
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnnulated carbamates are precursors for the ring contraction of the adamantane framework†
Radim Hrdina *a,
Oksana M. Holovko-Kamoshenkovaad,
Ivana Císařováb,
Filip Kouckýb and
Oldřich Machalickýc
*a,
Oksana M. Holovko-Kamoshenkovaad,
Ivana Císařováb,
Filip Kouckýb and
Oldřich Machalickýc
aCharles University, Faculty of Science, Department of Organic Chemistry, Hlavova 8, 12840 Praha, Czech Republic. E-mail: hrdina@natur.cuni.cz
bCharles University, Faculty of Science, Department of Inorganic Chemistry, Hlavova 8, 12840 Praha, Czech Republic
cUniversity of Pardubice, Faculty of Chemical Technology, Studentská 573, 53210 Pardubice, Czech Republic
dUzhhorod National University, Narodna Ploshcha 3, 88000 Uzhhorod, Ukraine
First published on 31st October 2022
Abstract
We report a protocol for the one-pot two-step synthesis of noradamantane methylene amines. The first step is the triflic acid-promoted decarboxylation of adamantane carbamates, which causes rearrangement of the adamantane framework to form noradamantane iminium salts, which are reduced to amines in the second separate step.
Introduction
Noradamantane (adamantane lacking one methylene unit) derivatives such as amines,1 acids,2 methylene alcohols,3 and others4 are useful building blocks in organic synthesis. Together with adamantane derivatives,5 they are typically used to make target molecules more lipophilic, bulky or conformationally rigid, which is exploited in drug6 and catalyst design7 or in materials science for preparation of polymers, dendrimers, and light emitting and other special devices.8 Noradamantane compounds can also serve as precursors for the synthesis of adamantane derivatives via ring expansion methods.9 Various simple noradamantane derivatives (e.g. containing methylene amine group) exhibit biological activity and were tested as inhibitors (Fig. 1).10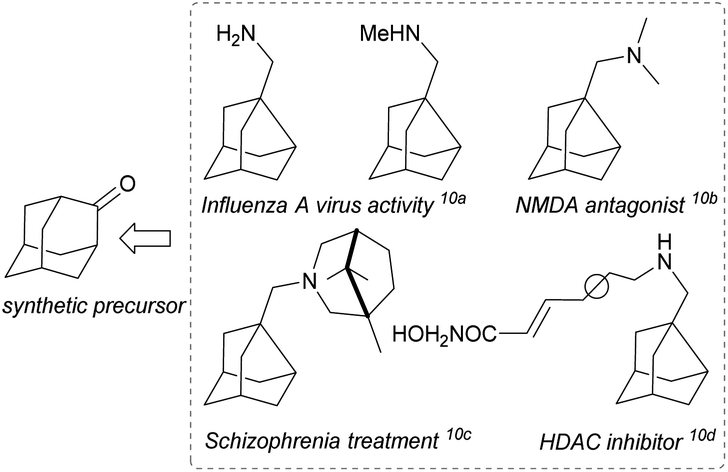 | ||
| Fig. 1 Bioactive noradamantane-3-methylene amine derivatives. | ||
Compared to adamantane (Td point group), the noradamantane skeleton (C2v) has a lower symmetry and exhibits different reactivity towards oxidation reactions, which complicates the synthesis of on cage-substituted derivatives.11 Noradamantane framework can be prepared by skeletal rearrangement of deltacyclane,11 from 4-protoadamantanone12 or from suitable adamantane precursors such as 1,3-adamantane diols.13 Most syntheses of noradamantane cage compounds start from adamantan-2-one.14 This substantially limits the preparation of (on cage)-substituted derivatives as there are not many C2 symmetric substrates available or easily synthesizable (Fig. 2).
 | ||
| Fig. 2 New approach to noradamantane-3-methylene amines from adamantan-1-ol. | ||
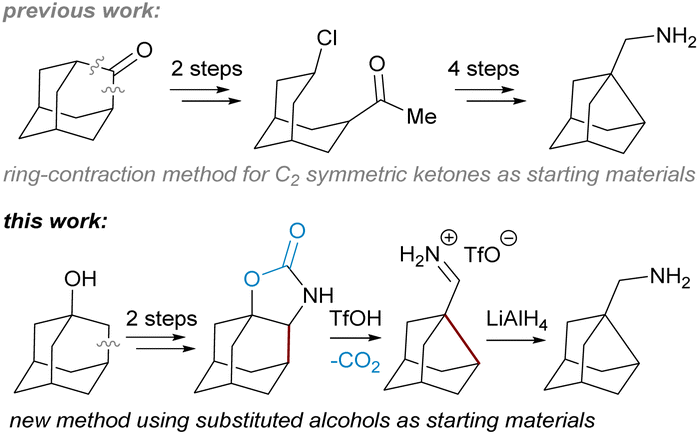
We have recently contributed with a new method for the preparation of noradamantane carbaldehydes based on the decarboxylation of annulated N-methylated adamantane carbamates followed by a nucleophilic 1,2-alkyl shift and hydrolysis of the resulting iminium salts.15
Results and discussion
In this work, we focus on the triflic acid promoted decarboxylation of N-unsubstituted carbamates, which leads to the ring contraction of the adamantane framework by nucleophilic 1,2-alkyl shift16 producing noradamantane carbaldiminium salts, and the one-pot reduction of the resulting iminium salts to noradamantane methylene amines was performed in the same pot subsequently using LiAlH4 (Fig. 2). The decarboxylation of the carbamate with triflic acid in the presence of HX was performed to get a halogen in the position next to an amino group on the adamantane framework to have possibility to study all steps of the cascade process (decarboxylation and 1,2-alkyl shift) separately. The ring contraction of the adamantane framework containing a leaving group in the position next to an amine is now described in acidic and basic media and is analogical to aziridine formation17 for non-bridged flexible aliphatic systems (Fig. 3).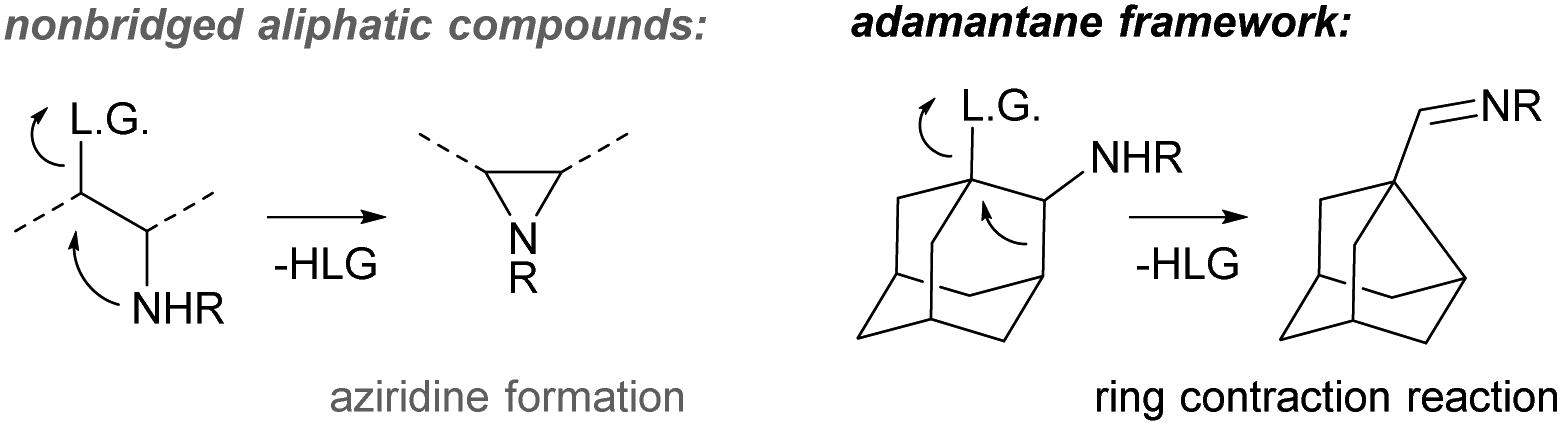 | ||
| Fig. 3 Nucleophilic rearrangement reaction of the adamantane framework containing a leaving group next to the amino group. | ||
The cyclic carbamate 3a was prepared in two steps from adamantan-1-ol 1a and was used as a model compound for optimisation of the reaction conditions.18 The optimal conditions; 2 equivalents of trifluoromethanesulfonic acid, 1,2,4-trichlorobenzene as a solvent, 0.06 M dilution, 120 °C and 4 h reaction time for the decarboxylation and rearrangement step.
The reduction of the in situ formed iminium salt (Int1-OTf, Fig. 5A) was performed by adding LiAlH4 and THF to the reaction mixture and stirring at 25 °C for three hours. This method was used for the rearrangement and reduction of four various starting materials substituted on the adamantane cage leading to compounds 5, 9, 11, 13 (Fig. 4). For the decarboxylation and reduction of N-substituted carbamates (Me and 2,2′-dichlorobenzyl) the reaction conditions (dilution 0.5 M, temperature 140 °C and reaction time 16 h) were changed.
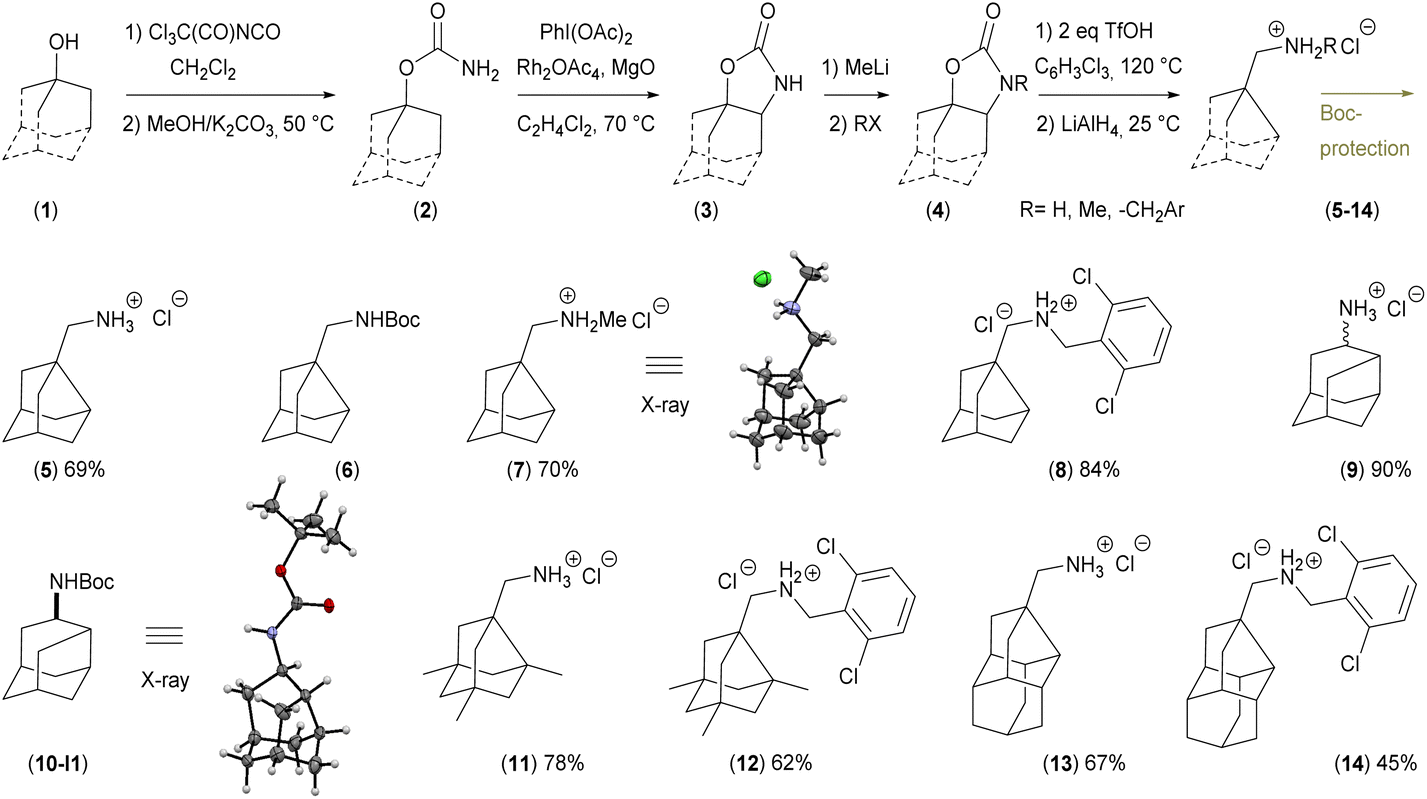 | ||
| Fig. 4 Scope of the new transformation. Yields of isolated products are stated. | ||
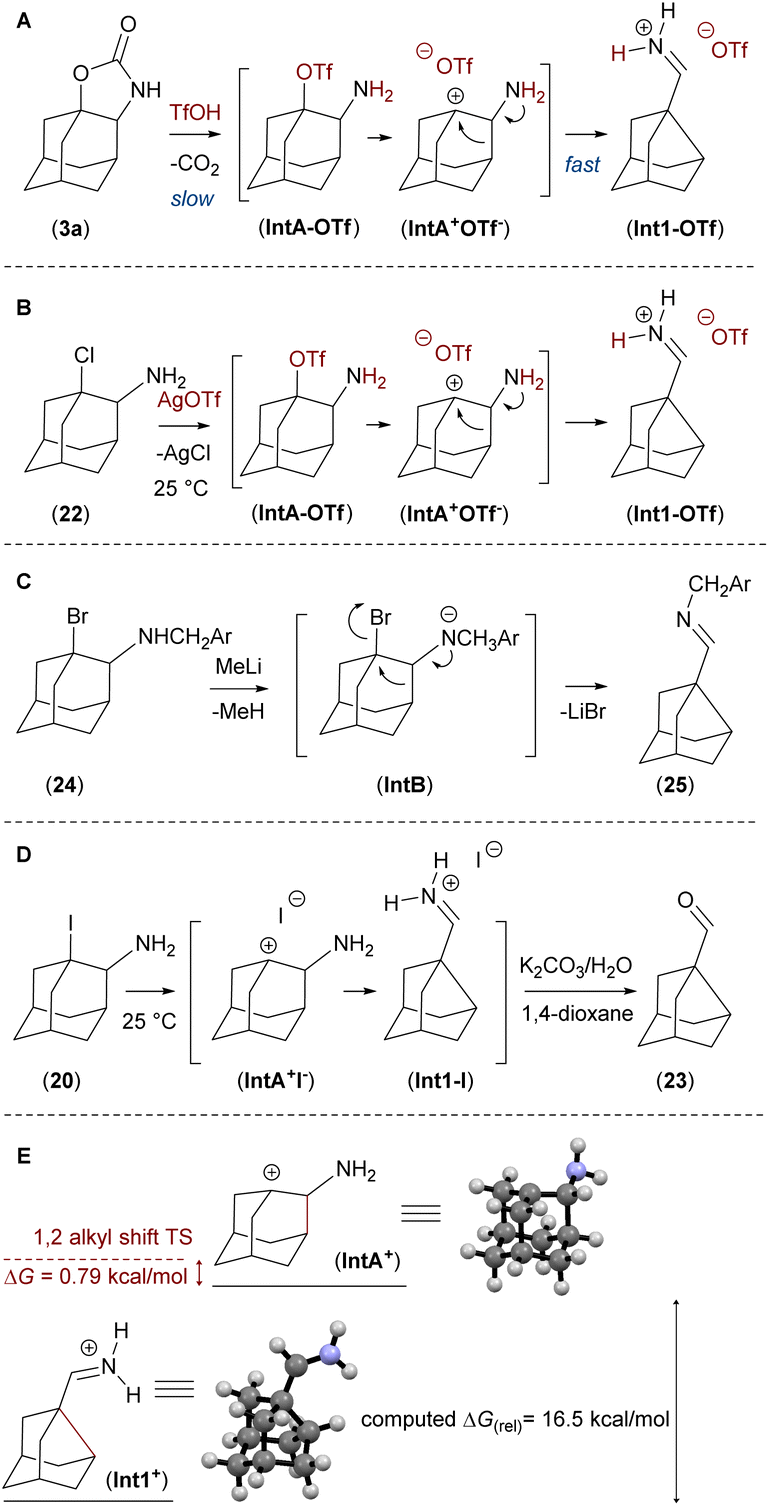 | ||
| Fig. 5 (A): Ring-contraction reaction of the adamantane framework in acidic medium; (B): generation of the reactive intermediate IntA-OTf from chloro-derivative 22; (C): ring-contraction reaction of 24 with strong base; (D): ring-contraction reaction of 20 in polar basic medium; (E): computed relative stability of intermediate IntA+ versus target structure Int1+ using DFT method B3LYP/deF2-TZVPP and computed ΔG for transition state of the 1,2-alkyl shift. | ||
Compound 7 was crystallised and characterised by X-ray crystallography (Fig. 4). Starting materials 3e, 4ea (N-Me carbamate), and 4eb (N-CH2Ar carbamate) undergo the reaction sequence, but the final products are not obtained in required purity and the final amine 19 was prepared by reductive amination from the aldehyde 18 (Scheme 1). The amines can be stored as hydrochloride salts or protected with e.g. Boc group (compounds 6 and 10). Diastereomers of 9 were resolved by fractional crystallisation of the Boc-protected compound 10 in pentane.19 Isomer 10-I1 was recrystallised and its configuration was determined using X-ray crystallography (Fig. 4).
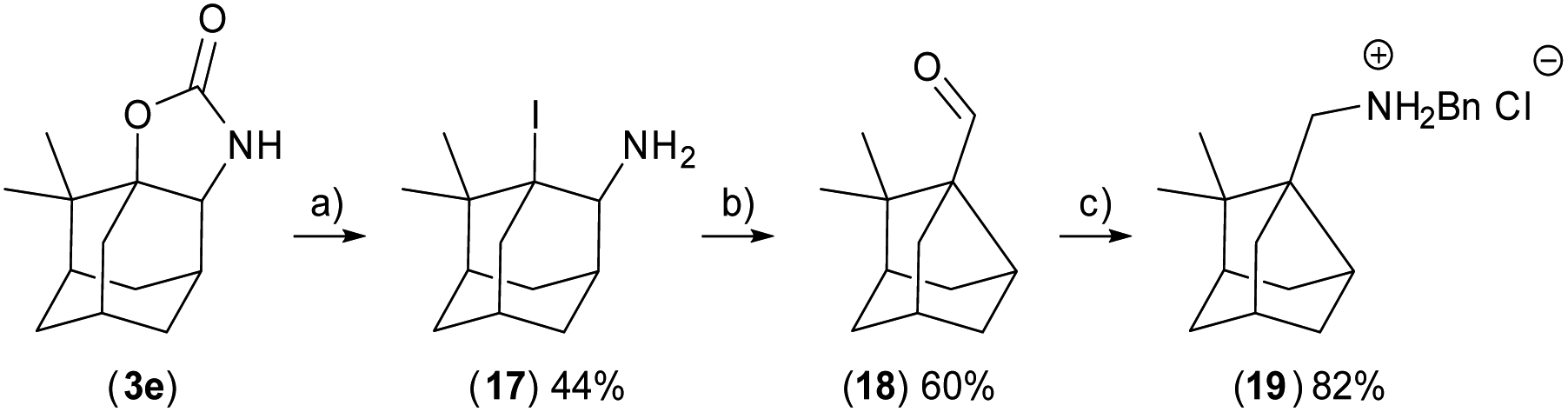 | ||
| Scheme 1 Rearrangement reaction of 17 at basic media to imine intermediate, which is in situ hydrolysed to aldehyde 18 and amination reaction of 18 to amine hydrochloride salt 19. (a) TfOH/KBr/CH2Cl2, 40 °C; (b) K2Co3/H2O, 100 °C; (c) (1). PhCH2NH2/MgSO4/THF, 25 °C; (2).LiAIH/THF/25 °C; (3) HCl/Et2O. | ||
The proposed mechanism of the ring contraction reaction cascade is the triflic acid promoted decarboxylation of 3 to form intermediate IntA-OTf, followed by a nucleophilic 1,2-alkyl shift to form the iminium salt Int1-OTf. The calculated (DFT method) difference in stability of the intermediate cations IntA+ and Int1+ is ΔG = 16.5 kcal mol−1 (Fig. 5E).
The iminium salt intermediate Int1-OTf is reduced in the next step using LiAlH4 to form the product 5 (Fig. 4) or it can be trapped by other nucleophiles such as a nitrile using the Strecker reaction with TMSCN to form compound 15, precursor for non-biogenic α-amino acids20 as demonstrated on the synthesis of compound 16 (Scheme 2).
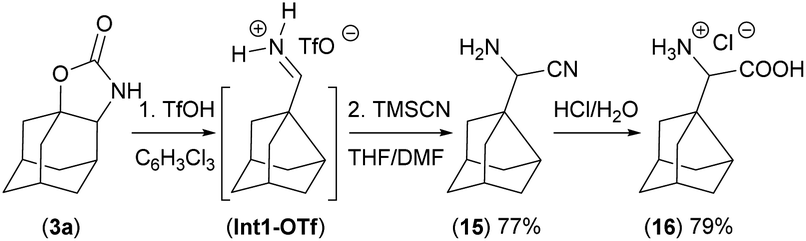 | ||
| Scheme 2 Trapping of the iminium intermediate (Int1-OTf) by TMSCN and preparation of the α-amino acid hydrochloride salt 16. | ||
The ring-contraction reaction was then studied using compounds containing halogens as a leaving group next to an amine group mimicking the reactive intermediate IntA-OTf, which is obtained after the decarboxylation step (Fig. 5A).
First, we performed the ring contraction of 24 using MeLi to deprotonate an amine group, to generate imine 25, which was isolated and characterised (Fig. 5C).
Then we prepared compounds 20, 21, 22 (ref. 18) and studied the ring contraction reaction in a polar solvent in analogy to SN1-substitution reactions. Thermal cleavage of 1-iodo derivative 20 in a polar solvent mixture (1,4-dioxane/K2CO3 water solution), provided the ring contracted imine Int1-I, which was in situ hydrolysed to aldehyde 23 (Fig. 5D).
Kinetics of the thermal rearrangements were studied with compounds 20, 21 and 22 in 1,4-dioxane/K2CO3 water solution mixture using 1H-NMR measurements at various temperatures and concentrations (Scheme 3).
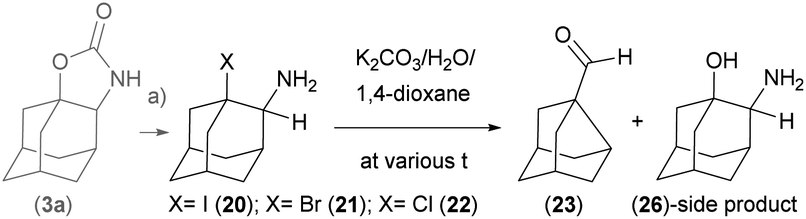 | ||
| Scheme 3 Kinetics of the thermal rearrangement reactions in a polar solvent mixture. (a) TfOH/KX/CH2Cl2, 40 °C and neutralisation; (b) K2CO3/H2O/1,4-dioxane, VT-exp. | ||
These reactions resemble SN1-substitution reactions following the same trend: iodo-derivative 20 undergoes the ring contraction reaction already at room temperature, slower ring-contraction reaction proceeds with bromo-derivative 21 and the slowest reaction occurs with chloro-derivative 22. The product of a substitution side reaction, 2-amino adamantane-1-ol 26 was detected, isolated and characterised (Scheme 3). Apparent first order kinetic constants estimated for solvolysis of 1-halogeno-2-amine adamantanes to the main product at 50 °C are within the range: k = (130 ± 20) × 10−5 s−1 for X = I; k = (125 ± 20) × 10−5 s−1 for X = Br; and k = (3 ± 0.2) × 10−5 s−1 for X = Cl. For determination of kinetic constants of solvolysis of individual 1-halogen-2-amine adamantanes see ESI.†
Taking all data into account, the rate limiting step of the ring contraction reaction is the decarboxylation of compound 3 to the intermediate IntA-OTf. The computed barrier of the next step 1,2-alkyl shift between IntA+ and Int1+ is only 0.79 kcal mol−1 (Fig. 5E).21 This is in line with an experiment where intermediate IntA-OTf was generated from compound 22 by adding AgOTf to the solution at room temperature and instantaneous formation of the imine Int1-OTf was observed (Fig. 5B).9
Conclusions
We have shown that adamantane based cyclic carbamates are suitable precursors for ring contraction of the adamantane framework and that the formed iminium moiety can be reduced or trapped by a nucleophile gaining access to noradamantane amino derivatives. The ring contraction reaction can be performed under acidic or basic conditions from suitable starting materials, and proceeds through imine intermediates. The ring contraction reaction of adamantane based 2-amino-1-alcohols as starting materials is part of our future studies.Author contributions
R. H. and O. H. performed the synthetic work. I. C. is responsible for X-ray crystallographic analysis, F. K. for NMR measurements and O. M. for kinetic studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Artur Mardyukov for DFT analyses and for manuscript proofreading.Notes and references
- (a) E. Torres, R. Fernandez, S. Miquet, M. Font-Bardia, E. Vanderlinden, L. Naesens and S. Vazquez, ACS Med. Chem. Lett., 2012, 3, 1065–1069 CrossRef CAS; (b) S. Codony, E. Valverde, R. Leiva, J. Brea, M. I. Loza, C. Morisseau, B. D. Hammock and S. Vázquez, Bioorg. Med. Chem., 2019, 27, 115078 CrossRef CAS; (c) C.-M. Chan, Q. Xing, Y.-C. Chow, S.-F. Hung and W.-Y. Yu, Org. Lett., 2019, 21, 8037–8043 CrossRef CAS; (d) F. Li, C. Ma, W. F. DeGrado and J. Wang, J. Med. Chem., 2016, 59, 1207–1216 CrossRef CAS PubMed.
- (a) S. Jin, G. C. Haug, V. T. Nguyen, C. Flores-Hansen, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9764–9774 CrossRef CAS; (b) L. Wang, V. S. Guillen, N. Sharma, K. Flessa, J. Min, K. E. Carlson, W. Toy, S. Braqi, B. S. Katzenellenbogen, J. A. Katzenellenbogen, S. Chandarlapaty and A. Sharma, ACS Med. Chem. Lett., 2018, 9, 803–808 CrossRef CAS PubMed; (c) Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS PubMed; (d) G.-T. Zhu, F. Liu, S. He, X.-M. He, S.-K. Zhu and Y.-Q. Feng, RSC Adv., 2018, 8, 19486–19493 RSC.
- R. A. Moss, F. Zheng, J.-M. Fedé, L. A. Johnson and R. R. Sauers, J. Am. Chem. Soc., 2004, 126, 12421–12431 CrossRef CAS.
- (a) K. i. Takeuchi, I. Kitagawa, F. Akiyama, T. Shibata, M. Kato and K. Okamoto, Synthesis, 1987, 612–615 CrossRef CAS; (b) I. R. Likhotvorik, E. L. Tae, C. Ventre and M. S. Platz, Tetrahedron Lett., 2000, 41, 795–796 CrossRef CAS.
- (a) R. C. Fort and P. v. R. Schleyer, Chem. Rev., 1964, 64, 277–300 CrossRef CAS; (b) R. Yasue and K. Yoshida, Adv. Synth. Catal., 2021, 363, 1662–1671 CrossRef CAS; (c) R. Hrdina, Synthesis, 2019, 51, 629–642 CrossRef CAS.
- (a) L. Wanka, K. Iqbal and P. R. Schreiner, Chem. Rev., 2013, 113, 3516–3604 CrossRef CAS; (b) E. De Clercq, Nat. Rev. Drug Discovery, 2006, 5, 1015–1025 CrossRef CAS PubMed; (c) C. Tzitzoglaki, K. McGuire, P. Lagarias, A. Konstantinidi, A. Hoffmann, N. A. Fokina, C. Ma, I. P. Papanastasiou, P. R. Schreiner, S. Vázquez, M. Schmidtke, J. Wang, D. D. Busath and A. Kolocouris, ACS Chem. Biol., 2020, 15, 2331–2337 CrossRef CAS PubMed; (d) R. Hrdina, F. M. Metz, M. Larrosa, J. P. Berndt, Y. Y. Zhygadlo, S. Becker and J. Becker, Eur. J. Org. Chem., 2015, 2015, 6231–6236 CrossRef CAS; (e) V. V. Bakhonsky, A. A. Pashenko, J. Becker, H. Hausmann, H. J. De Groot, H. S. Overkleeft, A. A. Fokin and P. R. Schreiner, Dalton Trans., 2020, 49, 14009–14016 RSC.
- (a) K. A. Agnew-Francis and C. M. Williams, Adv. Synth. Catal., 2016, 358, 675–700 CrossRef CAS; (b) J.-P. Berndt, Y. Radchenko, J. Becker, C. Logemann, D. R. Bhandari, R. Hrdina and P. R. Schreiner, Chem. Sci., 2019, 10, 3324–3329 RSC; (c) A. Zapf, A. Ehrentraut and M. Beller, Angew. Chem., Int. Ed., 2000, 39, 4153–4155 CrossRef CAS.
- (a) H. Nasrallah and J.-C. Hierso, Chem. Mater., 2019, 31, 619–642 CrossRef CAS; (b) R. Ma, Z. Ma, X. Wang, Z. Si, Q. Duan and S. Shao, Chem. Eng. J., 2022, 47, 137517 CrossRef; (c) J. Belz, J. Haust, M. J. Müller, K. Eberheim, S. Schwan, S. Gowrisankar, F. Hüppe, A. Beyer, P. R. Schreiner, D. Mollenhauer, S. Sanna, S. Chatterjee and K. Volz, J. Phys. Chem. C, 2022, 126(23), 9843–9854 CrossRef CAS.
- B. Zonker, E. Duman, H. Hausmann, J. Becker and R. Hrdina, Org. Biomol. Chem., 2020, 18, 4941–4945 RSC.
- (a) G. Zoidis, N. Kolocouris, L. Naesens and E. De Clercq, Bioorg. Med. Chem., 2009, 17, 1534–1541 CrossRef CAS; (b) P. Camps, M. D. Duque, S. Vázquez, L. Naesens, E. DeClercq, F. X. Suredac, M. López-Querol, A. Camins, M. Pallàs, S. R. Prathalingam, J. M. Kelly, V. Romero, D. Ivorra and D. Cortés, Bioorg. Med. Chem., 2008, 16, 9925–9936 CrossRef CAS; (c) N. Takahashi and D. Mochizuki, Daisuke, Preparation and formulation of azepine derivatives as central nervous system agents,WO Patent WO9406773, 1994; (d) B. Gopalan, T. Ponpandian, V. Kachhadia, K. Bharathimohan, R. Vignesh, V. Sivasudar, S. Narayanan, B. Mandar, R. Praveen, N. Saranya, S. Rajagopal and S. Rajagopal, Bioorg. Med. Chem. Lett., 2013, 23, 2532–2537 CrossRef CAS.
- J. S. Wishnok, P. v. R. Schleyer, E. Funke, G. D. Pandit, R. O. Williams and A. Nickon, J. Org. Chem., 1973, 38, 539 CrossRef CAS.
- Z. Majerski, R. Šarac-Arneri, D. Škare and B. Lončar, Synthesis, 1980, 74 CrossRef CAS.
- M. Eakin, J. Martin and W. Parker, Chem. Commun., 1965, 206 RSC.
- R. M. Black and G. B. Gill, Chem. Commun., 1970, 972 RSC.
- B. Zonker, J. Becker and R. Hrdina, Org. Biomol. Chem., 2021, 19, 4027–4031 RSC.
- For Wagner-Meerwein reaction see: (a) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Angew. Chem., Int. Ed., 2013, 52, 9266–9270 CrossRef CAS PubMed; (b) D. J. Cram, J. Am. Chem. Soc., 1949, 71, 3863–3870 CrossRef CAS; (c) Z. Pakulski, P. Cmoch, A. Korda, R. Luboradzki, K. Gwardiak and R. Karczewski, J. Org. Chem., 2021, 86, 1084–1095 CrossRef CAS; (d) V. A. Shadrikova, E. V. Golovin, V. B. Rybakov and Y. N. Klimochkin, Chem. Heterocycl. Compd., 2020, 56, 898–908 CrossRef CAS; (e) H. A. Sharma, K. M. Mennie, E. E. Kwan and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 16090–16096 CrossRef CAS; (f) Y.-Y. Liu, Z. Ao, G.-M. Xue, X.-B. Wang, J.-G. Luo and L.-Y. Kong, Org. Lett., 2018, 20, 7953–7956 CrossRef CAS ; for Demjanov–Tiffeneau reaction see:; (g) S. M. Kohlbacher, V.-S. Ionasz, L. Ielo and V. Pace, Monatsh. Chem., 2019, 1–9 Search PubMed ; for Meinwald reaction see:; (h) D. Ma, C.-B. Miao and J. Sun, J. Am. Chem. Soc., 2019, 141, 13783–13787 CrossRef CAS; (i) R. Hrdina, C. E. Müller, R. C. Wende, K. M. Lippert, M. Benassi, B. Spengler and P. R. Schreiner, J. Am. Chem. Soc., 2011, 133, 7624–7627 CrossRef CAS ; for semi-pinacol reaction see:; (j) B. Wang and Y. Q. Tu, Acc. Chem. Res., 2011, 44, 1207–1222 CrossRef CAS PubMed.
- (a) A. Weissberger and H. Bach, Ber. Dtsch. Chem. Ges. B, 1931, 64B, 1095–1108 CrossRef CAS; (b) M. S. Kharasch and H. M. Priestley, J. Am. Chem. Soc., 1939, 61, 3425–3431 CrossRef CAS.
- R. Hrdina, M. Larrosa and C. Logemann, J. Org. Chem., 2017, 82, 4891–4899 CrossRef CAS.
- M. Horvat, L. Uzelac, M. Marjanovic, N. Cindro, O. Frankovic, K. Mlinaric-Majerski, M. Kralj and N. Basaric, Chem. Biol. Drug Des., 2012, 79, 497–506 CrossRef CAS PubMed.
- J. Müller, R. A. Kirschner, J. P. Berndt, T. Wulsdorf, A. Metz, R. Hrdina, P. R. Schreiner, A. Geyer and G. Klebe, ChemMedChem, 2019, 14, 663–672 CrossRef PubMed.
- D. T. Stoelting and V. J. Shiner Jr, J. Am. Chem. Soc., 1993, 115, 1695–1705 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2194850–2194852. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06402b |
| This journal is © The Royal Society of Chemistry 2022 |