
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow silica-coated porous carbon with embedded iron oxide particles for effective methylene blue degradation
Kai Wanga,
Kang Zhao *ab,
Qingnan Mengab,
Qian Baia,
Xin Lia,
Huating Hua,
Hua Jiaoab and
Yufei Tangab
*ab,
Qingnan Mengab,
Qian Baia,
Xin Lia,
Huating Hua,
Hua Jiaoab and
Yufei Tangab
aDepartment of Materials Science and Engineering, Xi'an University of Technology, Xi'an 710048, PR China. E-mail: kzhao@xaut.edu.cn
bShaanxi Province Key Laboratory of Corrosion and Protection, Xi'an University of Technology, Xi'an 710048, PR China
First published on 12th December 2022
Abstract
A novel catalyst, consisting of hollow silica-coated porous carbon with embedded iron oxide particles (FeOx@C/SiO2), was synthesized by the extended Stöber method. Iron ions were incorporated in a resorcinol–formaldehyde resin in the presence of citric acid to form a template, which was then coated with a silica layer. The iron oxide-embedded porous carbon and hollow silica were simultaneously formed during calcination under N2 atmosphere. Through this process, silica endowed the iron oxide with low crystallinity and small size, resulting in a higher catalytic activity in the heterogeneous Fenton system for the decolorization of a methylene blue (MB) solution within 25 min. Moreover, the sample maintained 78.71% of its catalytic activity after three cycles.
1. Introduction
Water pollution has a serious impact on human health and is one of the most urgent tasks that need to be solved.1–4 The Fenton reaction is one of the effective ways to degrade refractory organic pollutants in industrial wastewater.5 The classical Fenton reagent consists of a homogeneous solution of iron ions and hydrogen peroxide.6,7 However, the corresponding degradation process usually produces iron hydroxide sludge as a byproduct, causing secondary environmental pollution.8 This has an adverse impact on the environment and limits the application scope of the catalysts. These problems can be solved using a heterogeneous Fenton catalyst.9,10 Among these catalysts, iron oxide has attracted much attention due to its abundance, cost-effectiveness, and eco-friendliness. Xiang et al. reported a solvent-free preparation of α-Fe2O3 nanoparticles as photo-Fenton catalysts for the degradation of methyl orange.11 Zhou et al. produced porous α-Fe2O3 rods by thermal dehydration of α-FeOOH precursors.12 They found that α-Fe2O3 nanoparticles possessed more active sites for dye degradation than micrometer-sized ones. However, iron oxide nanoparticles tend to undergo to serious aggregation in the process; hence, it is essential to provide a carrier (e.g., clay, carbon materials, silica, etc.) to ensure dispersion and efficient catalytic performance. Wang and co-workers used a reactive template method to prepare Fe3O4@Prussian blue core/shell composites as catalysts to enhance the degradation of RhB.13 Under optimal conditions, the rate constant (k) of the Fe3O4@Prussian blue core/shell composites was ∼1.5 times higher than that of Fe3O4 nanoclusters.Recently, catalysts with hollow structures have attracted increasing attention, due to their large surface area and high exposure of active sites for heterogeneous Fenton reactions.14–16 Miao et al. investigated the photocatalytic activity of Fe2O3/HPS materials calcined at different temperatures.17 They concluded that the sample calcined at a lower temperature had higher catalytic activity, which was attributed to the lower crystallinity of iron oxide. Currently, there are two common methods to prepare hollow particles of support catalysts. The first involves the preparation of the hollow particles followed by the loading of the catalyst. Wu et al. used hollow silica spheres as catalyst carrier and Fe(NO3)3·9H2O as iron source to obtain Fe3O4/SiO2 double-mesoporous-shelled hollow spheres by solvothermal method.18 In the second method, the preparation of the catalyst is followed by the fabrication of the hollow structure. Cheng et al. prepared hollow Fe3O4@SiO2 microspheres by a template-etching approach.19 This method consisted of four steps: preparation of Fe2O3, coating with the SiO2 shell, etching, and heating in reducing atmosphere. However, both methods are time-consuming, which hinders their wide application.
In light of the above findings, hollow particles of the supported catalyst were prepared by a one-pot method. We prepared hollow silica-coated porous carbon embedded with iron oxide (FeOx@C/SiO2) composites by an extended Stöber method. During the annealing process, iron oxide-intercalated porous carbon and hollow silica were obtained simultaneously. In addition, silica promoted the formation of iron oxide particles with low crystallinity and small size, and the resulting composite exhibited superior performance in the degradation of methylene blue (MB) than the FeOx@C counterpart.
2. Experimental
2.1. Material
All chemicals were of analytical reagent grade and used without purification. FeCl3·6H2O, NH3·H2O (25%), H2O2 (30%), citric acid monohydrate (C6H8O7·H2O), resorcinol (C6H4(OH)2), tetraethoxysilane (Si(OC2H5)4), formaldehyde (HCHO), and ethanol (C2H5OH) were obtained from Sinopharm Chemical Reagent Company.2.2. Synthesis of FeOx@C/SiO2 hollow nanoparticles
The FeOx@C/SiO2 hollow nanoparticles were synthesized by a one-pot two-step method. First, 0.5 mM FeCl3·6H2O and 71.4 mg C6H8O7·H2O were added to a mixture solution of 15 mL H2O and 12 mL C2H5OH, stirring until complete dissolution, respectively. Next, 0.8 mL NH3·H2O, 0.2 g C6H4(OH)2, and 0.28 mL HCHO were sequentially added to the above solution to synthesize Fe-RF nanoparticles by stirring for 10 h. Finally, the silica shell was prepared by adding 40 mL C2H5OH, 0.8 mL NH3·H2O, and 0.75 mL Si(OC2H5)4, followed by stirring overnight. The precipitates formed in the solution were collected by centrifugation and denoted as Fe-RF@SiO2. The final FeOx@C/SiO2 samples (denoted as 0.5-FCS-500) were obtained by annealing for 2 h at 500 °C in N2 atmosphere. The products prepared by adding 1.0 mM and 1.5 mM FeCl3·6H2O were labeled 1.0-FCS-500 and 1.5-FCS-500, respectively. For comparison, pure FeOx@C nanoparticles (1.0-FC-500) were also prepared by direct calcination of Fe-RF nanoparticles.2.3. Characterization
The crystal structure of FeOx@C/SiO2 was determined by X-ray diffraction (XRD, Bruker D8) with Cu Kα radiation at 40 kV and 40 mA. The measurements were carried out in the 2θ range of 20–80° with a scanning step of 8° min−1. Thermogravimetric analysis (TGA, DTG-60AH) was performed to monitor the pyrolysis carbonization process of the sample. Under N2 atmosphere, the TG curve was obtained in the temperature range of 30–1000 °C at a rate of 10 °C min−1. Fourier transform infrared (FTIR) spectroscopy (Nicolet Avatar 360) was used to identify the molecular vibration modes; the measurements were carried out by the KBr method in the range of 4000 to 400 cm−1. The morphology of the samples was inspected by scanning electron microscopy (SEM, JSM-6700F). The inner structure of the nanoparticles was examined by transmission electron microscopy (TEM, JEM-3010). In addition, the average diameter size was averaged over 100 nanoparticles. The specific surface area and pore structure of the samples were analyzed by N2 adsorption–desorption at 77 K using an ASAP 2020 system. In the test, 250 mg of the samples were kept at the degassing temperature of 200 °C for 10 h. X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha) was used to determine the valence states of the elements. High-resolution spectra were recorded using a monochromatic 150 W Al Kα radiation source and a pass energy of 50 eV. Binding energies were referenced to the standard C 1s signal at 284.80 eV. The amount of iron in solution was determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES), using an ICAP 6500 Thermo Scientific device. The catalytic degradation of MB was monitored with a 7600 MC UV-Vis spectrophotometer (BILON).2.4. Catalytic activity
The heterogeneous Fenton catalytic activities of hollow FeOx@C/SiO2 were evaluated by studying the catalytic degradation of MB. Typically, 15 mg of catalyst was poured into a 100 mL MB solution (c0 = 0.1 mM, pH = 3, T = 30 °C) under magnetic stirring. Afterward, the mixed solution was stirred for 30 min to reach the adsorption–desorption equilibrium, followed by the addition of 60 μL H2O2. At intervals of 3 min, 1 mL solution aliquots were extracted and filtered through a membrane (0.22 μm). The concentration of MB was determined based on the absorption intensity at 664 nm, as follows:| Degradation rate = C/C0 |
To evaluate the stability of the catalyst, the nanoparticles were collected from the mixed solution by centrifugation, washed with deionized water, and then redispersed into fresh MB solution for the next cycle. In the iron ion leaching experiment, the solution obtained after centrifugation was directly used for ICP measurements.
Free radical elimination tests were performed under the same conditions as the catalytic experiments. When the catalyst reached the adsorption–desorption equilibrium, H2O2 and a scavenger [tert-butyl alcohol (TBA) or p-benzoquinone (BQ)] were added at the same time, and then 1 mL solution aliquots were collected at intervals of 3 min for testing.
3. Results and discussion
Fig. 1a shows the pyrolysis process of Fe-RF@SiO2. The first stage (25–200 °C) corresponded to a weight loss of 12%, which was attributed to the evaporation of adsorbed water and the evolution of unreacted oligomers.20 The second weight loss stage (24%) in the range of 200–800 °C was ascribed to the formation of FeOx and carbonization of Fe-RF during pyrolysis.21 In the final stage (800–900 °C), the weight loss was due to the structural ordering of the carbon matrix. The TG curve of Fe-RF (Fig. 1b) also included three processes: evaporation of adsorbed water (25–200 °C), carbonization of Fe-RF (200–600 °C), and ordering of carbon atoms (600–900 °C). Compared with Fe-RF@SiO2, the FeOx formation temperature of Fe-RF was lower, which was attributed to the silica shell preventing grain growth.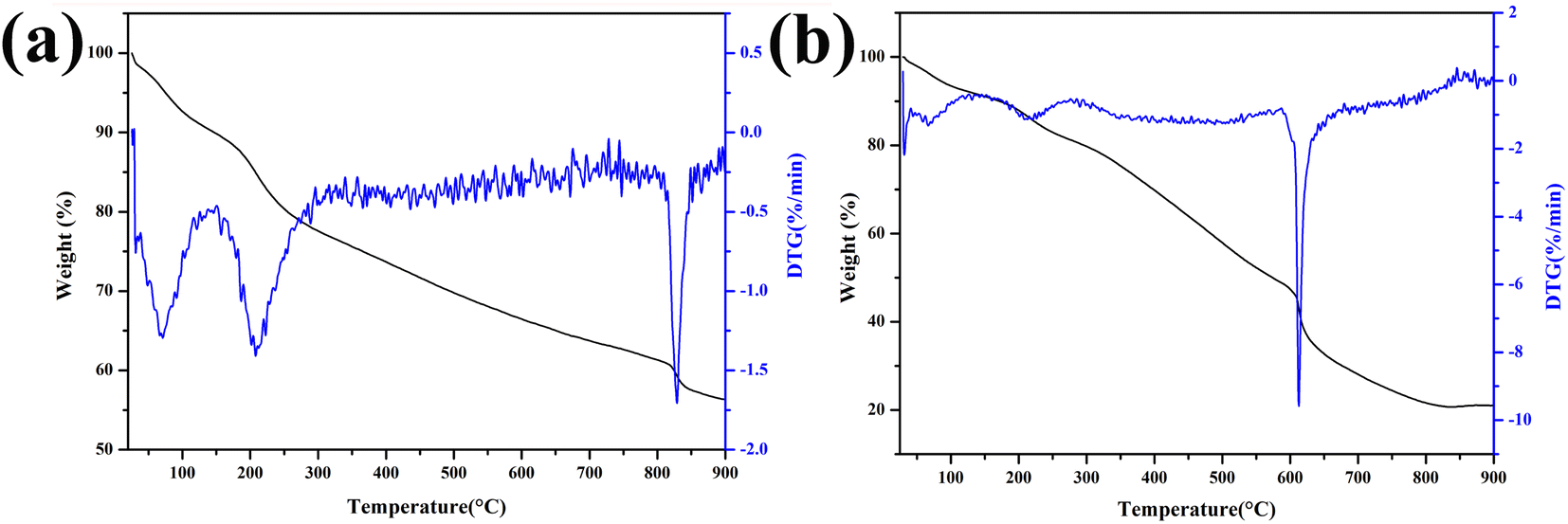 | ||
| Fig. 1 TG curves of Fe-RF@SiO2 (a) and Fe-RF (b). | ||
The XRD patterns of 1.0-FCS-500 samples prepared at different temperatures are shown in Fig. 2a. No distinct diffraction peaks were observed in the curves after pyrolysis at 400 and 500 °C, indicating that the FeOx particles embedded in silica were either in amorphous form or of a size too small to be detected by X-ray diffraction. When the temperature increased to 600 °C, four peaks appeared at 2θ = 30.2°, 35.6°, 43.2°, and 62.9°, corresponding to the (220), (311), (400), and (440) crystal faces of Fe2O3 (PDF #39-1346),22 respectively. These results show that increasing the temperature is beneficial to grain growth. For comparison, 1.0-FC-500 also exhibited four peaks corresponding to Fe2O3 (Fig. 2b). This demonstrates that the silica shell layer can effectively prevent the crystallization of grains.23 A higher temperature is believed to favor the formation of stable carbon structures by resorcinol–formaldehyde pyrolysis. Therefore, to achieve a lower crystallinity and stable porous carbon structure of the products, the Fe-RF@SiO2 samples were calcined at 500 °C to prepare 1.0-FCS-500.
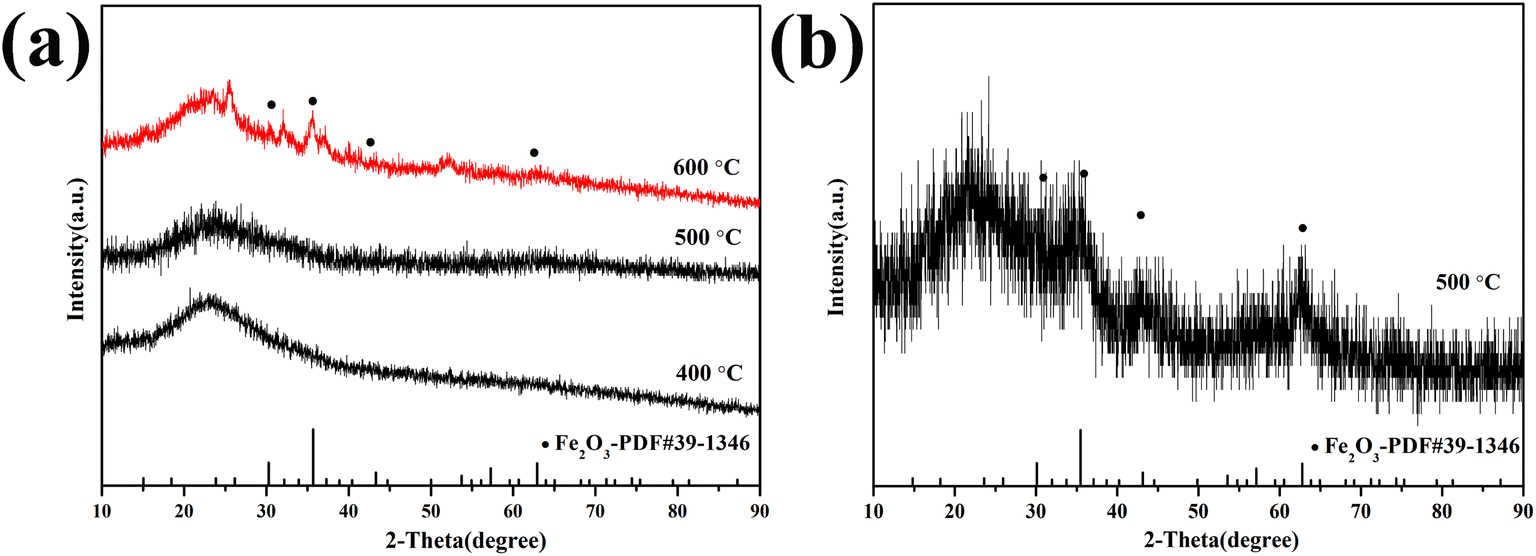 | ||
| Fig. 2 XRD patterns of 1.0-FCS-500 (a) and 1.0-FC-500 (b) samples prepared at different temperatures. | ||
The iron content and valence state of the samples prepared with different dosages were analyzed by XPS, as shown in Fig. 3. As shown in the high-resolution Fe 2p XPS spectrum (Fig. 3a), two peaks at 710.44 and 724.16 eV were attributed to the Fe 2p3/2 and Fe 2p1/2 states of Fe(II), respectively. In addition, the two peaks at 711.77 and 726.41 eV were attributed to Fe(III) Fe 2p3/2 and Fe 2p1/2 signals, respectively.24,25 The Fe 2p3/2 and Fe 2p1/2 signals did not shift with increasing Fe content (Fig. 3b and c). Table 1 shows that the iron content of the final sample increased with increasing FeCl3·6H2O content. When the added amount of FeCl3·6H2O was 1.0 mM, the Fe(II) content was the highest, which was beneficial for the Fenton reaction.26
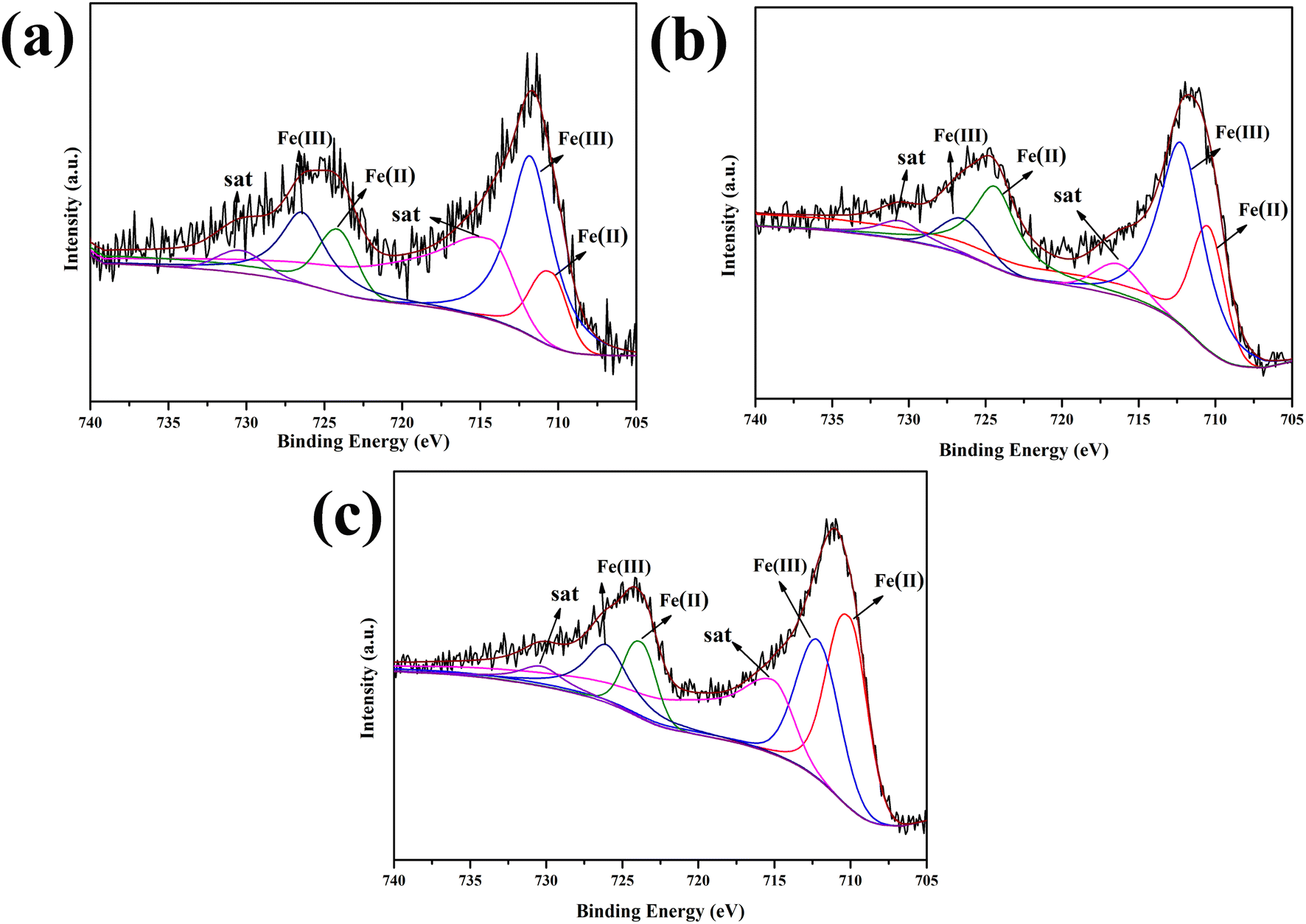 | ||
| Fig. 3 XPS spectra of 0.5-FCS-500 (a), 1.0-FCS-500 (b), and 1.5-FCS-500 (c). | ||
| Sample | Fe (Fe(II)/Fe)/at% | C/at% | O/at% | Si/at% |
|---|---|---|---|---|
| 0.5-FCS-500 | 1.09 (32.95%) | 16.73 | 54.06 | 28.11 |
| 1.0-FCS-500 | 1.74 (54.09%) | 12.05 | 57.98 | 28.22 |
| 1.5-FCS-500 | 2.1 (50.21%) | 11.88 | 58.64 | 27.38 |
The chemical structure of 1.0-FCS-500 was analyzed by FTIR spectroscopy. The spectrum in Fig. 4 shows a broad band at 3463 cm−1, corresponding to the –OH groups of surface water molecules. The peaks located at 1088, 794, and 467 cm−1 were attributed to asymmetric and symmetric stretching vibrations of Si–O–Si bonds and bending vibrations of Si–O bonds, respectively.27 These results confirmed the presence of SiO2 in the sample. The peak at 562 cm−1 was assigned to Fe–O flexural vibrations, indicating the successful formation of FeOx.28 The bands at 1088 and 1637 cm−1 were attributed to C–O–C stretching and aromatic ring vibrations, respectively. The band at 1579 cm−1 confirmed the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, corresponding to the normal stretching of carbonaceous molecules. Moreover, the peak at 1407 cm−1 was assigned to C–C bond vibrations, related to the carbonaceous structure of the composites. In the spectrum of 1.0-FC-500, two distinct peaks appeared at 3463 and 1597 cm−1, corresponding to the absorptions of –OH and CC groups, respectively. In addition, the spectrum displayed an absorption peak corresponding to Fe–O bonds at 562 cm−1, but no SiO2 absorption peaks.
C bonds, corresponding to the normal stretching of carbonaceous molecules. Moreover, the peak at 1407 cm−1 was assigned to C–C bond vibrations, related to the carbonaceous structure of the composites. In the spectrum of 1.0-FC-500, two distinct peaks appeared at 3463 and 1597 cm−1, corresponding to the absorptions of –OH and CC groups, respectively. In addition, the spectrum displayed an absorption peak corresponding to Fe–O bonds at 562 cm−1, but no SiO2 absorption peaks.
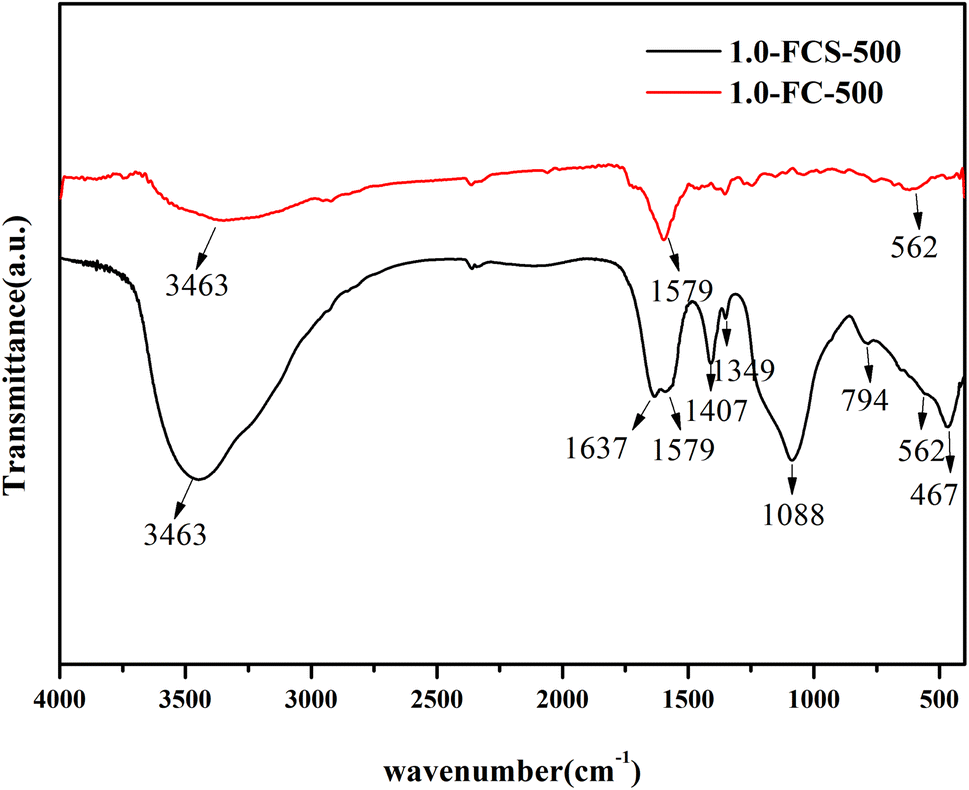 | ||
| Fig. 4 FTIR spectra of 1.0-FCS-500 and 1.0-FC-500 samples. | ||
Fig. 5 shows the morphology and structure of 1.0-FCS-500. The sample displayed uniform cavity and a worm-like morphology, implying the successful production of a hollow structure (Fig. 5a and b). The shell thickness was 9.9 ± 1.1 nm, depending on the amount of tetraethoxysilane added. The diameters of the inner cavity and of the nanoparticles embedded in the shell formed simultaneously during the calcination process, were approximately 18.4 ± 3.1 and 2.2 ± 0.5 nm, respectively. The corresponding SEM images show an irregular morphology, which was attributed to the interaction between Fe3+ and the resorcinol–formaldehyde resin to form a worm-like template under the action of citric acid (Fig. 5c and d). The energy-dispersive spectroscopy (EDS) mapping images of 1.0-FCS-500 (Fig. 5e–i) show that the Fe/Si/C/O elements were evenly distributed in the samples, indicating their good dispersion, which is beneficial for MB degradation.
 | ||
| Fig. 5 TEM (a and b), SEM (c and d), and EDS (e–i) images of 1.0-FCS-500. | ||
The TEM images are shown in Fig. 6a and b. In all cases, many nanoparticles were uniformly dispersed over the porous carbon support and no agglomerates were detected. The mean particle size of the nanoparticles was approximately 4.8 nm, which was slightly larger than that of the 1.0-FCS-500 particles. These data confirm that the silica shell prevented the grain growth and crystallization.18,21 Finally, 1.0-FC-500 exhibited an irregular morphology, similar to that of 1.0-FCS-500 (Fig. 6c and d).
 | ||
| Fig. 6 TEM (a and b) and SEM (c and d) images of 1.0-FC-500. | ||
Fig. 7 shows the TEM images of 1.0-FCS-500 after NaOH etching. As shown in the figure, the core–shell structure of the sample disappeared, resulting in a uniform worm-like porous structure. Compared with the 1.0-FCS-500 sample before etching, the sample showed a roughened surface, indicating that the SiO2 shell was completely etched. Moreover, the hollow structure was also destroyed, which was attributed to the low strength of porous carbon. These results indicate that 1.0-FCS-500 contained silica and carbon in its hollow structure, which is beneficial for organic dye degradation.29
 | ||
| Fig. 7 TEM images of 1.0-FCS-500 after NaOH etching (a) low magnification (b) high magnification. | ||
The N2 adsorption–desorption isotherms of 1.0-FCS-500 and 1.0-FC-500 are shown in Fig. 8; both samples showed type IV isotherms. The hysteresis loops (type H3) in the isotherms suggest the presence of mesopores in both materials.30 The Brunauer–Emmett–Teller (BET) surface areas of 1.0-FCS-500 and 1.0-FC-500 were 191.03 and 306.53 m2 g−1, respectively. The corresponding pore size distributions were estimated using Barrett–Joyner–Halenda (BJH) analysis (Fig. 8b).
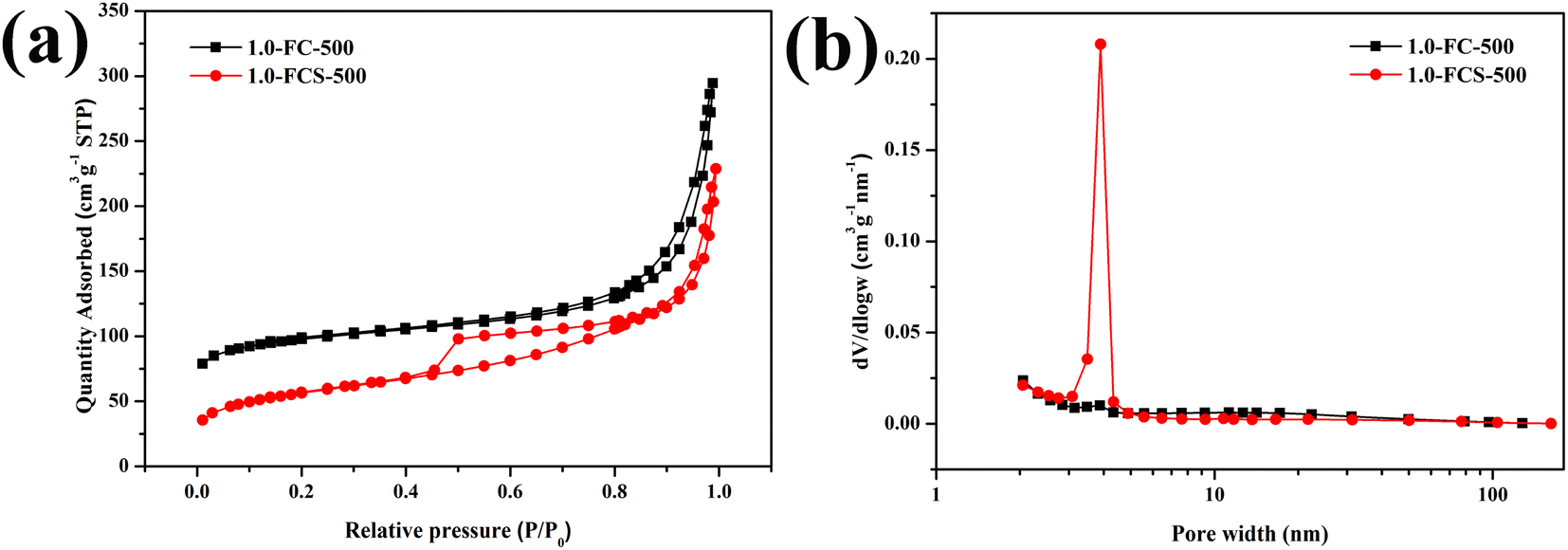 | ||
| Fig. 8 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of 1.0-FCS-500 and 1.0-FC-500. | ||
Fig. 9a shows the degradation efficiency of catalysts with different iron contents. The degradation efficiency of the catalyst prepared with 0.5 mM FeCl3·6H2O fluctuated around a constant value. As the FeCl3·6H2O content increased to 1.0 mM, the degradation rate reached 100% in 25 min, indicating that increasing the iron content can significantly improve the degradation efficiency. Upon continuing to increase the FeCl3·6H2O content to 1.5 mM, the time required for the complete MB degradation was the same as that of the sample with 1.0 mM content. Based on their combination of economy and degradation efficiency, the samples prepared with 1.0 mM content were selected for further characterization after pyrolysis at different temperatures. After pyrolysis at 400 °C, the degradation efficiency of the product was reduced to 55%, which was attributed to its low specific surface area, providing fewer reaction sites. However, when the temperature increased to 600 °C, the degradation efficiency remained almost constant, due to the higher crystallinity. To study the catalytic mechanism, Fig. 9b shows the degradation efficiency evaluated in comparative experiments. In the absence of H2O2, 1.0-FCS-500 only exhibited adsorption capacity. In contrast, in absence of 1.0-FCS-500, the MB solution remained stable upon adding H2O2. Moreover, the degradation rate of 1.0-FC-500 fluctuated around a constant value, likely due to the higher crystallinity and larger size of FeOx nanoparticles, leading to fewer and less active Fe ions exposed on the surface.19,21 Fig. 9c shows the first-order kinetic model for MB degradation under different conditions. The MB degradation rate constants of 1.0-FCS-500 and 1.0-FC-500 were 0.11507 ± 0.01396 and 0.00127 ± 0.00017 min−1, respectively. Compared with other studies, 1.0-FCS-500 showed a superior MB degradation ability (see comparison in Table 2). Free radical scavenging experiments were employed to explore the radical species produced by degradation. In particular, TBA and BQ were used to scavenge hydroxyl radicals (·OH) and superoxide (O2˙−) radicals, respectively. Compared to the reaction without scavenger, the MB degradation efficiency was slightly reduced with the addition of BQ (Fig. 9d).31 In the presence of TBA, the MB degradation efficiency was significantly reduced, indicating that ·OH was the main active species for MB degradation.
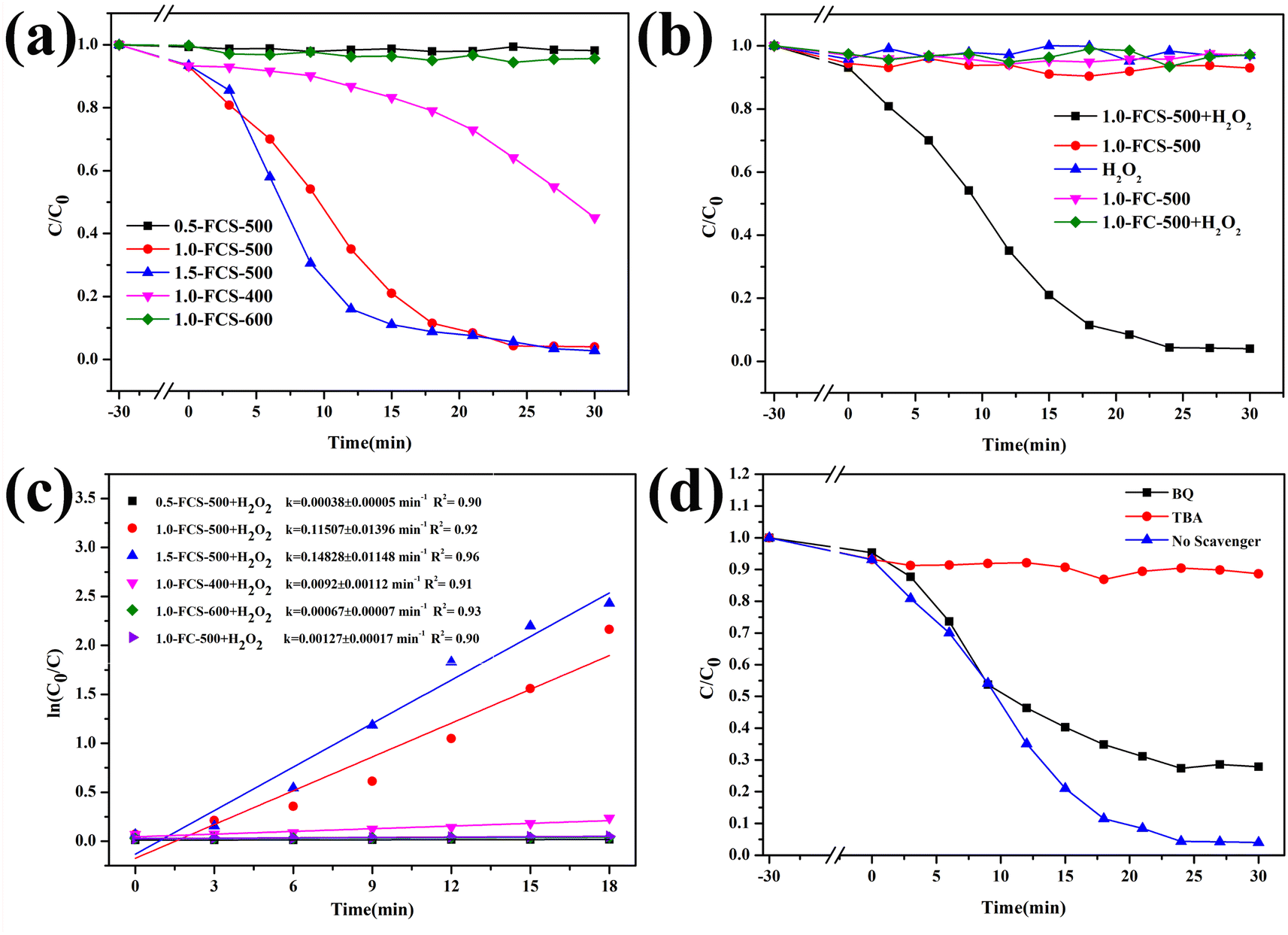 | ||
| Fig. 9 Removal efficiency of MB (a, b and d) and ln(C0/C) vs. reaction time plots (c) under different conditions. | ||
| Catalyst | Catalyst dosage | H2O2 dosage | Organic species | Degradation time and rate | Reference | |
|---|---|---|---|---|---|---|
| 1 | Fe2O3/TiO2-BC | 0.2 g L−1 | 2 mL (H2O2) | 20 mL, 50 mg L−1 MB | 60 min/100% | 32 |
| 2 | Mesoporous Fe2O3·SiO2 | 0.5 g L−1 | 12 mM (H2O2) | 100 mL, 50 mg L−1 tartrazine | 80 min/98.5% | 33 |
| 3 | MIL100(Fe)/GO | 0.5 g L−1 | 8 mM (H2O2) | 40 mL, 50 mg L−1 MO | 80 min/98% | 34 |
| 4 | Mesoporous Fe2O3–SiO2 | 2.0 g L−1 | 2 mL (H2O2) | 10 mL, 0.6 g L−1 MO | 20 min/98.5% | 35 |
| 5 | Fe2O3@mesoSiO2 | 0.5 g L−1 | 9.0 g L−1 (H2O2) | 50 mg L−1 MB | 120 min/99% | 36 |
| 6 | Fe2O3@mesoSiO2 | 0.5 g L−1 | 18 g L−1 (H2O2) | 50 mg L−1 MB | 400 min/90% | 37 |
| 7 | FeOx@GC-NBC | 0.06 g L−1 | 1 mM PDS | 2 mg L−1 BPA | 30 min/100% | 38 |
| 8 | FeOx@g-BC | 0.2 g L−1 | 0.5 mL (H2O2) | 100 mL, 20 mg L−1 TC | 15–60 min/100% | 39 |
| 9 | Fe3O4–Cs | 0.5 g L−1 | 10 mM H2O2 | 0.1 mM TC | 20 min/96.2% | 40 |
| 10 | FeOx/SiO2 | 10.0 g L−1 | 2 mL H2O2 | 50 mL, 1 g L−1 MB | 72 h/99.94% | 41 |
| 11 | 1.0-FCS-500 | 0.15 g L−1 | 60 μL (H2O2) | 100 mL, 37.3 mg L−1 MB | 30 min/100% | This work |
The stability and reusability of 1.0-FCS-500 were investigated by recycling reactions under the same conditions (Fig. 10a). We found that the degradation rate of 1.0-FCS-500 only decreased from 95.16% to 92.07% after two cycles, mainly attributed to the loss of small-sized iron ions under acidic conditions. In the third cycle, the MB degradation rate was only 78.71%, but the iron ion concentration was 0.85 mg mL−1, which was lower than that of the two previous cycles. The second reason could be that there are fewer activation sites, due to the coverage area of the intermediate products. The Fe 2p XPS spectrum of 1.0-FCS-500 after three cycles is shown in Fig. 10b. The Fe 2p3/2 and Fe 2p1/2 peaks were located at 711.9 and 725.1 eV, respectively; these values were consistent with the binding energies of the fresh sample. According to the fitting results, the atomic ratio of Fe(III) increased from 45.91% to 63.76%, indicating that the surface Fe(II) species were only slightly oxidized.42 In conclusion, the 1.0-FCS-500 catalyst possessed good stability and recyclability.
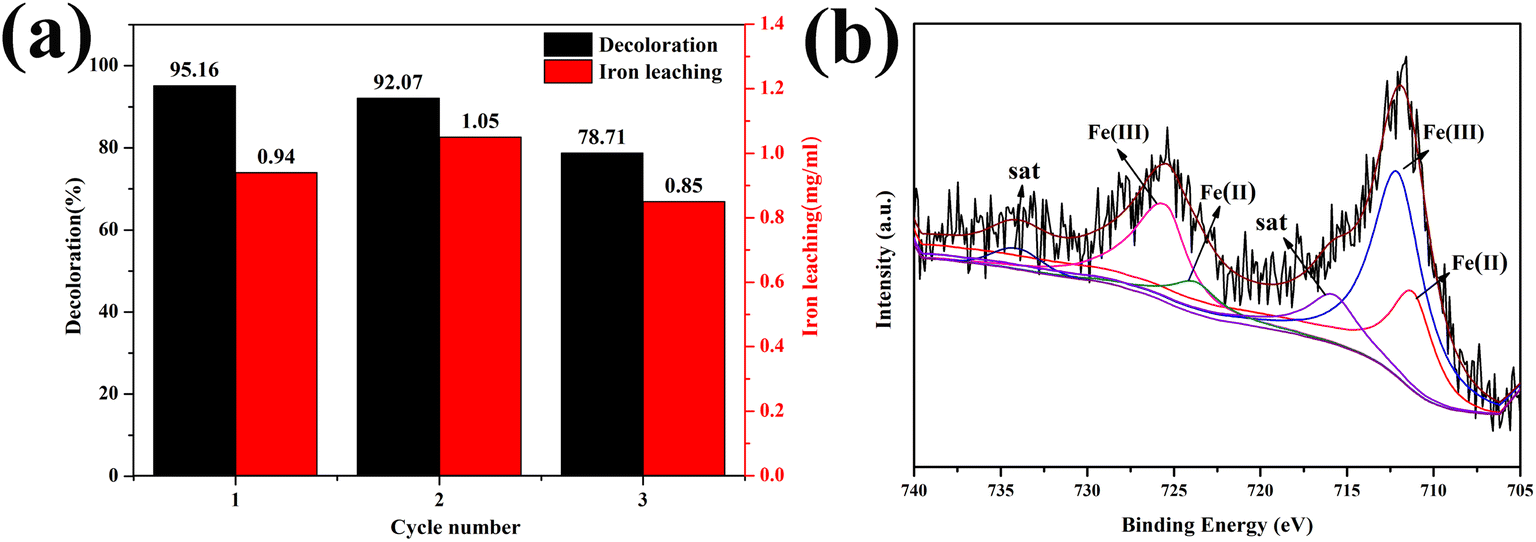 | ||
| Fig. 10 (a) Recycling performance and (b) Fe 2p XPS spectrum of 1.0-FCS-500 after three cycles. | ||
4. Conclusion
In this study, a novel catalyst (1.0-FCS-500) was synthesized by the extended Stöber method. Structural analysis indicated that hollow-structure and small-sized FeOx particles were located within the silica shell. Compared to 1.0-FC-500, the iron oxide particles in 1.0-FCS-500 had lower crystallinity and smaller size. MB was almost completely degraded by 1.0-FCS-500 within 25 min, with a degradation rate constant of 0.11507 ± 0.01396 min−1. Free radical scavenging experiments showed that ·OH radicals were the main active species for MB degradation. After three cycles, the catalytic degradation rate gradually decreased to 78.71%, mainly due to the leaching of Fe ions.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The research was financially supported by the National Natural Science Foundation of China (No. 52172074) and the Doctoral Innovation Fund of Xi'an University of Technology (No. 310-252072005).References
- K. A. M. Ahmed, H. Peng, K. B. Wu and K. X. Huang, Chem. Eng. J., 2011, 172, 531–539 CrossRef CAS.
- M. Z. Shahid, L. Yu and R. Mehmood, Catal. Sci. Technol., 2020, 10, 2242–2253 RSC.
- M. Z. Shahid, R. Mehmood, M. Athar, J. Hussain, Y. W. Wei and A. Khaliq, ACS Appl. Nano Mater., 2021, 4, 746–758 CrossRef CAS.
- M. Z. Shahid, R. Mehmood, U. Farooq and F. P. Han, Adv. Powder Technol., 2020, 31, 1708–1719 CrossRef CAS.
- M. N. Han, X. G. Duan, G. L. Cao, S. S. Zhu and S. H. Ho, Process Saf. Environ. Prot., 2020, 139, 230–240 CrossRef CAS PubMed.
- S. R. Pouran, A. R. A. Aziz and W. M. A. W. Daud, J. Ind. Eng. Chem., 2015, 21, 53–69 CrossRef.
- M. Punzi, B. Mattiasson and M. Jonstrup, J. Photochem. Photobiol., A, 2012, 248, 30–35 CrossRef CAS.
- A. Babuponnusami and K. Muthukumar, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- Q. N. Meng, L. L. Du, J. Yang, Y. F. Tang, Z. H. Han, K. Zhao and G. J. Zhang, Colloids Surf., A, 2018, 548, 142–149 CrossRef CAS.
- M. E. Hassan, Y. B. Chen, G. L. Liu, D. W. Zhu and J. B. Cai, J. Water Process. Eng., 2016, 12, 52–57 CrossRef.
- H. L. Xiang, G. K. Ren, X. S. Yang, D. H. Xu, Z. Y. Zhang and X. L. Wang, Ecotoxicol. Environ. Saf., 2020, 200, 110744 CrossRef CAS PubMed.
- X. M. Zhou, H. C. Yang, C. X. Wang, X. B. Mao, Y. S. Wang, Y. L. Yang and G. Liu, J. Phys. Chem. C, 2010, 114, 17051–17061 CrossRef CAS.
- Q. Q. Wang, Y. Yang, S. C. Ma, J. Wu and T. J. Yao, Colloids Surf., A, 2020, 606, 125416 CrossRef CAS.
- H. F. Zhou, J. Yang, W. Cao, C. Chen, C. Y. Jiang and Y. P. Wang, Appl. Surf. Sci., 2022, 590, 153120 CrossRef CAS.
- R. R. Ding, W. Q. Li, C. S. He, Y. R. Wang, X. C. Liu, G. N. Zhou and Y. Mu, Appl. Catal., B, 2021, 291, 120069 CrossRef CAS.
- Y. N. Guan, G. Fu, Q. Q. Wang, S. C. Ma, Y. Yang, B. F. Xin, J. X. Zhang, J. Wu and T. J. Yao, Sep. Purif. Technol., 2022, 299, 121699 CrossRef CAS.
- Z. H. Miao, S. Y. Tao, Y. C. Wang, Y. X. Yu, C. G. Meng and Y. L. An, Microporous Mesoporous Mater., 2013, 176, 178–185 CrossRef CAS.
- X. P. Wu and Z. D. Nan, Mater. Chem. Phys., 2019, 227, 302–312 CrossRef CAS.
- L. Cheng, Y. Y. Liu, B. F. Zou, Y. Yu, W. M. Ruan and Y. Q. Wang, Mater. Sci. Eng., C, 2017, 75, 829–835 CrossRef CAS.
- Q. N. Meng, K. Wang, X. B. Xu, Y. F. Tang, Z. H. Han, K. Zhao, G. J. Zhang, J. N. Cui and Y. Y. Ji, ChemistrySelect, 2018, 3, 8919–8925 CrossRef CAS.
- Q. N. Meng, K. Wang, Y. F. Tang, K. Zhao, G. J. Zhang and L. Zhao, J. Alloys Compd., 2017, 722, 8–16 CrossRef CAS.
- X. Y. Hou, J. L. Kang, G. Zhou and J. T. Wang, Mater. Lett., 2022, 313, 131736 CrossRef CAS.
- K. Wang, Q. N Meng, L. L. Du, Y. F. Tang and K. Zhao, J. Nanopart. Res., 2021, 23, 27 CrossRef CAS.
- W. L. Zhao, Y. Fu, Y. Chen, J. Sun, Q. Tang, B. H. Jiang, H. H. Kong, F. Xu and Q. M. Ji, Microporous Mesoporous Mater., 2021, 312, 110704 CrossRef CAS.
- J. C. Wang, B. B. Wang, W. N. Shi, X. Qiao, X. X. Yang, L. F. Zhang, W. Q. Zhang, R. L. Li and Y. X. Hou, J. Environ. Chem. Eng., 2022, 10, 108565 CrossRef CAS.
- S. L. Orozco, E. R. Bandala, C. A. Arancibia-Bulnes, B. Serrano, R. Suárez-Parra and I. Hernández-Pérez, J. Photochem. Photobiol., A, 2008, 198, 144–149 CrossRef CAS.
- Q. N. Meng, S. Y. Xiang, W. Cheng, Q. N. Chen, P. F. Xue, K. Zhang, H. C. Sun and B. Yang, J. Colloid Interface Sci., 2013, 405, 28–34 CrossRef CAS PubMed.
- S. Pirsa and F. Asadzadeh, Adv. Powder Technol., 2021, 32, 1233–1246 CrossRef CAS.
- Q. N. Meng, K. Wang, Y. F. Tang, K. Zhao, G. J. Zhang, Z. H. Han and J. Yang, ChemistrySelect, 2017, 2, 10442–10448 CrossRef CAS.
- Q. N. Meng, J. N. Cui, K. Wang, Y. F. Tang, K. Zhao and L. Huang, Mol. Catal., 2020, 493, 111107 CrossRef CAS.
- Z. W. Xie, J. Yang, K. Wang, Q. N Meng, Y. F. Tang and K. Zhao, Ceram. Int., 2022, 48, 9114–9123 CrossRef CAS.
- X. L. Chen, F. Li, H. Y. Chen, H. J. Wang and G. G. Li, J. Environ. Chem. Eng., 2020, 8, 103905 CrossRef CAS.
- A. T. Vu, T. N. Xuan and C. H. Lee, J. Water Process. Eng., 2019, 28, 169–180 CrossRef.
- J. T. Tang and J. L. Wang, RSC Adv., 2017, 7, 50829 RSC.
- N. Panda, H. Sahoo and S. Mohapatra, J. Hazard. Mater., 2011, 185, 359–365 CrossRef CAS.
- Z. M. Cui, J. Hao, J. Liu and W. G. Song, RSC Adv., 2016, 6, 74545–74549 RSC.
- Z. M. Cui, Z. Chen, C. Y. Cao, L. Jiang and W. G. Song, Chem. Commun., 2013, 49, 2332 RSC.
- Y. J. Wang, L. Wang, F. Ma and Y. Q. You, Chem. Eng. J., 2022, 438, 135552 CrossRef CAS.
- X. Zhang, H. Y. Wu, M. Chen and Z. L. Li, Int. J. Biol. Macromol., 2022, 205, 329–340 CrossRef CAS PubMed.
- X. Y. Li, K. P. Cui, Z. Guo, T. T. Yang, Y. Cao, Y. P. Xiang, H. H. Chen and M. F. Xi, Chem. Eng. J., 2020, 379, 122324 CrossRef CAS.
- L. Liu, S. Q. Liu, S. B. Mishra and L. P. Sheng, Ceram. Int., 2019, 45, 15475–15481 CrossRef CAS.
- D. B. Jiang, X. Y. Liu, X. Xu and Y. X. Zhang, J. Phys. Chem. Solids, 2018, 112, 209–215 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |