DOI:
10.1039/D2RA05593G
(Paper)
RSC Adv., 2022,
12, 34797-34807
Colour-tuneable solid-state fluorescence of crystalline powders formed from push–pull substituted 2,5-diphenyl-stilbenes†
Received
5th September 2022
, Accepted 18th November 2022
First published on 8th December 2022
Abstract
The solid-state fluorescence (SSF) of eight DPA–DPS-EWG derivatives (DPA = diphenylamino, DPS = 2,5-diphenyl-stilbene building block, EWG = electron withdrawing group) was studied. Varying the strength of the EWG enabled the tuning of the LUMO energy within a range broader than 1 eV, while the simultaneous changes of HOMO energy were less than 0.1 eV, according to cyclic voltammetry. The fluorescence maxima in dichloromethane laid between 483 and 752 nm and exhibited monoexponential decay and a photoluminescence quantum yield (PLQY) always higher than 35%. Six derivatives with a SSF PLQY higher than 10% in polycrystalline powder form continuously covered the range from 475 to 733 nm. Three components of SSF multiexponential decay, obtained by time-resolved fluorescence spectroscopy, were ascribed to exciton migration to nonfluorescent traps, and monomer-like and aggregate fluorescence. The character of the emitting aggregates was evaluated by quantum chemical modelling based on time-dependent density functional theory computations, carried out on the dimer arrangements obtained by X-ray diffractometry of the single crystals.
Introduction
Luminescent materials based on all-organic conjugated molecules are usually used in solid physical form in technological fields, such as biological probes, chemical sensing, optoelectronic systems and stimuli-responsive materials.1 Solid-state fluorescence (SSF) has been studied on single crystals, polycrystalline powders, thin films and nanostructures.2 Typical applications of single-crystal emission are in lasing and organic light-emitting field-effect transistors,3–6 fluorescent powders are common in stimuli-responsive research,7,8 thin films/layers form a key component of organic light-emitting diodes9–13 and water dispersions of nanoparticles dominate biomedical studies.14–19 The efficiency of SSF depends on the combination of nonradiative intramolecular deactivation processes, intermolecular arrangement and morphology, the net effect of which can cause either quenching or enhancement of the photoluminescence quantum yield (PLQY) compared to solution.20 Intramolecular nonradiative pathways occur either with a conserved spin (internal conversion, IC) or with a spin-flip (intersystem crossing, ISC), both of which are often supported by large-amplitude excited-state structural motions.20,21 Solidification is, thus, a way to improve PLQY by limiting these vibrations/rotations via various mechanisms.20,22 The mutual arrangement of the molecules in crystals determines their photophysics through excitonic coupling, forming physical J- or H-aggregates unambiguously defined by the bright (oscillator strength fosc > 0) or dark (fosc ∼ 0) character of the lowest excited state, respectively.23,24 True H-aggregates, determined by a decrease in the radiative rate of a solid with respect to a solution, i.e. not only by geometrical factors, are more common than J-aggregates, and it is believed that their SSF is enabled by static and/or dynamic symmetry breaking enhancing the fosc of the H-aggregate lowest state.20 Excitons formed during photon absorption are either localised by self-trapping or delocalised over several molecules, and they may migrate in crystals and fall into various traps, either in bulk or on the surface.25 This trapping is generally considered to be the main source of SSF quenching in solids, and a higher concentration of traps on the surface and a shorter distance from the excited site to the surface lead to a common decrease in the PLQY when transitioning from single crystals to polycrystalline powders, films and nanoparticles.20 Consequently, reducing exciton migration is one of the few general ways to obtain highly luminescent poly- and nanocrystalline organic solids.20
Exciton traps are often connected with cofacial H-aggregates.26 If they both fulfil the conditions of limited delocalisation on only two molecules and show a pronounced charge-transfer (CT) character, they can be called excimers.27 On the other hand, an excimer-induced enhanced emission28 phenomenon has been observed on crystals, formed by discrete pairwise π–π stacked dimers of polycyclic aromatic hydrocarbons, such as perylene28 or anthracene,29,30 suitably substituted by bulk groups. Excimer-type SSF can also originate from crystals with alternating (e.g. stacked/unstacked) dimers in columns;31 however, relatively small changes in the molecular structure, saving the same building block, may transform this unique arrangement into uniform columns with either H- or J-type aggregation.32,33 We have recently observed that one of the polymorphs of DPA-DPS-DCV (Chart 1) forms a crystal arrangement with such alternating dimers and consequent intense infrared excimer-type emission was observed.34 Research on solid-state emitters has paid special attention to the series of derivatives based on the same building block with SSF maxima tuned by a variation of simple substituents.35–37 An impressive interval of SSF maxima from 383 to 631 nm, i.e. covering a range of 248 nm (1.27 eV), obtained by varying the electron withdrawing group (EWG) on simple triphenylamines,38 shows the potential of this approach, e.g. for multicomponent white-light-emitting materials.39
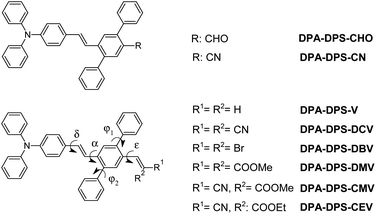 |
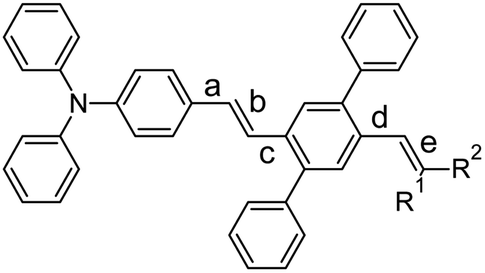
| | Chart 1 Notations of the compounds under study. | |
Recently, we synthesised34 DPA-DPS-CHO (Chart 1), a synthetic precursor of DPA-DPS-DCV, with a push–pull substituted stilbene π-system decorated at the acceptor part by a pair of side-phenyls. Both polymorphs of DPA-DPS-CHO have shown green monomer-type SSF with only moderate PLQY. Side-phenyl interactions (CH–π or π-stacking) in DPA-DPS-CHO and DPA-DPS-DCV, respectively, were found to be vital for an overall crystalline arrangement of molecules and the consequent SSF ability. Here we present further research into additional DPA-DPS-EWG derivatives, synthesised from DPA-DPS-CHO, in order to cover a much broader area of SSF colours by tuning the EWG strength within a series of emitters based on the same donor (DPA) and the same building block (DPS). Attention was paid to the relationship between the acceptor strength and SSF spectra (energy), and between the crystal structure and photophysics (PLQY and fluorescence decay), in order to better understand the nature of the SSF phenomenon in polycrystalline solids. The series was designed to answer, at minimum, the following questions. (1) Is the presence of side-phenyl substituents a universal way of achieve SSF of at least moderate intensity? (2) Does the intensity of SSF follow the so-called “energy gap law”, usually indicating intramolecular internal conversion as a dominant deactivation pathway, as sometimes found in isolated molecules?20 (3) Is the crystal arrangement consisting of alternating stacked/unstacked dimers, as observed for DPA-DPS-DCV,34 driven by side-phenyl specific interactions which lead to excimer-induced fluorescence unique or quite common in a series of isostructural compounds differing only by EWG substituent?
Experimental
Synthesis
All reagents and solvents were purchased from commercial sources (Sigma-Aldrich, Fluorochem, Acros Organics, TCI Europe, Merck, and Lach-Ner). Commercial grade reagents were used without further purification. Reactions were monitored using thin layer chromatography (TLC) plates coated with 0.2–0.5 mm silica gel (60 F254, Merck). TLC plates were visualised using UV irradiation (254 nm). All melting points were determined using a melting point B-540 apparatus (Büchi, Switzerland) and are given in their uncorrected form. Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet iS50 using the ATR technique. The NMR spectra were measured in CDCl3 and CD2Cl2-d2 solvents at ambient temperature using either a Bruker Avance™ III 400 spectrometer at frequencies of 400 MHz (1H) and 100.26 MHz (13C), or a Bruker Ascend™ 500 spectrometer at frequencies of 500.13 MHz (1H) and 125.76 MHz (13C{1H}). The chemical shifts (δ) reported in the ESI† are given in ppm and are related to the following residual solvent peaks: −4.79 (D2O-d2), −5.32 (CD2Cl2-d2) and −7.27 (CDCl3). Tetramethylsilane (TMS) was used as an internal standard. The coupling constants (J) are reported in Hz. Elemental analyses (C, H, and N) were performed on an automatic microanalyser (Flash 2000 Organic elemental analyser). High-resolution mass spectrometry was performed by the “dried droplet” method using the MALDI mass spectrometer LTQ Orbitrap XL (Thermo Fisher Scientific) equipped with a nitrogen UV laser (337 nm, 60 Hz). Spectra were measured in positive ion mode and in regular mass extent with a resolution of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 at a mass-to-charge ratio (m/z) of 400, with 2,5-dihydrobenzoic acid (DBH) used as the matrix.
000 at a mass-to-charge ratio (m/z) of 400, with 2,5-dihydrobenzoic acid (DBH) used as the matrix.
Single-crystal X-ray diffraction (XRD)
Full-sets of diffraction data for DPA-DPS-CMV and DPA-DPS-DMV were collected at 150(2) K with a Bruker D8-Venture diffractometer equipped with Cu (Cu/Kα radiation; λ = 1.54178 Å) or Mo (Mo/Kα radiation; λ = 0.71073 Å) microfocus X-ray (IμS) sources, a Photon CMOS detector and an Oxford Cryosystems cooling device. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. Data were corrected for absorption effects using the Multi-Scan method (SADABS). The obtained data were treated by XT-version 2017/1 and SHELXL-2014/7 software implemented in the APEX3 v2017.1-0 (Bruker AXS) system. CCDC 2118685 and 2118686 contains the ESI† crystallographic data for this paper.
Electrochemical measurements
Electrochemical measurements were carried out in acetonitrile containing 0.1 M Bu4NPF6 in a three-electrode cell by cyclic voltammetry (CV) and rotating disk voltammetry (RDV). For CV and RDV experiments, the working electrode was a glassy carbon disk (3 mm in diameter). A saturated calomel electrode (SCE) separated by a bridge filled with supporting electrolyte and Pt wire were used as the reference and auxiliary electrodes. All potentials are given vs. SCE. Voltametric measurements were performed using a potentiostat PGSTAT 128N (AUTOLAB, Metrohm Autolab B.V., Utrecht, The Netherlands) operated via NOVA 1.11 software.
Spectral and photophysical measurements
UV-vis absorption was measured by employing a Varian Cary Probe 50 UV-Vis spectrometer (Agilent Technologies Inc.). Steady-state photoluminescence was captured using an SF5 spectrofluorometer (Edinburgh Instruments). Photoluminescence quantum yields (PLQYs) were determined by the absolute method employing an integrating sphere SC-30 (Edinburgh Instruments). PLQYs of powdered samples were determined by a combination of direct and indirect excitation. Time-resolved fluorescence was recorded by means of the time-correlated single photon counting method (TCSPC) using a Horiba Jobin Yvon Fluorocube.
Quantum chemical calculations
Monomer geometries in the ground and the lowest excited state were optimised by density functional theory (DFT) and time-dependent (TD) DFT calculations, respectively. Either ωB97X-D51 or CAM-B3LYP52 xc functionals, and either 6-31G(d,p) or 6-311G(d,p) basis sets were used. The solvent effect was introduced by the polarised continuum model (PCM). Excitation energies of the dimers (and monomers, for comparison) were calculated by the TD DFT ωB97X-D/6-311G(d,p) level in vacuum on the geometries obtained from single-crystal XRD. All calculations were carried out with Gaussian 09 software.40
Results and discussion
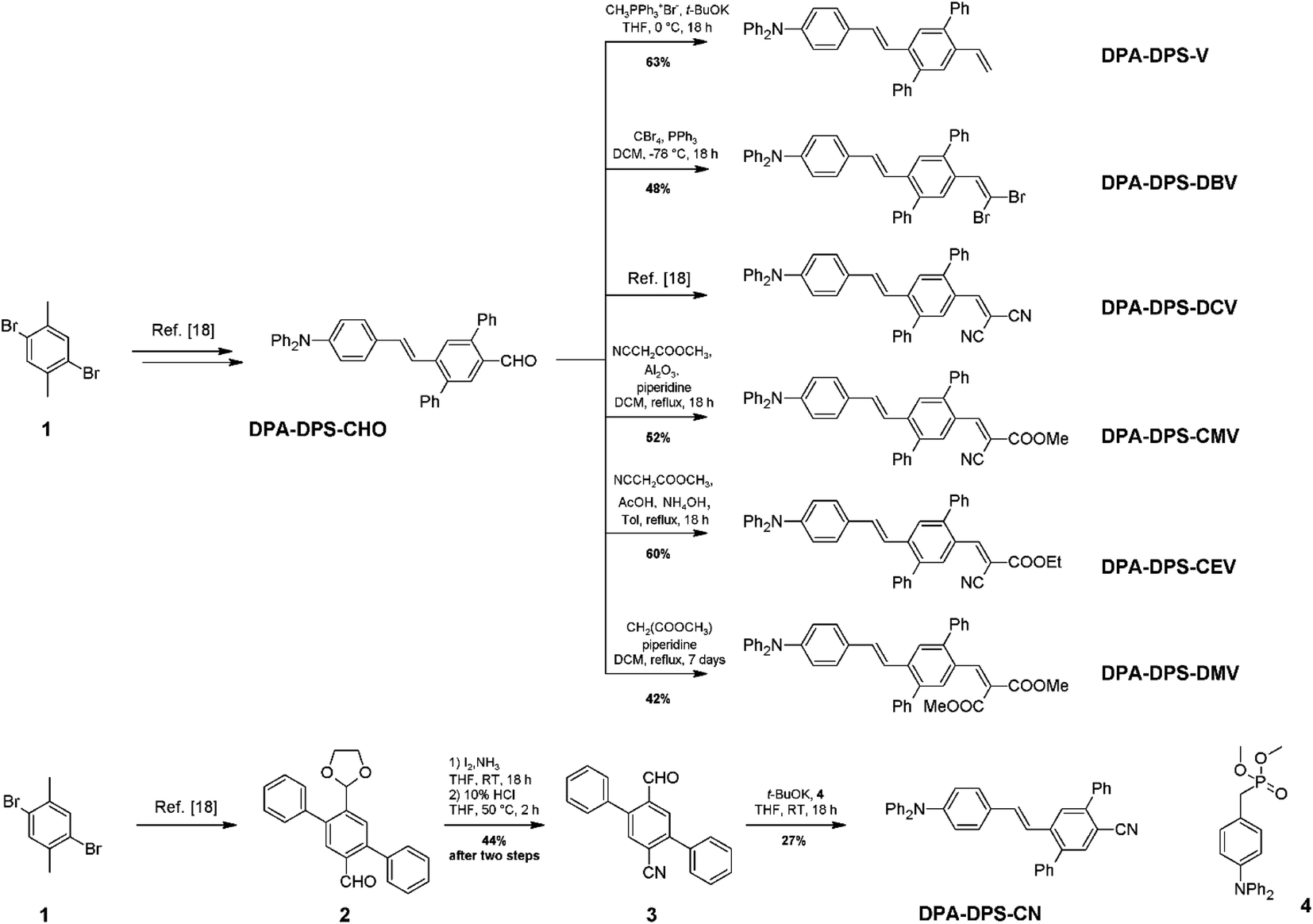
The synthesis of new compounds was carried out according to Scheme 1. In most cases, the previously reported34 DPA-DPS-CHO was used as the starting precursor. The only exception were the syntheses of DPA-DPS-CN, for which an independent synthesis according to Scheme 1 gave a considerably better yield and purity compared to the oxidation of DPA-DPS-CHO with iodine in NH4OH, which provided an inseparable mixture of substances. Final powders for SSF measurements were obtained by crystallisation from ethyl acetate (DPA-DPS-CN, -V and -DBV), DCM (DPA-DPS-CMV) or an EtOAc/MeOH/n-hexane/DCM mixture (DPA-DPS-DMV and -CEV), only dried, without any physical treatment.
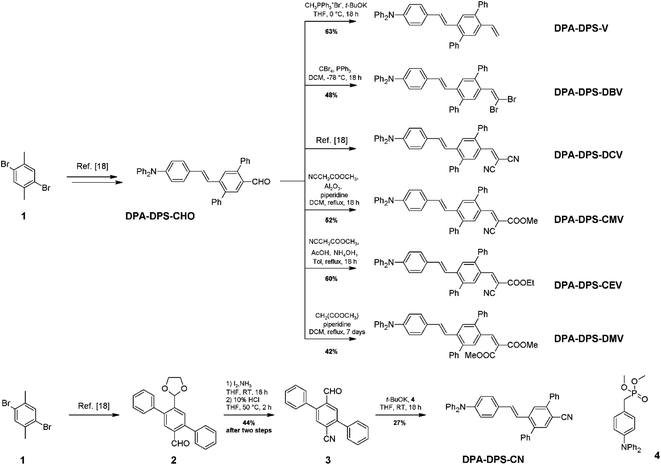 |
| | Scheme 1 Synthesis of DPA-DPS-EWGs. Compound 4 = dimethyl (4-(diphenylamino)benzyl)phosphonate. | |
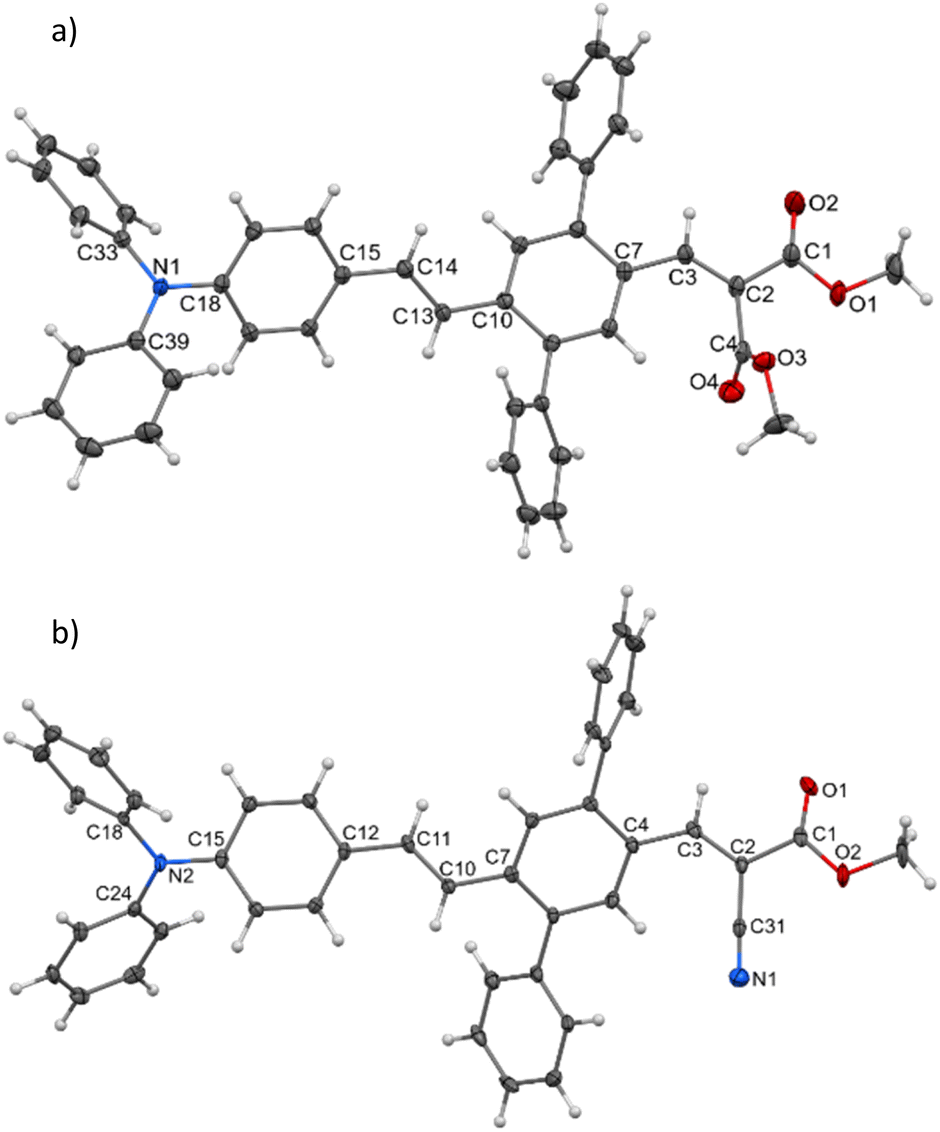
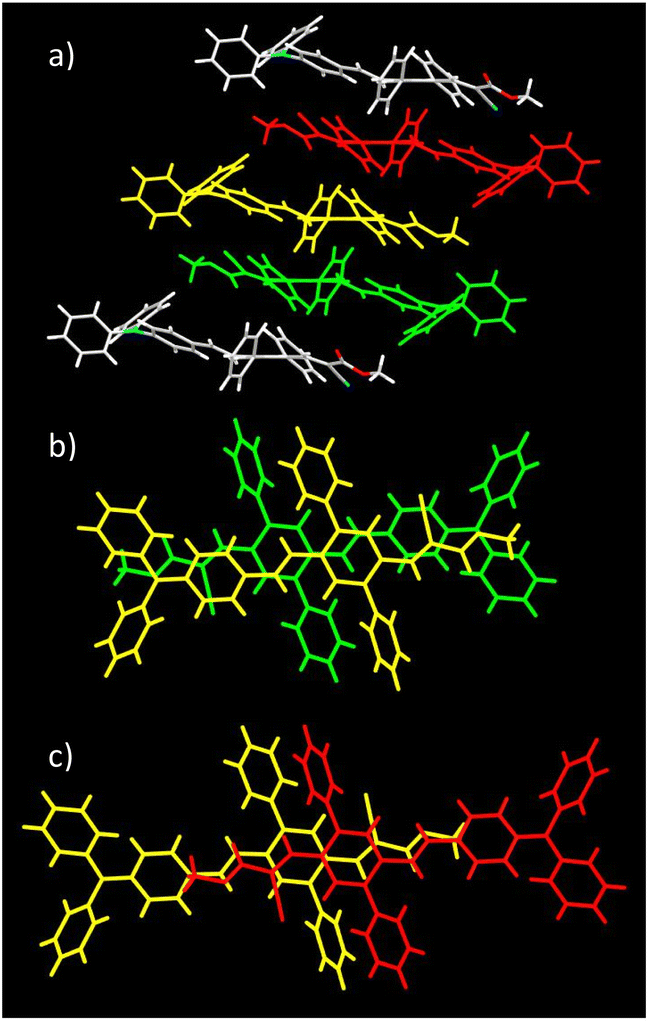
Single crystals of DPA-DPS-DMV and DPA-DPS-CMV, obtained by crystallisation from solution, were prepared in a quality suitable for X-ray diffraction (XRD) analysis. The former crystallises in a non-centrosymmetrical P212121 space group (such as DPA-DPS-CHO),34 and the latter in a centrosymmetrical P21/c group (DPA-DPS-DCV crystallizes in a different centrosymmetrical P1 space group).34 A unit cell of all four derivatives consists of four molecules (Z = 4) and contains only one independent molecule. The molecular geometries of both DPA-DPS-DMV and DPA-DPS-CMV in crystals are mutually similar (Fig. 1). Contrary to -CHO and -DCV derivatives, which have nearly coplanar side-phenyls, i.e. out-of-plane rotated in the same manner (e.g. φ1 = −53.5° and φ2 = +46.0° for -DCV), the phenyls in -DMV and -CMV derivatives are rotated out-of-plane in a mutually opposite manner (φ1 = +61.2° and φ2 = +46.2° for CMV and φ1 = −65.0° and φ2 = −52.5° for DMV). As for -CHO and -DCV compounds,34 the stilbene units in both derivatives are non-planar (α = +29.9°, δ = −1.6° and ε = −24.4° for -CMV, and α = −29.2°, δ = −10.5° and ε = +6.0° for -DMV), i.e. considerably more distorted than in analogous compounds without side-phenyls.41,42
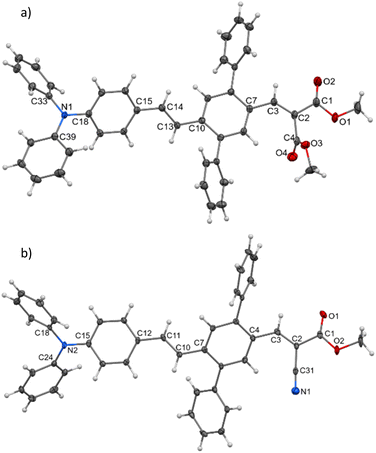 |
| | Fig. 1 Molecular structure of DPA-DPS-DMV (a) and DPA-DPS-CMV (b), ORTEP view, 50% probability level. | |
Molecules of DPA-DPS-DMV form infinite stacked columns consisting of the same type of non-centrosymmetrical dimer with parallel dipole moments (Fig. 2). The key intermolecular interactions that form the columns are of the edge-to-face C–H⋯π type. Contrary to DPA-DPS-CHO, where y-shaped C–H⋯π interactions connected the side-phenyls of adjacent molecules,34 in DPA-DPS-DMV, these interactions are of a T-shape43 and form a network with multiple contacts either between the side-phenyls and the phenyls of a DPA group, or between the phenyls of triphenylamine groups mutually.
 |
| | Fig. 2 Crystal structure of DPA-DPS-DMV. Overall view (a) of the stacked column. Detailed view of a stacked dimer with PP = 3.445 Å (b). | |
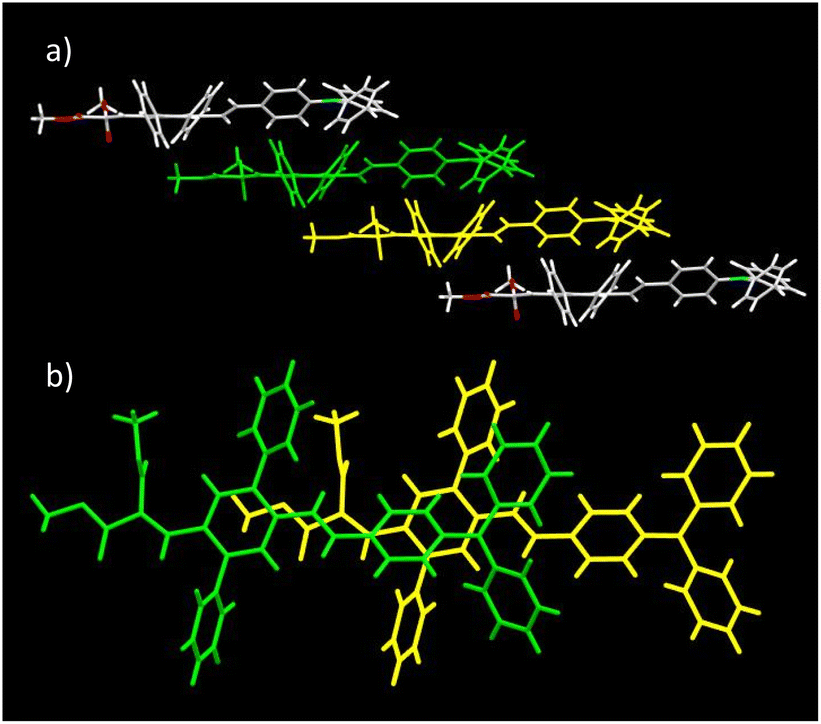
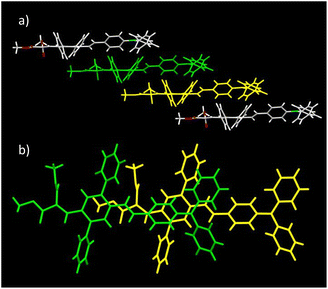
As for DPA-DPS-DCV,34 the molecules of DPA-DPS-CMV form slipped infinite columns with two alternating (stacked/unstacked) types of centrosymmetrical dimer with antiparallel dipole moments (Fig. 3). In contrast to DPA-DPS-DCV, the stacked dimer 1 is shorter (NN = 16.636 Å) than the unstacked dimer 2 (NN = 21.221 Å). The dimer forming an interaction between adjacent molecules in DPA-DPS-CMV columns is created by T-shape C–H⋯π type connections exclusively between side-phenyls. Such a decisive role of side-phenyls was also observed in DPA-DPS-DCV, in which case the side-phenyls were coupled by parallel offset face-to-face stacks.34
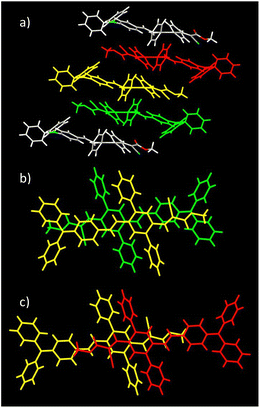 |
| | Fig. 3 Crystal structure of DPA-DPS-CMV. Overall view (a) of the alternation of the dimers in a slipped stacked column. Dimer 1 consists of yellow and green monomers, and dimer 2 of yellow and red monomers. Detailed view of stacked dimer 1 (b) with PP = 3.449 Å. Detailed view of unstacked dimer 2 (c) with PP = 4.291 Å. | |
The geometry of both alternating dimers 1 and 2 of DPA-DPS-CMV (Fig. 3) was recomputed from crystal structures by DFT (ωB97X-D/6-31G(d,p))40 and compared with the experimental data. The computed structure of dimer 1 matches the experiment quite well and dimer 1 can, thus, be considered as a standalone dimer. The overall length of a conjugated system of this dimer, characterised by a distance between the nitrogens of TPA groups (NN), was found to be 16.501 Å (exp. NN = 16.636 Å). The plane-to-plane (PP) distance, defined by the central benzene rings of the 2,5-diphenyl-1,4-divinyl-benzene (PVB) unit, was computed to be 3.286 Å (exp. PP = 3.448 Å), signalising a slightly overestimated dispersion attractive force with respect to repulsive electron exchange in the ωB97X-D xc functional, as in the previously studied case of DPA-DPS-DCV.33 However, the experimental arrangement of the unstacked dimer 2 could not be well reproduced, because (1) the optimised geometry always converged to a dimer with coplanar stacked side-phenyls, and (2) the NN distances were always significantly shorter (by more than 0.5 Å) than the experimental ones. The computations of the stacked dimer of DPA-DPS-DMV (Fig. 2) failed entirely as it was impossible to reproduce the coplanarity of the central benzene rings of adjacent molecules. Consequently, all reported energies of the dimers (vide infra) were obtained by (time-dependent) density functional theory [(TD) DFT] calculations, carried out on non-optimised geometries obtained from XRD single-crystal structures.
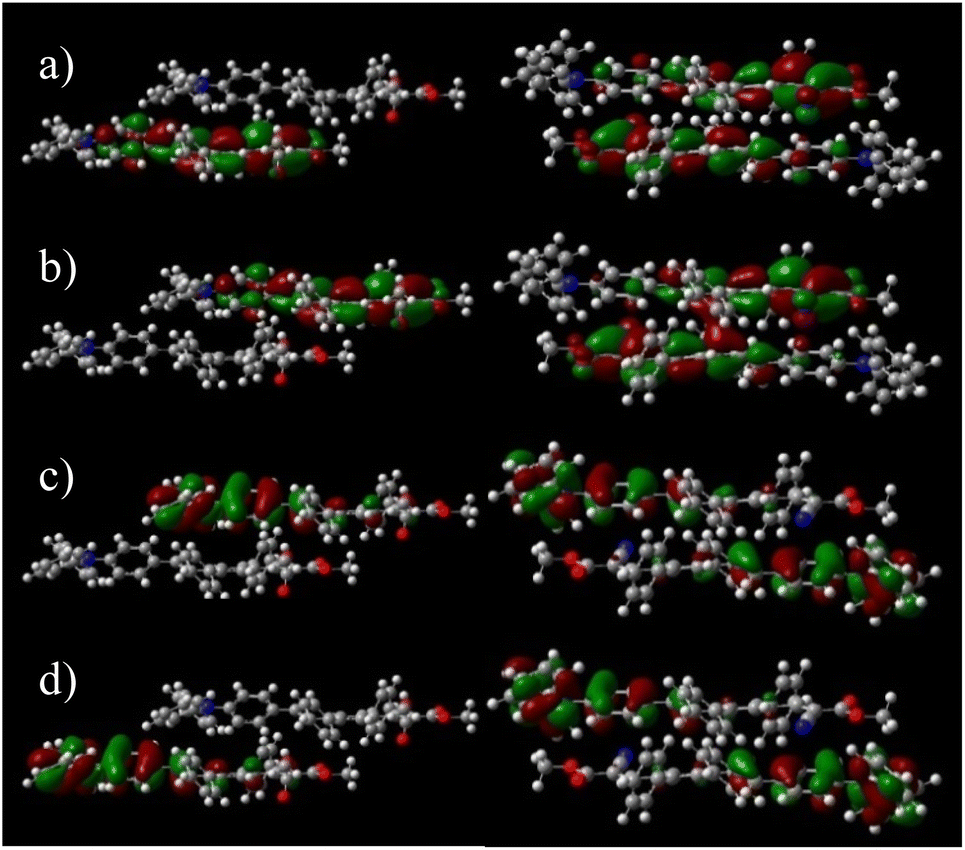
Although DPA-DPS-CMV forms columns of stacked/unstacked alternating centrosymmetrical dimers such as DPA-DPS-DCV, the more compact arrangement of a stacked dimer 1 for the former derivative significantly affects the electronic properties expressed in terms of split frontier Kohn–Sham orbitals (KS FMOs). First, the symmetries of the HOMO (au) and HOMO−1 (ag) are the same for both derivatives, but the symmetries of the LUMO (ag) and LUMO+1 (au) of DPA-DPS-CMV (Fig. 4) are opposite to the FMOs of DPA-DPS-DCV, i.e. during the slipping, longitudinal geometrical shift virtual orbitals pass a node.44
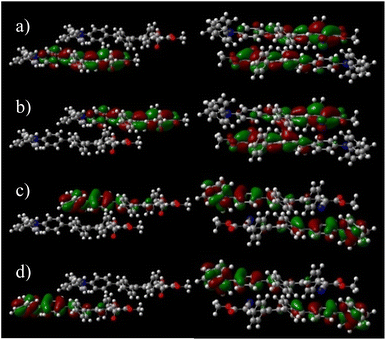 |
| | Fig. 4 Split orbitals LUMO+1 (a), LUMO (b), HOMO (c) and HOMO−1 (d) in a dimer of DPA-DPS-DMV (left) and dimer 1 of DPA-DPS-CMV (right). DFT calculations were carried out on the wB97X-D/6-31G(d,p) level. An isovalue of 0.02 was used for the drawing. | |
Second, the virtual orbital energy splitting (ΔLUMO = 108 meV), relating to electron transfer, is lower, while the occupied orbital splitting (ΔHOMO = 73 meV), relating to hole transfer, is higher than for DPA-DPS-DCV, i.e. both transfer integrals are better balanced for DPA-DPS-CMV. Third, FMOs in an antiparallel arrangement of DPA-DPS-CMV are delocalised over both molecules in a dimer with a significant overlap in the LUMO, contrary to parallelly packed DPA-DPS-DMV (Fig. 4), in which all four KS orbitals are localised on only one molecule, with a consequent low orbital energy splitting (ΔHOMO = 24 meV and ΔLUMO = 19 meV).
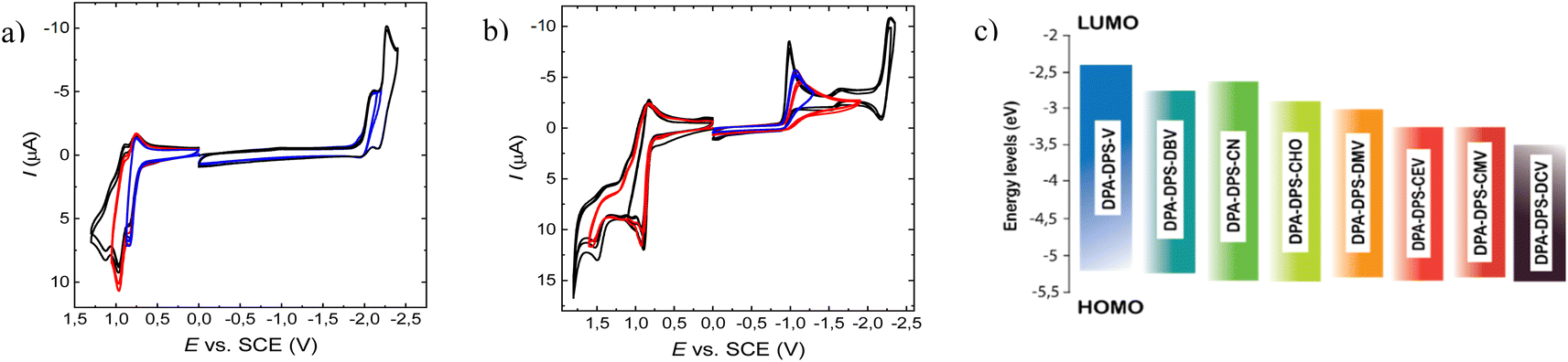
The strength of acceptor substituents in push–pull substituted derivatives was related to the LUMO energy obtained from cyclic voltammetry (Table 1). The representative CV curves of the compounds with the highest (DPA-DPS-V) and the lowest (DPA-DPS-DCV) LUMO are shown in Fig. 5 together with a visualised HOMO and LUMO dependence on the EWG. CV curves for further compounds can be found in the ESI.† HOMO energies in the set under study are only slightly affected by the EWG, within 0.07 eV, while the effect on the LUMO is dramatic. The double cyanation of DPA-DPS-V to DPA-DPS-DCV decreases its energy by more than 1.08 eV. The difference between the electrochemical gaps of these compounds is (consequently) also higher than 1 eV, i.e. the set based on the same donor (DPA) and building block (DPS) is suitable for wide-range tuning of the spectral properties.
Table 1 Results of cyclic voltammetry. E(ox1) (E(red1)) stands for the potentials of the first oxidation (reduction)a
| Material |
E(ox2) [V] |
E(ox1) [V] |
E(red1) [V] |
HOMO [eV] |
LUMO [eV] |
ΔHL [eV] |
| HOMO/LUMO energies were computed from oxidation/reduction potentials according to the formula −EHOMO/LUMO [eV] = E(ox1/red1) [V] + 4.429 (ref. 45). |
| DPA-DPS-V |
0.929 |
0.794 |
−2.032 |
−5.223 |
−2.397 |
2.83 |
| DPA-DPS-DBV |
0.947 |
0.807 |
−1.682 |
−5.236 |
−2.747 |
2.49 |
| DPA-DPS-CN |
0.944 |
0.855 |
−1.741 |
−5.284 |
−2.688 |
2.60 |
| DPA-DPS-CHO |
0.977 |
0.854 |
−1.564 |
−5.283 |
−2.866 |
2.42 |
| DPA-DPS-DMV |
0.982 |
0.813 |
−1.444 |
−5.242 |
−2.985 |
2.26 |
| DPA-DPS-CEV |
0.994 |
0.838 |
−1.133 |
−5.267 |
−3.296 |
1.97 |
| DPA-DPS-CMV |
0.984 |
0.819 |
−1.126 |
−5.248 |
−3.303 |
1.95 |
| DPA-DPS-DCV |
1.005 |
0.862 |
−0.949 |
−5.291 |
−3.480 |
1.81 |
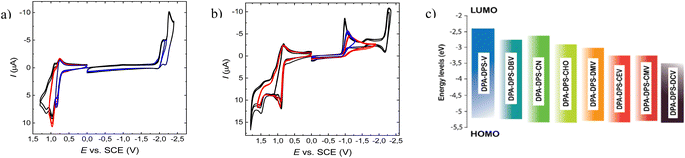 |
| | Fig. 5 Representative CV curves of the oxidation and reduction of DPA-DPS-V (a) and DPA-DPS-DCV (b) compounds at the glassy carbon electrode in acetonitrile containing 0.1 M Bu4NPF6.; v = 100 mV s−1. Visualisation of HOMO and LUMO levels (c) from Table 1. | |
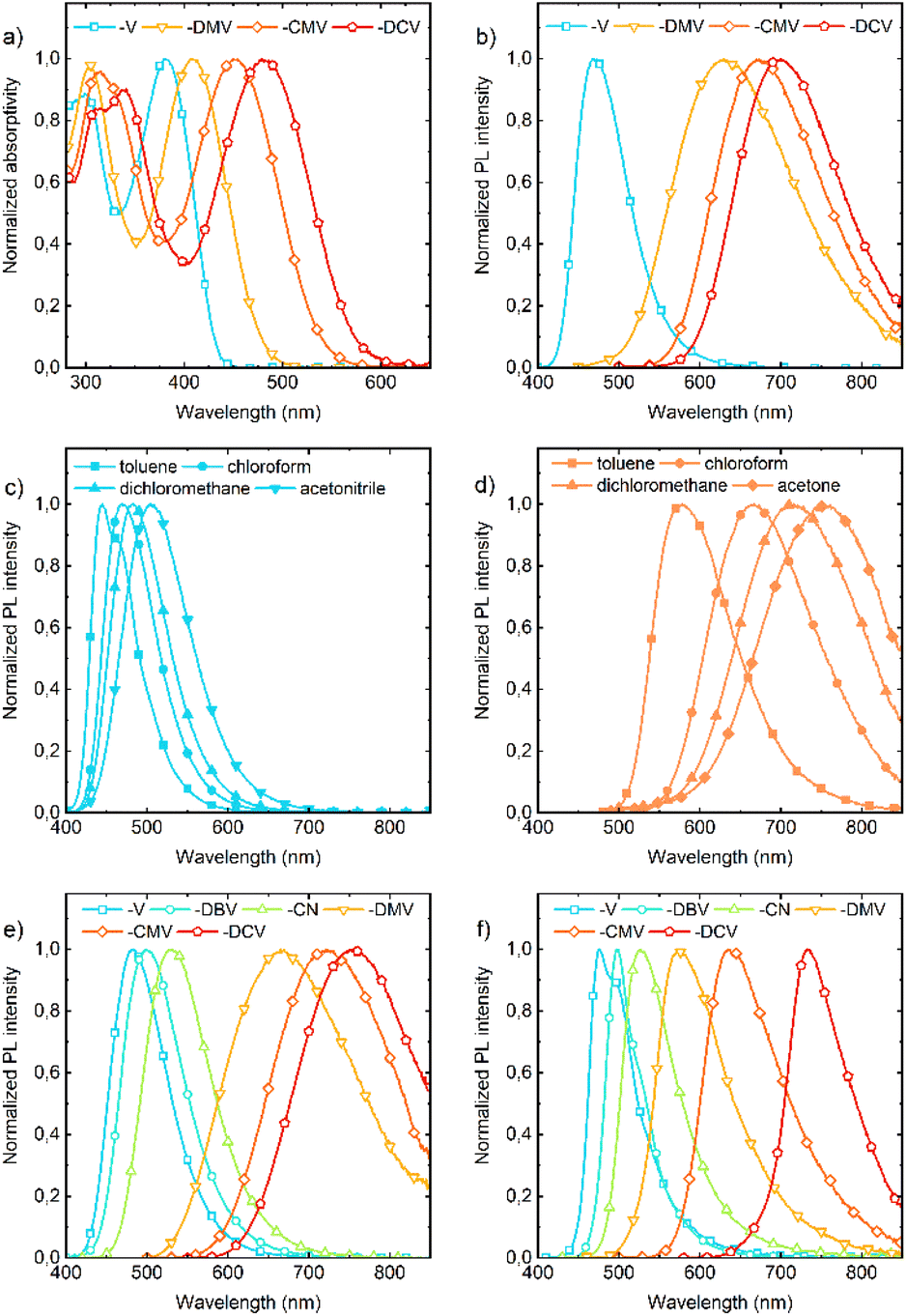
Absorption and fluorescence spectra in solution show dependencies that are clear and expected for push–pull compounds (Tables S3–S9†). They show a bathochromic shift (Fig. 6a) and a more pronounced bathofluoric (Fig. 6b) shift with an increasing electron-acceptor strength of the EWG. The effect of side-phenyls on the spectra of DPA-DPS-CN (with respect to DPA-stilbene-CN) is qualitatively similar to the reported shifts for an analogous substituted pair with EWG = CHO,34 i.e. DPA-DPS-CN shows a small red shift in absorption (5 nm) and a moderate shift in fluorescence (33 nm), both in acetonitrile.46 Positive fluorescence solvatochromism is more distinct for the compounds with a strong EWG; the effect is given both by a stronger solute–solvent interaction and the redistribution of a vibronic pattern from a 0–0 (typically DPA-DPS-V in less polar solvents, Fig. 6c) to a 0–1 (typically DPA-DPS-CMV in more polar solvents, Fig. 6d) maximum. This effect relates to the solvent-dependent structural relaxation in the S1 (HOMO–LUMO) state (Table 2). DFT calculated exocyclic bond lengths in S0 are independent of solvent within 0.001 Å and show a remarkable agreement with experimental values (computed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths in toluene are only 0.003 Å longer than those obtained from single-crystal XRD). Irrespective of the solvent, TD DFT calculated excitation to S1 is accompanied by the planarization of a main conjugated chain on account of a more pronounced side-phenyl torsion. However, the bond length alternation (BLA) of exocyclic bonds is considerably dependent on the solvent, e.g. the terminal CC bond (bond e in Table 2) of DPA-DPS-DCV is lengthened by 0.011 Å when changing the solvent from toluene to acetonitrile. This change in BLA causes a solvent-dependent shift of the S1 hypersurface with respect to S0 along the CC coordinate, responsible for the main vibronic progressions in conjugated compounds,47 and consequent modifications of Franck–Condon factors, responsible for vibronic band intensities. Such a redistribution of vibronic intensities affects, of course, the Stokes shift in solvents of moderate polarity (Table 3). Its highest value in all solvents was found for DPA-DPS-DMV, both experimentally and theoretically, due to the more energetic geometrical relaxation of the ester with respect to the cyano group. While the differences between the computed and measured values are relatively consistent for the three compounds with the strongest EWG, this difference found for DPA-DPS-V is about 2000 cm−1 higher in all solvents. This discrepancy arises from the vibronic structure, i.e. the Stokes shift for all three DPA-DPS-DMV, -CMV and -DCV compounds is estimated between 0–1 maxima both in absorption and fluorescence, while for DPA-DPS-V, the Stokes shift is the difference between the 0–1 absorption maximum and the 0–0 fluorescence maximum, consequently differing by a frequency of CC stretching from the other three compounds.
C bond lengths in toluene are only 0.003 Å longer than those obtained from single-crystal XRD). Irrespective of the solvent, TD DFT calculated excitation to S1 is accompanied by the planarization of a main conjugated chain on account of a more pronounced side-phenyl torsion. However, the bond length alternation (BLA) of exocyclic bonds is considerably dependent on the solvent, e.g. the terminal CC bond (bond e in Table 2) of DPA-DPS-DCV is lengthened by 0.011 Å when changing the solvent from toluene to acetonitrile. This change in BLA causes a solvent-dependent shift of the S1 hypersurface with respect to S0 along the CC coordinate, responsible for the main vibronic progressions in conjugated compounds,47 and consequent modifications of Franck–Condon factors, responsible for vibronic band intensities. Such a redistribution of vibronic intensities affects, of course, the Stokes shift in solvents of moderate polarity (Table 3). Its highest value in all solvents was found for DPA-DPS-DMV, both experimentally and theoretically, due to the more energetic geometrical relaxation of the ester with respect to the cyano group. While the differences between the computed and measured values are relatively consistent for the three compounds with the strongest EWG, this difference found for DPA-DPS-V is about 2000 cm−1 higher in all solvents. This discrepancy arises from the vibronic structure, i.e. the Stokes shift for all three DPA-DPS-DMV, -CMV and -DCV compounds is estimated between 0–1 maxima both in absorption and fluorescence, while for DPA-DPS-V, the Stokes shift is the difference between the 0–1 absorption maximum and the 0–0 fluorescence maximum, consequently differing by a frequency of CC stretching from the other three compounds.
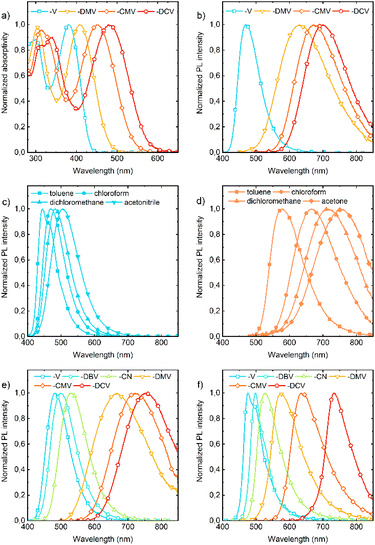 |
| | Fig. 6 Absorption (a) and fluorescence (b) spectra of DPA-DPS-V, -DMV, -CMV and -DCV in chloroform. Fluorescence of DPA-DPS-V (c) and -CEV (d) in toluene, chloroform, dichloromethane and acetonitrile (-V) or acetone (-CEV). Fluorescence of DPA-DPS-V, -DBV, -CN, -DMV, -CMV and -DCV in dichloromethane (e) and the solid-state (f). | |
Table 2 Experimental (XRD) and PCM (TD) DFT (ωB97X-D/6-31G(d,p)) optimised exocyclic CC/C–C bond lengths of a monomer in S0 and S1 states (SS = solid-state, all values in Å)
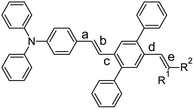
|
| R1 = R2 |
State |
Solvent |
Bond length [Å] |
| a |
b |
c |
d |
e |
| H |
S0 |
SS (exp.) |
— |
— |
— |
— |
— |
| CH3Ph |
1.469 |
1.340 |
1.472 |
1.477 |
1.334 |
| S1 |
CH3Ph |
1.404 |
1.407 |
1.407 |
1.459 |
1.342 |
| CHCl3 |
1.402 |
1.410 |
1.405 |
1.459 |
1.343 |
| CH2Cl2 |
1.400 |
1.412 |
1.403 |
1.458 |
1.343 |
| CH3CN |
1.399 |
1.414 |
1.402 |
1.458 |
1.344 |
| COOMe |
S0 |
SS (exp.) |
1.458 |
1.338 |
1.465 |
1.463 |
1.342 |
| CH3Ph |
1.467 |
1.341 |
1.470 |
1.466 |
1.344 |
| S1 |
CH3Ph |
1.409 |
1.401 |
1.405 |
1.429 |
1.369 |
| CHCl3 |
1.406 |
1.404 |
1.402 |
1.426 |
1.372 |
| CH2Cl2 |
1.404 |
1.406 |
1.400 |
1.423 |
1.374 |
| CH3CN |
1.402 |
1.408 |
1.397 |
1.421 |
1.376 |
| CN |
S0 |
SS (exp.) |
1.462 |
1.338 |
1.465 |
1.454 |
1.351 |
| CH3Ph |
1.467 |
1.341 |
1.469 |
1.457 |
1.357 |
| S1 |
CH3Ph |
1.412 |
1.396 |
1.408 |
1.417 |
1.393 |
| CHCl3 |
1.408 |
1.400 |
1.404 |
1.412 |
1.398 |
| CH2Cl2 |
1.405 |
1.403 |
1.401 |
1.409 |
1.401 |
| CH3CN |
1.402 |
1.406 |
1.398 |
1.405 |
1.404 |
Table 3 Comparison of the experimental and theoretical Stokes shift dependence on EWG and solventa
| Compound |
Experiment/theory |
Toluene |
Chloroform |
Dichloromethane |
| A |
F |
S |
A |
F |
S |
A |
F |
S |
| Theoretical excitation energies were obtained by PCM TD DFT (CAM-B3LYP/6-311G(d,p)) calculations on the S0 and S1 geometries obtained on the same PCM (TD) DFT level [A = absorption, F = fluorescence and S = Stokes shift (all values in cm−1)]. |
| DPA-DPS-V |
Experiment |
25840 |
22520 |
3320 |
26040 |
21320 |
4720 |
26040 |
20700 |
5340 |
| Theory |
28570 |
22120 |
6450 |
28570 |
21320 |
7250 |
28570 |
20830 |
7740 |
| DPA-DPS-DMV |
Experiment |
24630 |
18800 |
5830 |
24450 |
15950 |
8500 |
24630 |
15080 |
9550 |
| Theory |
27470 |
20530 |
6940 |
27400 |
19460 |
7940 |
27320 |
18800 |
8520 |
| DPA-DPS-CMV |
Experiment |
22370 |
17240 |
5130 |
22220 |
14860 |
7360 |
22370 |
13720 |
8650 |
| Theory |
25710 |
19420 |
6290 |
25580 |
18290 |
7290 |
25510 |
17540 |
7970 |
| DPA-DPS-DCV |
Experiment |
21280 |
16530 |
4750 |
20920 |
14240 |
6680 |
21100 |
13300 |
7800 |
| Theory |
24690 |
18970 |
5720 |
24570 |
17760 |
6810 |
24570 |
17040 |
7530 |
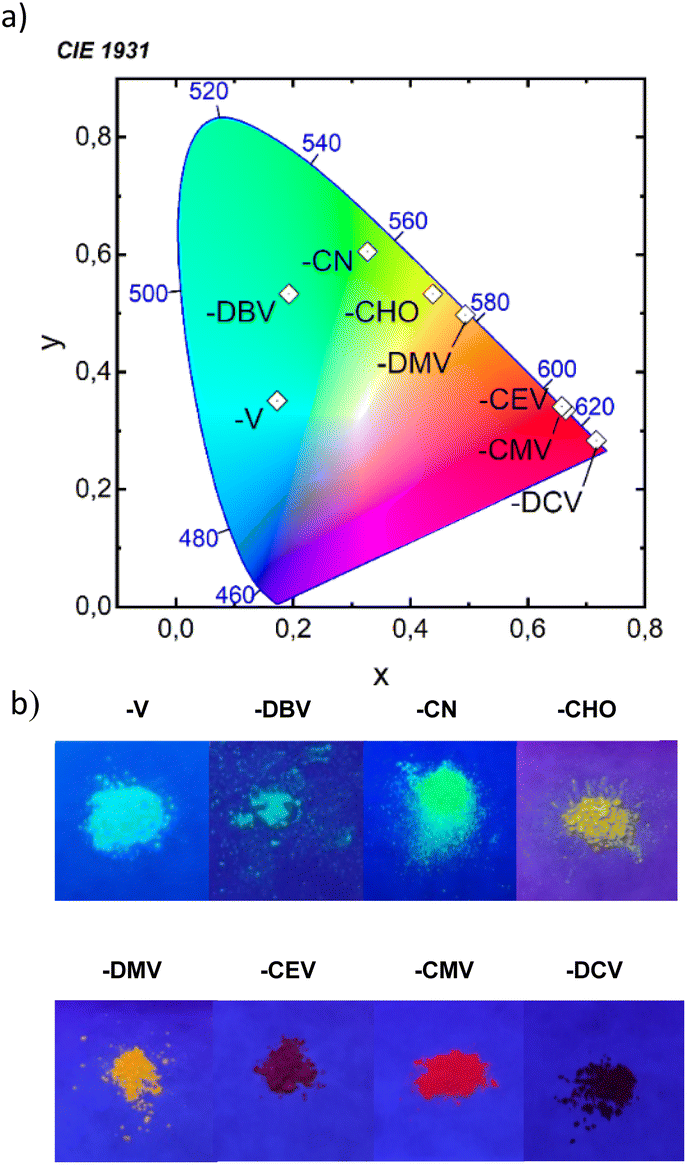
All eight compounds show strong fluorescence with monoexponential decay in solution, except the low PLQY of most polar compounds in acetonitrile, where some quenching by excited-state twist may play a role. Moreover, only six of the eight compounds have a PLQY of SSF higher than 10% (Table 4), usually with multiexponential decay. Their SSF maxima relate quite well to those in DCM (Fig. 6e) for the compounds with weaker EWGs, while for compounds with stronger EWGs, the SSF spectra show a considerable blue shift with respect to fluorescence in DCM (around 90 nm for DPA-DPS-DMV, -CMV and -CEV), because of a limited excited-state relaxation in a rigid environment. The only exception is DPA-DPS-DCV with an excimer character of the solid-state emission,34 for which only a small blue (21 nm) shift compared to DCM is observed. Otherwise, while DPA-DPS-DMV, -CEV and -CMV show a blue shift of the fluorescence maxima (36–53 nm, Tables S7–S9†) when going from chloroform to the solid state, DPA-DPS-DCV shows a red shift of 31 nm.34 SSF maxima cover a range from 475 nm (DPA-DPS-V) to 733 nm (IR polymorph of DPA-DPS-DCV),34 thus covering a large area of fluorescence colours in the CIE diagram (Fig. 7a). Such a range of SSF maxima, tuned only by an acceptor substituent—i.e. keeping the same donor and building block—ranks among the biggest ever reported36,37 if expressed in wavelength scale (258 nm). Furthermore, in energy units (0.975 eV), this interval is lower than was found for simple acceptor-substituted triphenylamines (248 nm, but 1.273 eV).38 The reason is that the conjugation extension of a bridge between the donor and acceptor from benzene to stilbene dramatically shifts the emission range from violet–red to blue–infrared (Fig. 7b), i.e. by about 90 nm.
Table 4 Absorption and fluorescence maxima and PLQY values in DCM and the solid state
| Material |
DCM |
Solid |
| λA [nm] |
ε [l mol−1 cm−1] |
λF [nm] |
Stokes [cm−1] |
PLQY [%] |
τF [ns] |
λF [nm] |
PLQY [%] |
| Polymorph G. polymorph IR. |
| DPA-DPS-V |
378 |
29400 |
483 |
5751 |
83 |
1.96 |
475 |
12 |
| DPA-DPS-DBV |
386 |
40000 |
502 |
5986 |
48 |
1.64 |
498 |
11 |
| DPA-DPS-CN |
393 |
18000 |
530 |
6577 |
62 |
1.98 |
529 |
18 |
| DPA-DPS-CHOa |
408 |
24800 |
587 |
7474 |
86 |
2.96 |
549 |
5 |
| DPA-DPS-DMV |
406 |
29900 |
663 |
9548 |
46 |
1.45 |
574 |
20 |
| DPA-DPS-CEV |
442 |
27900 |
716 |
8658 |
58 |
2.03 |
627 |
9 |
| DPA-DPS-CMV |
445 |
28200 |
729 |
8754 |
44 |
1.85 |
637 |
35 |
| DPA-DPS-DCVb |
473 |
34000 |
752 |
7844 |
35 |
1.47 |
733 |
32 |
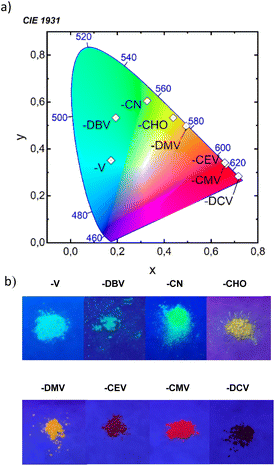 |
| | Fig. 7 Position of SSF colours in the CIE diagram (a) and photographs of powders under UV (365 nm) irradiation (b). We note that the IR SSF of DPA-DPS-DCV was outside of the detection limits of the camera. | |
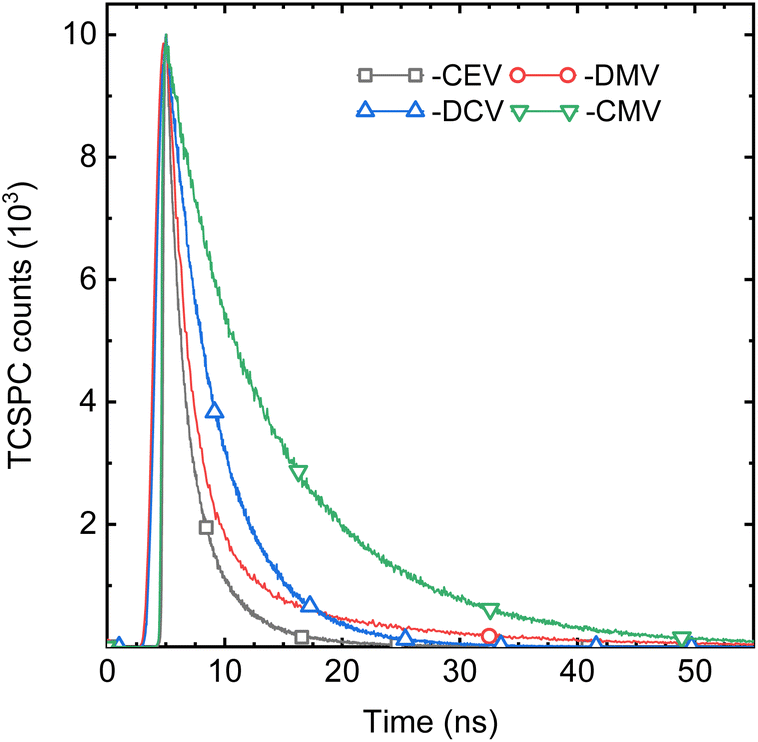
While the spectral properties are mainly a function of the molecular structure, here of the EWG substituent, the photophysics is much more complicated and is driven mainly by intermolecular arrangement. An illustrative example is an almost isomorphic, π-isoelectronic DPA-DPS-CMV and -CEV pair. The SSF spectrum of the former is slightly (10 nm) red-shifted with respect to the latter, as is the case in chloroform (9 nm, Tables S8 and S9†), but its SSF PLQY is almost four times higher (Table 4) and the weighted average SSF lifetime τ is more than five times longer (Fig. 8 and Table 5). The deconvolution of SSF multiexponential decay curves (Table 5) enables the division of the set of eight compounds into three groups. The only member of the first of these groups is DPA-DPS-DBV, which shows an anomalous ultrashort monoexponential decay (0.42 ns), much shorter than in any solvent, and with a relatively low SSF PLQY (11%). We consider that intersystem crossing, enhanced by the heavy atom effect of terminal bromines, may play a significant role,21 but we have no experimental or theoretical evidence, so this compound was not considered. The second group contains four compounds (DPA-DPS-V, -CHO, -CN and -CEV) and is characterised by a PLQY lower than 20% (Table 4) and a significant percentage (B1 in Table 5) of the short-lived (0.80–0.91 ns) component. The overall PLQY is the lowest (5%) for the compound (aldehyde) with the highest proportion of fast decay constant, B1 = 83%. By contrast, the highest PLQY (18%) was found for the nitrile with the lowest proportion of B1 (46%). Thus, we consider the presence of the process, represented by this fast component, as the dominating quenching pathway. We ascribe it to energy migration, which is the most frequent reason for SSF quenching in poly- and nanocrystalline solids20 and which manifests itself by a shortening of the lifetime with respect to monomer fluorescence, proportional to the concentration of quenching sites.48 The dominant (B2 = 51%) portion of DPA-DPS-CN emission, thus, originates from monomer-like fluorescence with τ2 = 2.24 ns, which is only slightly longer than in DCM (1.98 ns, Table 4). Three compounds of the third group show a PLQY of 20% (DPA-DPS-DMV) and higher (DPA-DPS-CMV and -DCV), and the fast component is completely absent in their multiexponential decay (Table 5). We consider this PLQY enhancement to be a result of exciton self-trapping,25 avoiding detrimental migration, and longer lifetimes (as compared to a monomer in solution) as a signature of aggregate emission.
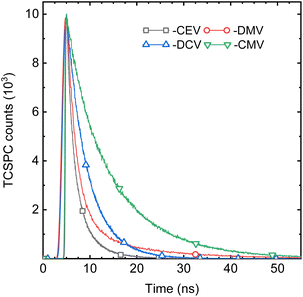 |
| | Fig. 8 Fluorescence decay curves of DPA-DPS-CEV, -DMV, -DCV and -CMV in the solid state. | |
Table 5 Deconvolution of a multiexponential fluorescence decay in the solid statea
| Material |
τ [ns] |
τ1 [ns] |
τ2 [ns] |
τ3 [ns] |
τ4 [ns] |
B1 [%] |
B2 [%] |
B3 [%] |
B4 [%] |
χ2 |
| Lifetimes with the highest expansion coefficients are set in bold for each compound. The weighted average fluorescence lifetime was computed according to τ = ΣBiτi. |
| DPA-DPS-V |
2.20 |
0.80 |
2.93 |
5.77 |
— |
61 |
19 |
20 |
— |
1.0701 |
| DPA-DPS-DBV |
0.42 |
0.42 |
— |
— |
— |
100 |
— |
— |
— |
1.0126 |
| DPA-DPS-CN |
2.42 |
0.80 |
2.24 |
9.62 |
— |
40 |
51 |
10 |
— |
1.0896 |
| DPA-DPS-CHO |
1.84 |
0.84 |
— |
6.75 |
— |
83 |
— |
17 |
— |
1.1551 |
| DPA-DPS-DMV |
3.12 |
— |
2.25 |
— |
14.73 |
— |
93 |
— |
7 |
0.9863 |
| DPA-DPS-CEV |
1.88 |
0.91 |
2.25 |
5.05 |
— |
53 |
35 |
12 |
— |
1.0050 |
| DPA-DPS-CMV |
10.03 |
— |
— |
5.29 |
11.53 |
— |
— |
24 |
76 |
1.1670 |
| DPA-DPS-DCV |
4.51 |
— |
2.76 |
4.82 |
— |
— |
15 |
85 |
— |
1.0536 |
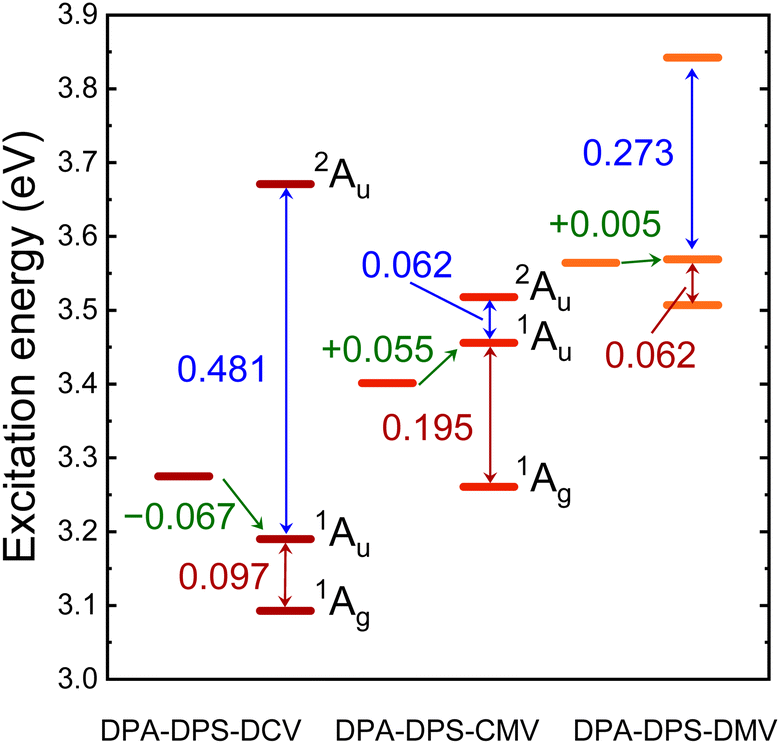
TD DFT calculations on monomers and stacked dimers of the third-group compounds, with the geometries extracted from single-crystal XRD, were carried out in order to understand the character of the fluorescent traps. Directly calculated excitation energies (Table 6) were recomputed to the energy difference values (Fig. 9). All three dimers exhibit a dark lowest excited state S1, so they can be classified as H-aggregates, even for DPA-DPS-DMV with a parallel arrangement of the molecules in a stack.23,24 DPA-DPS-DCV is the only compound which can be considered a red-shifted H-aggregate24 according to the monomer-to-dimer energy difference. If one considers an energy difference between bright (S2) and dark (S1) FR states as the depth of a trap, these values (0.062 eV, 0.097 eV and 0.195 eV, Fig. 9) for DPA-DPS-DMV, -DCV and -CMV, respectively, qualitatively correlate to the fluorescence lifetimes with a dominating participation in SSF decay deconvolution (2.25 ns, 4.82 ns and 11.53 ns, Table 5), respectively. The energy difference between allowed FR (S2) and CT (S3) states, originating from TD DFT calculations, relates to the FR/CT interaction, i.e. the larger this difference is, the more CT character can be expected for the bright FR state.49
Table 6 TD DFT (ωB97X-D/6-311G(d,p)) excitation energies of stacked dimers in three lowest statesa
| Compound |
Aggregate |
S1 |
S2 |
S3 |
| Eexc |
fosc |
Eexc |
fosc |
Eexc |
fosc |
| The excitation energies of bright monomer and dimer FR states are set in bold. |
| DPA-DPS-DCV |
Monomer |
3.275 |
1.521 |
|
|
|
|
| Dimer |
3.093 |
0.000 |
3.190 |
2.868 |
3.671 |
0.003 |
| DPA-DPS-CMV |
Monomer |
3.401 |
1.641 |
|
|
|
|
| Dimer |
3.261 |
0.000 |
3.456 |
2.615 |
3.518 |
0.286 |
| DPA-DPS-DMV |
Monomer |
3.564 |
1.701 |
|
|
|
|
| Dimer |
3.507 |
0.000 |
3.569 |
3.179 |
3.842 |
0.010 |
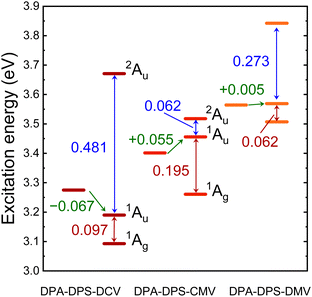 |
| | Fig. 9 Excitation energy levels of monomer S1 state (left) and dimer S1, S2 and S3 states (right) of DPA-DPS-DCV, -CMV and -DMV, as computed by TD DFT (ωB97X-D/6-311G(d,p)) on experimental XRD geometries. Monomer-to-dimer excitation energy shift between the lowest allowed transition is expressed in green, twice the excitonic coupling between the lower and upper Frenkel (FR) states of the dimers (S1 and S2) in expressed in red and the splitting between allowed FR (S2) and charge-transfer (CT) states (S3) is expressed in blue. | |
Consequently, the pronounced CT character of an emitting state can be expected for DPA-DPS-DCV (ΔE = 0.481 eV), relating well to the red spectral shift and excimer character of SSF, while for DPA-DPS-CMV (ΔE = 0.062 eV), the emitting state is of almost pure FR character with a slight blue shift with respect to monomer, as is usual for H-aggregates. In summary, the most intense fluorescent DPA-DPS-CMV derivative is, thus, quite unique, as its SSF spectrum appears to be monomer-like, but fluorescence is dramatically delayed as it originates from a deep-trap neutral H-aggregate. Thanks to the delocalisation of KS FMOs (Fig. 4 and ref. 34), the traps in DPA-DPS-CMV and -DCV crystals are localised on stacked dimers, while in DPA-DPS-DMV, the monomers can be considered as self-trapping sites, as both transitions to the S1 and S2 states are formed by symmetrical or antisymmetrical combinations of intramolecular (Fig. 4) HOMO to LUMO and HOMO−1 to LUMO+1 monoexcitations in a configurational interaction. Promoting a J-type coupling is one of the ways to enhance the PLQY of polycrystalline solids through a reduction of exciton lifetime.20 An inclined (staircase) packing of push–pull conjugated organics may lead to abnormal photophysics, prolonging an exciton lifetime, as is the case in H-aggregates.50 This resembles the photophysical behaviour observed in DPA-DPS-DMV (Fig. 2 and Table 5). We consider its moderate PLQY to be the result of an on-monomer self-trapping and small electronic coupling in terms of low values of the transfer integrals, both of which reduce the migration of an exciton, and we ascribe the slight increase of exciton lifetime to a shallow trap in a dimer approximation (Fig. 9).
Conclusions
All eight DPA-DPS-EWG derivatives with the same donor and building block show fluorescence both in solution and in the solid state. The SSF maximum (colour) in powder form can be tuned by the EWG strength over a broad (from blue to infrared) range of 258 nm. Excluding intramolecular internal conversion as a decisive nonradiative deactivation channel in the solid state within the set, no significant relation between the band gap energy of the compounds and the SSF intensity was found. Exciton localisation either on a monomer (in J-like packing) or a stacked dimer (for H-aggregates) was found to be effective for improving the overall SSF PLQY through an avoidance of exciton migration to the quenching sites by a self-trapping process. The highest SSF PLQY (35%) was found for the red emission of DPA-DPS-CMV with the deepest trap, localised on an electronically isolated stacked dimer, causing delayed fluorescence with a lifetime more than five times longer compared to the monomer fluorescence in solution. The anomalous (with respect to other push–pull derivatives in a set) spectral shift of the SSF maximum (with respect to fluorescence in solvents) and the partial CT character of its bright state in a stacked dimer arrangement brought further evidence of an excimer character of the previously reported IR emission of DPA-DPS-DCV. The role of side-phenyls in the formation of beneficial structural traps in crystals is crucial, but their effect on packing is not universal, as detrimental exciton migration takes place in at least half of the derivatives under study. A relatively marginal (with respect to the molecular fluorescence in solution) change in molecular structure, such as an exchange of a methyl ester group with an ethyl ester group as an EWG substituent, causes changes in packing that are hardly predictable, switching the photophysics in the solid state from an absence to a dominance of exciton migration. We have shown here that the formation of crystal packing enabling exciton self-trapping is an efficient way to improve the SSF intensity of polycrystalline solids, but a significant amount of work on the design, syntheses and crystal engineering remains to be undertaken in order to make this method more universal.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors express their gratitude for financial support from the Czech Science Foundation, grant No. 19-22783S. Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA CZ LM2018140) supported by the Ministry of Education, Youth and Sports of the Czech Republic.
Notes and references
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- M. Martínez-Abadía, R. Giménez and M. B. Ros, Adv. Mater., 2018, 30, 1704161 CrossRef PubMed.
- H. H. Fang, J. Yang, J. Feng, T. Yamao, S. Hotta and H. B. Sun, Laser Photonics Rev., 2014, 8, 687–715 CrossRef.
- Y. Jiang, Y. Y. Liu, X. Liu, H. Lin, K. Gao, W. Y. Lai and W. Huang, Chem. Soc. Rev., 2020, 49, 5885–5944 RSC.
- J. Zaumseil, Adv. Funct. Mater., 2020, 30, 1905269 CrossRef CAS.
- V. M. Angela, A. Anjali, D. Harshini and S. Nagarajan, ACS Appl. Electron. Mater., 2021, 3, 550–573 CrossRef.
- P. Gayathri, M. Pannipara, A. G. Al-Sehemi and S. P. Anthony, New J. Chem., 2020, 44, 8680–8696 RSC.
- J. Zhang, B. He, Y. Hu, P. Alam, H. Zhang, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2021, 33, 2008071 CrossRef CAS PubMed.
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- Ch. Adachi and A. S. D. Sandanayaka, CCS Chem., 2020, 2, 1203–1216 CrossRef CAS.
- H. Nakanotani, Y. Tsuchiya and C. Adachi, Chem. Lett., 2021, 50, 938–948 CrossRef CAS.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- Z. Zheng, T. Zhang, H. Liu, Y. Chen, R. T. K. Kwok, C. Ma, P. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, K. S. Wong and B. Z. Tang, ACS Nano, 2018, 12, 8145–8159 CrossRef CAS PubMed.
- J. Qi, X. Duan, Y. Cai, S. Jia, C. Chen, Z. Zhao, Y. Li, H. Q. Peng, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, Chem. Sci., 2020, 11, 8438–8477 RSC.
- W. L. Algar, M. Massey, K. Rees, R. Higgins, K. D. Krause, G. H. Darwish, W. J. Peveler, Z. Xiao, H. Y. Tsai, R. Gupta, K. Lix, M. V. Tran and H. Kim, Chem. Rev., 2021, 121, 9243–9358 CrossRef CAS PubMed.
- J. Wang, L. Zhang and Z. Li, Adv. Healthcare Mater., 2021, 10, 2101169 CrossRef CAS PubMed.
- Y. Wang, B. Xia, Q. Huang, T. Luo, Y. Zhang, P. Timashev, W. Guo, F. Li and X. J. Liang, Adv. Healthcare Mater., 2021, 10, 2100945 CrossRef CAS PubMed.
- K. Li, T. B. Ren, S. Huan, L. Yuan and X. B. Zhang, J. Am. Chem. Soc., 2021, 143, 21143–21160 CrossRef CAS PubMed.
- J. Gierschner, J. Shi, B. Milián-Medina, D. Roca-Sanjuán, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2021, 9, 2002251 CrossRef CAS.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- C. Zheng, C. Zhong, C. J. Collison and F. C. Spano, J. Phys. Chem. C, 2019, 123, 3203–3215 CrossRef CAS.
- O. P. Dimitriev, Chem. Rev., 2022, 122(9), 8487–8593 CrossRef CAS PubMed.
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- L. M. Ibele, P. A. Sánchez-Murcia, J. J. Nogueira and L. González, J. Phys. Chem. Lett., 2020, 11, 7483–7488 CrossRef CAS PubMed.
- Y. Shen, Z. Zhang, H. Liu, Y. Yan, S. Zhang, B. Yang and Y. Ma, J. Phys. Chem. C, 2019, 123, 13047–13056 CrossRef CAS.
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC.
- Y. Shen, H. Liu, S. Zhang, Y. Gao, B. Li, Y. Yan, Y. Hu, L. Zhao and B. Yang, J. Mater. Chem. C, 2017, 5, 10061–10067 RSC.
- Q. Luo, L. Li, H. Ma, C. Lv, X. Jiang, X. Gu, Z. An, B. Zou, C. Zhang and Y. Zhang, Chem. Sci., 2020, 11, 6020–6025 RSC.
- Z. Sun, Q. Zang, Q. Luo, C. Lv, F. Cao, Q. Song, R. Zhao, Y. Zhang and W. Y. Wong, Chem. Commun., 2019, 55, 4735–4738 RSC.
- C. Lv, W. Liu, Q. Luo, H. Yi, H. Yu, Z. Yang, B. Zou and Y. Zhang, Chem. Sci., 2020, 11, 4007–4015 RSC.
- K. Pauk, S. Luňák Jr, A. Růžička, A. Marková, A. Mausová, M. Kratochvíl, K. Melánová, M. Weiter, A. Imramovský and M. Vala, Chem.–Eur. J., 2021, 27, 4341–4348 CrossRef CAS PubMed.
- M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed.
- M. K. Bera, P. Pal and S. Malik, J. Mater. Chem. C, 2020, 8, 788–802 RSC.
- M. Shimizu, Chem. Rec., 2021, 21, 1489–1505 CAS.
- E. Ishow, A. Brosseau, G. Clavier, K. Nakatani, P. Tauc, C. Fiorini-Debuisschert, S. Neveu, O. Sandre and A. Léaustic, Chem. Mater., 2008, 20, 6597–6599 CrossRef CAS.
- S. Kundu, B. Sk, P. Pallavi, A. Giri and A. Patra, Chem.–Eur. J., 2020, 26, 5557–5582 CrossRef CAS PubMed.
- M. J. Frisch, W. G. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. G. Mennucci, A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox and C. T. Wallingford, Gaussian 09, Revision D.01, Gaussian, Inc., 2009 Search PubMed.
- Y. D. Lin, Y. S. Pen, W. Su, K. L. Liau, Y. S. Wen, C. H. Tu, C. H. Sun and T. J. Chow, Chem.–Asian J., 2012, 7, 2864–2871 CrossRef CAS PubMed.
- C. Coluccini, A. K. Sharma, M. Caricato, A. Sironi, E. Cariati, S. Righetto, E. Tordin, C. Botta, A. Forni and D. Pasini, Phys. Chem. Chem. Phys., 2013, 15, 1666–1674 RSC.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- N. J. Hestand and F. C. Spano, Acc. Chem. Res., 2017, 50, 341–350 CrossRef CAS PubMed.
- A. A. Isse and A. Gennaro, J. Phys. Chem. B, 2010, 114, 7894–7899 CrossRef CAS PubMed.
- B. Kumari, A. Singh, P. Jana, M. Radhakrishna and S. Kanvah, New J. Chem., 2019, 43, 11701–11709 RSC.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed.
- O. V. Mikhnenko, P. W. M. Blom and T. Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867–1888 RSC.
- S. Canola, G. Bagnara, Y. Dai, G. Ricci, A. Calzolari and F. Negri, J. Chem. Phys., 2021, 154, 124101 CrossRef CAS PubMed.
- J. Xue, Q. Liang, R. Wang, J. Hou, W. Li, Q. Peng, Z. Shuai and J. Qiao, Adv. Mater., 2019, 31, 1808242 CrossRef PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Aneta Markováb,
Kateřina Teichmanováa,
Anna Mausováa,
Matouš Kratochvílb,
Rastislav Smolkab,
Tomáš Mikysekd,
Martin Weiterb,
Aleš Imramovský*a and
Martin Vala
c,
Aneta Markováb,
Kateřina Teichmanováa,
Anna Mausováa,
Matouš Kratochvílb,
Rastislav Smolkab,
Tomáš Mikysekd,
Martin Weiterb,
Aleš Imramovský*a and
Martin Vala