
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-stage one-pot synthetic strategy for the key triazone-triazole intermediate of ensitrelvir (S-217622), an oral clinical candidate for treating COVID-19†
Wei Hua,
Xiang Zhanga,
Yuanchang Liua,
Teng Liua,
Jiale Wena,
Xiaopeng Pengab,
Xin Xie*ab and
Weiming Chen *ab
*ab
aSchool of Pharmaceutical Sciences, Gannan Medical University, Ganzhou 341000, P. R. China. E-mail: wader_chen@163.com
bKey Laboratory of Prevention and Treatment of Cardiovascular and Cerebrovascular Diseases of Ministry of Education, Gannan Medical University, Ganzhou 341000, P. R. China
First published on 6th December 2022
Abstract
Herein, the preparation of the key triazone-triazole intermediate of ensitrelvir (S-217622) via sequential cyclization and alkylation reaction is described. Firstly, chloromethyl triazole was synthesized through a one-pot tandem process (condensation and cyclization reaction) from commercially available chloroacetamide in a 72% yield. Then, the key triazone-triazole intermediate was obtained in a second one-pot process by N-alkylation with triazone followed by highly selective N1-methylation with iodomethane in a 54% yield. In addition, two of the main process impurities were synthesized and identified. This novel alternative two-stage one-pot strategy for synthesizing the key triazone-triazole intermediate opens a new avenue for further research and development of ensitrelvir analogs.
Introduction
COVID-19 is an acute respiratory infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1 that spread rapidly since it was first identified in January 2020. As of Nov. 30th, 2022, it had infected over 638 million people and claimed over 6.61 million lives.2 Paxlovid, a peptide covalent 3C-like protease inhibitor developed by Pfizer Inc.,3 was the first new oral drug approved by the FDA for the treatment of COVID-19,4 reducing the risk of hospitalization or death by as much as 89 percent.3b However, peptide covalent inhibitors exhibit several disadvantages, such as the intrinsic nature of the reactivity, low membrane permeability and low metabolic stability.5 Ensitrelvir (S-217622, 1) developed by Shionogi Co., Ltd, Japan, was the first orally active, noncovalent, nonpeptidic SARS-CoV-2 3C-like protease inhibitor.6 This approach solved the shortcomings of peptide covalent inhibitors and had favorable drug metabolism and pharmacokinetic (DMPK) profiles for the oral agents.7 Phase IIb clinical trials have demonstrated that ensitrelvir can rapidly reduce viral titers and significantly improve clinical symptoms. As of September 2022, Shionogi announced achievement of the primary endpoint for ensitrelvir fumaric acid (S-217622) in the phase 3 part of the phase 2/3 clinical trial in Asia.8As shown in Scheme 1, triazone-triazole 4 is considered a key intermediate for ensitrelvir synthesis. In the Shionogi's synthetic strategy, it was obtained from N-alkylation with triazone 2 and chloromethyl triazole 3 in a 45% yield. Triazone 2 could be efficiently prepared from 3-(tert-butyl)-6-(ethylthio)-1,3,5-triazine-2,4(1H,3H)-dione in an exciting yield of 90.2%.6b However, efficient synthesis of chloromethyl triazole 3 has, to date, proved challenging, so a novel synthetic strategy for chloromethyl triazole 3 and a further N-alkylation reaction to triazone-triazole 4 is needed.
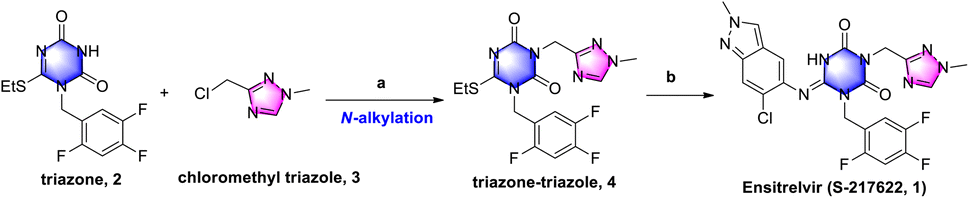 | ||
| Scheme 1 Shionogi's synthetic strategy for ensitrelvir. Reagents and conditions: (a) K2CO3, DMF, 60 °C, 3.5 h, 45%; and (b) 6-chloro-2-methyl-2H-indazol-5-amine, LiHMDS, THF, 0 °C to r.t., 25%. | ||
Chloromethyl triazole 3 was obtained by three strategies (Scheme 2). Starting from toxic chloroacetonitrile, the 1,2,4-triazole ring was constructed in three complicated steps with a 54% yield, while N-methyl-N-formylhydrazine was not commercially available (Scheme 2A).9 Three redundant multi-steps (hydroxyl-protection, de-protection, and chlorination) were applied in strategy 2B,10 while two steps (ester-reduction and chlorination) were necessary for the functional group transformation from an ester to a chloromethyl group in strategy 2C.11 Although these methods provide several pathways to chloromethyl triazole 3, major drawbacks, such as the need for multiple steps and poor atom economy, have limited their application. Therefore, exploring practical and efficient strategies for accessing chloromethyl triazole 3 is desirable.
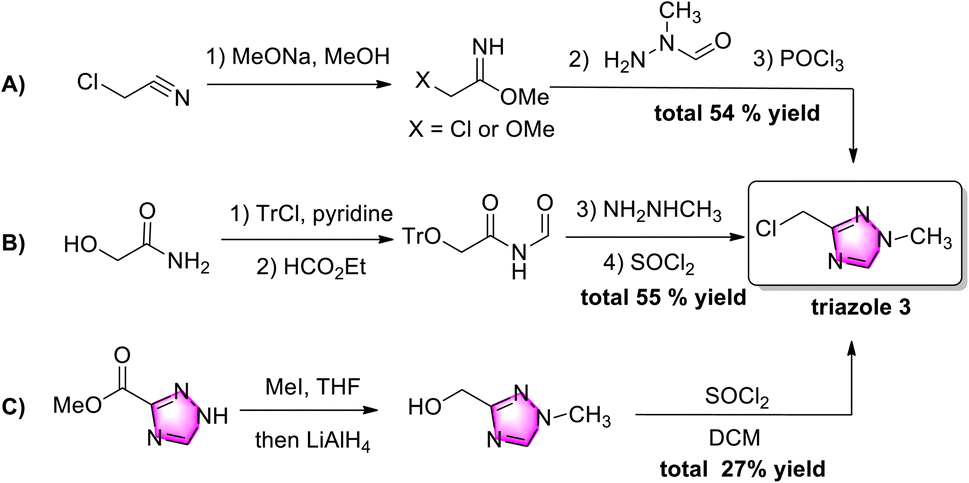 | ||
| Scheme 2 The three existing synthetic strategies for preparing chloromethyl triazole 3. | ||
Herein we report a facile and practical strategy for synthesizing the 1,2,4-triazole derivative from commercially available chloroacetamide and use the material to prepare the key triazone-triazole intermediate of clinical candidate drug ensitrelvir.
Results and discussion
Investigation into the preparation of 1,2,4-triazole derivatives
Considering atom economy and seeking to avoid chlorination of a hydroxyl group, for the initial trials we directly employed starting materials incorporating the chloromethyl group (Scheme 3). To explore a similar cyclization strategy as Scheme 2B, an attempt was made to prepare N-formylacetamide 5 from chloroacetyl chloride and chloroacetamide using formamide and ethyl formate, respectively, but the no N-formylacetamide could be subsequently detected by GC-MS.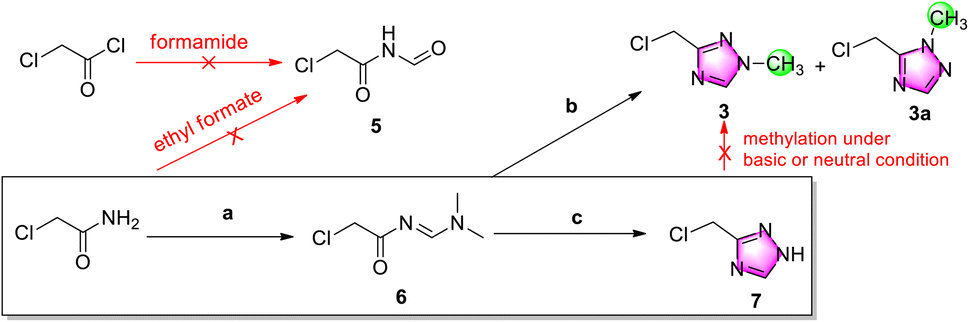 | ||
Scheme 3 Investigation into the preparation of 1,2,4-triazole derivatives. Reagents and conditions: (a) DMF·DMA, 80 °C, 1.5 h; (b) methylhydrazine sulfate, acetic acid: THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), r.t., overnight, 7.4% of 3 and 18.3% of 3a; and (c) hydrazine hydrate, acetic acid, 50 °C, overnight, 49% for two steps. :1), r.t., overnight, 7.4% of 3 and 18.3% of 3a; and (c) hydrazine hydrate, acetic acid, 50 °C, overnight, 49% for two steps. | ||
Consequently, chloroacetamide was instead condensated with N,N-dimethylformamide dimethyl acetal (DMF-DMA) to give active imine 6, and then cyclized with methylhydrazine sulfate.12 After easy purification by column chromatography, N1-methylation target product 3 was isolated in a 7.4% yield accompanied by N2-methylation side-product 3a in an 18.3% yield, which indicated low yield and position selectivity for cyclization. Further research revealed that chloromethyl triazole was stable in neutral, acidic and weak alkaline solution but unstable in stronger alkaline solution, such as aqueous potassium carbonate solution.
The poor selectivity and low yield of cyclization of imine 6 and methylhydrazine demonstrated that this was not a good protocol for the preparation of chloromethyl triazole 3. Therefore, the hydrazine hydrate was replaced to obtain de-methyl triazole 7 in a 49% yield for two steps. Further research indicated that 7 has a weak UV absorption peak at 200–400 nm (c = 1 and 10 mg mL−1 in ethanol) and was unstable in alkaline solution accompanied by complicated impurities.
After de-methyl triazole 7 was successfully obtained, its N1-methylation to chloromethyl triazole 3 was explored. Because of its instability in alkaline solution, the N1-methylation product was not observed using methylation reagents MeI and Me2SO4 (see ESI†). The neutral methylation reagent Me3O+BF4− showed a similar failure.13 In addition, when Ph3P and methanol were used for a Mitsunobu reaction, no methylated product 3 was generated.14
Investigation of N-alkylation and N1-methylation to triazone-triazole 4
The results of the previous investigation showed that de-methyl triazole 7 was a good choice to use in the following research.De-methyl triazole 7 could not be methylated in alkaline solution, but it could be N-alkylated with triazone 2 to de-methyl triazone-triazole 8 under the conditions of 1.5 equiv. chloromethyl triazole and potassium carbonate in N,N-dimethylformamide at 60 °C in an isolated yield of 25% (Scheme 4). This may be the result of the different pKb values of the 1-position nitrogen atom of 7 and the 3-position nitrogen atom of 8. After de-methyl triazone-triazole 8 was obtained, N1-methylation with iodomethane under potassium carbonate was performed and triazone-triazole 4 was isolated by column purification in an 80% yield. To accurately determine the substituted position of the key triazone-triazole 4, a single crystal obtained in volatile ethyl acetate was subjected to X-ray analysis (CCDC 2212798).
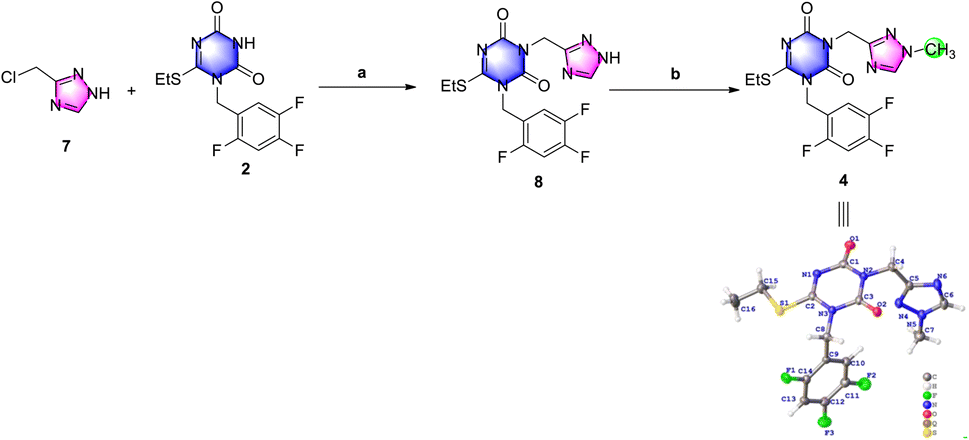 | ||
| Scheme 4 Investigation of N-alkylation and N1-methylation to prepare triazone-triazole. Reagents and conditions: (a) K2CO3, DMF, 60 °C, 3.5 h, 25%; and (b) MeI, K2CO3, DMF, r.t., overnight, 80%. | ||
Preliminary optimization
Based on the above synthetic strategy for the key intermediate triazone-triazole 4 from chloroacetamide, a preliminary optimization of the preparation of de-methyl triazole 7 was performed.To reduce material consumption and concentration temperature, 1.05 equiv. DMF·DMA in ethyl acetate was used for the condensation reaction. Considering its instability under aqueous conditions of amine 6, hydrazine hydrate was replaced by hydrazine acetate for the following cyclization. At the same time, in place of acetic acid, a combination of 3.0 equiv. acetic acid in ethanol gave only a 20% yield in the product. However, when the cyclization was performed in 1,4-dioxane and tetrahydrofuran, the isolated yields rose to 70% and 60%, respectively. By a tandem one-pot process, including condensation and cyclization, de-methyl triazole 7 was obtained as a white solid in a 74% yield (Scheme 5) after column purification, which was more efficient than previously reported methods.9b,15
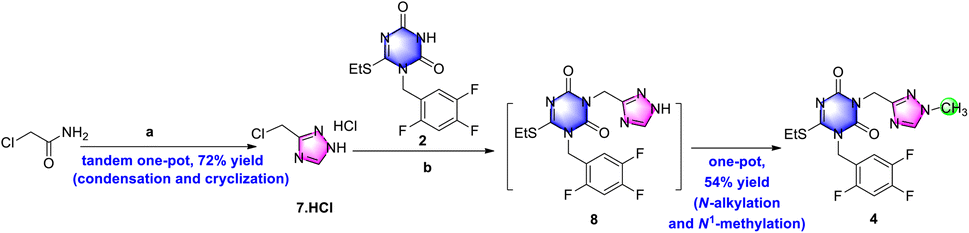 | ||
| Scheme 5 Two-stage one-pot synthetic strategy for the preparation of the key ensitrelvir intermediate. Reagents and conditions: , (a) DMF·DMA, ethyl acetate, 50 °C, 1.5 h; then hydrazine acetate, acetic acid, 1,4-dioxane, 50 °C, overnight, HCl/ethyl acetate for workup, 72% for two steps. and (b) K2CO3, DMF, 40 °C, 5 h; then MeI, r.t., 54% for two steps. | ||
Next, because of the instability of de-methyl triazole 7 under basic media, different bases were tried for N-alkylation with 2 as shown in Table 1. When several weak bases, including TEA, DIPEA, and NaHCO3 were used (entries 1–3), the TLC results were poor. When strong bases such as DBU, CH3ONa, t-BuOK, and aqueous NaOH were adopted (entries 4–7), a below 10% yield in the product was observed. Based on the better result with K2CO3, solvent screening for K2CO3 was carried out, which indicated that DMF was more efficient than acetonitrile (entries 8–9).
| Entry | Base (equiv.) | Solvent | T (°C) | 7 (equiv.) | Yieldb |
|---|---|---|---|---|---|
| a The reaction was performed according to the conditions unless otherwise mentioned. A mixture of triazone 2 (100 mg) and base was added into 2 mL of solvent. After 30–60 min, de-methyl triazole 7 was added into the mixture and stirred for 5 h with hourly monitoring by TLC. If necessary, the crude product was obtained by common workup and purified by column purification.b TLC was used to monitor the reaction unless otherwise mentioned.c Not reacted.d Isolated yield. | |||||
| 1 | TEA (2.2) | DMF | 60 | 1.5 | N. R.c |
| 2 | DIPEA (2.2) | DMF | 60 | 1.5 | N. R. |
| 3 | NaHCO3 (2.2) | DMF | 60 | 1.5 | Trace |
| 4 | DBU (2.2) | DMF | 60 | 1.5 | Trace |
| 5 | CH3ONa (1.1) | DMF | 60 | 1.5 | <10% |
| 6 | t-BuOK (1.1) | DMF | 60 | 1.5 | <10% |
| 7 | NaOH (1.1) | H2O | 60 | 1.5 | Trace |
| 8 | K2CO3 (2.2) | DMF | 60 | 1.5 | 25% |
| 9 | K2CO3 (2.2) | ACN | 60 | 1.5 | <15% |
| 10 | K2CO3 (2.2) | DMF | −20 | 1.5 | Trace |
| 11 | K2CO3 (2.2) | DMF | 0 | 1.5 | Trace |
| 12 | K2CO3 (2.2) | DMF | R. T. | 1.5 | 40%d |
| 13 | K2CO3 (2.2) | DMF | 40 | 1.5 | 51%d |
| 14 | K2CO3 (2.2) | DMF | 40 | 2.0 | 60%d |
Based on the combination of K2CO3 and DMF, the reaction temperature was further explored. Comparing the yields at different temperatures, a better-isolated yield (51%) was obtained at 40 °C (entry 13) than at −20 °C, 0 °C, r.t. (entry 10–12), and 60 °C (entry 8), which may be due to low reaction activity at lower temperatures (−20 °C, 0 °C, and R.T. entry 10–12) and poor thermodynamic stability at higher temperatures (entry 8). In addition, when de-methyl triazole 7 was increased from 1.5 equiv. to 2.0 equiv. (entry 14), a large amount of de-methyl triazone-triazole 8 was observed. After column purification, the target product was isolated as a white solid in a 60% yield. No further research into controlling the quantity of de-methyl triazole was performed.
Because of similar basic conditions for N1-methylation of 8, iodomethane was added directly into the reaction mixture of de-methyl triazone-triazole 8 to obtain triazone-triazole 4. By the one-pot process, the key intermediate was isolated as a base in a 54% isolated yield on a gram scale, which decreased the complexity of the operation and maintained similar results as the two individual steps. However, using triethyl amine as base, poor conversion rate was monitored and no desired product 4 was isolated.
Having identified the reaction conditions of 7 and 4, the workup procedure was further investigated. The hydrochloride salt of 7 was obtained by extracting with ethyl acetate and salt-forming using HCl gas in ethyl acetate. The scale-up up a five-gram scale gave a similar result in a 72% yield as a stable hydrochloride salt. In the further optimization of intermediate 4, when triethyl amine was used to basify 7·HCl before adding into the reaction mixture of 2 and potassium carbonate in N,N-dimethylformamide, only viscous residue was obtained with a large of impurities. Intermediate 4 was obtained in a 54% yield using potassium carbonate as base via extraction with ethyl acetate followed by slurrying in tert-butyl methyl ether. Ultimately, as shown in Scheme 5, the two-stage one-pot strategy was successful in the preparation of the key ensitrelvir intermediate.
Subsequently, an impurity assay was performed, including the two potential impurities N-methyl triazone 9 and N2-methyl impurity 10 in triazone-triazole 4, which were generated from unconsumed 2 and probably from de-methyl triazone-triazole 8 with successive with iodomethane in N-methylation reaction, respectively.
To confirm and control the impurities, impurity 9 was easily obtained by triazone 2 with iodomethane as shown in Scheme 6, which was generated in the reaction mixture and removed easily during the workup procedure. By HPLC monitoring, 23% of impurity 9 in reaction mixture was observed, which was reduced to below 1.0% by slurrying in tert-butyl methyl ether. To investigate the selectivity of N2-methylation on the 1,2,4-triazole group, N2-methylation impurity 10 was prepared from triazone 2 and N2-methyl-triazole 3a, which was observed below 5% in the reaction mixture under potassium carbonate in N,N-dimethylformamide and not detected after purification by HPLC. This indicated a high selectivity of N1-methylation using iodomethane and potassium carbonate to triazone-triazole 4.
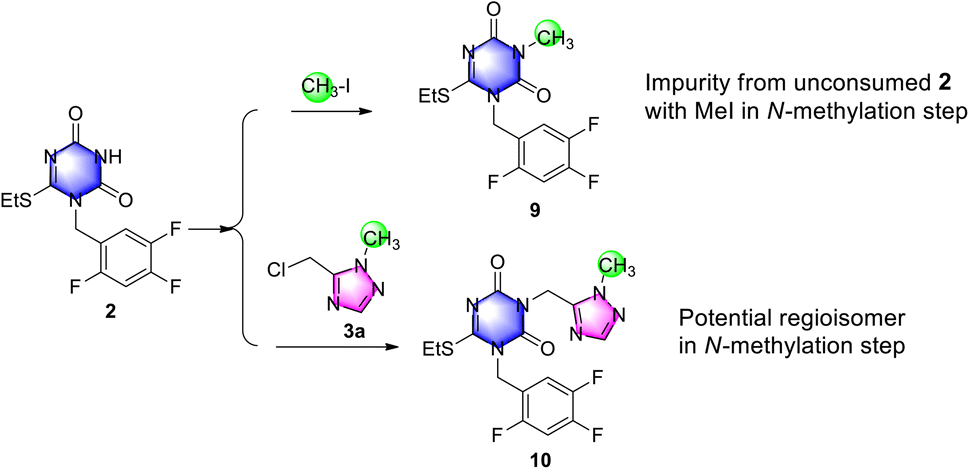 | ||
| Scheme 6 Generation and preparation of the two main impurities. | ||
Finally, due to the high energy of the 1,2,4-triazole scaffold, the thermodynamic stability of compounds 7, 3, 2, and 4 containing this triazole core was investigated by differential scanning calorimetry (DSC). The results indicated that all four compounds were stable below 100 °C (see ESI†), and safe under the conditions of the reaction and workup procedure.
Conclusion
Starting from commercially available chloroacetamide, a two-stage one-pot strategy was pursued to prepare the key ensitrelvir intermediate triazone-triazole. Firstly, 3-(chloromethyl)-1H-1,2,4-triazole hydrochloride was isolated as a solid in a 72% yield by a tandem one-pot “condensation and cyclization” process. Then, the key intermediate of ensitrelvir was isolated as a solid in a 54% yield by a one-pot “N-alkylation and N1-methylation” process. The results show that this strategy is suitable for further research and instructive for future drug discovery.Experimental section
All commercially available materials and solvents were used directly without further purification unless otherwise noted. Thin-layer chromatography (TLC) was performed on 0.25 mm silica gel F-254 plates. 1H NMR and 13C NMR data were recorded with a Bruker spectrometer (400 MHz) using TMS as internal standard and reported relative to residual solvent signals as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz), and integration. The multiplicities are denoted as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. The ESI mass spectra were determined on a THERMO LTQ. Single Crystal X-ray diffraction data were collected using a Bruker D8 Quest diffractometer (MoKα, λ = 0.71073 Å). DSC was recorded with Netzsch DSC214. HPLC was performed on Waters e2695 Separation Module.Preparation of 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride (3) and 5-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride (3a)
2-Chloroacetamide (1.0 g, 10.69 mmol, 1.0 equiv.) was suspended in DCM (10 mL) and N,N-dimethyformamide dimethyl acetal (1.911 g, 16.04 mmol, 1.5 equiv.) was added. The resulting mixture was stirred under 45 °C for 1.5 hours, then concentrated under reduced pressure. The resulting white solid was dissolved in a mixture of THF (6 mL) and acetic acid (6 mL). Methylhydrazine sulfate (1.85 g, 12.83 mmol, 1.2 equiv.) was added and then was stirred under 50 °C overnight.The reaction mixture was concentrated under reduced pressure, and then diluted with aqueous Na2CO3 (20 mL), extracted with DCM (50 mL × 3). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford a yellow oil. The residue was purified by silica gel column chromatography (PE to PE/EA = 4:1) to give the free bases of the title compounds.
The free bases were added a solution of 1 M HCl in EA respectively, and then evaporated to dryness to give two products. Via TLC using phosphomolybdic acid or bromocresol green as a chromogenic agent, side-product 3a returned a higher Rf value (Rf = 0.4) than chloromethyl triazole 3 (Rf = 0.3). 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride 3 (132 mg, 0.78 mmol) was obtained as a white solid, yield: 7.4%. 1H NMR (DMSO-d6, 400 MHz): δ (ppm) 8.61 (s, 1H), 4.71 (s, 2H), 3.86 (s, 3H). The 1H NMR data met with the publication.16 13C NMR (DMSO-d6, 100 MHz): δ (ppm) 159.41, 145.94, 38.04, 36.56. HRMS (ESI+) m/z calcd for C4H7ClN3+ [M + H]+: 132.0323, found 132.0324. 5-(Chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride 3a (328 mg, 1.95 mmol) was obtained as a white solid, yield: 18.3%. 1H NMR (DMSO-d6, 400 MHz): δ (ppm) 8.01 (s, 1H), 4.99 (s, 2H), 3.89 (s, 3H). The 1H NMR data met with the publication.17 13C NMR (DMSO-d6, 100 MHz): δ (ppm) 151.38, 150.27, 36.02, 34.41. HRMS (ESI+) m/z calcd for C4H7ClN3+ [M + H]+: 132.0323, found 132.0324.
Preparation of 6-(ethylthio)-1-(2,4,5-trifluorobenzyl)-1,3,5-triazine-2,4-(1H,3H)-dione (2)6b
To a solution of 6-(ethylthio)-3 t-butyl-1,3,5-triazine-2,4(1H,3H)-dione (5.0 g, 21.81 mmol, 1.0 equiv.) and 1-(bromomethyl)-2,4,5-trifluorobenzene (5.4 g, 23.99 mmol, 1.1 equiv.) in MeCN (20 mL), K2CO3 (3.62 g, 26.17 mmol, 1.2 equiv.) was added. The resulting mixture was stirred at 80 °C for 2 hours and then cooled to room temperature. The cooled mixture was diluted with EA (50 mL). The precipitate was filtered, and the filtrate was concentrated under reduced pressure to afford a yellow oil.The yellow oil was mixed with TFA (10 mL) and stirred at room temperature overnight. After concentration under reduced pressure, the residue was triturated with MTBE to afford 2 (5.1 g, 16.07 mmol) as a white solid, yield: 74%. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.91 (s, 1H), 7.10–6.93 (m, 2H), 5.15 (s, 2H), 3.22 (q, J = 7.4 Hz, 2H), 1.37 (t, J = 7.4 Hz, 3H). The 1H NMR data met with the publication.6b 13C NMR (DMSO-d6, 100 MHz): δ (ppm) 170.02, 154.71 (ddd, J = 244.5, 10.1, 2.6 Hz), 152.49, 150.13, 150.04–144.75 (m), 119.56 (ddd, J = 16.0, 6.0, 4.0 Hz), 116.53 (dd, J = 20.7, 5.1 Hz), 106.18 (dd, J = 28.3, 21.4 Hz), 40.70 (d, J = 4.4 Hz), 26.43, 13.77. HRMS (ESI+) m/z calcd for C12H11F3N3O2S+ [M + H]+: 318.0519, found 318.0516.
Preparation of 3-(chloromethyl)-1H-1,2,4-triazole (7) as a base form
2-Chloroacetamide (20.0 g, 213.88 mmol, 1.0 equiv.) was suspended in EA (200 mL) and N,N-dimethyformamide dimethyl acetal (26.76 g, 224.57 mmol, 1.05 equiv.) was added. The resulting mixture was stirred under 50 °C for 1.5 hours, then removed the solvent under reduced pressure. The resulting white solid was dissolved in a mixture of 1,4-dioxane (120 mL) and acetic acid (38.53 g, 641.64 mmol, 3.0 equiv.). Hydrazine acetate (23.64 g, 256.66 mmol, 1.2 equiv.) was added and then the mixture was stirred under 50 °C overnight. When hydrazine acetate was added, the reaction system immediately turned into a white suspension, which was clarified and turned pink during the heating process. Surprisingly, it proved difficult to monitor the reaction by TLC using common chromogenic methods except for bromocresol green.The reaction mixture was concentrated under reduced pressure, and then diluted with water (400 mL), extracted with EA (400 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to obtained a yellow oil. The residue was purified by silica gel column chromatography (PE to PE/EA = 1:1) to give 3-(chloromethyl)-1H-1,2,4-triazole 7 (18.60 g, 158.27 mmol) as a white solid, yield: 74%. 1H NMR (DMSO-d6, 400 MHz) δ (ppm) 14.05 (br s, 1H), 8.47 (s, 1H), 4.76 (s, 2H). The 1H NMR data met with the publication.9b 13C NMR (DMSO-d6, 100 MHz) δ (ppm) 158.84, 146.20, 37.93. HRMS (ESI+) m/z calcd for C3H5ClN3+ [M + H]+: 118.0167, found 118.0181.
Preparation of 3-[(1H-1,2,4-triazol-3-yl)methyl]-6-(ethylthio)-1-(2,4,5- trifluorobenzyl)-1,3,5-triazine-2,4(1H,3H)-dione (8)
To a solution of material 2 (1.0 g, 3.15 mmol, 1.0 equiv.) in DMF (5 mL), K2CO3 (958 mg, 6.93 mmol, 2.2 equiv.) was added. The resulting mixture was stirred under 40 °C for 0.5 hour, and then 3-(chloromethyl)-1H-1,2,4-triazole 7 (741 mg, 6.30 mmol, 2.0 equiv.) was added. The mixture was stirred for 5 hours.The reaction mixture was diluted with water (100 mL), extracted with DCM (300 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford a residue. The residue was purified by silica gel column chromatography (PE to PE/EA = 1:2) to give 8 (753 mg, 1.89 mmol) as a white solid, yield: 60%. 1H NMR (DMSO-d6, 400 MHz): δ (ppm) 8.26 (s, 1H), 7.73–7.57 (m, 1H), 7.56–7.43 (m, 1H), 5.11 (s, 2H), 5.05 (s, 2H), 4.59 (s, 1H), 3.15 (q, J = 7.4 Hz, 2H), 1.28 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ (ppm) 169.73, 155.89, 155.21 (ddd, J = 245.1, 10.0, 2.5 Hz), 151.81, 150.67, 150.57–145.08 (m), 147.24, 119.61 (dt, J = 16.3, 4.9 Hz), 116.66 (dd, J = 20.7, 5.1 Hz), 106.75 (dd, J = 28.2, 21.6 Hz), 56.09, 49.05, 42.06 (d, J = 4.4 Hz), 26.96, 14.19. HRMS (ESI+) m/z calcd for C15H14F3N6O2S+ [M + H]+: 339.0846, found 339.0854.
Preparation of 6-(ethylthio)-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluoro-benzyl)-1,3,5-triazine-2,4-(1H,3H)-dione (4)
To a solution of 8 (100 mg, 0.251 mmol, 1.0 equiv.) and MeI (54 mg, 0.377 mmol, 1.5 equiv.) in DMF (2 mL) was added K2CO3 (76 mg, 0.552 mmol, 2.2 equiv.). The mixture was stirred overnight then diluted with aqueous NH4Cl (20 mL) and extracted with EA (20 mL × 2). The combined organic layer was dried with anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by silica gel column chromatography (PE to PE/EA = 1:2) to give 4 (83 mg, 0.201 mmol) as a white solid, yield: 80%. Single crystal was obtained by slowly evaporating a mixture of 100 mg compound 4 in 5 mL ethyl acetate at ambient temperature and tested by X-ray analysis. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.94 (s, 1H), 7.17–7.09 (m, 1H), 6.99–6.90 (m, 1H), 5.23 (s, 2H), 5.16 (s, 2H), 3.85 (s, 3H), 3.20 (q, J = 7.4 Hz, 2H), 1.33 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 169.56, 159.16, 155.09 (ddd, J = 246.7, 9.6, 2.9 Hz), 151.65, 150.51, 151.21–145.64 (m), 144.30, 118.00 (dt, J = 16.3, 4.7 Hz), 116.13 (dd, J = 20.0, 5.4 Hz), 105.74 (dd, J = 27.6, 21.1 Hz), 41.05 (d, J = 4.4 Hz), 39.83, 36.09, 27.24, 13.49. The 1H NMR and 13C NMR data met with the publication.6b HRMS (ESI+) m/z calcd for C16H16F3N6O2S+ [M + H]+: 413.1002, found 431.1001.
Preparation of 3-(chloromethyl)-1H-1,2,4-triazole hydrochloride (7·HCl) by tandem one-pot process
2-Chloroacetamide (5.0 g, 53.47 mmol, 1.0 equiv.) was suspended in EA (50 mL) and N,N-dimethyformamide dimethyl acetal (6.7 g, 56.14 mmol, 1.05 equiv.) was added. The resulting mixture was stirred under 50 °C for 1.5 hours., then removed the solvent under reduced pressure to obtained a white solid, and resulting white solid was dissolved in a mixture of 1,4-dioxane (10 mL) and acetic acid (9.6 g, 160.41 mmol, 3.0 equiv.). Hydrazine acetate (5.9 g, 64.16 mmol, 1.2 equiv.) was added and then the resulting mixture was stirred under 50 °C overnight.The mixture was concentrated under reduced pressure. The residue was diluted with water (100 mL), extracted with EA (100 mL × 3). The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to obtained a yellow oil. The residue was dissolved in EA (10 mL) and then HCl/EA (1 M, 10 mL) was added dropwise to obtain a white suspension, which was filtered and dried to give 3-(chloromethyl)-1H-1,2,4-triazole hydrochloride 7·HCl (5.9 g, 38.50 mmol) as a white solid, yield: 72%. 1H NMR (DMSO-d6, 400 MHz) δ (ppm) 8.75 (s, 1H), 7.30 (br s, 2H), 4.80 (s, 2H). 13C NMR (DMSO-d6, 100 MHz) δ (ppm) 157.16, 145.66, 37.05.
Preparation of 6-(ethylthio)-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluoro-benzyl)-1,3,5-triazine-2,4-(1H,3H)-dione (4) by one-pot process
To a solution of compound 2 (1.0 g, 3.15 mmol, 1.0 equiv.) in DMF (5 mL) was added K2CO3 (958 mg, 6.39 mmol, 2.2 equiv.). The mixture was stirred under 40 °C for 0.5 hour. Then 3-(chloromethyl)-1H-1,2,4-triazole 7 (741 mg, 6.30 mmol, 2.0 equiv.) prepared via basification from hydrochloride salt using weak base sodium bicarbonate in ethyl acetate was added and stirred for 5 hours. After no improvement of this reaction was monitored by TLC, MeI (984 mg, 6.93 mmol, 2.2 equiv.) was added. The resulting mixture was stirred overnight.The reaction mixture was diluted with water (100 mL), extracted with EA (300 mL × 3). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford a residue. The residue was slurried by tert-butyl methyl ether to give 4 (703 mg, 1.70 mmol) as a solid, yield: 54%.
Preparation of 6-(ethylthio)-3-methyl-1-(2,4,5-trifluorobenzyl)-1,3,5-triazine-2,4(1H,3H)-dione (9)
To a solution of material 2 (100 mg, 0.351 mmol, 1.0 equiv.) and MeI (67 mg, 0.47 mmol, 1.5 equiv.) in MeCN (1 mL), K2CO3 (87 mg, 0.63 mmol, 2.0 equiv.) was added. The mixture was stirred at room temperature overnight, and then diluted with water (20 mL) and extracted with EA (20 mL × 2). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE to PE/EA = 5:1) to give 9 (94 mg, 0.28 mmol) as a white solid, yield: 90%. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.08–6.93 (m, 2H), 5.14 (s, 2H), 3.39 (d, J = 1.0 Hz, 3H), 3.22 (q, J = 7.4 Hz, 2H), 1.36 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ (ppm) 169.20, 155.31 (ddd, J = 247.0, 9.5, 2.8 Hz), 152.37, 150.63, 151.61–145.31 (m), 117.99 (dt, J = 16.0, 4.9 Hz), 116.43 (ddd, J = 20.4, 5.0, 1.5 Hz), 106.00 (dd, J = 27.6, 21.0 Hz), 41.51 (d, J = 3.9 Hz), 29.16, 27.29, 13.64. HRMS (ESI+) m/z calcd for C13H13F3N3O2S+ [M + H]+: 332.0675, found 332.0674.
Preparation of 6-(ethylthio)-3-[(1-methyl-1H-1,2,4-triazol-5-yl)methyl]-1-(2,4,5-trifluorobenzyl)-1,3,5-triazine-2,4-(1H,3H)-dione (10)
To a solution of material 2 (100 mg, 0.35 mmol, 1.0 equiv.) and 5-(chloromethyl)-1H-1,2,4-triazole HCl 3a (80 mg, 0.47 mmol, 1.5 equiv.) in DMF (2 mL) was added K2CO3 (131 mg, 0.945 mmol, 3.0 equiv.). The mixture was stirred at 60 °C for 4 hours and then cooled to room temperature. The cooled mixture was diluted with aqueous NH4Cl (20 mL) and extracted with EA (20 mL × 2). The combined organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE to PE/EA = 1:1) to give impurity 10 (104 mg, 0.25 mmol) as a grey viscous oil, yield: 80%. 1H NMR (CDCl3, 400 MHz) δ (ppm) 7.74 (s, 1H), 7.19–7.04 (m, 1H), 7.01–6.83 (m, 1H), 5.17 (s, 2H), 5.11 (s, 2H), 3.93 (s, 3H), 3.15 (q, J = 7.4 Hz, 2H), 1.29 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 170.32, 155.21 (ddd, J = 246.8, 9.5, 2.8 Hz), 151.43, 150.55, 150.35, 149.93, 151.31–145.51 (m), 117.72 (dt, J = 16.1, 4.8 Hz), 116.50 (dd, J = 19.8, 4.8 Hz), 105.90 (dd, J = 27.6, 21.0 Hz), 41.56 (d, J = 4.0 Hz), 36.94, 35.54, 27.44, 13.52. HRMS (ESI+) m/z calcd for C16H16F3N6O2S+ [M + H]+: 413.1002, found 413.1001.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Talent Project of Jiangxi Province (No. jxsq2019101065) and Ganzhou Science and Technology Project (No. 2020-60). The Start-up Fund of Gannan Medical University is gratefully acknowledged.Notes and references
- (a) WHO Solidarity Trial Consortium, Repurposed antiviral drugs for COVID-19 - interim WHO solidarity trial results, N. Engl. J. Med., 2021, 384, 497–511 CrossRef PubMed; (b) J. L. McKimm-Breschkin, A. J. Hay, B. Cao, R. J. Cox, J. Dunning, A. C. Moen, D. Olson, A. Pizzorno and F. G. Hayden, COVID-19, influenza and RSV: surveillance-informed prevention and treatment – meeting report from an isirv-WHO virtual conference, Antiviral Res., 2022, 197, 105227 CrossRef CAS PubMed; (c) L. Pan, P. Huang, X. Xie, J. Xu, D. Guo and Y. Jiang, Metabolic associated fatty liver disease increases the severity of COVID-19: A meta-analysis, Dig. Liver Dis., 2021, 53, 153–157 CrossRef CAS PubMed; (d) Z. Huang, P. Huang, B. Du, L. Kong, W. Zhang, Y. Zhang and J. Dong, Prevalence and clinical outcomes of cardiac injury in patients with covid-19: a systematic review and meta-analysis, Nutr., Metab. Cardiovasc., 2021, 31, 2–13 CrossRef CAS PubMed; (e) B. Hu, H. Guo, P. Zhou and Z.-L. Shi, Characteristics of SARS-CoV-2 and COVID-19, Nat. Rev. Microbiol., 2021, 19, 141–154 CrossRef CAS PubMed; (f) L. Lin, K. Hu, S. Cai, X. Deng, X. Shao, Y. Liang, J. Wang, T. Zhong, Z. Hu and M. Lei, Hypoproteinemia is an independent risk factor for the prognosis of severe COVID-19 patients, J. Clin. Biochem. Nutr., 2020, 67, 126–130 CrossRef CAS PubMed; (g) X. Hu, L. Zhu, Y. Luo, Q. Zhao, C. Tan, X. Chen, H. Zhang, X. Hu, L. Lu, Y. Xiao, S. Huang, Y. He, J. X. L. Sim, S. Su, F. Wang, Y. Peng, J. Wang, Y. Guo and T. Zhong, Evaluation of the clinical performance of single-, dual-, and triple-target SARS-CoV-2 RT-qPCR methods, Clin. Chim. Acta, 2020, 511, 143–148 CrossRef CAS PubMed.
- From WHO website: https://www.who.int/emergencies/diseases/novel-coronavirus-2019.
- (a) L. Riva, S. Yuan, X. Yin, L. Martin-Sancho, N. Matsunaga, L. Pache, S. Burgstaller-Muehlbacher, P. D. D. Jesus, P. Teriete, M. V. Hull, M. W. Chang, J. F.-W. Chan, J. Cao, V. K.-M. Poon, K. M. Herbert, K. Cheng, T.-T. H. Nguyen, A. Rubanov, Y. Pu, C. Nguyen, A. Choi, R. Rathnasinghe, M. Schotsaert, L. Miorin, M. Dejosez, T. P. Zwaka, K.-Y. Sit, L. Martinez-Sobrido, W.-C. Liu, K. M. White, M. E. Chapman, E. K. Lendy, R. J. Glynne, R. Albrecht, E. Ruppin, A. D. Mesecar, J. R. Johnson, C. Benner, R. Sun, P. G. Schultz, A. I. Su, A. García-Sastre, A. K. Chatterjee, K.-Y. Yuen and S. K. Chanda, Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing, Nature, 2020, 586, 113–119 CrossRef CAS PubMed; (b) B. Halford, The path to paxlovid, ACS Cent. Sci., 2022, 8, 405–407 CrossRef CAS PubMed; (c) D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- K. S. Yang, S. Z. Leeuwon, S. Xu and W. R. Liu, Evolutionary and structural insights about potential SARS-CoV-2 evasion of nirmatrelvir, J. Med. Chem., 2022, 65, 8686–8698 CrossRef CAS PubMed.
- H. Tan, Y. Hu, P. Jadhav, B. Tan and J. Wang, Progress and challenges in targeting the SARS-CoV-2 papain-like protease, J. Med. Chem., 2022, 65, 7561–7580 CrossRef CAS PubMed.
- (a) J. D. A. Tyndall, S-217622, a 3CL protease inhibitor and clinical candidate for SARS-CoV-2, J. Med. Chem., 2022, 65, 6496–6498 CrossRef CAS PubMed; (b) Y. Unoh, S. Uehara, K. Nakahara, H. Nobori, Y. Yamatsu, S. Yamamoto, Y. Maruyama, Y. Taoda, K. Kasamatsu, T. Suto, K. Kouki, A. Nakahashi, S. Kawashima, T. Sanaki, S. Toba, K. Uemura, T. Mizutare, S. Ando, M. Sasaki, Y. Orba, H. Sawa, A. Sato, T. Sato, T. Kato and Y. Tachibana, Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19, J. Med. Chem., 2022, 65, 6499–6512 CrossRef CAS PubMed.
- M. Sasaki, K. Tabata, M. Kishimoto, Y. Itakura, H. Kobayashi, T. Ariizumi, K. Uemura, S. Toba, S. Kusakabe, Y. Maruyama, S. Iida, N. Nakajima, T. Suzuki, S. Yoshida, H. Nobori, T. Sanaki, T. Kato, T. Shishido, W. W. Hall, Y. Orba, A. Sato and H. Sawa, Oral administration of S-217622, a SARS-CoV-2 main protease inhibitor, decreases the viral load and accelerates recovery from clinical aspects of COVID-19, bioRxiv, 2022 DOI:10.1101/2022.02.14.480338.
- From Shionogi's Website: https://www.shionogi.com/global/en/news/2022/09/20220928.html.
- (a) S. N. Owen, C. J. Swain and B. J. Williams, Substituted morpholine derivative and its use as a therapeutic agent, 1999 Search PubMed; (b) U. Bechstein, A. Rumler and J. Liebscher, Efficient syntheses of ω-chloro-, ω-imido- and ω-aminoalkyl-1,2,4-triazoles from N-acyl-formamidinium salts or N-acylformamides and hydrazines, Arch. Pharm., 1992, 325, 519–529 CrossRef CAS; (c) M. Xi, C. Feng, K. Du, W. Lv, C. Du, R. Shen and H. Sun, Design, synthesis, biological evaluation and molecular modeling of N-isobutyl-N-((2-(p-tolyloxymethyl)thiazol-4-yl)methyl)benzo[d][1,3] dioxole-5-carboxamides as selective butyrylcholinesterase inhibitors, Bioorg. Med. Chem. Lett., 2022, 61, 128602 CrossRef CAS PubMed.
- F. Gao, S. Zeng, J. Jiang and X. Zhang, Method for preparing (1-methyl-1H-[1,2,4]triazole-3-yl)-methyl alcohol, Invention Pat., CN107879992A, 2017 Search PubMed.
- T. Harit, R. Bellaouchi, Y. Rokni, A. Riahi, F. Malek and A. Asehraou, Synthesis, characterization, antimicrobial activity and docking studies of new triazolic tripodal ligands, Chem. Biodiversity, 2017, 14, e1700351 CrossRef PubMed.
- Y. Xu, M. McLaughlin, E. N. Bolton and R. A. Reamer, Practical synthesis of functionalized 1,5-disubstituted 1,2,4-triazole derivatives, J. Org. Chem., 2010, 75, 8666–8669 CrossRef CAS PubMed.
- Y. Hoashi, T. Takai, Y. Kosugi, M. Nakashima, M. Nakayama, K. Hirai, O. Uchikawa and T. Koike, Discovery of a potent and orally bioavailable melatonin receptor agonist, J. Med. Chem., 2021, 64, 3059–3074 CrossRef CAS PubMed.
- S. Shen, M. Hadley, K. Ustinova, J. Pavlicek, T. Knox, S. Noonepalle, M. T. Tavares, C. A. Zimprich, G. Zhang, M. B. Robers, C. Bařinka, A. P. Kozikowski and A. Villagra, Discovery of a new isoxazole-3-hydroxamate-based histone deacetylase 6 inhibitor ss-208 with antitumor activity in syngeneic melanoma mouse models, J. Med. Chem., 2019, 62, 8557–8577 CrossRef CAS PubMed.
- R. G. Jones and C. Ainsworth, 1,2,4-Triazole-3-alanine, J. Am. Chem. Soc., 1955, 77, 1538–1540 CrossRef CAS.
- A. Tasaka, K. Teranishi, Y. Matsushita, N. Tamura, R. Hayashi, K. Okonogi and K. Itoh, Optically active antifungal azoles. III. Synthesis and antifungal activity of sulfide and sulfonamide derivatives of (2R,3R)-2-(2,4-difluorophenyl)-3- mercapto-1-(1H-1,2,4-triazol-1-yl)-2-butanol, Chem. Pharm. Bull., 1994, 42, 85–94 CrossRef CAS PubMed.
- K. J. Wildonger and R. W. Ratcliffe, Heteroaryliumthio substituted carbapenem derivatives: Synthesis and in vitro activity of 1β-methyl-2-(dihydropyrrolotriazoliumthio)carbapenems, J. Antibiot., 1993, 46, 1866–1882 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2212798. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06841a |
| This journal is © The Royal Society of Chemistry 2022 |