is beneficial for the sufficient contact of reactants with catalytic sites; the adjustable pore structures including pore size, hydrophilic/hydrophobic property, electron-deficient/electron-rich nature, chirality and so on endow MOFs with different affinity for various reactant molecules, which distinctly improve the catalytic selectivity.43,44 Therefore, the rational design/synthesis and post-synthetic modification of MOFs for heterogeneous catalysis have gained particular attention recently and MOFs have been demonstrated effective as the pre, co, cooperative, or promotor catalysts in various reactions like cyanosilylation, Knoevenagel condensation, Heck and Fischer–Tropsch reactions. In terms of cyanosilylation, the most studied is the transformation from aldehydes or ketones with TMSCN but using imines as the reactants have been scarcely reported.45–55
In this work, 3,3′,5,5′-azobenzene tetracarboxylic acid (H4abtc), 4,4′-azobipyridine (azpy), and Zn(II)/Cd(II) were employed as the organic linkers and metal nodes to elaborate novel MOFs to serve as heterogeneous catalysts for the cyanosilylation of imines. The choice of such linkers and metal nodes is attributed to the following factors: (a) H4abtc has four carboxyl groups and can generate a variety of structures via complete or partial deprotonation; (b) the azo bond is a Lewis base group and could easily reverse its cis or trans configuration, which may enable the synthesized MOFs more structural diversity and modulate the catalytic performance; (c) the π-conjugated aromatic linkers may be beneficial to the accessibility of active sites and transport to the substrate/products in catalysis; (d) the d10 transition metal nodes (Zn(II)/Cd(II)) could afford various Lewis acid catalytic sites.56–62 By the reaction of H4abtc, azpy, and Zn(II)/Cd(II) under solvothermal conditions, two novel MOFs with the formula of {[Zn2(abtc)(azpy)(H2O)2]·4H2O}n (1) and {[Cd(abtc)0.5(azpy)0.5(H2O)]·3H2O}n (2) were obtained. Single-crystal X-ray diffraction (SC-XRD) results revealed that these two MOFs were isostructural although complex 1 and 2 were respectively constructed from 2D and 3D coordination networks. Due to the larger radius of the metal cation, the central Cd(II) cations in 2 could form coordination bonds between the central nodes from two adjacent 2D coordination networks to form a 3D coordination framework and such a phenomenon has been scarcely reported. Furthermore, the Lewis acid nature of synthesized MOFs has been examined with their substrate scopes by heterogeneously catalyzed imine cyanosilylation reactions and experimental results showed that both complexes 1 and 2 possessed effective catalytic activity for imine cyanosilylation with favorable reusability.
Experimental
Materials and methods
The organic linkers H4abtc and azpy were synthesized according to the previously reported procedures.63,64 All other commercially available chemicals and solvents were of the reagent grades and used as received without further purification. The powder X-ray diffraction (PXRD) data was collected using an X-ray diffractometer equipped with Cu Kα (λ = 1.54184 Å; 20 mA, 40 kV) radiation in the 2-theta range of 5–50° on a Bruker D8 Advance X-ray diffractometer. Thermogravimetric analysis (TGA) was conducted on a Mettler-Toledo (TGA/DSC1) thermal analyzer with a heating rate of 10 °C min−1 under an N2 atmosphere in the temperature range of 25–800 °C. Fourier transform infrared-attenuated total internal reflectance (FTIR-ATR) spectra were measured on a Bruker Tensor II spectrophotometer equipped with a diamond ATR module using the mid-IR region (4000–400 cm−1). PerkinElmer's 240C elemental analyzer was used to carry out the elemental analysis (EA) of C, H, and N. The reactions were monitored with thin-layer chromatography (TLC) on silica gel plates (HSGF254). 1H and 13C NMR spectra were recorded at room temperature on Bruker-DRX 400 MHz instrument and the chemical shifts were reported in parts per million (ppm) with tetramethyl silane or the residual solvent peak as an internal reference. High-resolution mass (HRMS) spectra were performed on a Thermo Fischer QExactive quadrupole-Orbitrap mass spectrometer.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) as the developing solvent. After the reaction was completed, the reaction mixture was filtered carefully to separate the catalyst as residue, and the filtrate containing the final product was then evaporated under a vacuum, which then was neutralized by adding 5 mL of 1% NaHCO3 solution and extracted with ethyl acetate. The ethyl acetate layer was dried over anhydrous Na2SO4, and the crude product was obtained by evaporating the ethyl acetate under vacuum and then purified by using column chromatography (silica gel, 100–200 mesh) with ethyl acetate/n-hexane (1:12 v/v) as the eluent. In addition, the separated catalyst residue was washed with DCM multiple times, air-dried for about four hours, and then reused for the following run-up to 5 cycles.
:9) as the developing solvent. After the reaction was completed, the reaction mixture was filtered carefully to separate the catalyst as residue, and the filtrate containing the final product was then evaporated under a vacuum, which then was neutralized by adding 5 mL of 1% NaHCO3 solution and extracted with ethyl acetate. The ethyl acetate layer was dried over anhydrous Na2SO4, and the crude product was obtained by evaporating the ethyl acetate under vacuum and then purified by using column chromatography (silica gel, 100–200 mesh) with ethyl acetate/n-hexane (1:12 v/v) as the eluent. In addition, the separated catalyst residue was washed with DCM multiple times, air-dried for about four hours, and then reused for the following run-up to 5 cycles.X-ray crystallography
Suitable single crystals of 1 and 2 were mounted on a cryoloop using Paratone N oil, and then the crystallographic data collections were carried out on a Bruker D8 Venture diffractometer equipped with graphite-monochromated Ga Kα radiation (λ = 1.34139 Å) and a Bruker D8 Venture Photon 100 diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) using the φ–ω scan technique, respectively. SAINT program was used to integrate diffraction data and the intensity corrections for Lorentz and polarization effects.65 Semi-empirical absorption corrections were applied using SADABS program.66 The structures were solved by direct methods with SHELXT-2014, expanded by subsequent Fourier-difference synthesis, and all the non-hydrogen atoms were refined anisotropically on F2 using the full-matrix least-squares technique using the SHELXL-2018 crystallographic software package.67 Hydrogen atoms except those of water molecules were introduced at the calculated positions and refined isotopically using a riding model. The hydrogen atoms of water molecules were located based on the Fourier difference maps. The free solvent molecules in the unit cell of 2 were taken into account using SQUEEZE option of the PLATON program.68 The final formula of 2 was calculated based on volume/count electron analysis, TGA, and EA, and the results were attached to the CIF file. Structural refinement details and crystallographic data for 1 and 2 are given in Table 1 and the selected bond lengths and angles are given in Table S1.†| 1 | 2 | |
|---|---|---|
| a R1 = ∑‖Fo| − |Fc‖/∑|Fo|.b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2, where w = 1/[σ2(Fo)2 + (aP)2 + bP]. | ||
| Empirical formula | C26H26N6O14Zn2 | C13H9CdN3O5 |
| Formula mass | 777.2 | 399.64 |
| Crystal system | Triclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a (Å) | 7.8726 (3) | 7.5893 (8) |
| b (Å) | 9.1910 (3) | 9.8529 (11) |
| c (Å) | 11.7294 (4) | 12.2846 (13) |
| α (°) | 96.2350 (10) | 100.780 (4) |
| β (°) | 104.8540 (10) | 99.296 (3) |
| γ (°) | 112.3330 (10) | 109.467 (4) |
| V (Å3) | 738.39 (5) | 825.58 (16) |
| Z | 1 | 2 |
| T/K | 193 (2) | 296 (2) |
| ρcalcd (g cm−3) | 1.748 | 1.608 |
| μ (mm−1) | 1.794 | 1.346 |
| λ (Å) | 1.34139 (Ga Kα) | 0.71073 (Mo Kα) |
| θmin (°) | 4.650 | 2.270 |
| θmax (°) | 53.880 | 27.478 |
| Reflection collected | 7625 | 7402 |
| Unique reflections | 2661 | 3745 |
| Rint | 0.0331 | 0.0310 |
| R indexes [I > 2σ(I)] | R1 = 0.0377, wR2 = 0.1033 | R1 = 0.0280, wR2 = 0.0697 |
| R indexes (all data) | R1 = 0.0401, wR2 = 0.1053 | R1 = 0.0316, wR2 = 0.0716 |
| F(000) | 396 | 392 |
| GOF on F2 | 1.028 | 1.047 |
Results and discussion
Crystal structure description
space group and the asymmetric unit was comprised of a crystallographically independent Zn(II) cation, one-half completely deprotonated carboxylate linker abtc4−, half an azpy linker, one coordinated and two lattice H2O molecules. As shown in Fig. 1a, atom Zn1 exhibited a seesaw geometry (τ4 = 0.78),69 furnished by two carboxylate oxygen (O1, O3#1) atoms from two discrete abtc4− linkers, one nitrogen atom (N2) belonging to azpy linker, and one coordinated H2O molecule (O5). The Zn–N bond length was 2.024(2) Å, and the bond lengths observed between Zn–O were in the range of 1.9309(18)–2.049(2) Å (Table S1†). Additionally, the coordination bond angles surrounding the Zn(II) nodes vary in the range of 93.75(8)–128.77(9)° (Table S1†). The complete deprotonation of the carboxylic groups of the organic linker H4abtc was also demonstrated by the absence of the infrared absorption characteristic peaks of –COOH around 1700 cm−1 in FTIR-ATR spectra (Fig. S3†). The Zn(II) nodes were bridged by the carboxylate groups of abtc4− linkers to form 1-dimensional (1D) ladder-like coordination chains (Fig. 1b), which were further linked by azpy linker to generate 2-dimensional (2D) coordination networks with two kinds of windows with the Zn⋯Zn distances of 9.564(3) × 12.911(3) Å and 9.564(3) × 16.413(4) Å (Fig. 1c). Furthermore, these 2D networks could be stacked in a staggered mode to form a 3-dimensional (3D) supramolecular framework (Fig. 1d and S1a and S1b†) with 1D channels along a-axis through π⋯π interactions between the benzene ring of abtc4− and the pyridine ring of azpy and the hydrogen bonds among the lattice waters, coordinated waters and organic linkers from the 2D networks (Fig. S1c†). According to the result of PLATON, the porosity of complex 1 was calculated to be 15.7%. In addition, the unusual bond length between the metal centre and carboxylate oxygen (Zn1–O1#4 = 2.7810(1), Fig. S1d†) from adjacent layer suggested a non-negligible weak interaction. Hence, the geometry around the Zn(II) centre in 1 may also be regarded as a distorted trigonal bipyramid and bridged by the carboxylate oxygen atom to form binuclear Zn(II) centre. Therefore, taking the weak Zn–O interaction into consideration, each binuclear Zn(II) cluster was bound to six organic linkers and thus could be regarded as a six-connected node, while the organic linker abtc4− and azpy could be similarly treated as four-connected and two-connected nodes respectively from the point view of topology. Hence, the structure of complex 1 could be simplified as a (2,4,6)-connected framework with the topological symbol of {44·610·8} {44·62} (Fig. S1e†) using the TOPOS program,70 which was also reported in some other MOFs.71
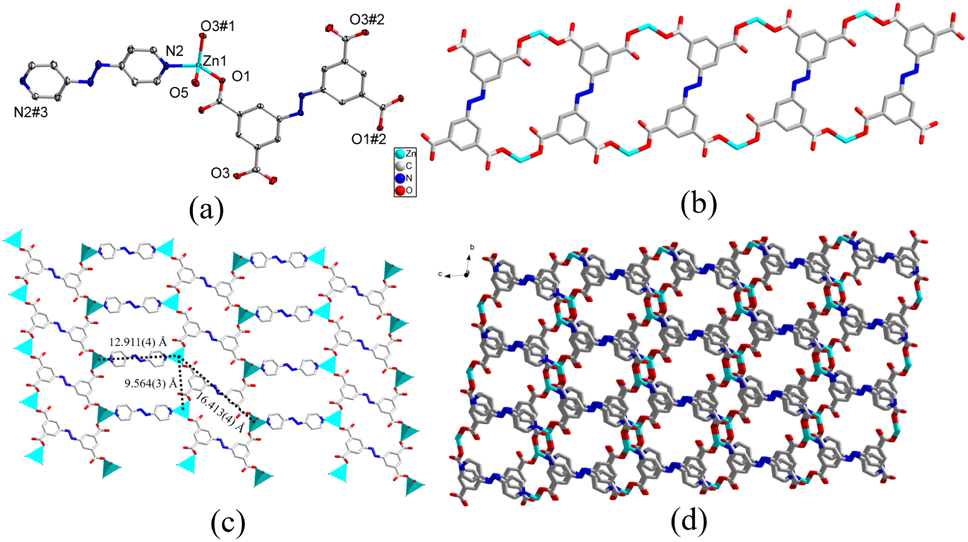 | ||
| Fig. 1 (a) Coordination environment of Zn(II) atoms in 1 with the ellipsoid drawn at the 30% probability level. Symmetry code: #1 −x + 1, −y + 1, z; #2 −x + 1, −y + 1, −z + 2; #3 −x, −y − 1, −z. (b) 1D coordination ladders constructed from Zn(II) and abtc4−, (c) 2D coordination networks, (d) 3D supramolecular framework along a-axis in 1. Hydrogen atoms and free H2O molecules are omitted for clarity. | ||
space group and each asymmetric unit of 2 was comprised of a crystallographically independent Cd(II) node, half one abtc4− organic anion, half one azpy molecule, and one coordinated H2O molecule. Structural analysis revealed that the structure of complex 2 was almost identical to that of 1 except for the coordination modes of central metal cations. Compared to complex 1, due to the large ionic radius of Cd(II) than Zn(II) cation, the central Cd(II) cations could also coordinate with the carboxylate groups from the adjacent network to form binuclear clusters (Fig. 2b) and thus the coordination geometry of central Cd1 atoms was a six-coordinated octahedron surrounded by one nitrogen atom (N1) from the pyridine ring of azpy ligand, four carboxylate oxygen atoms (O2, O3, O4#1, O5#2) from two different abtc4− linkers and one coordinated water (O1) molecule (Fig. 2a). The Cd–N bond length was 2.303(2) Å, and the Cd–O bond lengths were observed in a typical range of 2.269(2)–2.596(2) Å (Table S1†). The O–Cd–O and O–Cd–N bond angles varied in the range of 53.04(7)–171.15(9)° and 84.98(9)–138.80(9)°, respectively (Table S1†). The coordination between Cd(II) cations and organic ligands abtc4− led to the formation of 2D coordination networks possessing the windows with the Cd1–Cd1 distances of 10.239(1) × 12.284(6) Å (Fig. 2c and S2a†), which were bridged by linker azpy to generate 3D coordination frameworks with 1D channels along a-axis through the connection between Cd(II) cations and pyridine of azpy (Fig. 2d). The porosity of complex 2 could be calculated to be 23.6% using PLATON software. Furthermore, according to the topological analysis, the structure of complex 2 had the same topological structure as that of 1 with the topological symbol of {44·610·8} {44·62} (Fig. S2b†).
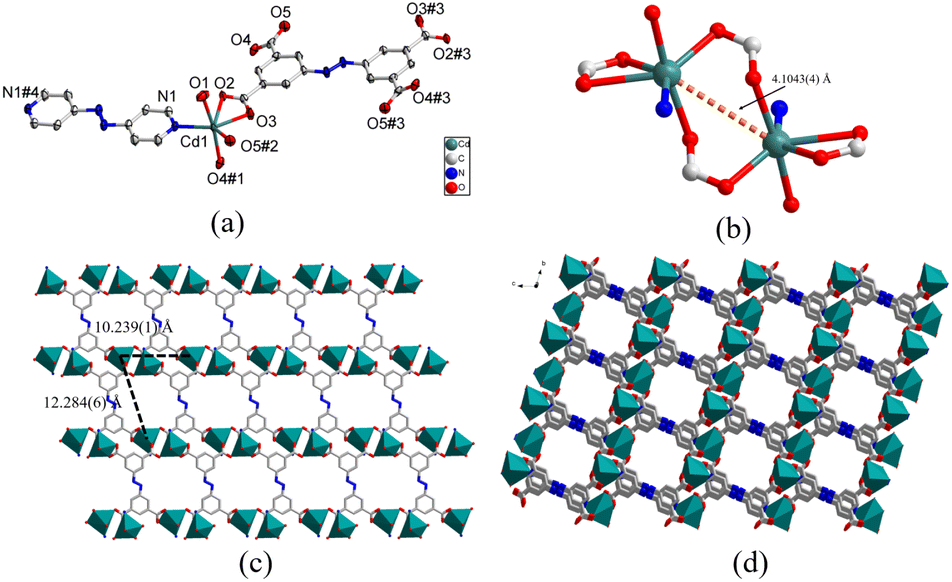 | ||
| Fig. 2 (a) Coordination environment of Cd(II) atoms in 2 with the ellipsoid drawn at the 30% probability level. Symmetry code: #1 −x, −y + 1, −z + 1; #2 x + 1, y + 1, z; #3 −x, −y + 1, −z + 2; #4 −x + 1, −y + 1, −z. (b) The structure of binuclear Cd2 node in 2. (c) 2D coordination networks in 2 constructed from Cd(II) cations and ligand abtc4−. (d) 3D coordination framework of 2 along the a-axis. Hydrogen atoms were omitted for clarity. | ||
Chemical and thermal stability
PXRD was employed to check the phase purities and investigate the chemical stability of synthesized complexes 1 and 2. As shown in Fig. S4,† the PXRD patterns of the as-synthesized samples of 1 and 2 were almost identical to the simulated ones obtained from the single-crystal data, demonstrating the phase purities and crystallinity of synthesized complexes 1 and 2. Furthermore, the PXRD patterns of complexes 1 and 2 remained unchanged after exposing to air for one month, immersed in different organic solvents for one week including dichloromethane (DCM), ether (Et2O), tetrahydrofuran (THF), acetone, acetonitrile (MeCN), methanol (MeOH), ethanol (EtOH), n-hexane, ethyl acetate (EA), DMA, N,N-dimethylformamide (DMF) and dimethyl sulfoxide (DMSO), and soaking in water within the pH range from 1 to 13 and boiling water for at least one day (Fig. S5 and S6†), which demonstrated the ultrahigh chemical stability of complexes 1 and 2. Furthermore, the thermal stability of complexes 1 and 2 was also investigated by thermogravimetric (TG) measurements. As shown in Fig. S7a,† complex 1 exhibited a weight loss of 9.6% in the range of 25–110 °C and then 4.6% before 170 °C, corresponding to the release of the lattice water molecules (calcd. 9.3%) and coordinated water molecules (calcd. 4.6%), respectively. Further weight loss revealed that its framework could retain up to 310 °C. As for complex 2 shown in Fig. S7b,† the initial weight loss in the temperature range of 25–135 °C was about 11.1%, which could be attributed to the removal of lattice water molecules (calcd. 11.9%). The second weight loss before 220 °C was about 3.0%, corresponding to the release of the coordinated water (calcd. 4.0%). And then the structure of complex 2 started to decompose until 315 °C. These results demonstrated that complexes 1 and 2 not only possessed outstanding chemical stability but also had fairly high thermal stability.Gas adsorption behaviors
Prior to gas adsorption, as-synthesized porous frameworks 1 and 2 were activated by heating at 170 and 180 °C under dynamic vacuum overnight, respectively. The TG curves and the PXRD patterns of the activated 1 and 2 indicated the removal of solvents and the retainment of their framework structure (Fig. S7 and S8†). The adsorption isotherms of the activated samples of 1 and 2 for N2 were monitored at 77 K. The results revealed that they both displayed the typical reversible type-II N2 adsorption isotherm and the maximum N2 sorption capacity for 1 and 2 was both 0.5 mmol g−1 under 0.99 bar. The Brunauer–Emmett–Teller (BET) surface area based on the N2 sorption data was calculated to be 6.5 m2 g−1 and 15.2 m2 g−1 for 1 and 2, respectively (Fig. S9a and b†). Furthermore, the CO2 adsorption isotherms were collected at 273 K and 298 K for the guest-free frameworks 1 and 2 as well. The obtained adsorption isotherms exhibited the maximum CO2 sorption capacity was 0.15, 1.52 mmol g−1 at 273 K and 0.040, 0.77 mmol g−1 at 298 K for 1 and 2, respectively under 800 mmHg (Fig. S9c and d†). This indicate that the pores in the structures are very small, and the catalytic reactions occur on the surface of the catalysts.Catalytic activity for the cyanosilylation of imines
In consideration of the presence of coordinated water molecules enabling the central metal nodes as the potential vacant metal coordination metal sites, complexes 1 and 2 may be used as a Lewis acid heterogeneous catalyst and their catalytic performance for the cyanosilylation of imines was investigated. First of all, benzylideneaniline was chosen as the model substrate to check the catalytic activity of complexes 1 and 2 for the cyanosilylation with TMSCN and optimize the reaction conditions. As shown in Table 2, it could be clearly found that the yields of the cyanosilylation reaction improved distinctly with time and reach up to 95% for 1 and 98% for 2 in 4 h at room temperature with DCM as the solvent (entry 1–4 and 8–11). More, the influence of solvent and temperature were also checked and it could be found that the yields of the cyanosilylation reaction in the ice-water bath (0 °C) and in THF or ethyl ether as solvent were all obviously lower than those at room temperature in DCM (entry 5–7 and 12–14). Therefore, the catalytic cyanosilylation reactions were carried out at room temperature in DCM. In addition, it could be also found that the reaction yields were much lower without any catalyst or with the metal salts Zn(NO3)2/CdCO3 or the organic ligands H4abtc and azpy as the catalyst (entry 15–19), which indicated the catalytic activity of complexes 1 and 2 for the cyanosilylation of imines with TMSCN and the significance of the formation of MOFs for the catalytic reactions.
|
|||||
|---|---|---|---|---|---|
| Catalyst | Solvent | Temperature (°C) | Time (h) | Yield (%) | TON |
| a Reaction conditions: N-benzylideneaniline (0.5 mmol), Me3SiCN (0.75 mmol), dry DCM (4 mL), catalysts 1 and 2, linkers H4abtc, azpy (0.1 mmol), blank reaction, 4 h. | |||||
| 1 | DCM | RT | 4 | 95 | 4.75 |
| DCM | RT | 3 | 75 | 3.75 | |
| DCM | RT | 2 | 59 | 2.95 | |
| DCM | RT | 1 | 43 | 2.15 | |
| DCM | 0 | 4 | 61 | 3.05 | |
| THF | RT | 4 | 63 | 3.15 | |
| (C2H5)2O | RT | 4 | 69 | 3.45 | |
| 2 | DCM | RT | 4 | 98 | 4.90 |
| DCM | RT | 3 | 79 | 3.95 | |
| DCM | RT | 2 | 62 | 3.10 | |
| DCM | RT | 1 | 49 | 2.45 | |
| DCM | 0 | 4 | 66 | 3.30 | |
| THF | RT | 4 | 71 | 3.55 | |
| (C2H5)2O | RT | 4 | 93 | 4.65 | |
| Zn(NO3)2·6H2O | DCM | RT | 4 | 23 | 1.15 |
| CdCO3 | DCM | RT | 4 | 13 | 0.65 |
| H4abtc | DCM | RT | 4 | 7 | 0.35 |
| azpy | DCM | RT | 4 | 6 | 0.30 |
| Blank | DCM | RT | 4 | 6 | 0.30 |
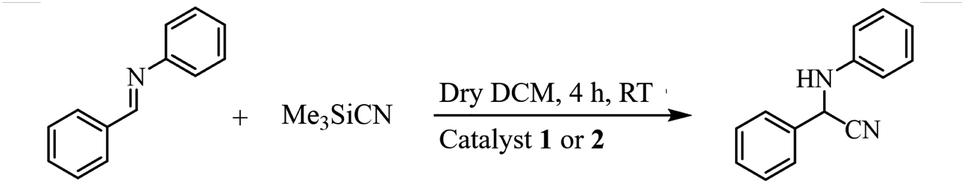
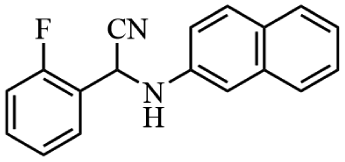
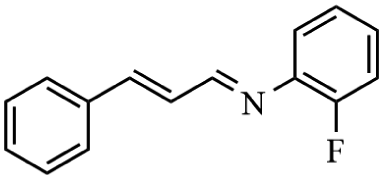
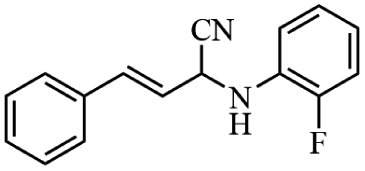
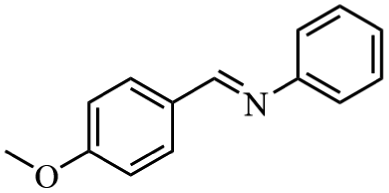
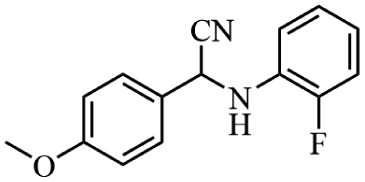
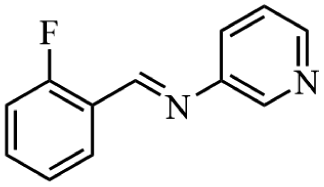
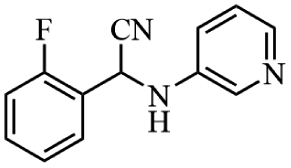
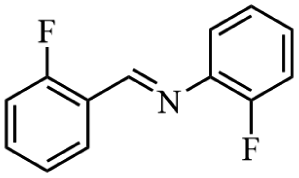
Next, the catalytic versatility of complexes 1 and 2 for the cyanosilylation reaction of other aromatic imines were explored. As shown in Table 3, varieties of aromatic imines bearing different electron-withdrawing or electron-donating groups were chosen to react with TMSCN under similar conditions using complexes 1 and 2 as the catalysts, and the results clearly revealed that all of these compounds could be successfully transformed into the corresponding nitrile compounds with the yields higher than 85%. Meanwhile, it could be also found that the yields with these compounds as the substrate were all lower than those of benzylideneaniline and electron-withdrawing groups afforded higher yields than electron-donating groups on the reactions because they could help the reaction activate more electrophilic sites on the reactants. In addition, it could be observed that the catalytic performance of complex 1 and 2 towards the cyanosilylation of all the selected substrates was nearly the same and the reason may be attributed to the similarity in their structure.
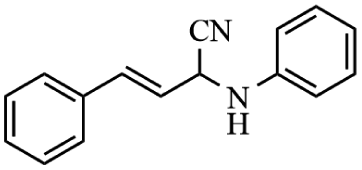
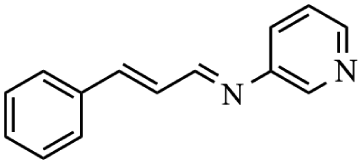
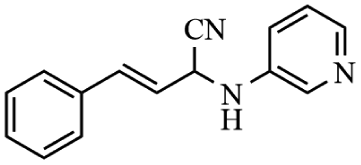
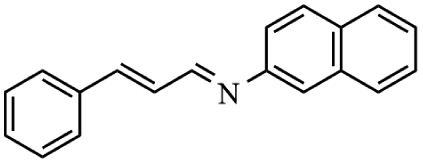
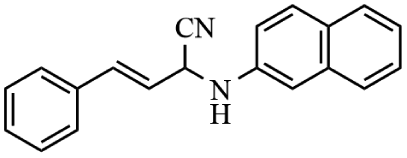
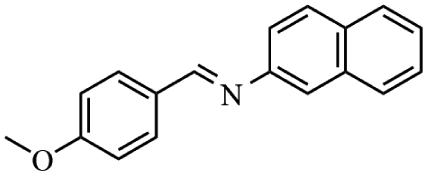
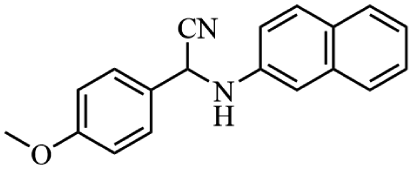
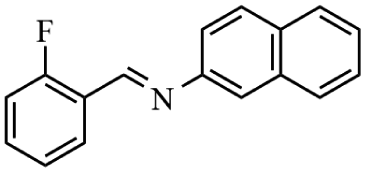
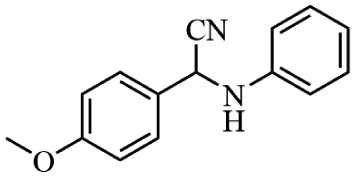
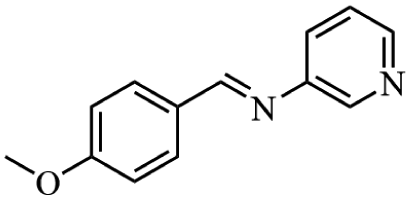
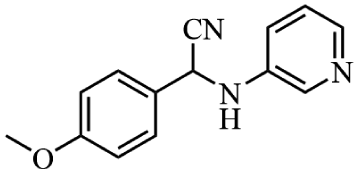
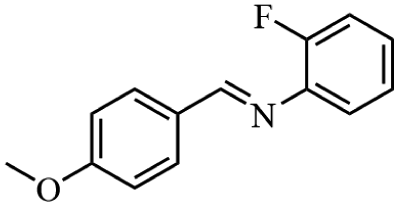
Furthermore, filtration tests were also conducted to examine whether complexes 1 and 2 worked in a heterogeneous manner to promote the cyanosilylation reaction of imines. Taking the cyanosilylation of benzylideneaniline as the model reaction, the reaction mixture was filtrated to remove the solid catalyst after the reaction time of 2 h, and then it could be observed that the conversion of benzylideneaniline almost remained unchanged and significantly lower than the reactions with catalyst, which obviously verified the heterogeneous nature of catalyst 1 and 2 (Fig. 4). Finally, recycling experiments were carried out to explore the reusability of complexes 1 and 2 as the catalysts for the cyanosilylation of imines. PXRD measurements revealed that PXRD patterns of 1 and 2 after catalytic reactions matched well with those simulated ones obtained from the SC-XRD data, which suggested their structural stability in the process of catalytic reactions and thus afforded the possibility of reusability (Fig. S10†). After the catalytic reactions, the solid catalysts 1 and 2 were collected by centrifugation, washed with ethanol several times and then dried in an oven at 60 °C. Afterward, the recovered catalysts 1 and 2 were directly introduced into the reaction system again to catalyze the reaction of benzylideneaniline with TMSCN and the yields showed no significant decay even after five cycles (Fig. 3), indicating the satisfactory reusability of complexes 1 and 2 as the catalysts for the cyanosilylation of imines.
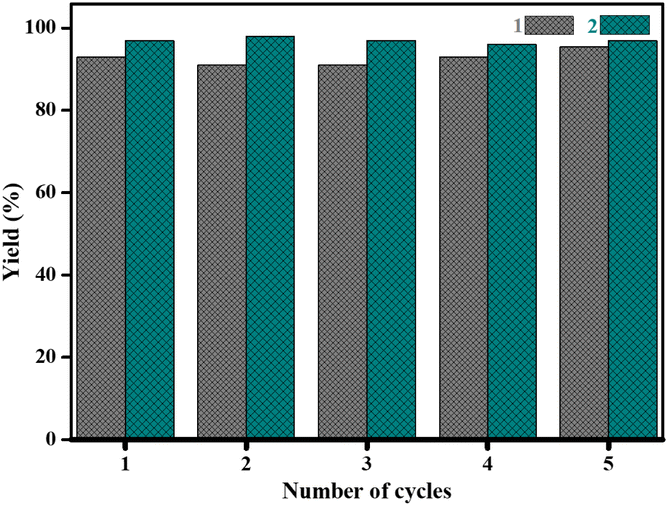 | ||
| Fig. 3 Recyclability of complexes 1 and 2 as the catalysts during cyanosilylation reaction of benzylideneaniline up to 5 consecutive cycles. | ||
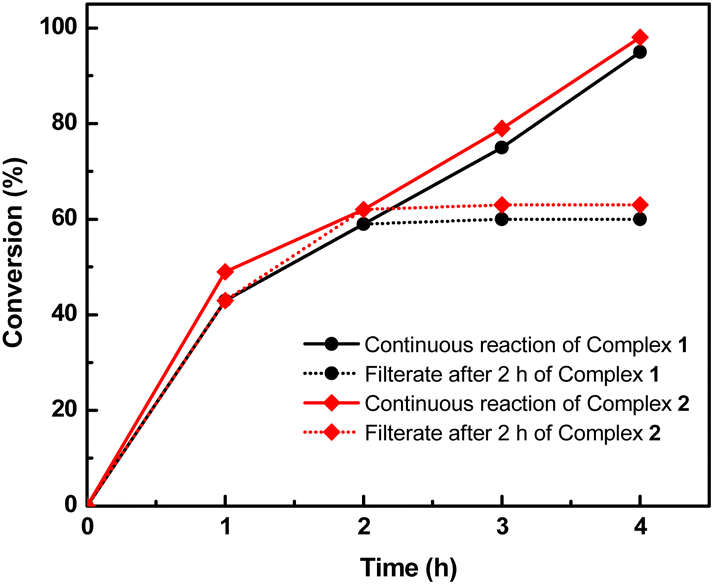 | ||
| Fig. 4 The filtration test of the cyanosilylation of benzylideneaniline catalyzed by complexes 1 and 2 determined by NMR. | ||
Based on the experimental results and the comparison with previously reported results (Table S3†), a possible reaction mechanism was proposed to illustrate the catalytic process of complexes 1 and 2 for the cyanosilylation of imines. In consideration of the existence of lone pairs at the nitrogen atoms in imines and Lewis acid metal nodes in complexes 1 and 2, a weak connection may be formed between the nitrogen atoms of imines and metal nodes. Meanwhile, the coordinated oxygen atoms could act as an electron-rich nucleophilic moiety to facilitate the electron transfer to the trimethylsilyl groups of TMSCN, which resulted in the formation of weakly coordinated Si–O bonds. Meanwhile, the coordinated water molecules also may contribute to the silicon intermediate production that included the weaker C–Si link with the Lewis acid sites. As a final step, electrons from the nucleophilic –CN group were transferred to carbon to create the cyanosilylated species. The whole catalytic process was depicted in Scheme 1. Such an arrangement allows for more effective use of heterogeneous catalysts in subsequent cycles. Furthermore, the results of gas adsorption experiments and the catalytic performance for different substrates also revealed that the catalytic reaction occurred on the surface of catalyst but not in the pores of the synthesized MOFs.
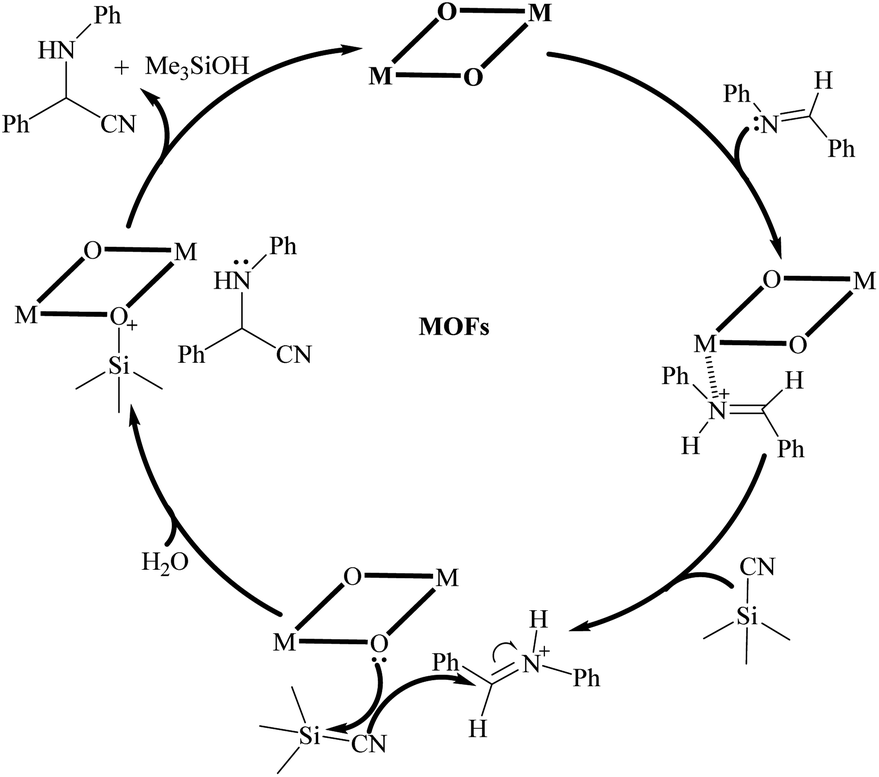 | ||
| Scheme 1 Lewis acid catalyzed imine cyanosilylation proposed mechanism for catalyst 1 and 2. | ||
Conclusions
In conclusion, two novel MOFs 1 and 2 have been synthesized by the solvothermal reaction of azobenzene-containing ligands H4abtc and azpy with Zn(NO3)2/CdCO3. Although the SC-XRD analysis showed that complexes 1 and 2 were isostructural, the difference in the cationic radius of Zn(II) and Cd(II) resulted in distinctly different coordination geometry, which led to the formation of 2D coordination networks for 1 but 3D coordination frameworks for 2. Complexes 1 and 2 both exhibited ultrahigh chemical and fairly high thermal stability. In addition, the catalytic experiments manifested that both complexes 1 and 2 had high catalytic activity as heterogeneous Lewis-acid catalysts for the imine cyanosilylation reactions under mild conditions with well-performed reusability. We believe this work will stimulate further efforts to develop new frameworks with multidentate ligands to explore new functional applications for various chemical transformations.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (grant nos. 21401099, 21671099 and 91753121) and the Shenzhen Basic Research Program (JCYJ20180508182240106).Notes and references
- J. J. Du, X. Zhang, X. P. Zhou and D. Li, Inorg. Chem. Front., 2018, 5, 2772–2776 RSC.
- W. Wang, M. Luo, J. Li, S. A. Pullarkat and M. Ma, Chem. Commun., 2018, 54, 3042–3044 RSC.
- F. Garnes-portolés, M. Á. Rivero-crespo and A. Leyva-pérez, J. Catal., 2020, 392, 21–28 CrossRef.
- J. M. Brunel and I. P. Holmes, Angew. Chem., Int. Ed., 2004, 43, 2752–2778 CrossRef CAS PubMed.
- D. H. Ryu and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 8106–8107 CrossRef CAS PubMed.
- M. North, D. L. Usanov and C. Young, Chem. Rev., 2008, 108, 5146–5226 CrossRef CAS.
- Z. Zhang, J. Chen, Z. Bao, G. Chang, H. Xing and Q. Ren, RSC Adv., 2015, 5, 79355–79360 RSC.
- G. K. Surya Prakash, H. Vaghoo, C. Panja, V. Surampudi, R. Kultyshev, T. Mathew and G. A. Olah, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3026–3030 CrossRef CAS PubMed.
- J. M. Pérez, S. Rojas, A. García-García, H. Montes-Andrés, C. R. Martínez, M. S. Romero-Cano, D. Choquesillo-Lazarte, V. K. Abdelkader-Fernández, M. Pérez-Mendoza, J. Cepeda, A. Rodríguez-Diéguez and I. Fernández, Inorg. Chem., 2022, 61, 1377–1384 CrossRef PubMed.
- G. Cainelli, D. Giacomini, A. Treré and P. Galletti, Tetrahedron: Asymmetry, 1995, 6, 1593–1600 CrossRef CAS.
- L. Mei, S. Jie and J. Ying, Res. Chem. Intermed., 2010, 36, 227–236 CrossRef CAS.
- S. Pahar, G. Kundu and S. S. Sen, ACS Omega, 2020, 5, 25477–25484 CrossRef CAS PubMed.
- S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 15872–15883 CrossRef CAS PubMed.
- S. Rawat, M. Bhandari, B. Prashanth and S. Singh, ChemCatChem, 2020, 12, 2407–2411 CrossRef CAS.
- H. Deka, N. Fridman and M. S. Eisen, Inorg. Chem., 2022, 61, 3598–3606 CrossRef CAS.
- D. E. Fuerst and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 8964–8965 CrossRef CAS PubMed.
- Z. Z. Weng, J. Xie, K. X. Huang, J. P. Li, L. S. Long, X. J. Kong and L. S. Zheng, Inorg. Chem., 2022, 61, 4121–4129 CrossRef CAS.
- M. Malmir and M. M. Heravi, Sci. Rep., 2022, 12, 11573 CrossRef CAS PubMed.
- C. Baleizão, B. Gigante, H. Garcia and A. Corma, J. Catal., 2004, 221, 77–84 CrossRef.
- S. Zhao, Y. Chen and Y. F. Song, Appl. Catal., A, 2014, 475, 140–146 CrossRef CAS.
- K. Iwanami, J. C. Choi, B. Lu, T. Sakakura and H. Yasuda, Chem. Commun., 2008, 2, 1002–1004 RSC.
- S. Zhang, B. Zhang, H. Liang, Y. Liu, Y. Qiao and Y. Qin, Angew. Chem., Int. Ed., 2018, 57, 1091–1095 CrossRef CAS.
- K. Yamaguchi, T. Imago, Y. Ogasawara, J. Kasai, M. Kotani and N. Mizuno, Adv. Synth. Catal., 2006, 348, 1516–1520 CrossRef CAS.
- Y. X. Wang, H. M. Wang, P. Meng, D. X. Song, J. J. Hou and X. M. Zhang, Dalton Trans., 2021, 50, 1740–1745 RSC.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS PubMed.
- S. Natarajan and P. Mahata, Chem. Soc. Rev., 2009, 38, 2304–2318 RSC.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 CrossRef CAS.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- R. Medishetty, J. K. Zarȩba, D. Mayer, M. Samoć and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 RSC.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- A. Gheorghe, M. A. Tepaske and S. Tanase, Inorg. Chem. Front., 2018, 5, 1512–1523 RSC.
- P. C. Rao and S. Mandal, Chem.–Asian J., 2019, 14, 4087–4102 CrossRef CAS PubMed.
- M. Sadakiyo, Nanoscale, 2022, 14, 3398–3406 RSC.
- N. R. Habib, E. Asedegbega-Nieto, A. M. Taddesse and I. Diaz, Dalton Trans., 2021, 50, 10340–10353 RSC.
- M. Liu, J. Wu and H. Hou, Chem.–Eur. J., 2019, 25, 2935–2948 CAS.
- A. Yadav and P. Kanoo, Chem.–Asian J., 2019, 14, 3531–3551 CrossRef CAS.
- C. De Wu and M. Zhao, Adv. Mater., 2017, 29, 1605446 CrossRef PubMed.
- J. Z. Gu, S. M. Wan, W. Dou, M. V. Kirillova and A. M. Kirillov, Inorg. Chem. Front., 2021, 8, 1229–1242 RSC.
- C. Zhu, Q. Xia, X. Chen, Y. Liu, X. Du and Y. Cui, ACS Catal., 2016, 6, 7590–7596 CrossRef CAS.
- D. Dang, P. Wu, C. He, Z. Xie and C. Duan, J. Am. Chem. Soc., 2010, 132, 14321–14323 CrossRef CAS.
- M. Gustafsson, A. Bartoszewicz, B. Martiín-Matute, J. Sun, J. Grins, T. Zhao, Z. Li, G. Zhu and X. Zou, Chem. Mater., 2010, 22, 3316–3322 CrossRef CAS.
- F. Z. Jin, C. C. Zhao, H. C. Ma, G. J. Chen and Y. Bin Dong, Inorg. Chem., 2019, 58, 9253–9259 CrossRef CAS PubMed.
- W. Jiang, J. Yang, Y. Y. Liu, S. Y. Song and J. F. Ma, Inorg. Chem., 2017, 56, 3036–3043 CrossRef CAS PubMed.
- L. Hu, G. X. Hao, H. D. Luo, C. X. Ke, G. Shi, J. Lin, X. M. Lin, U. Y. Qazi and Y. P. Cai, Cryst. Growth Des., 2018, 18, 2883–2889 CrossRef CAS.
- N. Kumar, T. Rom, V. Singh and A. K. Paul, Cryst. Growth Des., 2020, 20, 5277–5288 CrossRef CAS.
- N. Kumar and A. K. Paul, Inorg. Chem., 2020, 59, 1284–1294 CrossRef CAS PubMed.
- D. Sarma, K. V. Ramanujachary, N. Stock and S. Natarajan, Cryst. Growth Des., 2011, 11, 1357–1369 CrossRef CAS.
- J. Chai, P. Zhang, J. Xu, H. Qi, J. Sun, S. Jing, X. Chen, Y. Fan and L. Wang, Inorg. Chim. Acta, 2018, 479, 165–171 CrossRef CAS.
- Y. P. Li, L. J. Zhang and W. J. Ji, J. Mol. Struct., 2017, 1133, 607–614 CrossRef CAS.
- B. Ma, Y. Fan, L. Wang, J. Xu and J. Zhao, Inorg. Chim. Acta, 2018, 480, 166–172 CrossRef CAS.
- W. Liu, L. Ye, X. Liu, L. Yuan, J. Jiang and C. Yan, CrystEngComm, 2008, 10, 1395–1403 RSC.
- B. Parmar, P. Patel, V. Murali, Y. Rachuri, R. I. Kureshy, N. U. H. Khan and E. Suresh, Inorg. Chem. Front., 2018, 5, 2630–2640 RSC.
- R. A. Agarwal and D. De, Polyhedron, 2020, 185, 114584 CrossRef CAS.
- H. Chen, L. Fan and X. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 54884–54892 CrossRef CAS PubMed.
- B. B. Lu, W. Jiang, J. Yang, Y. Y. Liu and J. F. Ma, ACS Appl. Mater. Interfaces, 2017, 9, 39441–39449 CrossRef CAS PubMed.
- S. Ameerunisha and P. S. Zacharias, J. Chem. Soc., Perkin Trans. 2, 1995, 1679–1682 RSC.
- G. Agustí, S. Cobo, A. B. Gaspar, G. Molnár, N. O. Moussa, P. Á. Szilágyi, V. Pálfi, C. Vieu, M. Carmen Muñoz, J. A. Real and A. Bousseksou, Chem. Mater., 2008, 20, 6721–6732 CrossRef.
- SAINT, Program for Data Extraction and Reduction, Bruker AXS, Inc., Madison, WI, 2001 Search PubMed.
- (a) G. M. Sheldrick, SADABS, Program for Empirical Adsorption Correction of Area Detector Data, University of Gottingen, Gottingen, Germany, 2003 Search PubMed; (b) G. M. Sheldrick, SHELXL-2018, Program for the Crystal Structure Solution, University of Gottingen, Gottingen, Germany, 2018 Search PubMed.
- (a) A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef; (b) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. R. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS; (b) V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- (a) L. L. Gao, Q. N. Zhao, M. M. Li, L. M. Fan, X. Y. Niu and X. Q. Wang, CrystEngComm, 2017, 19, 6651–6659 RSC; (b) S. L. Zhang, S. Gao, X. Wang, X. He, J. Zhao and D. R. Zhu, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 1060–1068 CrossRef CAS PubMed; (c) S. Geranmayeh and A. Abbasi, J. Inorg. Organomet. Polym. Mater., 2013, 23, 1138–1144 CrossRef CAS; (d) L. Heidari, M. Ghassemzadeh, D. Fenske, O. Fuhr, F. Mohsenzadeh and V. Bon, J. Solid State Chem., 2021, 296, 122011 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD, NMR, TG, IR spectra and additional figures. CCDC 2205084 and 2205086. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06858c |
| This journal is © The Royal Society of Chemistry 2022 |