
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective α-addition of vinylogous α-ketoester enolate in organocatalytic asymmetric Michael reactions: enantioselective synthesis of Rauhut–Currier type products†
Zhibing Weng‡
ab,
Ying Zhou‡c,
Xin Yuec,
Feng Jiangc and
Wengang Guo *c
*c
aSchool of Chemical and Pharmaceutical Engineering, Changzhou Vocational Institute of Engineering, Changzhou, China
bSchool of Pharmaceutical Sciences, Nanjing Tech University, Nanjing, China
cSchool of Petrochemical Engineering, Changzhou University, Changzhou, China. E-mail: wgguo@cczu.edu.cn
First published on 9th November 2022
Abstract
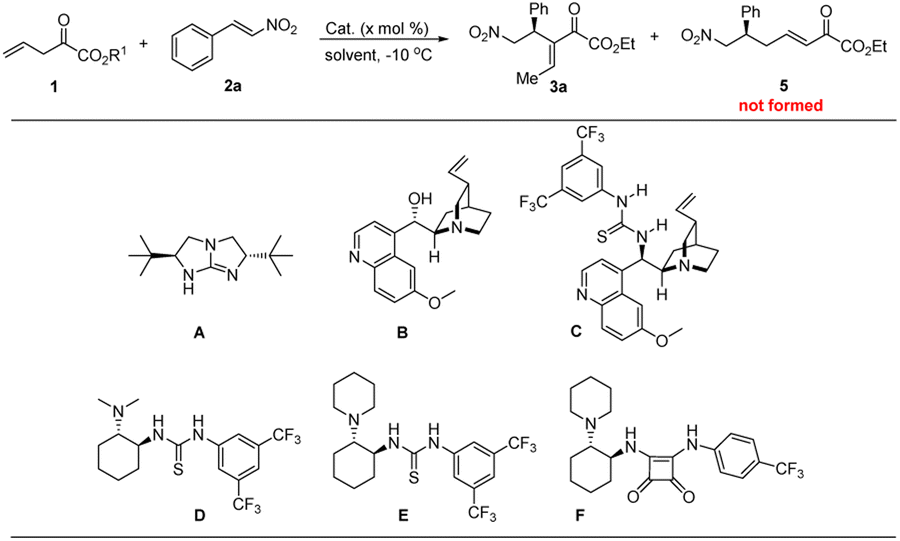
Catalytic asymmetric α-regioselective Michael additions of vinylogous α-ketoester enolate are described herein. With 0.1–1.0 mol% loadings of a chiral bifunctional organocatalyst, the addition of a deconjugated α-keto ester to a series of nitroolefins, including the challenging β-alkylnitroalkenes, efficiently proceed, providing the Rauhut–Currier type products after isomerization of the terminal double bond in good yields (60–88%) with excellent regio- and enantioselectivities (94–99% ee, TON up to 160 with 0.5 mol% of the catalyst).
Introduction
The enolate-based Michael reaction is fundamentally important in synthetic chemistry and could provide highly functionalized valuable products for natural and non-natural product synthesis. Its synthetic power has been dramatically improved by combing with the principle of vinylogy, thus increasing product complexity.1 Since the landmark report of vinylogous Mukaiya–Michael reaction of 2-silyloxyfurans and enals on use of iminium activation,2 tremendous asymmetric vinylogous Michael additions, employing 5-member ring heterocycles, such as 2-silyloxyfurans,3 lactone,4 and lactam,5 cyclic,6 or acyclic carbonyl derivatives7 as the nucleophiles,8 have been developed (Scheme 1a), furnishing valuable enantioenriched products with γ-site regioselectivity predominantly as a result of orbital coefficients and electrophilic susceptibility.9 | ||
| Scheme 1 Introduction to asymmetric vinylogous Michael addition reactions with dienolates and its equivalent dienamines. | ||
Alternatively, Shibasaki and co-workers disclosed a chiral barium complex catalyzed Mannich reaction of a β,γ-unsaturated ester, providing aza-Morita–Baylis–Hillman-type products after base-promoted isomerization of the terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of products.10a C–C bond formation at the α-position of “dienolates” or their equivalent “dienamine” intermediate has also been achieved.10 In this context, several methods were established for taming the issue of regioselectivity in synthetic chemistry, especially in asymmetric organocatalysis. For example, steric shielding of Cγ-position of unsaturated aldehydes is one of the used strategies for achieving α-regioselectivity of vinylogous Michael reaction through dienamine catalysis (Scheme 1b, left).10b–g On the other hand, bifunctional Lewis base/hydrogen bonding catalysis is also found to be competent. The attack site of the in situ generated dienolate could be controlled via the carefully selected organocatalysts through “anchoring effect” (Scheme 1b, right).10h–o However, after extensive literature research, it can be found that the elaborated catalyst, substrate, and other reaction conditions are often required for switching the regioselectivity. Inspired by the elegant works of Barbas,10g Palomo,10h,i and Alemán10o in catalytic asymmetric vinylogous Michael additions for the synthesis of RC (Rauhut–Currier)-type products, we questioned whether vinylogous α-keto esters could be employed in vinylogous asymmetric Michael reaction in the control of regioselectivity under low catalyst loading11 and mild reaction conditions.
C bond of products.10a C–C bond formation at the α-position of “dienolates” or their equivalent “dienamine” intermediate has also been achieved.10 In this context, several methods were established for taming the issue of regioselectivity in synthetic chemistry, especially in asymmetric organocatalysis. For example, steric shielding of Cγ-position of unsaturated aldehydes is one of the used strategies for achieving α-regioselectivity of vinylogous Michael reaction through dienamine catalysis (Scheme 1b, left).10b–g On the other hand, bifunctional Lewis base/hydrogen bonding catalysis is also found to be competent. The attack site of the in situ generated dienolate could be controlled via the carefully selected organocatalysts through “anchoring effect” (Scheme 1b, right).10h–o However, after extensive literature research, it can be found that the elaborated catalyst, substrate, and other reaction conditions are often required for switching the regioselectivity. Inspired by the elegant works of Barbas,10g Palomo,10h,i and Alemán10o in catalytic asymmetric vinylogous Michael additions for the synthesis of RC (Rauhut–Currier)-type products, we questioned whether vinylogous α-keto esters could be employed in vinylogous asymmetric Michael reaction in the control of regioselectivity under low catalyst loading11 and mild reaction conditions.
α-Dicarbonyl compounds have rarely been used as nucleophiles in Brønsted base catalyzed asymmetric reactions due to their intrinsic potential for self-condensation.12 α-Ketoamides with a higher pKa value of α-C–H Bond were usually employed to tame the reactivity in catalytic asymmetric Michael reactions.13 To this end, Sodeoka and coworkers developed the first example of asymmetric conjugate addition of α-ketoesters to nitroalkenes by elaborating a strategy of “endogenous and exogenous bases effect”.14 Rodriguez and co-workers developed the highly stereoselective 1,4-addition of α-keto esters to nitroolefins by using Takemoto's thiourea organocatalyst.15 Our continuous interest in developing new organocatalytic asymmetric reactions,16 and inspiration from the work of Rodriguez and others prompted our investigation into the vinylogous Michael addition reaction of deconjugated α-keto esters. Herein, we report the successful realization of this, affording valuable enantioenriched Rauhut–Currier type products with high enantioselectivities (94–99%) upon in situ isomerization of the terminal CC double bond, which could not be accessed by direct Rauhut–Currier reaction (Scheme 1c).
Results and discussion
Firstly, our investigations focused on the synthesis of vinylogous α-keto esters. It was reported that vinylogous α-keto esters could exist as stable dienol forms, depending on the properties of substituents on the terminal double bond.17 Thus, it persuaded us to prepare two types of vinylogous α-ketoesters to investigate their reactivities (Scheme 2, compounds 1 and 1′, eqn (1). | ||
| Scheme 2 Synthesis of vinylogous α-keto esters. | ||
Secondly, with the desired α-ketoester derivatives in hand, we proceeded to screen chiral organic base catalysts for achieving the asymmetric version of this cascade reaction. Guanidine A, which was reported as an excellent organocatalyst for isomerization of 3-alkanoates,18 was found to be effective in promoting the cascade reaction between 1 and 2a. However, the corresponding Rauhut–Currier type product 3a is obtained with moderate enantioselectivity (Table 1, entry 1). Similar enantioselectivities were achieved when evaluating Cinchona alkaloid derived organocatalysts (Table 1, entries 2–3). We thus shifted our attentation to Takemoto's thioureas and Rawal's squaraines. General information shows that steric hindrance of tertiary amino moiety exerts a significant effect on the enantioselectivity. In this context, the enantioselectivity of product 3a was greatly improved to 86%, albeit in somewhat lower yield (Table 1, entry 5 versus entry 4) when using catalyst E. In view of the structural differences between thiourea and squaramide, and particularly the pKa and distance between the two donor N–H atoms,19 Rawal's squaramide catalyst F was investigated. As was expected, the reactivity and enantioselectivity were significantly increased (97% ee, Table 1, entry 6).20 A survey of solvent shows that toluene is the best solvent for this reaction (Table 1, entry 6 versus entries 7–9). It is should be noted that α-selectivity of vinylogous Michael reaction is predominant in all of the reactions, the corresponding isomer 5 could not be observed by TLC or crude 1H NMR. The efficiency of this catalytic asymmetric reaction is demonstrated by reducing the catalyst loading to 1.0 mol% without any negative effect in yield and enantioselectivity (Table 1, entries 10–12). Changing the ethyl group in R1 to methyl shows a negligible effect on the enantioselectivity (Table 1, entry 13). However, an inferior result was obtained when further increasing the steric bulk of the ester group (Table 1, entry 14). Finally, we found the reaction temperature has a pronounced influence on the isolated yield of the product (Table 1, entries 15–16), a better yield was obtained when the reaction was conducted at −10 °C.21
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | R1 | x | Solvent | Yieldb (%) | eec (%) |
| a Unless otherwise specified, all reactions were performed by employing 0.2 mmol 2a with 0.4 mmol 1, and the amount of corresponding catalyst in solvent (1.0 mL, 0.2 M) for 24 h.b Isolated yield of 3a.c Determined by chiral HPLC analysis of 3a.d Reaction at −5 °C.e Reaction at 0 °C. | ||||||
| 1 | A | Et | 10 | Toluene | 64 | 32 |
| 2 | B | Et | 10 | Toluene | 83 | −25 |
| 3 | C | Et | 10 | Toluene | 91 | 38 |
| 4 | D | Et | 10 | Toluene | 70 | 74 |
| 5 | E | Et | 10 | Toluene | 62 | 86 |
| 6 | F | Et | 10 | Toluene | 80 | 97 |
| 7 | F | Et | 10 | PhCF3 | 80 | 95 |
| 8 | F | Et | 10 | DCM | 79 | 90 |
| 9 | F | Et | 10 | EtOAc | 75 | 91 |
| 10 | F | Et | 5 | Toluene | 84 | 98 |
| 11 | F | Et | 2.5 | Toluene | 79 | 97 |
| 12 | F | Et | 1.0 | Toluene | 77 | 97 |
| 13 | F | Me | 1.0 | Toluene | 70 | 95 |
| 14 | F | tBu | 1.0 | Toluene | 50 | 85 |
| 15d | F | Et | 1.0 | Toluene | 74 | 97 |
| 16e | F | Et | 1.0 | Toluene | 69 | 97 |
With the optimized reaction conditions in hand (Table 1, entry 12), we examined the substrate scope of this vinylogous Michael addition/isomerization cascade reactions (Scheme 3). Firstly, we investigated β-aryl-substituted nitroalkenes 2a–l with diverse electronic and steric properties. It was found that nitroalkenes with both electron-donating and electron-withdrawing substituents at different positions on the phenyl ring give the corresponding products 3a–j with excellent enantioselectivities (97–99% ee). Notably, substrates 2k–l bearing 2-thienyl and 2-furan rings were also tolerated. β-Alkyl-substituted nitroalkenes 2m–2p are also tested. To our delight, this type of substrates was also amenable to the reaction protocol, giving rise to the desired products with 95–99% ee. However, β-disubstitutednitroolefins, such as 2q–2s, are still inactive under the current reaction conditions. Besides, we also tried β,γ-unsaturated α-ketoester as the activated alkene partner (for example 2t), however, unfruitful results were obtained. The absolute configuration of the product 3b were unambiguously determined to be (E, S) by X-ray crystallography (Scheme 4). It needs to be emphasized that only E-isomer of RC-type product 3 could be detected. The other products were presumed to have the same absolute configuration as that of 3b, considering a uniformly stereocontrol mode was maintained during the reactions.
 | ||
| Scheme 3 Substrate scope. Reaction conditions: 0.4 mmol 2a with 0.8 mmol 1 (2.0 equiv.), and 1.0 mol% F (0.004 mmol) in toluene (2.0 mL, 0.2 M) at −10 °C for 24 h. Yields are for the isolated products based on 2 after purification by silica gel flash column chromatography. The ee values were determined by chiral HPLC analysis. | ||
 | ||
| Scheme 4 X-ray structure of 3b. | ||
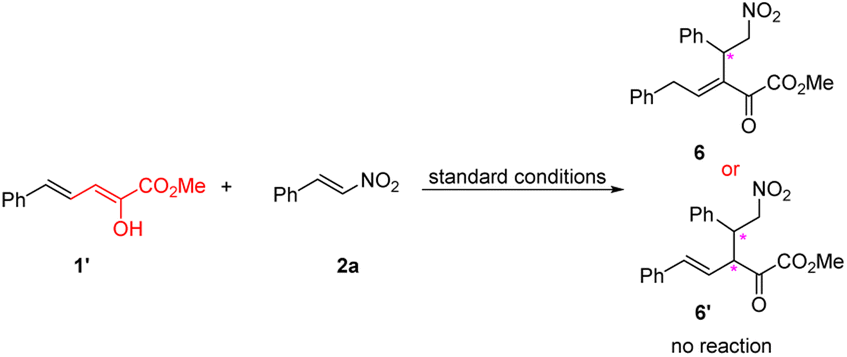
Finally, we study the reactivity of a known stable dienolate of vinylogous α-ketoester 1′,17c conceiving that the scope of vinylogous α-ketoester could be further expanded if it's workable for the current reaction (eqn (3)). However, after many trials, we found this kind of substrate is totally inactive in the current reaction conditions, and the enol form 1′ keeps intact.
To prove the practicality of this methodology, gram-scale synthesis of 3o was carried out by employing 20 mmol 2o and 1.5 equiv of 1. To our delight, when the temperature rised to 0 °C, the amount of catalyst loading could be reduced to 0.5 mol% without significantly affecting the catalytic efficiency as well as the ee of the product (Scheme 5, TON = 160 based on the isolated yield, the amount of substrate, and the catalyst loading).22 The highly functionalized Rauhut–Currier type products in this study could be utilized as useful synthetic intermediates in organic synthesis. For example, the terminal CC double in 3p can be selectively transformed into aldehyde 7 by using ruthenium-catalyzed oxidative cleavage of alkene or internal alkene 8 by using intermolecular olefin metathesis.
 | ||
| Scheme 5 Gram-scale synthesis and synthetic applications. | ||
Control experiments were conducted to clarify the reactive intermediate and pathway of this reaction. We performed the reaction of 4 with 2a under standard reaction conditions and found that there was no product detected. This fact not only indicates that β,γ-unsaturated α-ketoester 4 is not the reactive intermediate for this formal RC-reaction (Scheme 6), but also suggests deprotonation of vinylogous α-ketoester 1 is more feasible under the action of bifunctional catalyst F. In addition, the intermediate of α-selective vinylogous Michael addition INT (see Scheme 1c) could be detected at an early stage of reaction. On the basis of the above experiments, a sequential of α-vinylogous Michael addition, followed by isomerization of the terminal double bond is involved, and these two steps are all catalyzed by the same bifunctional Lewis base/hydrogen bonding organocatalyst F.
 | ||
| Scheme 6 Control experiment. | ||
Conclusions
In summary, a highly efficient cascade reaction by using vinylogous α-keto esters as the nucleophiles was developed in the presence of the low loading squaramide organocatalyst. Rauhut–Currier type products were obtained with excellent enantioselectivities (94–99% ee). Control experiments prove that the reaction proceeds via α-selectivity of vinylogous Michael addition, followed by isomerization of the terminal double bond.Author contributions
W. Guo: supervision, conceptualization, methodology, investigation, data curation, and writing–review & editing. Z. Weng: methodology, investigation, data curation, and writing–original draft. Y. Zhou: methodology, investigation, data curation, and writing–original draft. X. Yue, and F. Jiang: resources. Z. Weng, and Y. Zhou contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the start-up funding of Changzhou University (ZMF21020030). We also thank the analysis and testing centre, NERC Biomass of Changzhou University for the assistance in NMR analysis. The authors also thank Prof. H. Huang at Changzhou university for sharing the chiral HPLC instrument.Notes and references
- For selected reviews: (a) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076–3154 CrossRef CAS PubMed; (b) G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929–1972 CrossRef CAS PubMed; (c) J. Christoffers, Synlett., 2001, 723–732 CrossRef CAS.
- (a) S. P. Brown, N. C. Goodwin and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192–1194 CrossRef CAS PubMed ; For seminal work on chiral Lewis acid catalyzed vinylogous Mukaiya−Michael reaction of 2-silyloxyfurans with activated alkenes:; (b) H. Kitajima, K. Ito and T. Katsuki, Tetrahedron, 1997, 53, 17015–17028 CrossRef CAS; (c) H. Kitajima and T. Katsuki, Synlett., 1997, 568–570 CrossRef CAS; (d) G. Desimoni, G. Faita, S. Filippone, M. Mella, M. G. Zampori and M. Zema, Tetrahedron, 2001, 57, 10203–10212 CrossRef CAS.
- For selected examples: (a) A. Takahashi, H. Yanai and T. Taguchi, Chem. Commun., 2008, 2385–2387 RSC; (b) Q. Zhang, X. Xiao, L. Lin, X. Liu and X. Feng, Org. Biomol. Chem., 2011, 9, 5748–5754 RSC; (c) T. A. Qin, R. P. Johnson and J. A. Porco, J. Am. Chem. Soc., 2011, 133, 1714–1717 CrossRef CAS; (d) E. K. Kemppainen, G. Sahoo, A. Valkonen and P. M. Pihko, Org. Lett., 2012, 14, 1086–1089 CrossRef CAS PubMed.
- For selected examples: (a) B. M. Trost and J. Hitce, J. Am. Chem. Soc., 2009, 131, 4572–4573 CrossRef CAS PubMed; (b) M. Terada and K. Ando, Org. Lett., 2011, 13, 2026–2029 CrossRef CAS PubMed; (c) A. Quintard, A. Lefranc and A. Alexakis, Org. Lett., 2011, 13, 1540–1543 CrossRef CAS PubMed; (d) J. Wang, C. Qi, Z. Ge, T. Cheng and R. Li, Chem. Commun., 2010, 46, 2124–2126 RSC; (e) M. S. Manna, V. Kumar and S. Mukherjee, Chem. Commun., 2012, 48, 5193–5195 RSC; (f) M. S. Manna and S. Mukherjee, Chem. Sci., 2014, 5, 1627–1633 RSC.
- For selected examples: (a) H. Huang, Z. Jin, K. Zhu, X. Liang and J. Ye, Angew. Chem., Int. Ed., 2011, 50, 3232–3235 CrossRef CAS PubMed; (b) L. Lin, J. Zhang, X. Ma, X. Fu and R. Wang, Org. Lett., 2011, 13, 6410–6413 CrossRef CAS PubMed.
- For the selected examples using α,α-dicyanoalkene as the nucleophile: (a) J.-W. Xie, W. Chen, R. Li, M. Zeng, W. Du, L. Yue, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Angew. Chem., Int. Ed., 2007, 46, 389–392 CrossRef CAS PubMed; (b) D. Xue, Y.-C. Chen, Q.-W. Wang, L.-F. Cun, J. Zhu and J.-G. Deng, Org. Lett., 2005, 7, 5293–5296 CrossRef CAS PubMed; (c) J.-W. Xie, L. Yue, D. Xue, X. L. Ma, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Chem. Commun., 2006, 1563–1565 RSC; (d) J. Alemán, C. B. Jacobsen, K. Frisch, J. Overgaard and K. A. Jørgensen, Chem. Commun., 2008, 632–634 RSC ; for the selected examples using 3-Alkylidene oxindoles as the nucleophile:; (e) C. Curti, G. Rassu, V. Zambrano, L. Pinna, G. Pelosi, A. Sartori, L. Battistini, F. Zanardi and G. Casiraghi, Angew. Chem., Int. Ed., 2012, 51, 6200–6204 CrossRef CAS PubMed; (f) Q. Chen, G. Wang, X. Jiang, Z. Xu, L. Lin and R. Wang, Org. Lett., 2014, 16, 1394–1397 CrossRef CAS PubMed; (g) G. Rassu, V. Zambrano, L. Pinna, C. Curti, L. Battistini, A. Sartori, G. Pelosi, G. Casiraghi and F. Zanardi, Adv. Synth. Catal., 2014, 356, 2330–2336 CrossRef CAS ; for the selected examples using β-substituted cyclohexenone as the nucleophile:; (h) G. Bencivenni, P. Galzerano, A. Mazzanti, G. Bartoli and P. Melchiorre, Proc. Natl. Acad. Sci., 2010, 107, 20642–20647 CrossRef CAS PubMed.
- For selected examples: (a) J. Wang, J. Chen, C. W. Kee and C.-H. Tan, Angew. Chem., Int. Ed., 2012, 51, 2382–2386 CrossRef CAS PubMed; (b) V. Gupta, V. S. Sudhir, T. Mandal and C. Schneider, Angew. Chem., Int. Ed., 2012, 51, 12609–12612 CrossRef CAS PubMed; (c) D. Yang, L. Wang, F. Han, D. Zhao, B. Zhang and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 6739–6742 CrossRef CAS PubMed; (d) Y. Gu, Y. Wang, T.-Y. Yu, Y.-M. Liang and P.-F. Xu, Angew. Chem., Int. Ed., 2014, 53, 14128–14131 CrossRef CAS PubMed; (e) G. Zhan, Q. He, X. Yuan and Y.-C. Chen, Org. Lett., 2014, 16, 6000–6003 CrossRef CAS PubMed.
- For other selected examples using the in situ generated nuceophiles in vinylogoues Michael reactions: (a) G. R. Boyce, S. B. Liu and J. S. Johnson, Org. Lett., 2012, 14, 652–655 CrossRef CAS PubMed; (b) G. R. Boyce and J. S. Johnson, Angew. Chem., Int. Ed., 2010, 49, 8930–8933 CrossRef CAS PubMed.
- S. E. Denmark, J. R. Heemstra and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682–4698 CrossRef CAS PubMed.
- For selected examples: (a) A. Yamaguchi, N. Aoyama, S. Matsunaga and M. Shibasaki, Org. Lett., 2007, 9, 3387–3390 CrossRef CAS PubMed; (b) J. Stiller, E. Marqués-López, R. P. Herrera, R. Fröhlich, C. Strohmann and M. Christmann, Org. Lett., 2011, 13, 70–73 CrossRef CAS PubMed; (c) B. Han, Y.-C. Xiao, Z.-Q. He and Y.-C. Chen, Org. Lett., 2009, 11, 4660–4663 CrossRef CAS PubMed; (d) D. Enders, X. Yang, C. Wang, G. Raabe and J. Runsik, Chem.–Asian J., 2011, 6, 2255–2259 CrossRef CAS PubMed; (e) B. Han, Z.-Q. He, J.-L. Li, R. Li, K. Jiang, T.-Y. Liu and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5474–5477 CrossRef CAS PubMed; (f) Marqués-López, R. P. Herrera, T. Marks, W. C. Jacobs, D. Könning, R. M. De Figueiredo and M. Christmann, Org. Lett., 2009, 11, 4116–4119 CrossRef PubMed; (g) N. Utsumi, H. Zhang, F. Tanaka and C. F. Barbas III, Angew. Chem., Int. Ed., 2007, 46, 1878–1880 CrossRef CAS PubMed; (h) I. Iriarte, O. Olaizola, S. Vera, I. Gamboa, M. Oiarbide and C. Palomo, Angew. Chem., Int. Ed., 2017, 56, 8860–8864 CrossRef CAS PubMed; (i) O. Olaizola, I. Iriarte, G. Zanella, E. Gomez-Bengoa, I. Ganboa, M. Oiarbide and C. Palomo, Angew. Chem., Int. Ed., 2019, 58, 14250–14254 CrossRef CAS; (j) H. Zhang, J. He, Y. Chen, C. Zhuang, C. Jiang, K. Xiao, Z. Su, X. Ren and T. Wang, Angew. Chem., Int. Ed., 2021, 60, 19860–19870 CrossRef CAS PubMed; (k) B. Wu, Z. Yu, X. Gao, Y. Lan and Y.-G. Zhou, Angew. Chem., Int. Ed., 2017, 56, 4006–4010 CrossRef CAS PubMed; (l) J. A. Griswold, M. A. Horwitz, L. V. Leiva and J. S. Johnson, J. Org. Chem., 2017, 82, 2276–2280 CrossRef CAS PubMed; (m) G. Tong, B. Zhu, R. Lee, W. Yang, D. Tan, C. Yang, Z. Han, L. Yan, K.-W. Huang and Z. Jiang, J. Org. Chem., 2013, 78, 5067–5072 CrossRef CAS PubMed; (n) J. Guang, S. Rout, M. Bihani, A. J. Larson, H. D. Arman and J. C.-G. Zhao, Org. Lett., 2016, 18, 2648–2651 CrossRef CAS PubMed; (o) M. Frias, R. Mas-Ballesté, S. Arias, C. Alvarado and J. Alemán, J. Am. Chem. Soc., 2017, 139, 672–679 CrossRef CAS PubMed . For selected examples about employing other catalytic methods to switch the conventional γ- to unusual α-regioselectivity: via NHC catalysis,; (p) Y.-M. Zhao, M. S. Cheung, Z. Lin and J. Sun, Angew. Chem., Int. Ed., 2012, 51, 10359–11036 CrossRef CAS PubMed ; via water-containing solvent,; (q) M. Woyciechowska, G. Forcher, S. Buda and J. Mlynarski, Chem. Commun., 2012, 48, 11029–11031 RSC; (r) T. E. Campano, I. Iriarte, O. Olaizola, J. Etxabe, A. Mielgo, I. Ganboa, J. M. Odriozola, J. M. García, M. Oiarbide and C. Palomo, Chem.–Eur. J., 2019, 25, 4390–4397 CrossRef CAS PubMed.
- F. Giacalone, M. Gruttadauria, P. Agrigento and R. Noto, Chem. Soc. Rev., 2012, 41, 2406–2447 RSC.
- For selected examples: (a) P. Dambruoso, A. Massi and A. Dondoni, Org. Lett., 2005, 7, 4657–4660 CrossRef CAS PubMed; (b) P. Chen, K. Wang, B. Zhang, W. Guo, Y. Liu and C. Li, Chem. Commun., 2019, 55, 12813–12816 RSC . The recent examples using α-ketoesters as the nuclephiles in catalytic asymmetric reactions:; (c) S. Mondal, R. D. Aher, V. Bethi, Y.-J. Lin, T. Taniguchi, K. Monde and F. Tanaka, Org. Lett., 2022, 24, 1853–1858 CrossRef CAS PubMed; (d) P. V. Chouthaiwale, R. D. Aher and F. Tanaka, Angew. Chem., Int. Ed., 2018, 57, 13298–13301 CrossRef CAS PubMed.
- For leading examples: (a) Y. Xu, S. Matsunaga and M. Shibasaki, Org. Lett., 2010, 12, 3246–3249 CrossRef CAS PubMed; (b) O. Basle, W. Raimondi, M. M. Sanchez Duque, D. Bonne, T. Constantieux and J. Rodriguez, Org. Lett., 2010, 12, 5246–5249 CrossRef CAS PubMed.
- (a) A. Nakamura, S. Lectard, D. Hashizume, Y. Hamashima and M. Sodeoka, J. Am. Chem. Soc., 2010, 132, 4036–4037 CrossRef CAS PubMed; (b) D. Shi, Y. Xie, H. Zhou, C. Xia and H. Huang, Angew. Chem., Int. Ed., 2012, 51, 1248–1251 CrossRef CAS PubMed.
- W. Raimondi, O. Basle, T. Constantieux, D. Bonne and J. Rodriguez, Adv. Synth. Catal., 2012, 354, 563–568 CrossRef CAS.
- (a) W. Guo, Y. Luo, H. H. Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed; (b) W. Guo, M. Wang, Z. Han, H. Huang and J. Sun, Chem. Sci., 2021, 12, 11191–11196 RSC; (c) W. Guo, F. Jiang, S. Li and J. Sun, Chem. Sci., 2022, 13, 11648–11655 RSC.
- (a) N. V. Shymanska and J. G. Pierce, Org. Lett., 2017, 19, 2961–2964 CrossRef CAS PubMed; (b) N. P. Massaro and J. G. Pierce, Org. Lett., 2020, 22, 5079–5084 CrossRef CAS PubMed; (c) The enol-form of α-keto ester 1′ is a known compound, for its synthesis and ulitlity, see ref. 17a..
- H. Liu, D. Leow, K.-W. Huang and C.-H. Tan, J. Am. Chem. Soc., 2009, 131, 7212–7213 CrossRef CAS PubMed.
- J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem.–Eur. J., 2011, 17, 6890–6899 CrossRef PubMed.
- Organocatalyst F was firstly developed by Rawal's group: Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153–156 CrossRef CAS PubMed.
- Indeed, some unclear impurities could be observed by TLC analysis, however, the isomer 4 and regio-isomer 5 could not be detected by crude 1H NMR. Moreover, we found vinylous α-ketoester 1 is unstable at room temperature, the “abnormal” higher yield at lower reaction temperature may be ascribed to the more stability of 1 at the indicated reaction temperature..
- For a more scientific understanding the concept of TON, see: S. Kozuch and J. M. L. Martin, ACS Catal., 2012, 2, 2787–2794 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1536616. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06416b |
| ‡ Z. Weng and Y. Zhou contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |