
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic and thermodynamic impact of L94A, W100A, and W100L mutations on the D2 dopamine receptor bound to risperidone†
Faizul Azam a and
Martiniano Bello*b
a and
Martiniano Bello*b
aDepartment of Pharmaceutical Chemistry and Pharmacognosy, Unaizah College of Pharmacy, Qassim University, Unaizah, 51911, Saudi Arabia
bLaboratorio de Diseño y Desarrollo de Nuevos Fármacos e Innovación Biotecnológica, Escuela Superior de Medicina, Instituto Politécnico Nacional, Plan de San Luis y Diaz Mirón, S/N, Col. Casco de Santo Tomas, Ciudad de México 11340, Mexico. E-mail: bellomartini@gmail.com; mbellor@ipn.mx
First published on 30th November 2022
Abstract
DRD2 is an important receptor in the mediation of antipsychotic drugs but also in Parkinson medication, hyperprolactinemia, nausea and vomiting. Recently, crystallographic studies of the DRD2–risperidone complex have provided important information about risperidone recognition in wild-type and different stabilizing DRD2–risperidone residues. Using the crystallographic structure of the DRD2–risperidone complex as a starting point, we undertook molecular dynamics (MD) simulations to investigate the structural and thermodynamic basis of molecular recognition by risperidone at the ligand-binding sites of wild-type and mutant DRD2. A solvated phospholipid bilayer was used to construct DRD2–risperidone complexes, which were then subjected to several microsecond (μs) MD simulations in order to obtain realistic receptor–ligand conformations under the equilibrated simulation time. Risperidone had a higher affinity for wild-type and L94A mutant DRD2 than the W100L and W100A mutants, according to binding free energy calculations using the Molecular Mechanics Generalized-Born Surface Area (MMGBSA) method, explaining the experimental differences in ligand residence times. Principal component (PC) analysis revealed important conformational mobility upon molecular recognition of risperidone for the L94A mutant compared to the wild type, indicating an unfavorable entropic component that may contribute to improving risperidone affinity in the L94A DRD2 mutant.
1. Introduction
Numerous brain regions, including the basolateral amygdala and the ventral subiculum of the hippocampus, regulate the complex dopaminergic activity. Dopamine is a neurotransmitter1 produced in the central and peripheral nervous systems and exerts its effects via binding to G protein-coupled receptors. Dopamine receptors (DRs) play important roles in different neuronal processes, such as reward and addiction.2–4 Dysfunction of the dopamine system leads to several pathological conditions, such as Parkinson's disease, schizophrenia, and attention deficit and hyperactivity disorder.1 DRs exert their biological functions by linking to and activating different G protein complexes. These DRs may be divided into two categories, D1-like, and D2-like receptors. Both classes have different physiological effects and signal transduction mechanisms.5 D1 and D5 belong to the class of D1-like receptors, which are primarily coupled to stimulatory Gs-proteins and increase adenylyl cyclase activity. In contrast, D2, D3, and D4 belong to the class of D2-like receptors, primarily coupled to inhibitory Gi-proteins, decreasing adenylate cyclase activity.5Although the D2 dopamine receptor (DRD2) was cloned almost three decades ago6,7 and has been subject to vast experimental8,9 and theoretical10 studies, the lack of experimental evidence of DRD2 in complex with ligands has limited our knowledge of its molecular recognition. Recently, Shen et al. cocrystallized DRD2 in complex with risperidone (a typical antipsychotic).11 DRD2 is an important protein in the mediation of antipsychotic drugs but also Parkinson medication, hyperprolactinemia, nausea and vomiting. DRD2 has also been related to drug abuse, including amphetamines, cocaine and opioids. Different from its analogs from the same dopamine receptor group, DRD2 displays substantial structural differences in extracellular loop 1 (EL1) and 2 (EL2) but also in extracellular ends from the transmembrane (TM) helices (TMVI and TMVII).11
The cocrystallized DRD2–risperidone revealed an inactive conformation. Comparison between DRD2–risperidone with the active and inactive β2 adrenergic receptor or adenosine A2A receptor structures did not reveal important structural movement of the intracellular TMVI.11 However, the ‘ionic lock’ between the conserved Arg3.50 and Glu6.30 (ref. 12–14) is preserved in the DRD2–risperidone complex. Mutational studies around residues stabilizing the DRD2–risperidone complex showed that W100A and W100L mutations decreased risperidone residence time, whereas L94A mutation increased it at the binding pocket with respect to the wild-type DRD2. The objective of the current study was to investigate the structural and energetic basis of the implication of W100A, W100L, and L94A mutations on the DR2D–risperidone complex. For this, we conducted microsecond molecular dynamics (MD) simulations combined with the MMGBSA approach starting from crystallographic information of the DRD2–risperidone complex. As DRD2 is a membrane protein, our MD simulations were run in an all-atom palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer membrane, mimicking the plasma membrane lipid environment, allowing us to obtain protein–ligand conformations in close proximity to one biological environment.
2. Methods
2.1 Structural modeling
The structure of DRD2 was taken from the protein data bank (PDB entry 6CM4, chain A). The missing regions of the DRD2 structure were constructed using Modeler 9.17 (ref. 15) and the human DRD2 sequence (UniProt, P14416). The best human DRD2 model was obtained based on the DOPE score of Modeler 9.17. The quality of the model was evaluated with MolProbity.16 The protonation state of charged residues at neutral pH was determined with the PROPKA server.17 The protonation state of risperidone was established at neutral with Avogadro software.182.2 Anchoring of the receptor–ligand complex into the membrane
The Orientations of Proteins in Membranes (OPM) server was used to determine the orientation of the receptor–ligand complex with respect to the membrane.19 A rectangular preequilibrated POPC membrane was generated for each system using the membrane-builder tool of CHARM20,21 with dimensions of 106.9 × 107.2 × 134.0 Å (xyz). The receptor–ligand complex was placed into the POPC membrane using the replacement method. The POPC membrane was composed of 290 POPC phospholipids. The protein–receptor-membrane system was solvated with 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 068 TIP3P water molecules22 and neutralized with 0.15 M NaCl using the ion placing method.
068 TIP3P water molecules22 and neutralized with 0.15 M NaCl using the ion placing method.
2.3 MD simulations
Using the Amber 16 program, MD simulations of the protein–receptor-membrane systems were performed.23 The Generalized Amber Force Field (GAFF), which uses the AM1-BCC technique and GAFF to assign atomic charges, was used to derive the ligand parameters. The Leap module was used to build the topology for the systems using ff14SB24 for protein, Lipid14 (ref. 25) for POPC lipids, and GAFF26 for small molecule. Positional constraints on the atoms of the protein, receptor, and membrane allowed the systems to be energy minimized while allowing the solvent to relax. The systems were gradually heated from 0 to 310 K for 1 ns while with protein–receptor-membrane system's heavy atoms being restrained to produce NVT ensembles. The systems were equilibrated for 5 ns with the completely unconstrained system after 1 ns under the NPT ensemble at 310 K and 1 bar pressure with confined heavy atoms. The temperature was maintained using Langevin dynamics,27,28 and pressure was controlled using an anisotropic Berendsen weak coupling barostat.29 Using an NPT ensemble at 310 K and 1 bar pressure, three independent MD simulations were run for 1 microseconds (μs) under periodic boundary conditions (PBCs). With a 10 Å cutoff for van der Waals interactions, long-range electrostatic interactions were handled using the particle mesh Ewald.30 Bond lengths at equilibrium were constrained using the SHAKE method.312.4 MD trajectory analysis
With the help of AmberTools16, the time-dependent Cα root mean squared deviation (RMSD), radius of gyration (Rg), and clustering analysis were calculated. A single joined trajectory created by concatenating the equilibrated portions of each simulation was used to assess the PC analysis, clustering analysis, and binding free energy analysis. PyMOL was used to generate the figures.322.5 Binding and per-residue free energy
Using the MMGBSA technique, the binding free energies of receptor–ligand interactions were determined.33 During the equilibrated simulation time, the binding free energy was calculated while preserving a total of 2000 receptor–ligand conformations. Using implicit solvent models34 and an ionic strength of 0.15 M, the solvation free energy was calculated. The MMGBSA technique was used to estimate the binding free energy (ΔGbind) and per-residue free values, as reported previously.352.6 Energy minimization of wild-type and mutant DRD2–risperidone complexes
Most populated wild-type and mutant DRD2–risperidone complexes obtained through clustering analysis were minimized using the YASARA-minimization server.36 This server performs minimization using the YASARA force field, which can optimize the harm of the mutant proteins and precisely determine the energy that is reliable.3. Results
3.1 Stability of the simulated complexes
Different geometrical parameters, such as the area per lipid, root mean squared deviation (RMSD), and radius of gyration (Rg), were evaluated before structural and energetic analyses. The area per lipid analysis showed that wild-type and mutant DRD2–risperidone systems exhibited higher area per lipid values at the commencement of the simulations, decreasing to a converged behavior in the time scale of 0.1 to 0.2 μs, but reaching constant values after 0.8 μs with area per lipid values of approximately 64.0 to 67.0 Å2 for free and bound systems (Fig. S1A†), in agreement with other protein–POPC-membrane systems.37 Fig. S1B† shows that wild-type and mutant DRD2–risperidone systems reached a first equilibrium between 0.1 and 0.2 μs, with a final equilibrium observed after 0.8 μs, with RMSD values between 3.5 and 8.0 Å. Fig. S1C† shows that similar to that observed for RMSD, all the systems achieved a first equilibrium between 0.1 and 0.2 μs, with a final convergence observed after 0.8 μs with Rg values between 28.5 and 30 Å. Based on these values, only the last 0.2 μs were employed for further analysis.3.2 RMSF analysis of the simulated systems
RMSF analysis over the equilibrated simulation time showed that the DRD2L94A–risperidone complex exhibited reduced mobility in the second part (residues 311–365) of the cytoplasmic region between TMV and TMVI (cytoplasmic TMV–VI loop) and the intracellular TMIII–IV loop (residues 138–147) compared with DRD2WT–risperidone (Fig. S2A†). The DRD2W100L–risperidone complex exhibited reduced mobility at the first (residues 257–268) and second parts (residues 311–365) of the cytoplasmic TMV–VI loop compared with DRD2WT–risperidone (Fig. S2B†), except for a small fraction (residues 347–353) of the cytoplasmic TMV–VI loop, which showed an increased mobility for W100L compared to wild-type DRD2. DRD2W100A–risperidone displays increased mobility at TMIV (residues 152–174), EL2 (residues 175–193), and two regions in the cytoplasmic TMV–VI loop (residues 227–274 and 339–351) compared with DRD2WT–risperidone (Fig. S2C†). In contrast, reduced mobility was observed for the TMIII–IV loop (residues 136–145) and the cytoplasmic TMV–VI loop (residues 319–333) with respect to DDR2WT–risperidone.3.3 Protein–ligand interactions
In the cocrystallized DRD2–risperidone complex (PDB entry 6CM4), the benzisoxasole group of risperidone interacts with seven hydrophobic and two polar residues at TMIII (F110, D114, V115, C118, T119 and I122), TMV (S193, S197 and F198), and TMVI (F382, W386 and F389). The tetrahydropyridopyrimidinone moiety of risperidone interacts with residues EL1 (W100), TMVI (F390), and TMVII (Y408, T412 and Y416) (Fig. S3†).Clustering analysis over the equilibrated simulation time provided the representative conformation for wild-type and mutant DRD2–risperidone systems. Structural analysis of the most populated conformers showed differences at the entrance to the binding pocket (Fig. 1). A more open entrance was observed for DRD2W100L–risperidone (Fig. 1C) and DRD2W100A–risperidone (Fig. 1D) than for DRD2WT–risperidone (Fig. 1A) and DRD2W100A–risperidone (Fig. 1B).
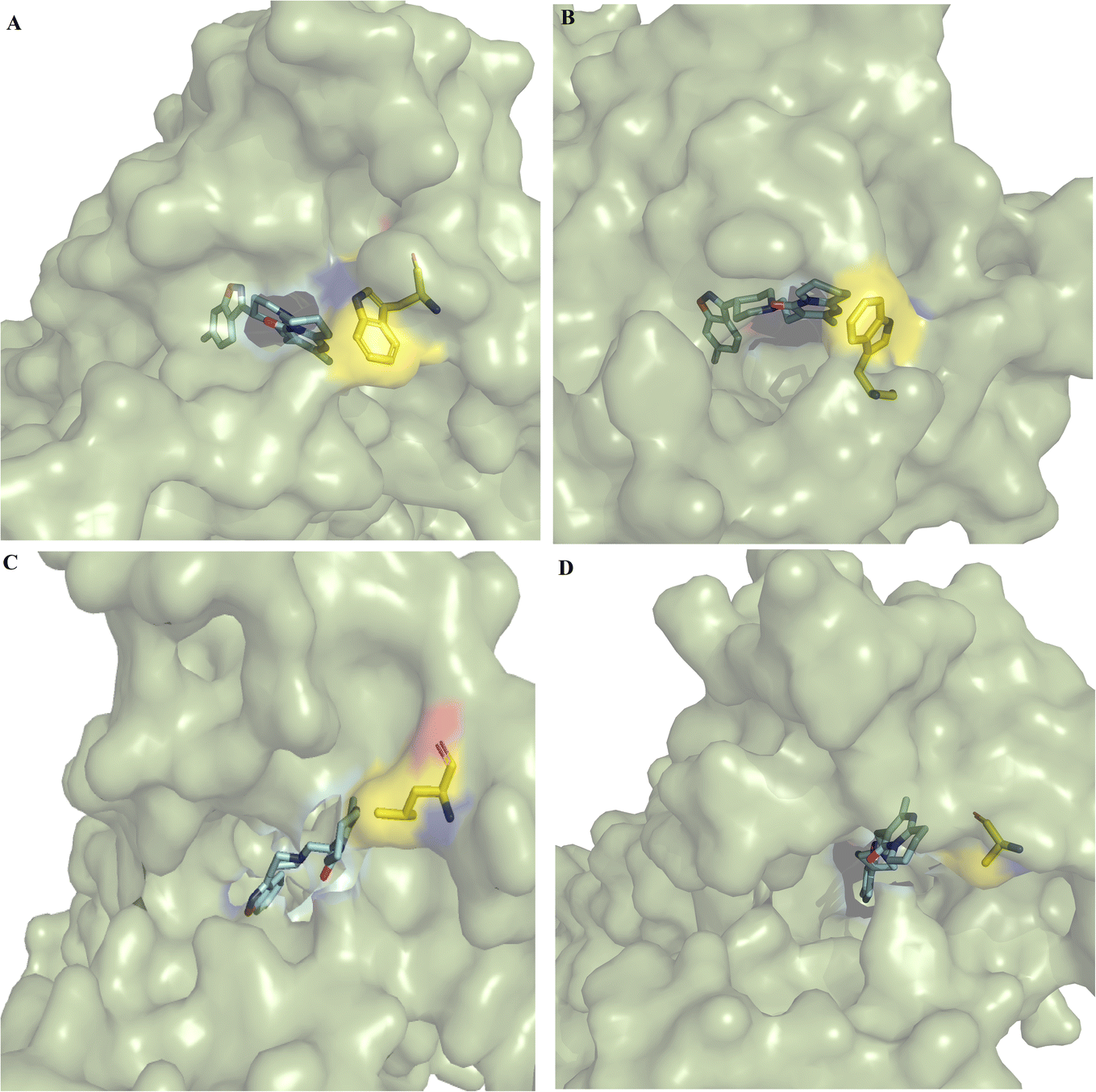 | ||
| Fig. 1 Entrances to the binding pocket of wild-type and mutant DRD2. (A) DRD2WT–risperidone, (B) DRD2L94A–risperidone, (C) DRD2W100L–risperidone, and (D) DRD2W100A–risperidone. Proteins are depicted as green surfaces, risperidone is depicted as cyan sticks, and the residue at the 100 position is shown as yellow sticks. The figure illustrates the increase in the aperture of the entrance of the binding pocket cavity by conformational changes caused by the W100L and W100A mutations compared to the wild type and the L94A mutant. The complexes correspond to the most populated conformations taken from MD simulations. | ||
In the DRD2WT–risperidone complex, MD simulations showed that the benzisoxasole group interacted with seven hydrophobic and two polar interactions with TMIII (V115, C118, T119, and I122), EL2 (F189, V190, and S193), and TMV (S197 and F198). The tetrahydropyridopyrimidinone portion of risperidone formed interactions with EL1 (W100), EL2 (I184), the TMVI–VII loop (P405), and TMVII (Y408, S409, and T412) (Fig. 2A). Comparison with the cocrystallized DRD2–risperidone complex11 shows that a high number of residues were preserved in both systems: EL1 (W100), TMIII (V115, C118, T119, and I122), TMV (S193, S197, and F198), and TMVII (Y408, and T412).
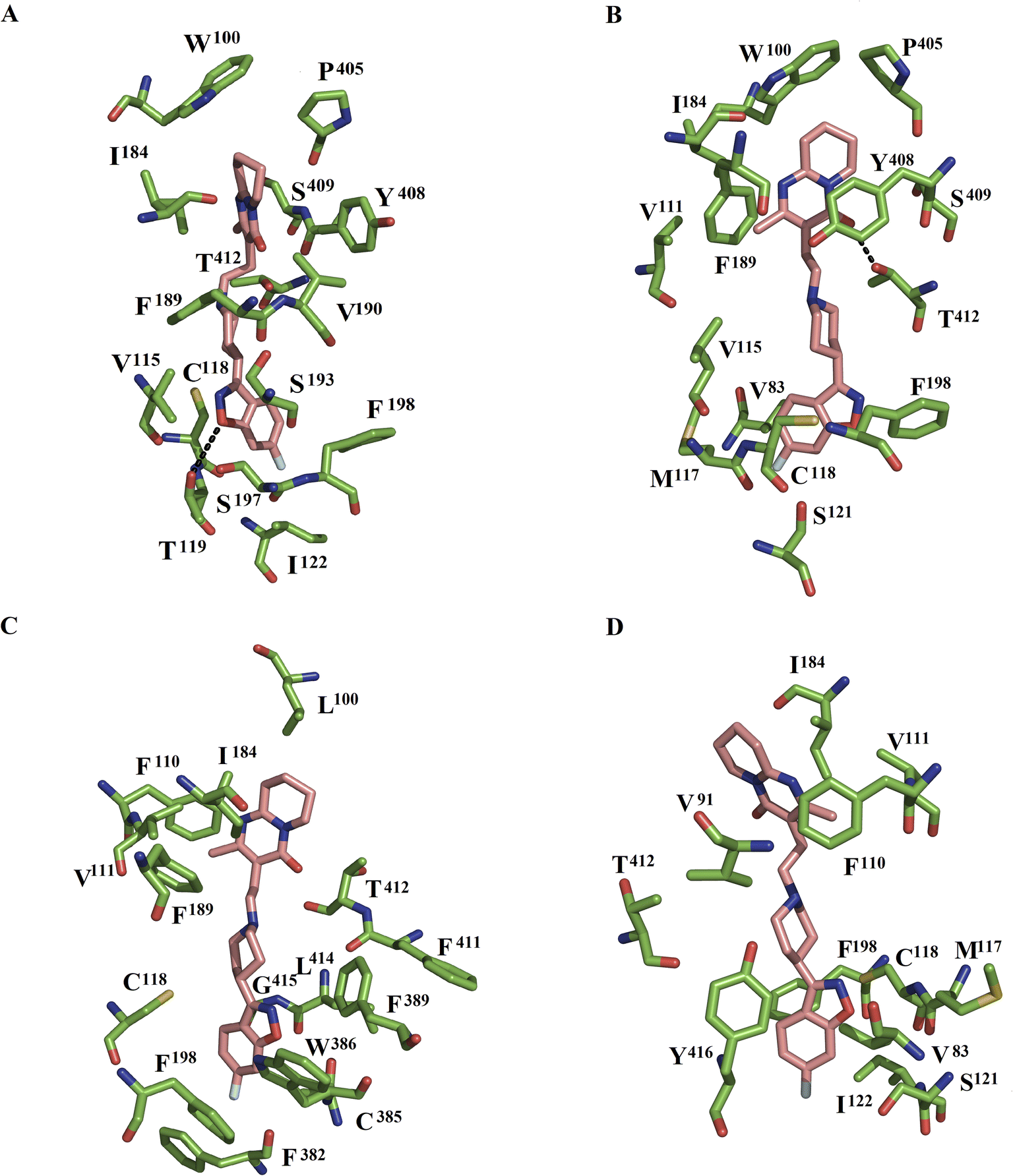 | ||
| Fig. 2 Protein–ligand interactions of risperidone in the wild-type and mutant DDR2–ligand systems. Interactions with risperidone at the ligand-binding sites of DDR2WT (A) and DDR2L94A (B), DDR2W100L (C) and DDR2W100A (D). These structures are representative of the first major populated complex. Dotted lines indicate HBs. | ||
In the DDR2L94A–risperidone complex, the benzisoxasole group was bounded by five hydrophobic and one polar contact with TMII (V83), TMIII (V115, M117, C118, and S121), and TMV (F198). The tetrahydropyridopyrimidinone portion of risperidone interacted with six hydrophobic and two polar residues at EL1 (W100 and V111), EL2 (I184 and F189), the TMVI–VII loop (P405), and TMVII (Y408, S409, and T412). Of these residues, T412 formed one hydrogen bond with the tetrahydropyridopyrimidinone portion (Fig. 2B). Comparison between the wild type and the L94A mutant showed that the benzisoxasole group was bound by residues of TMIII, EL2, and TMV in DDR2WT–risperidone, whereas residues at TMII, TMIII, and TMV participated in the stabilization of the benzisoxasole group in DDR2L94A–risperidone. The tetrahydropyridopyrimidinone portion was bound by residues at EL1, EL2, the TMVI–VII loop, and TMVII in DDR2WT–risperidone, while EL1, EL2, the TMVI–VII loop and TMVII were bound in DDR2L94A–risperidone.
In the DDR2W100L–risperidone complex, the benzisoxasole group of risperidone was bounded by eight hydrophobic residues and one neutral residue: TMIII (C118), TMV (F198), TMVI (F382, C385, W386, and F389), and TMVII (F411, L414, and G415). The tetrahydropyridopyrimidinone portion of risperidone formed interactions with EL1 (L100), TMIII (F110, and V111), EL2 (I184, and F189) and TMVII (T412) (Fig. 2C). Analysis of residues forming interactions in WT and W100L with the benzisoxasole group showed that only residues at TMIII and TMV were maintained in both systems. However, only residues EL1 (L100), EL2 (I184) and TM7 (T412) were present in the binding of the tetrahydropyridopyrimidinone portion.
In the DDR2W100A–risperidone complex, the benzisoxasole group of risperidone interacted with seven residues: TMII (V83), TMIII (M117, C118, I121, and I122), TMV (F198), and TMVII (Y416). The tetrahydropyridopyrimidinone portion of risperidone formed interactions with TMII (V91), TMIII (F110, and V111), EL2 (I184) and TMVII (T412) (Fig. 2D). Comparison with the wild type shows that only residues at TMIII (C118 and I122) and TMV (F198) were present in the binding of the benzisoxasole group in the wild type and the W100A mutant. For the tetrahydropyridopyrimidinone portion, only one residue at EL2 (I184) and TMVII (T412) was present in the coupling of this portion in the wild type and the W100A mutant.
3.4 Binding free energy calculations
Binding free energy analysis using the MMGBSA approach allowed us to determine the binding free energy (ΔGbind) value for each DRD2–ligand complex. Table 1 shows that all the complexes were energetically favorable, guided through nonpolar interactions (ΔEvdw + ΔGnpol,sol), whereas polar interactions (ΔEele + ΔGele,sol) opposed binding.| System | ΔEvdw | ΔEele | ΔGele,sol | ΔGnpol,sol | ΔEnonpolar | ΔEpolar | ΔGbind | ΔGexp |
|---|---|---|---|---|---|---|---|---|
| a ΔEpolar is equal to the summation of ΔEele + ΔGele,sol contributions, and ΔEnonpolar is the sum of ΔEvwd and ΔGnpol,sol terms. All the energies are averaged over 2000 snapshots and are in kcal mol−1 (standard deviation of the mean). *Values taken from Wang et al., 2018.11 | ||||||||
| DDR2–risperidone | −25.30 ± 1.6 | 36.94 ± 3.7 | −33.79 ± 3.6 | −4.17 ± 0.1 | −29.47 | 3.15 | −26.32 ± 1.5 | −4.65 |
| DDR2L94A–risperidone | −24.21 ± 1.5 | 26.31 ± 3.6 | −24.92 ± 3.0 | −4.38 ± 0.1 | −28.59 | 1.39 | −27.2 ± 1.4 | −6.40 |
| DDR2W100A–risperidone | −22.02 ± 1.6 | 35.03 ± 8.3 | −29.76 ± 6.2 | −3.99 ± 0.2 | −26.01 | 5.27 | −20.74 ± 2.5 | −3.80 |
| DDR2W100L–risperidone | −18.38 ± 2.5 | 26.93 ± 4.3 | −26.18 ± 3.8 | −3.63 ± 0.3 | −22.01 | 0.75 | −21.26 ± 2.2 | −3.84 |
The ΔGmmgbsa values were thermodynamically more favorable for DRD2WT–risperidone and DRD2L94A–risperidone than for DRD2W100L–risperidone and DRD2W100A–risperidone, implying a higher affinity of risperidone for DRD2WT and DRD2L94A, in line with the experimental ΔG value tendency (Table 1). In fact, a more negative ΔGbind value of risperidone by DRD2L94A was observed with respect to DRD2WT, in line with the experimental tendency;11 however, it is clear that another structural component may be involved in the differences in residence time at the binding pocket of the wild-type and L94A DRD2 systems.
3.5 Dissection of the per-residue free energy for DRD2–ligand complexes
Table 2 displays the per-residue energy for each residue participating in the protein–ligand interactions of DRD2–ligand systems. In the DRD2WT–risperidone, the residues responsible for the ΔGbind value were W100, V115, C118, T119, I122, I184, F189, V190, S193, S197, F198, P405, Y408, S409 and T412. Most of these residues established nonpolar interactions (Fig. 2A).| Residue | DRD2WT | DRD2L94A | DRD2W100L | DRD2W100A |
|---|---|---|---|---|
| V83 | −0.67 ± 0.27 | −0.456 ± 0.25 | ||
| V91 | −0.464 ± 0.15 | |||
| W100 | −0.564 ± 0.20 | −1.202 ± 0.40 | −0.383 ± 0.14 | |
| F110 | −0.587 ± 0.25 | −1.055 ± 0.50 | ||
| V111 | −0.458 ± 0.12 | −0.531 ± 0.30 | −0.523 ± 0.22 | |
| V115 | −1.774 ± 0.30 | −0.596 ± 0.15 | ||
| M117 | −0.592 ± 0.33 | −0.498 ± 0.13 | ||
| C118 | −2.281 ± 0.40 | −2.21 ± 0.37 | −0.941 ± 0.50 | −1.781 ± 0.40 |
| T119 | −0.879 ± 0.15 | |||
| S121 | −0.417 ± 0.20 | −0.476 ± 0.20 | ||
| I122 | −0.521 ± 0.20 | −0.614 ± 0.33 | ||
| I184 | −1.86 ± 0.40 | −1.264 ± 0.25 | −1.486 ± 0.57 | −1.354 ± 0.80 |
| F189 | −0.962 ± 0.30 | −0.549 ± 0.26 | −0.582 ± 0.28 | |
| V190 | −0.839 ± 0.20 | |||
| S193 | −0.478 ± 0.15 | |||
| S197 | −0.991 ± 0.40 | |||
| F198 | −1.894 ± 0.60 | −0.603 ± 0.19 | −0.48 ± 0.17 | −0.445 ± 0.24 |
| F382 | −0.704 ± 0.29 | |||
| C385 | −0.436 ± 0.15 | |||
| W386 | −1.827 ± 0.70 | |||
| F389 | −1.249 ± 0.57 | |||
| P405 | −0.828 ± 0.25 | −0.792 ± 0.29 | ||
| Y408 | −3.041 ± 0.50 | −1.939 ± 0.58 | ||
| S409 | −0.47 ± 0.15 | −0.531 ± 0.30 | ||
| F411 | −0.451 ± 0.25 | |||
| T412 | −0.983 ± 0.40 | −2.358 ± 0.56 | −1.078 ± 0.77 | −0.787 ± 0.30 |
| L414 | −0.768 ± 0.31 | |||
| G415 | −0.97 ± 0.40 | |||
| Y416 | −1.359 ± 0.70 |
In the DRD2L94A–risperidone complex, V83, W100, V111, V115, M117, C118, S121, I184, F189, F198, P405, Y408, S409, and T412 were the residues contributing to the affinity for risperidone. Of these residues, most of them established hydrophobic interactions, except T412, which formed one hydrogen bond with the tetrahydropyridopyrimidinone portion of risperidone (Fig. 2B).
In the DRD2W100L–risperidone system, the L00, F110, V111, C118, I184, F189, F198, F382, C385, W386, F389, F411, T412, L414 and G415 residues contributed to complex stabilization through nonpolar contacts (Fig. 2C). In the case of the DRD2W100A–risperidone system, the V83, V91, F110, V111, M117, C118, S121, I122, I184, F198, T412 and Y416 residues guided the affinity of the complex (Fig. 2D). This analysis shows that the affinity in wild-type and L94A was guided by a higher number of similar residues (W100, V115, C118, I184, F189, F198, P405, Y408, S409 and T412) than those observed between wild type and W100L (L100, C118, I184, F189, F198, and T412) or W100A (C118, I184, F198 and T412).
Comparison of the participation of interactions between risperidone and the residue at position 100 indicated that the tetrahydropyridopyrimidinone portion formed stronger interactions with the L94A mutant than with the wild type (Table 2), which may be responsible for the decrease in the entrance of the binding pocket cavity of L94A compared to the wild type (Fig. 1A and B). In contrast, W100L or W100A have an important impact on the stabilization of risperidone, which was more apparent for W100A, which even lost interactions with the tetrahydropyridopyrimidinone portion (Table 2 and Fig. 2D). These differences may be linked with the increases in the sizes of the cavity entrances for the DRD2W100L–risperidone (Fig. 1C) and DRD2W100A–risperidone systems (Fig. 1D) compared to the DRD2WT–risperidone (Fig. 1A) and DRD2L94A–risperidone systems (Fig. 1B).
3.6 PC analysis
PC analysis identified the main motions along the wild-type and mutant DRD2–ligand systems. The first two eigenvectors include the major eigenvalues; these contained 54.7, 45.4, 57.4, and 54.4% of the total flexibility of the DRD2WT–risperidone, DRD2L94A–risperidone, DRD2W100L–risperidone and DRD2W100A–risperidone systems, respectively. Projection over the phase space of the first and second PC (PC1 vs. PC2) eigenvectors shows that the DRD2W100A–risperidone system (Fig. 3D) covers a more extensive distribution in the essential subspace than the DRD2WT–risperidone systems (Fig. 3A), indicating a larger conformational entropy for ligand recognition for W100A with respect to wild-type DRD2. The DRD2W100L–risperidone (Fig. 3C) and DRD2WT–risperidone systems exhibit similar distributions along the essential subspace, indicating similar conformational behavior in risperidone recognition. However, DRD2L94A–risperidone systems indicate a more compact distribution in the essential subspace with respect to the wild type (Fig. 3B), suggesting an unfavorable entropy contribution upon ligand recognition compared with the other systems.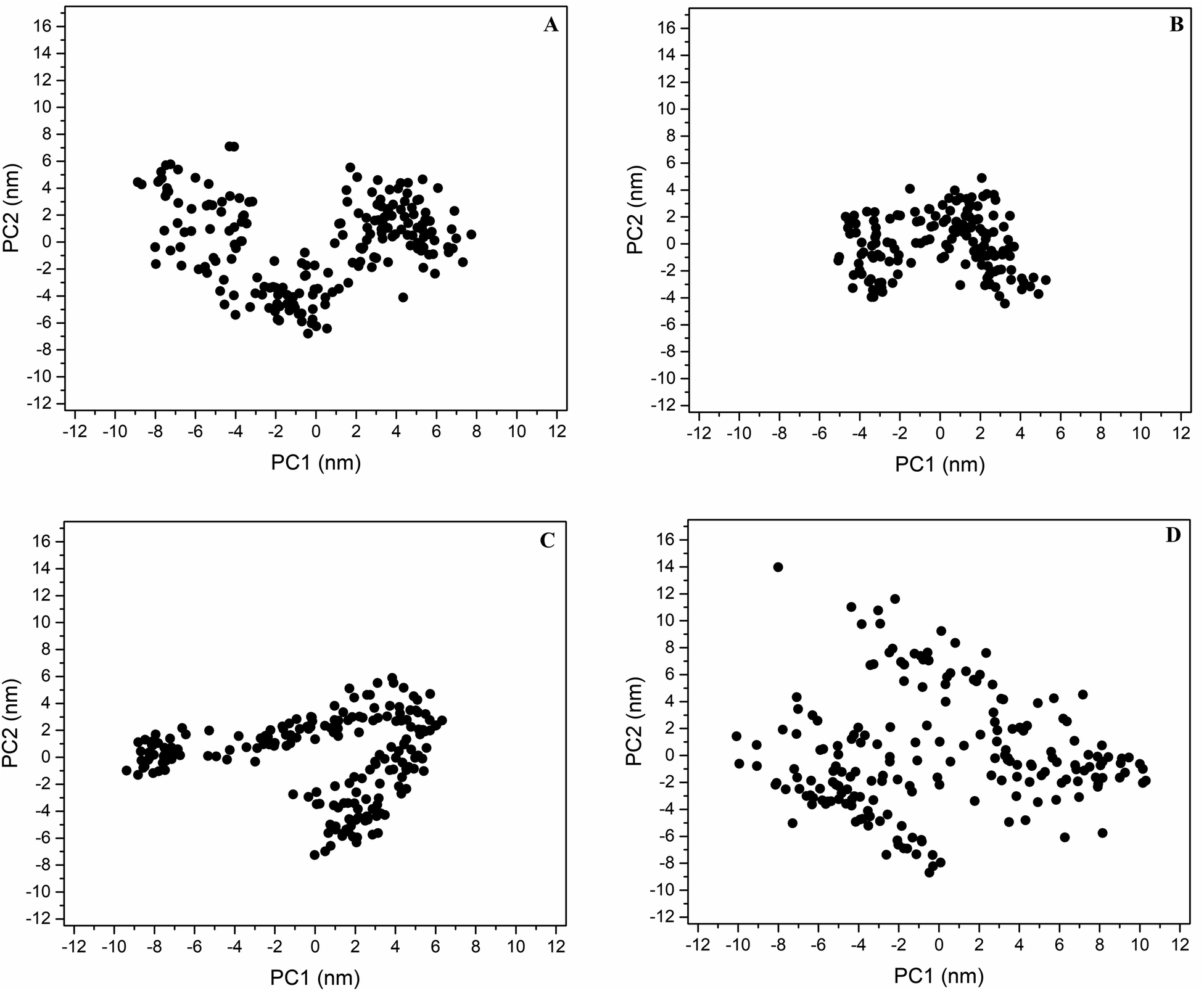 | ||
| Fig. 3 Projection of the wild-type and mutant DRD2–risperidone systems in phase space. Projection of motion in the phase space for DRD2WT–risperidone (A), DRD2L94A–risperidone (B), DRD2W100L–risperidone (C), and DRD2W100A–risperidone (D). | ||
The diagonalized covariance matrix over the backbone atoms provides the following values: DRD2WT–risperidone (53.7 nm2), DRD2L94A–risperidone (26.3 nm2), DRD2W100L–risperidone (50.6 nm2), and DRD2W100A–risperidone (81.2 nm2). These results suggest that the binding of risperidone to DRD2L94A reduces the number of conformational states regarding DRD2WT. The molecular recognition of risperidone on DRD2WT and DRD2W100L exhibits similar numbers of conformers. In contrast, the binding of risperidone to DRD2W100A increases the number of conformational states in solution compared to DRD2WT. Correlation of these covariance values with RMSF analysis indicated that the major contributor to the differences in mobility between the wild type and the mutants was the cytoplasmic TMV–VI loop for DRD2WT–risperidone, DDR2L94A–risperidone, and DDR2W100L–risperidone (Fig. S2†). For DDR2W100A–risperidone and DDR2WT–risperidone, the main contributors were the cytoplasmic TMV–VI loop, TMIV, and EL2.
Graphical depictions of the total mobility along PC1 vs. PC2 indicates that DDR2WT–risperidone (Fig. S4A†), DDR2L94A–risperidone (Fig. S4B†), and DDR2W100L–risperidone (Fig. S4C†), and DDR2W100A–risperidone (Fig. S4D†) exhibit the highest collective motions along the cytoplasmic TMV–VI loop that showed a breathing motion. However, this fluctuation was considerably higher for DDR2WT–risperidone (Fig. S4A†) and DDR2W100A–risperidone (Fig. 3D) with respect to DDR2L94A–risperidone and DDR2W100L–risperidone, supporting the high heterogeneity observed through visualization onto the essential space (Fig. 3).
3.7 Energy minimization of wild-type and mutant DRD2–risperidone complexes
The YASARA minimization server36 was used to minimize the most populated wild-type and mutant DRD2–risperidone complexes obtained during the equilibrated simulation time (i.e., the last 0.2 μs) through clustering analysis. Energy minimization findings revealed increases in the total energy for DRD2L94A–risperidone and DRD2W100L–risperidone with respect to DRD2WT–risperidone, whereas a decrease was observed for DRD2W100A–risperidone compared with DRD2WT–risperidone (Table 3). These results indicate that L94A and W100L optimize the structure, whereas W100A destabilizes the structure with respect to the wild type.| Systems | Energy after minimization (kJ mol−1) | RMSD (Å) |
|---|---|---|
| DDR2WT–risperidone | −235938.4 | |
| DDR2L94A–risperidone | −234063.4 | 2.18 |
| DDR2W100L–risperidone | −236213.3 | 3.70 |
| DDR2W100A–risperidone | −232787.4 | 3.50 |
These differences correlate with the significantly lower mobility of DRD2L94A–risperidone and the increased dynamic motion of DRD2W100A–risperidone compared to DRD2WT–risperidone, but not much with the slightly lower mobility DRD2W100L–risperidone compared to DRD2WT–risperidone. Analysis of RMSD between the four conformers showed that L94A less drastically impacted the structure of the protein compared with wild-type DDR2. In contrast, W100L and W100A suggest a critical change in the DRD2 structure compared to wild-type DDR2.
3.8 Structural analysis of W100 mutations in wild-type and mutant DRD2–risperidone complexes
Analysis of the most populated complexes showed that the side chain of W100 in DRD2WT–risperidone is stabilized by contacts of I184, L94 and P405, similar to the crystallographic structure (Fig. S5A†). The same interactions were also observed for the side chain of W100 in DRD2L94A–risperidone, together with the addition of interactions of the side chain with G98 (Fig. S5B†). In contrast, the side chain of L100 in DRD2W100L–risperidone only interacts with L94 (Fig. S5C†), and the side chain of A100 in DRD2W100A–risperidone only forms interactions with C107 (Fig. S5D†).4. Discussion
After the research of Paul Greengard's lab, it was proposed that DRD2 contains the molecular recognition site for antipsychotic drugs; other research groups also contributed to DRD2 identification through radiolabeled drug experiments.38 Experimental studies by X-ray crystallography have provided tridimensional information of DRD2 in complex with the atypical antipsychotic risperidone.11 Due to the lack of experimental information about DRD2 conformation in the active state, homology modeling studies have been implemented.39 This theoretical study showed that the main structural differences between active and inactive states are at the cytoplasmic loop between TMV and VI, where G protein coupling occurs. Meanwhile, comparison of DRD2–risperidone with the active and inactive adenosine A2A receptor structures did not show important structural changes at the intracellular TMVI,11 but the ‘ionic lock’ between (R131-E268 salt bridge) was preserved in the DRD2–risperidone complex. Recent experimental studies showed that W100A and W100L mutations decreased risperidone residence time at the ligand-binding site, whereas the L84A mutation increased the binding affinity compared with the wild-type DRD2. This contribution investigated the structural and energetic basis of the effect of W100A, W100L, and L84A mutations on the DR2D–risperidone complex. Initially, the cytoplasmic TMV–VI loop was modeled using homology modeling techniques exploiting the high-resolution DRD2–risperidone complex. The wild-type and mutant DRD2–risperidone complexes were constructed and subjected to microsecond (μs) MD simulations in an aqueous membrane system. Trajectory results were used for clustering analysis, binding free energies by MMGBSA technique, and principal component (PC) analysis.Comparison between the representative protein–ligand complexes obtained through clustering analysis showed that the L94A mutation impacts the type of residues contacting the benzisoxasole group of risperidone, in which only contacts at TMIII and TMV are preserved in the wild type and the L94A mutant. However, this chemical group still formed interactions with a similar number of residues, but these residues were predominantly hydrophobic for DRD2L94A–risperidone compared to DRD2WT–risperidone. In contrast, the tetrahydropyridopyrimidinone portion conserved similar numbers and types of hydrophobic and polar interactions at EL1, EL2, the TMVI–VII loop, and TMVII in the wild-type and the L94A mutant.
Comparison between DRD2WT–risperidone and DRD2W100L–risperidone shows that W100L markedly affects the number of types of residues coupling the benzisoxasole group, maintaining only similar interactions with two hydrophobic residues at TMIII (C118) and TMV (F198) from the ten residues stabilizing the benzisoxasole group. Similarly, as observed for the benzisoxasole group, the tetrahydropyridopyrimidinone portion was stabilized by a dissimilar type of residue, where only three interactions at EL1 (L100), EL2 (I184) and TMVII (T412) were preserved in the two systems.
Evaluation between DRD2WT–risperidone and DRD2W100A–risperidone indicates that only three residues at TMIII (C118 and I122) and TMV (F198) are maintained in both systems, stabilizing the benzisoxasole group. In addition, a reduction in the number of residues coordinating the benzisoxasole group was observed in the system containing the W100A mutation. A similar behavior was observed for the tetrahydropyridopyrimidinone portion, where only two residues at EL2 (I184) and TMVII (T412) were maintained in both systems.
In accordance with experimental results, ΔGbind values obtained using the MMGBSA method showed that the binding affinity of risperidone was energetically more favorable for L94A and WT than for W100L and W100A.11 However, the differences in affinity of risperidone to L94A and WT can not be explained by only determining the ΔGbind values; other factors, such as entropic contributions, may explain the higher affinity of risperidone to L94A than to WT.11
Per-residue free energy decomposition analysis indicated that the affinity in wild-type and L94A was guided by a higher number of similar residues (V115, C118, I184, F189, F198, P405, Y408, and T412) than those observed for W100L and W100A. Comparison among the systems showed that only C118 and I184 contributed to the affinity in the four systems, highlighting important roles for these residues in ligand molecular recognition. In fact, I184 is suggested to be an important residue in ligand stabilization through stabilization of interactions with W100 and L94.11 Comparative analysis of the per-residue energies of W100 showed stronger energies in DRD2L94A–risperidone (Table 2), which also impacted the entrance to the binding site, being closer to the L94A mutant than to the wild type.
PC analysis indicated that the higher heterogeneity of DRD2WT–risperidone compared to DDR2L94A–risperidone could be linked to an unfavorable entropy that may contribute to improving the predicted binding free energy (Table 1), explaining the higher experimentally reported affinity compared to DRD2L94A–risperidone. Since there are no significant conformational changes compared to DRD2WT–risperidone, no important entropic contributions should be anticipated for DRD2W100L–risperidone. However, the favorable entropic components for DRD2W100A–risperidone compared to DRD2WT–risperidone may contribute to a decrease in the predicted affinity.
Energy minimization of wild-type and mutant DRD2–risperidone complexes indicated that the L94A and W100L mutants have more optimized structures than wild-type DRD2, whereas W100A destabilizes the structure. Analysis of RMSD between mutants and wild-type DRD2 showed that the L94A mutant less drastically impacted the DRD2 structure than W100L and W100A. Structural analysis of the W100 mutation in wild-type and mutant DRD2–risperidone complexes indicated that the L94A mutant contributes to an increase in the number of contacts around the side chain of W100, which also contributes to improving the per-residue interactions with the tetrahydropyridopyrimidinone portion of risperidone.
5. Conclusions
To investigate the structural and energetic basis of the effects of the W100A, W100L, and L84A mutations in the DR2D–risperidone system in an aqueous membrane, high-resolution crystallographic structure information, homology modeling, and microsecond MD simulations studies were performed. Clustering analysis showed that L94A mutations did not markedly affect the binding mode or risperidone, indicating a similar affinity to that of DRD2WT. In contrast, W100L and W100A mutations experienced similar changes, decreasing the number of residues stabilizing risperidone, suggesting a lower affinity of risperidone for these mutations. In agreement with the experimental results, thermodynamic analysis showed that the binding affinity of risperidone is energetically more favorable for L94A and WT than for W100L and W100A. Per-residue free energy decomposition analysis indicated that the affinity in the wild type and the L94A mutant was guided by a higher number of similar residues than those observed in the W100L and W100A mutants, highlighting the participation of C118 and I184 in the ligand molecular recognition of DRD2–risperidone. Comparative analyses among the most populated conformers showed a more opened entrance to the binding pocket for the W100L and W100A mutants compared to the wild type and the L94A mutant. In fact, the L94A mutant showed a more closed entrance than the wild type and stronger interactions between risperidone and W100, supporting the higher residence time of risperidone at the binding pocket of L94A.The better affinity of risperidone by L94A compared to WT was explained using PC analysis, which revealed that the unfavorable entropy contribution observed for DRD2L94A–risperidone relative to DRD2WT–risperidone may have contributed to increasing the predicted binding affinity. In contrast with DRD2W100A–risperidone or DRD2W100L–risperidone, where favorable or non-conformational changes in the molecular recognition were observed compared with WT, which decreased the affinity observed for DRD2W100A–risperidone even more and did not affect the affinity for DRD2W100L–risperidone. Energy minimization on the most populated conformers showed that the L94A mutant reaches a more optimized structure compared to wild-type DRD2, which also contributes to reaching a more stabilized protein–ligand complex. Structural analysis around W100 showed a more favorable map of interactions for L94A, which contribute to making more favorable interactions with risperidone.
Data availability
The datasets supporting the conclusions of this research are contained within the paper and its additional files.Conflicts of interest
The authors declare they have no conflict of interest in terms of the content of this manuscript.Acknowledgements
The authors gratefully acknowledges the Deanship of Scientific Research, Qassim University, Saudi Arabia for funding the publication of this research.References
- C. Missale, S. R. Nash, S. W. Robinson, M. Jaber and M. G. Caron, Dopamine receptors: From structure to function, Physiol. Rev., 1998, 78, 189–225 CrossRef CAS PubMed.
- M. Y. S. Kalani, N. Vaidehi, S. E. Hall, R. J. Trabanino, P. L. Freddolino, M. A. Kalani, W. B. Floriano, V. W. T. Kam and W. A. Goddard III, The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3815–3820 CrossRef PubMed.
- B. Ja-Hyun, Dopamine signaling in reward-related behaviors, Front. Neural Circuits, 2013, 11(7), 152 Search PubMed.
- P. Jenner and C. D. Marsden, Chronic pharmacological manipulation of dopamine receptors in brain, Neuropharmacology, 1987, 26, 931–940 CrossRef CAS PubMed.
- J. M. Beaulieu and R. R. Gainetdinov, The physiology, signaling, and pharmacology of dopamine receptors, Pharmacol. Rev., 2011, 63, 182–217 CrossRef CAS.
- J. R. Bunzow, et al., Cloning and expression of a rat D2 dopamine receptor cDNA, Nature, 1988, 336, 783–787 CrossRef CAS PubMed.
- D. K. Grandy, et al., Cloning of the cDNA and gene for a human D2 dopamine receptor, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9762–9766 CrossRef CAS PubMed.
- J. A. Javitch, D. Fu, J. Chen and A. Karlin, Mapping the binding-site crevice of the dopamine D2 receptor by the substituted-cysteine accessibility method, Neuron, 1995, 14, 825–831 CrossRef CAS PubMed.
- J. A. Ballesteros, L. Shi and J. A. Javitch, Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors, Mol. Pharmacol., 2001, 60, 1–19 CrossRef CAS PubMed.
- E. Y. Chien, et al., Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist, Science, 2010, 330, 1091–1095 CrossRef CAS PubMed.
- S. Wang, T. Che, A. Levit, B. K. Shoichet, D. Wacker and B. L. Roth, Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone, Nature, 2018, 555(7695), 269–273 CrossRef CAS PubMed.
- D. A. Shapiro, K. Kristiansen, D. M. Weiner, W. K. Kroeze and B. L. Roth, Evidence for a model of agonist-induced activation of 5–HT2A serotonin receptors which involves the disruption of a strong ionic interaction between helices 3 and 6, J. Biol. Chem., 2002, 18, 11441–11449 CrossRef PubMed.
- J. A. Ballesteros, et al., Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6, J. Biol. Chem., 2001, 276, 29171–29177 CrossRef CAS PubMed.
- K. Palczewski, et al., Crystal structure of rhodopsin: a G protein-coupled receptor, Science, 2000, 289, 739–745 CrossRef CAS PubMed.
- B. Webb and A. Sali, Comparative Protein Structure Modeling Using Modeller, Current Protocols in Bioinformatics, John Wiley & Sons, Inc., 2016, vol. 54, pp. 5.6.1–5.6.37 Search PubMed.
- V. B. Chen, W. B. Arendall III, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(Pt 1), 12–21 CrossRef CAS PubMed.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: An advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- M. A. Lomize, A. L. Lomize, I. D. Pogozheva and H. I. Mosberg, OPM: Orientations of proteins in membranes database, Bioinformatics, 2006, 22, 623–625 CrossRef CAS PubMed.
- S. Jo, T. Kim and W. Im, Automated builder and database of protein/membrane complexes for molecular dynamics simulations, PLoS ONE, 2007, 2, e880 CrossRef PubMed.
- S. Jo, J. B. Lim, J. B. Klauda and W. Im, CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes, Biophys. J., 2009, 97, 50–58 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79(2), 926–935 CrossRef CAS.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- C. J. Dickson, B. D. Madej, A. A. Skjevik, R. M. Betz, K. Teigen, I. R. Gould and R. C. Walker, Lipid14: The amber lipid force field, J. Chem. Theory Comput., 2014, 10, 865–879 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. Izaguirre, D. Catarello, J. Wozniak and R. Skeel, Langevin stabilization of molecular dynamics, J. Chem. Phys., 2001, 114, 20902098 CrossRef.
- R. Pastor, B. Brooks and A. Szabo, An analysis of the accuracy of Langevin and molecular dynamics algorithms, Mol. Phys., 1988, 65, 14091419 CrossRef.
- H. J. Berendsen, J. V. Postma, W. F. Van Gunsteren, A. R. H. J. DiNola and J. R. Haak, Molecular dynamics with coupling to an external bath, J. Chem. Phys., 1984, 81(8), 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23(3), 327–341 CrossRef CAS.
- W. L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA, USA, 2002 Search PubMed.
- B. R. Miller III, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, MMPBSA.py: An Efficient Program for End-State Free Energy Calculations, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef PubMed.
- M. Feig, A. Onufriev, M. S. Lee, W. Im, D. A. Case and C. L. Brooks, Performance Comparison of Generalized Born and Poisson Methods in the Calculation of Electrostatic Solvation Energies for Protein Structures, J. Comput. Chem., 2004, 25, 265–284 CrossRef CAS PubMed.
- M. Bello, Binding mechanism of kinase inhibitors to EGFR and T790M, L858R and L858R/T790M mutants through structural and energetic analysis, Int. J. Biol. Macromol., 2018, 118, 1948–1962 CrossRef CAS PubMed.
- E. Krieger, et al., Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8, Proteins: Struct., Funct., Bioinf., 2009, 77(S9), 114–122 CrossRef CAS PubMed.
- M. G. Wolf, M. Hoefling, C. Aponte-Santamaría, H. Grubmuller and G. Groenhof, g_membed: Efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation, J. Comput. Chem., 2010, 31, 2169–2174 CrossRef CAS PubMed.
- B. K. Madras, History of the discovery of the antipsychotic dopamine D2 receptor: a basis for the dopamine hypothesis of schizophrenia, J. Hist. Neurosci., 2013, 22(1), 62–78 CrossRef.
- R. E. Salmas, M. Yurtsever and M. Stein, et al., Modeling and protein engineering studies of active and inactive states of human dopamine D2 receptor (D2R) and investigation of drug/receptor interactions, Mol. Diversity, 2015, 19, 321–332 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06694g |
| This journal is © The Royal Society of Chemistry 2022 |