
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Folic acid functionalization for targeting self-assembled paclitaxel-based nanoparticles†
Eleonora Colombo a,
Davide Andrea Coppinia,
Simone Maculana,
Pierfausto Senecia,
Benedetta Santinia,
Filippo Testab,
Lucia Salvionib,
Giovanni Maria Vanacorec,
Miriam Colombob and
Daniele Passarella*a
a,
Davide Andrea Coppinia,
Simone Maculana,
Pierfausto Senecia,
Benedetta Santinia,
Filippo Testab,
Lucia Salvionib,
Giovanni Maria Vanacorec,
Miriam Colombob and
Daniele Passarella*a
aDipartimento di Chimica, Università degli Studi di Milano, Via Golgi 19, 20133 Milano, Italy. E-mail: daniele.passarella@unimi.it
bDipartimento di Biotecnologie e Bioscienze, Università degli Studi di Milano Bicocca, Piazza della Scienza 2, 20126 Milano, Italy
cDipartimento di Scienza dei Materiali, Università degli Studi di Milano Bicocca, Via Roberto Cozzi 55, 20125 Milano, Italy
First published on 12th December 2022
Abstract
Hetero-nanoparticles self-assembled from a conjugate bearing folic acid as the targeting agent, and another bearing paclitaxel as the active agent are reported. Hetero-nanoparticles containing varying percentages of folic acid conjugates are characterised, and their biological activity is determined.
Introduction
The development of targeted drug delivery nanosystems is a challenging problem, aiming to provide an efficient transport of bioactive molecules and their site-specific release in the microenvironment of diseased tissues. Since several years we have been interested in modifying anticancer and neuroprotective drugs to obtain self-assembling nanoparticles (NPs) that could improve their therapeutic efficiency.Although traditional, carrier-based NPs have shown excellent progresses and promises in the field of cancer therapy, further improvements are still needed. For example, the drug-loading capacity of such carrier-based NPs is generally low (typically <10 wt%), which greatly reduces an efficient drug accumulation inside tumors and therapeutic efficacy of the released drugs.1 Additionally, meanwhile most reported nanocarriers are pharmaceutically inert, because of the sophisticated preparation procedures and excessive chemical treatments, application of these carriers raises concerns regarding their metabolisms, biodegradation, and potential long-term toxicities as well as serious inflammation.2 Because of that, self-assembling nanoparticles, which carry the therapeutic molecules by themselves instead of using other inert carriers, are a highly desirable alternative strategy for the development of NPs. They present in fact: (1) high drug-loading capacity; (2) precise control of drug loading because the nanostructures formed from customized individual molecular conjugates; (3) facile adjustment of the physicochemical features of the NPs by simply optimizing the molecular design; (4) avoiding tedious synthetic procedures for obtaining additional carriers and thus no carrier-induced potential cytotoxicity and immunogenicity; (5) enhancement of drug accumulation in tumors.3 With such advantages, we believe the development of this kind of nanoparticles will be the main trend in development of nanomedicine for drug delivery. During the years, we reported the synthesis of different lipid–drug conjugates, obtained by a covalent coupling of the drug to biocompatible lipid moieties through a linker. This kind of conjugates is capable to spontaneously assemble in water, forming NPs able to release a payload drug in cellular media.4–8 Additional modifications can be made on this simple design in order to obtain hetero-NPs bearing two different drugs (combining different conjugates that present the same lipidic self-assembling inducer);9,10 single and dual drug fluorescent hetero-NPs (where in one of the conjugates the drug is substituted by a fluorescent moiety)11,12 and NPs formed by self-assembling conjugate dual drugs (in which also the self-assembly inducer is pharmaceutically active).13,14 Recently, we focused on further improving these NPs, by exploiting targeted drug delivery through folate-containing hetero-NPs.
A wide variety of receptors, such as transferrin,15 death receptor (DR) complexes,16 epidermal growth factor receptor (EGFR),17 as well as tumor-specific antigens and folate ligands18 have been exploited to selectively target tumor tissues with nanodrugs. We focused our attention onto folate receptor α (FRα), which plays an important role in cancer biology. Firstly, this isoform has a limited physiological role in non-malignant tissues after embryogenesis, while it is overexpressed in a variety of cancer types.19–22 Moreover, FRα shows high affinity for non-physiological substrates, such as folic acid, compared with other folate transporters, enabling the development of folic acid nanoconjugates. In particular, folic acid has been intensively used for clinic applications,23–25 and even though no folate-based NPs have entered the clinic for cancer therapy yet, examples of folate-conjugated nanovehicles have been published in literature in the last years.26–28
The main objective of our work described here is to obtain hetero-NPs by the self-assembly of a conjugate based on folic acid (Fig. 1), to direct the hetero-NP to the tumor site, with another conjugate bearing the anticancer drug paclitaxel (PTX).
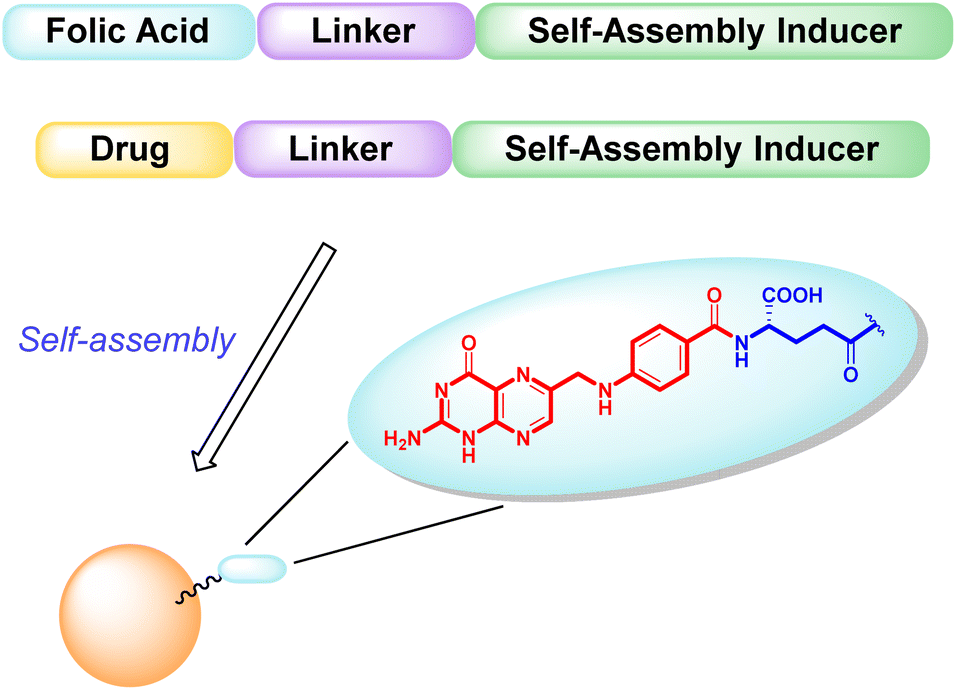 | ||
| Fig. 1 Schematic representation of targeted hetero-NPs, with folic acid highlighted in detail – pteroic acid in red, L-glutamic acid in blue. | ||
Both conjugates, shown in Fig. 2, consist of an active fragment (either folic acid or paclitaxel) linked to a lipophilic self-assembly inducer through a specific linker. The linker itself is designed to be quite stable for the folic acid conjugate (bottom structure, 2), to prevent the NP from losing its targeting agent; conversely, a labile linker is used for the drug conjugate (top structure, 1, Fig. 2), to release paclitaxel at its site of action. The synthesis of both conjugates, their ability to form hetero-self-assembled NPs, and their cytotoxicity against tumor cells are here reported.
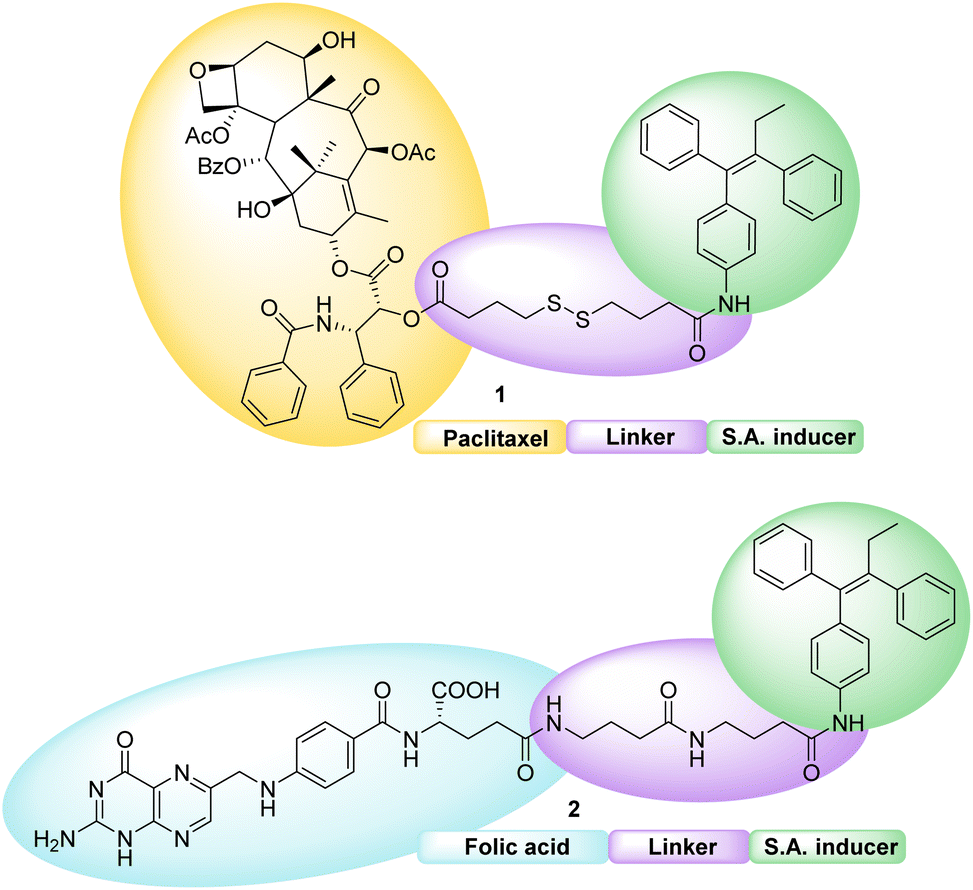 | ||
| Fig. 2 Chemical structure of paclitaxel (top, 1) and folic acid conjugates (bottom, 2). | ||
Results and discussion
As to the preparation of drug conjugate 1, the linker-self-assembly inducer moiety 5 was synthesized according to a previously reported strategy (Scheme 1).14 At last, target compound 1 was obtained through the condensation between said moiety 5 with PTX 6 in presence of EDC and DMAP. The conjugation occurred with complete regioselectivity, although in moderate yield.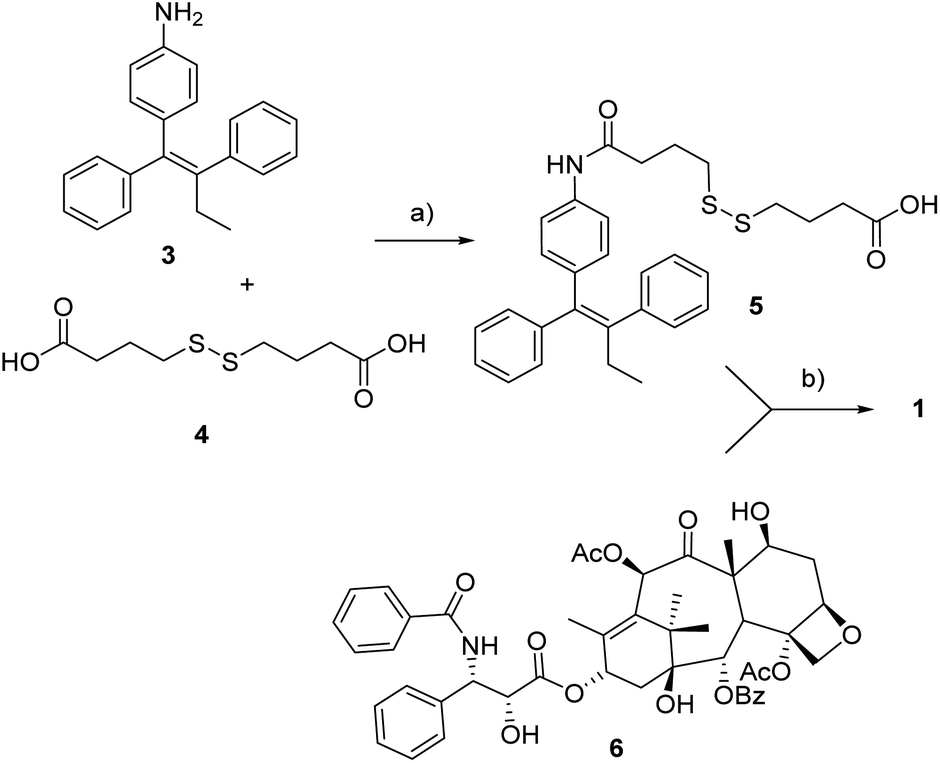 | ||
| Scheme 1 Synthesis of conjugate 1. a: DIPEA, HATU, dry THF, rt, 18 h, 30%, b: EDC·HCl, DMAP, rt, 22 h, 52%. | ||
A first attempted synthesis of the folic acid conjugate 2 resulted to be challenging (Scheme 2). The condensation of aniline 3 with N-Boc γ-aminobutyric acid 8 gave compound 9 that, after deprotection, was elongated with a second unit of N-Boc γ-aminobutyric acid, yielding Boc protected intermediate 11. After Boc removal, we obtained linker-self-assembly inducer conjugate 12 in overall good yields.
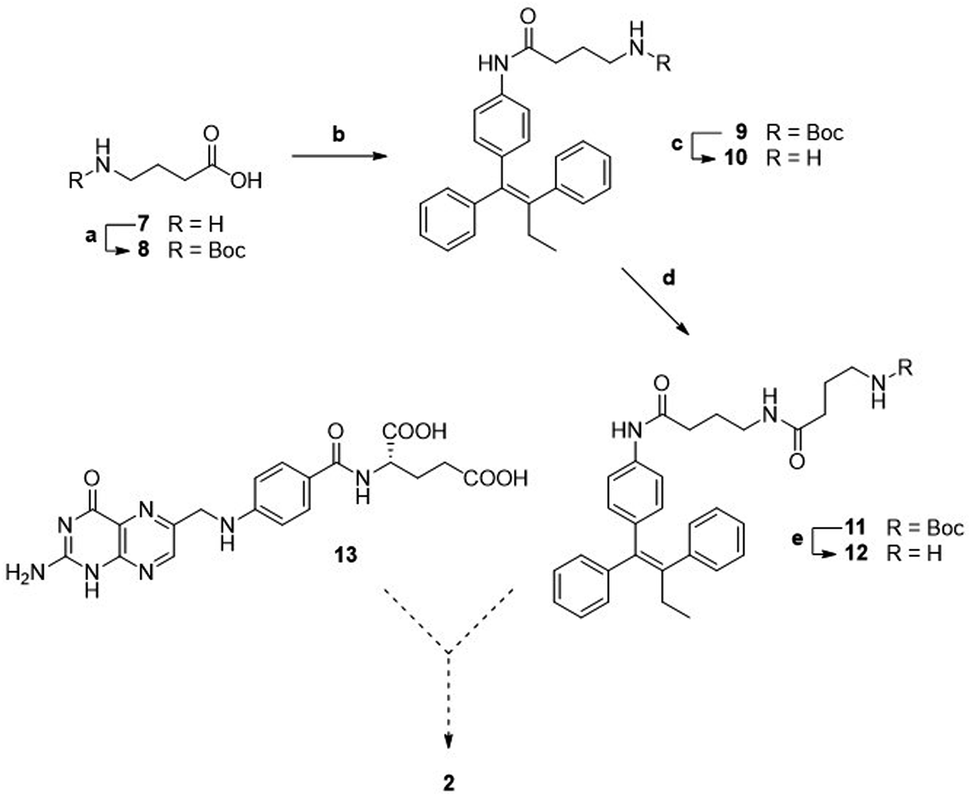 | ||
| Scheme 2 Synthesis of linker-self-assembly inducer 12. a: Boc2O, NaOH/diox, rt, 20 h, quant.; b: 3, HATU, DIPEA, dry THF, rt, 23 h, 94%; c: TFA, dry CH2Cl2, 0 °C to rt, 20 h, quant.; d: 8, HATU, DIPEA, dry THF, rt, 21 h, 73%; e: TFA, dry CH2Cl2, 0 °C to rt, 18 h, 90%. | ||
Unfortunately, despite multiple attempts performed in various reaction conditions, we could never obtain target folic acid conjugate 2 by direct condensation of folic acid 13 with intermediate 12. We attribute such failures to the scarce solubility of folic acid in organic solvents, leading to difficult reaction monitoring, to challenging recovery of the reaction products, and to unexpected side reactions.
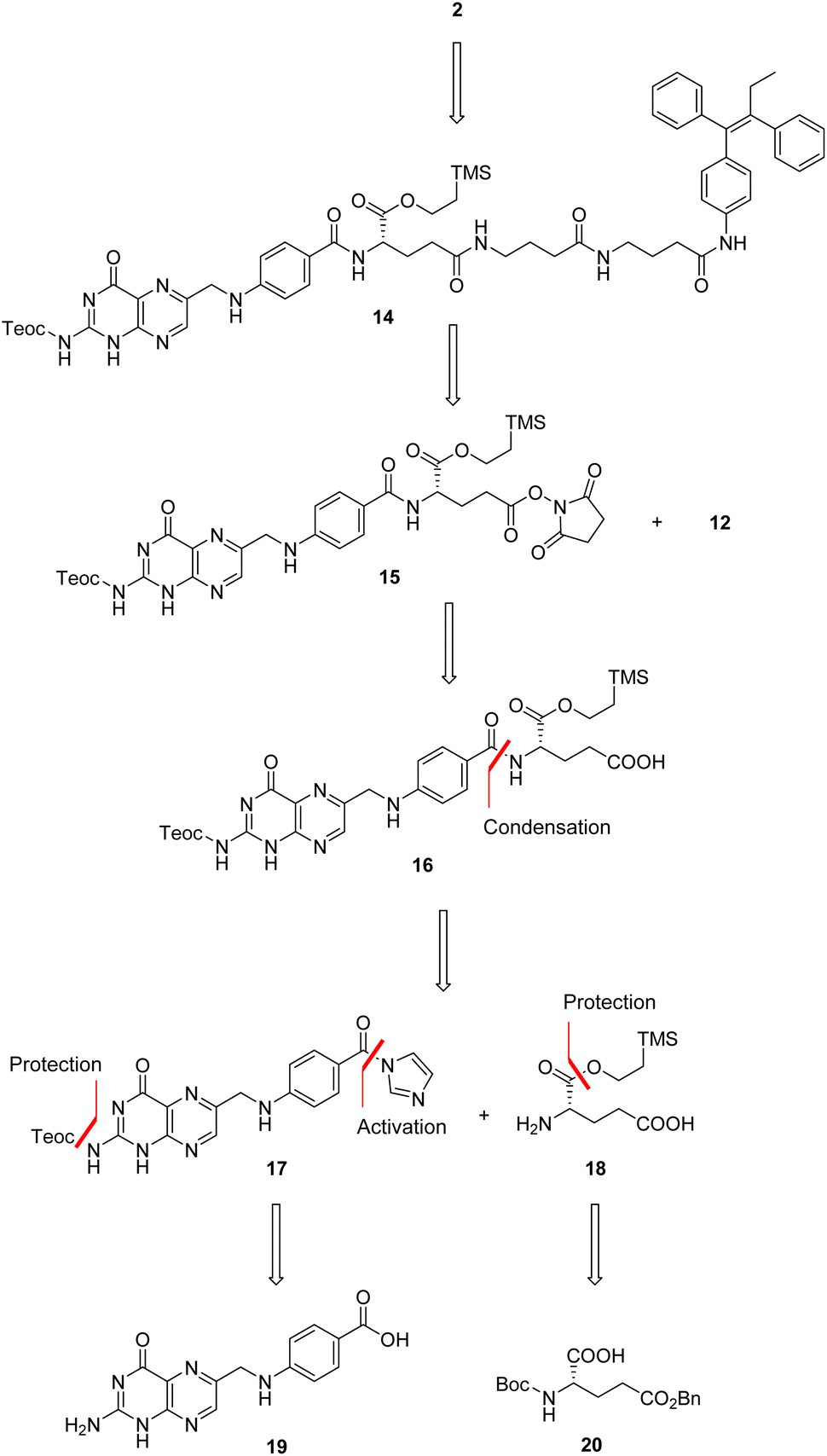
We thus conceived a different retrosynthetic strategy that, even though complex, should have led us to target folic acid conjugate 2.29 A stepwise assembly entailed at first to build folic acid from pteroic acid and L-glutamic acid (see also Fig. 1), proceeding through a sequential protection/activation cascade. The retrosynthetic pathway is shown in Scheme 3, ensuring for the α-carboxylic acid to remain protected throughout the synthesis, avoiding its involvement in side reactions but also improving the solubility of intermediates due to lipophilic protecting groups.
 | ||
| Scheme 3 Retrosynthetic pathway for target folic acid conjugate 2. | ||
In details, folic acid conjugate 2 should be obtained by deprotection of both amino and α-carboxyl groups of intermediate 14. Protected conjugate 14 should result from a coupling reaction between the linker-self-assembly inducer conjugate 12 and N,Cα-protected and Cγ-activated folic acid 15, to be obtained after N-hydroxysuccinimide (NHS)-activation of N,Cα-protected folic acid 16. The latter could be achieved by condensing N-protected and C-activated pteroic acid 17 with Cα-protected L-glutamic acid 18, which can be obtained through Cα-protection and Boc and Bn-deprotection of commercial compound 20. Finally, intermediate 17 could be obtained by protecting the amine and activating the carboxyl group of commercial pteroic acid 19.
Accordingly, we targeted first a suitably Cα-protected L-glutamic acid 18 to be coupled onto pteroic acid 19. Commercial Boc-L-glutamic acid 5-benzyl ester 20 was protected as a 2-(trimethylsilyl)ethyl (TMSE) ester (step a, Scheme 4) through a two-step, one pot procedure. Namely, compound 20 was first activated at its α-position with carbonyldiimidazole (CDI), and then treated with 2-(trimethylsilyl)ethanol to obtain fully protected diester 21. Then, selective deprotection of the Cγ-benzyl ester in 21 by hydrogenolysis led to intermediate 22 (step b). Aminoester 18 was finally obtained as a p-toluensulfonate salt by pTsOH-mediated Boc deprotection (step c, Scheme 4) in overall good yields.
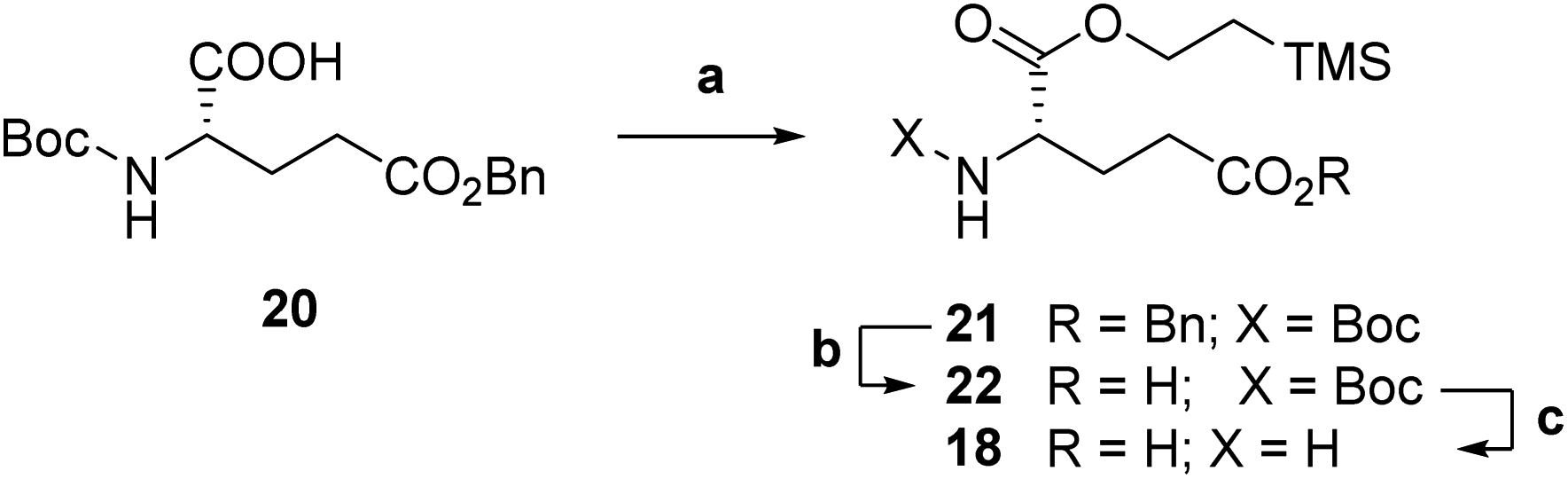 | ||
| Scheme 4 Synthesis of L-glutamic acid Cα-TMSE ester 18. a: (1) CDI, dry CH2Cl2, 0 °C to rt, 1 h; (2) TMSEtOH, rt, 19 h, 54%; b: H2, Pd/C, dry EtOH, rt, 3 h, 91%; c: pTsOH·H2O, diox/H2O, 60 °C, 3 h, quant. | ||
We focused then on the synthesis of suitably N-protected, C-activated pteroic acid derivative 17 in a two step, one pot procedure (step a, Scheme 5).
 | ||
| Scheme 5 Synthesis of N-protected, C-activated pteroic acid 17. a: (1) CDI, dry DMSO, 50 °C, 5 h; (2) TMSEtOH, 50 °C, 21 h, 51%. | ||
Treatment with CDI and TEA as a base in dry DMSO was meant to activate the carboxyl group as a carbonyl imidazole, and to convert the primary amine to a carbamate. Subsequently, 2-(trimethylsilyl)ethanol was to be added to carry out a nucleophilic acyl substitution on the carbamoyl imidazole, de facto providing a trimethylsilylethoxy carbamate (Teoc) protection for the amine group in compound 17.
The Teoc group was selected both to protect the amino group, decreasing the nitrogen's nucleophilicity and thus avoiding side reactions, and to improve the solubility of the resulting intermediate 17 in organic solvents due to its lipophilicity.
However, pteroic acid 19 showed limited solubility even in DMSO, complicating both reaction monitoring and purification. The first reaction attempts at rt were unsuccessful, as we only recovered starting material. To overcome poor solubility, following protocol published in literature,29 we heated the reaction to 50 °C, observing gradual solubilization of 19 and darkening of the reaction mixture. TLC monitoring after 26 hours showed a reaction product together with unreacted pteroic acid 19, but work-up and purification of the crude was affected by the poor solubility of 19 which diffused through both manual and automated flash chromatography. Thus, we recovered only 51% of pure N-protected, C-activated pteroic acid derivative 17 which, as expected, was now soluble in organic solvents and was thus used as such in the next reaction steps.
Then, N,Cα-protected folic acid 16 was synthesized by coupling α-protected aminoester 18 with activated pteroic acid derivative 17 (step a, Scheme 6). The coupling was carried out in dry DMSO, in presence of a strong guanidine base. Protected folic acid 16 was obtained in poor, unoptimized yield, and then activated with NHS in standard conditions to yield the activated ester 15 (step b, Scheme 6) in excellent yield.
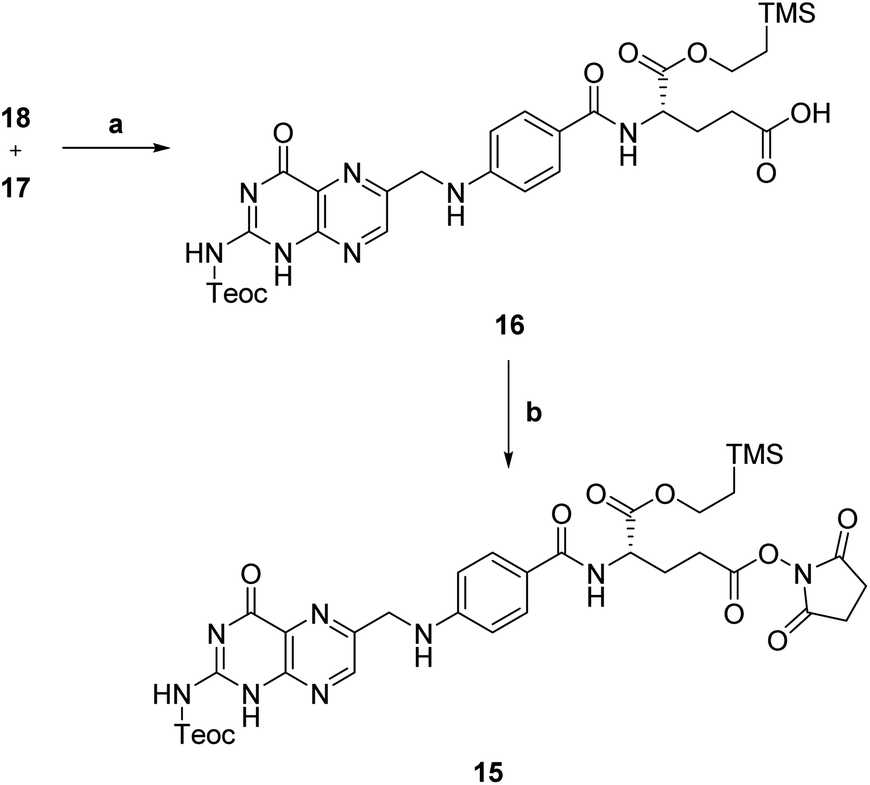 | ||
| Scheme 6 Synthesis of activated protected folic acid. a: MTBD, dry DMSO, rt, 21 h, 33%; b: NHS, EDC, dry DMF, rt, 19 h, 94%. | ||
The key synthetic step of our strategy entailed the coupling of Cγ-activated protected folic acid 15 with previously synthetized linker-self-assembly inducer conjugate 12 (step a, Scheme 7).
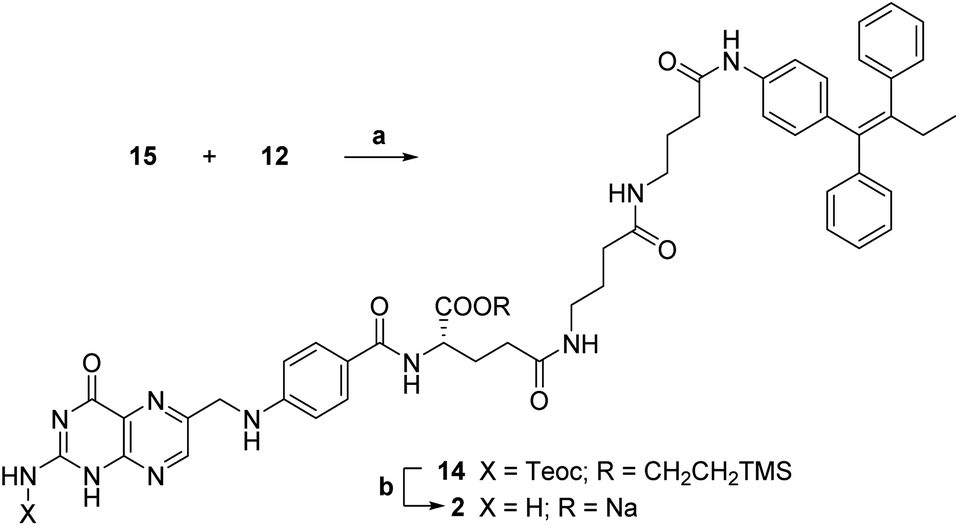 | ||
| Scheme 7 Synthesis of folic acid conjugate 2. a: TEA, dry DMSO, rt, 5 h, 40%; b: (1) 1 M TBAF, dry DMSO, rt, 19 h; (2) 0.2 M NaOAc, dry DMSO, rt, 10 min, 66%. | ||
This basic condensation was successfully completed in dry DMSO, affording N,C-protected folic acid conjugate 14 in moderate yield after flash chromatography purification.
At last, the target folic acid conjugate 2 was obtained as a sodium salt in 66% yield by simultaneous deprotection of both the amino and the α-carboxyl group of compound 14 (step b, Scheme 7). Namely, treatment with tetrabutylammonium fluoride (TBAF) removed both silyl groups; then, the addition of a NaOAc solution caused the precipitation of the desired folic acid conjugate 2 as a sodium salt in good yield and purity without further purification.
Conjugates 1 and 2 were then used to form self-assembled hetero-NPs. Four nanosuspensions were prepared using standard solvent evaporation protocol,30 combining the conjugates in different proportions – namely, 0% (pure 1, hNP1), 5% (hNP2), 10% (hNP3), and 15% (hNP4) of conjugate 2.
Hetero-NPs were characterized by dynamic light scattering (DLS, Table 1) and transmission electron microscopy (TEM, Fig. 3).
| Polydispersity index (PI) | Hydrodynamic diameter – intensity distribution (nm) | Zeta potential (mV) | |
|---|---|---|---|
| hNP1 | 0.221 ± 0.028 | 446.8 ± 58.8 | −37.1 ± 0.61 |
| hNP2 | 0.152 ± 0.044 | 325.8 ± 37.33 | −35.7 ± 0.83 |
| hNP3 | 0.188 ± 0.023 | 418.1 ± 36.57 | −38.7 ± 0.61 |
| hNP4 | 0.175 ± 0.024 | 364.9 ± 20.34 | −39.0 ± 1.4 |
 | ||
| Fig. 3 TEM micrographs of nanoparticles formed by self-assembly hNP2. All samples were stained with uranyl acetate solution. | ||
Self-assembly led to stable and monodisperse suspensions of all hetero-NPs, characterized by hydrodynamic diameters (HD) in the 320–450 nm range and a negative Z-potential (< −35.0 mV).
TEM images confirmed uniformity for each hetero-NP size and showed them to have a spherical shape (Fig. 3).
The effect on cell viability of each hetero-NP was evaluated by means of a colorimetric assay (MTS assay, Fig. 4).
 | ||
| Fig. 4 Cell viability for HeLa cells treated for 72 h with hNP1 to hNP4 at equivalent PTX concentrations (400 nM to 50 μM), and with self-assembly inducer 5 and free PTX 6 (312.5 nM to 5 μM) as control. Reported values are the mean ± SD (n = 3) normalized against untreated sample. | ||
For this purpose, cells were incubated with hetero-NPs at equivalent concentrations of PTX, ranging from 400 nM to 50 μM. In addition, free PTX 6 and self-assembly inducer 5 were used as controls, the former up to its solubility limit.
The results shown in Fig. 4 indicate that the self-assembly inducer 5 is not cytotoxic, while free PTX 6 and hetero-NPs hNP1–4 caused a significant reduction in cell viability (Table 2).
| Sample | IC50 (μM) |
|---|---|
| hNP1 | 4.495 ± 0.466 |
| hNP2 | 2.874 ± 0.109 |
| hNP3 | 5.635 ± 0.035 |
| hNP4 | 4.154 ± 0.088 |
Notably, hNP2 performed better than hNP1, made only of PTX conjugate 1, confirming that the inclusion of a targeting ligand may facilitate NPs' internalization.31 The reduced efficiency displayed by hNP3 and hNP4 suggests that an increased ligand density does not always correlate to an enhanced performance.32 It should also be noted that all hetero-NPs were less efficient than free PTX, as could be expected since PTX in hetero-NPs should be released only upon cytoplasmatic delivery and consequent reduction of the disulfide bridge.4
Conclusions
We report here the synthesis and characterization of conjugates 1 and 2, overcoming significant hurdles related to solubility and purification. Such conjugates were self-assembled in different proportions, to obtain targeted hetero-NPs hNP1 to hNP4. Their analytical characterization confirms their nature as suitable nanoformulations for cancer therapy. Their biological evaluation indicates modest cytotoxic efficacy for folic acid-containing hetero-NPs, depending on the ratio between PTX and folic acid conjugates. This will encourage us to further investigate this kind of nanosystems, and to better evaluate the NPs uptake in other cell lines, expressing different levels of folate receptors.Thus, by coupling a potent cytotoxic agent with an active targeting ligand in self-assembling hetero-NPs, we demonstrated a controlled release of PTX. We expect that, after further refinement, such approach will provide significant efficacy against cancer while mitigating the toxicity of the cytotoxic drug against non-cancer cells.
Experimental
Materials and methods
All reactions were carried out in oven-dried glassware and dry solvents under nitrogen atmosphere. Unless otherwise stated, all solvents were purchased from Sigma Aldrich and used without further purification. Substrates and reagents were purchased from Sigma Aldrich and used as received. Thin-layer chromatography (TLC) was performed on Merck precoated 60F254 plates. Reactions were monitored by TLC on silica gel, with detection by UV light (254 nm) or by charring with 1% permanganate solution. Flash chromatography was performed using silica gel (240–400 mesh, Merck). 1H-NMR spectra were recorded on Bruker DRX-400 and Bruker DRX-300 instruments and are reported relative to residual CDCl3. {1H}13C-NMR spectra were recorded on the same instruments (101 and 75 MHz) and are reported relative to CDCl3. Chemical shifts (δ) for proton and carbon resonances are quoted in parts per million (ppm) relative to tetramethylsilane (TMS), which was used as an internal standard. MS spectra were recorded using an electrospray ionization (ESI) technique on a Waters Micromass Q-Tof mass spectrometer.Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v n-hex/EtOAc) to obtain pure 3 (1.12 g, 3.75 mol, 74% yield) as a beige solid.
:2 v/v n-hex/EtOAc) to obtain pure 3 (1.12 g, 3.75 mol, 74% yield) as a beige solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 7.38–7.30 (m, 2H), 7.30–7.21 (m, 3H), 7.22–7.10 (m, 5H), 6.69 (d, J = 8.8 Hz, 2H), 6.42 (d, J = 8.8 Hz, 2H), 4.21 (bs, 2H), 2.44 (q, J = 7.6 Hz, 2H), 0.91 (t, J = 7.6 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 144.7, 144.0, 143.5, 141.4, 139.4, 134.7, 132.3 (2C), 130.4 (2C), 130.1 (2C), 128.6 (2C), 128.4, 127.0, 126.5 (2C), 115.2 (2C), 29.6, 14.0.
HRMS (ESI), m/z: calcd for C22H21N 299.1674, found 322.1580 (M + Na+).
:4 v/v n-hex/EtOAc) to obtain pure 5 (0.052 g, 0.100 mmol, 20% yield) as a yellow oil.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): 7.45–7.07 (m, 12H), 6.88–6.78 (m, 2H), 2.84–2.70 (m, 4H), 2.58–2.36 (m, 6H), 2.15–1.98 (m, 4H), 0.95 (t, J = 7.7 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 178.3, 170.6, 143.4, 142.2, 139.2, 138.1, 137.9, 135.4, 131.4 (2C), 129.7 (2C), 129.5 (2C), 128.2 (2C), 127.9, 126.7, 126.2 (2C), 118.8 (2C), 37.8, 37.5, 35.4, 32.3, 29.0, 24.5, 24.0, 13.5.
HRMS (ESI), m/z: calcd for C30H33NO3S 519.1902, found 542.1836 (M + Na+).
:1 v/v n-hex/AcOEt), 1 M HCl (30 mL) was added, and the mixture was extracted with CH2Cl2 (5 × 10 mL). The collected organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography (silicagel, eluent mixture 1:1 v/v n-hex/EtOAc) to obtain pure target 1 (70.2 mg, 0.0348 mmol, 52% yield) as a white solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 8.17–8.10 (m, 2H), 7.78–7.68 (m, 2H), 7.65–7.56 (m, 1H), 7.55–7.46 (m, 3H), 7.46–7.29 (m, 10H), 7.25–7.03 (m, 9H), 6.83–6.75 (m, 2H), 6.32–6.28 (m, 1H), 6.28–6.19 (m, 1H), 6.03–5.93 (m, 1H), 5.68 (d, J = 7.0 Hz, 1H), 5.52 (d, J = 3.5 Hz, 1H), 4.97 (dd, J = 9.7, 2.6 Hz, 1H), 4.44 (dd, J = 10.9, 6.6 Hz, 1H), 4.31 (d, J = 8.4 Hz, 1H), 4.20 (d, J = 8.5 Hz, 1H), 3.81 (d, J = 6.9 Hz, 1H), 2.82–2.41 (m, 14H), 2.42–2.25 (m, 4H), 2.25–2.08 (m, 5H), 2.09–1.78 (m, 8H), 1.68 (s, 3H), 1.23–1.17 (m, 3H), 1.13 (s, 3H), 0.93 (t, J = 7.4 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 204.0, 172.2, 171.4, 170.5, 170.1, 170.0, 168.3, 167.2, 142.8, 142.3, 142.1, 140.0, 138.6, 137.4, 137.0, 136.0, 135.6, 133.8, 133.7, 133.0, 132.2, 131.6 (2C), 130.4 (2C), 129.8 (2C), 129.6 (2C), 129.3 (2C), 129.2 (2C), 128.9 (2C), 128.3 (2C), 128.0 (2C), 127.3, 127.2, 126.8, 126.73 (2C), 126.66 (2C), 118.9, 118.7, 84.6, 81.2, 79.3, 76.7, 75.7, 75.2, 74.2, 72.3, 72.0, 58.7, 55.2, 45.8, 43.3, 37.7, 37.2, 35.8, 35.7, 35.6, 32.1, 29.2, 26.9, 24.5, 24.1, 22.8, 22.2, 21.0, 15.0, 13.7, 9.8.
HRMS (ESI), m/z: calcd for C77H82N2O16S2 1354.5106, found 1377.5018 (M + Na+).
:3 v/v n-hex/AcOEt) it was extracted with n-hexane (3 × 10 mL). The collected organic phases were washed with sat. NaHCO3 (2 × 8 mL); the combined aqueous phases were acidified with dil. HCl (5 mL) and extracted with EtOAc (7 × 20 mL). The reunited organic phases were then dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography (silicagel, eluent mixture 7:3 v/v n-hex/EtOAc) to obtain pure target 8 (1.97 g, 9.68 mmol, quantitative yield) as a beige solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 10.88 (bs, 1H), 4.73 (bs, 1H), 3.27–3.04 (m, 2H), 2.38 (t, J = 7.2 Hz, 2H), 1.81 (quint, J = 7.0 Hz, 2H), 1.43 (s, 9H).
13C-NMR (100 MHz, CDCl3): δ 178.5, 156.3, 79.6, 39.9, 31.4, 28.5, 25.3 (3C).
HRMS (ESI), m/z: calcd for C9H17NO4 203.1158, found 226.1058 (M + Na+).
:1 v/v n-hex/AcOEt) the solvent was evaporated under reduced pressure, 1 M HCl (15 mL) was added and the aqueous phase was extracted with CH2Cl2 (3 × 15 mL). The collected organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography (silicagel, eluent mixture 1:1 v/v n-hex/EtOAc) to obtain pure target 9 (1.12 g, 2.31 mmol, 94% yield) as a beige solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 8.45 (bs, 1H), 7.32–7.23 (m, 2H), 7.32–7.23 (m, 5H), 7.22–7.10 (m, 5H), 6.84 (d, J = 8.5 Hz, 2H), 4.76 (bs, 1H), 3.21 (q, J = 6.3 Hz, 2H), 2.51–2.46 (m, 2H), 2.31 (t, J = 6.6 Hz, 2H), 1.88–1.78 (m, 2H), 1.44 (s, 9H), 0.95 (t, J = 7.4 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 170.8, 150.4, 143.7, 142.3, 138.8, 137.6, 137.1, 131.9, 131.5 (2C), 129.8 (2C), 129.6 (2C), 128.2 (2C), 128.0 (2C), 126.7, 126.3, 118.7 (2C), 79.9, 39.4, 34.8, 29.2, 28.5 (3C), 23.2, 13.7.
HRMS (ESI), m/z: calcd for C31H36N2O3 484.2726, found 506.2629 (M + Na+).
:1 v/v n-hex/AcOEt) the solvent was evaporated under reduced pressure to obtain target trifluoroacetate 10 (1.01 g, 2.00 mmol, quant. yield) as a beige solid, without further purification.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 7.88 (bs, 1H), 7.38–7.30 (m, 2H), 7.27–7.17 (m, 5H), 7.14–7.01 (m, 5H), 6.82 (d, J = 8.5 Hz, 2H), 3.88 (bs, 3H), 3.11–2.86 (m, 2H), 2.45 (m, 4H), 2.00–1.77 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 171.6, 143.2, 142.7, 142.0, 140.2, 137.9, 134.4, 131.3 (2C), 129.6 (2C), 129.4 (2C), 128.2 (2C), 127.9 (2C), 126.8, 126.3, 119.6 (2C), 39.7, 34.1, 29.1, 22.6, 13.5.
HRMS (ESI), m/z: calcd for C28H29F3N2O2 482.2181, found 407.2110 (M-TFA + Na+).
:8 v/v n-hex/AcOEt). The solvent was evaporated under reduced pressure and the crude was purified by flash chromatography (silicagel, eluent mixture 2:8 v/v n-hex/EtOAc) to obtain pure target 11 (1.00 g, 1.75 mmol, 73% yield) as a beige solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 7.35–7.31 (m, 2H), 7.27–7.22 (m, 5H), 7.20–7.09 (m, 5H), 6.80 (d, J = 8.6 Hz, 2H), 4.72 (bs, 1H), 3.33 (q, J = 6.1 Hz, 2H), 3.14 (q, J = 6.4 Hz, 2H), 2.47 (q, J = 7.4 Hz, 2H), 2.30 (t, J = 6.6 Hz, 2H), 2.20 (t, J = 6.7, 2H), 1.90–1.81 (m, 2H), 1.81–1.72 (m, 2H), 1.42 (s, 9H), 0.93 (t, J = 7.4 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 173.8, 171.1, 159.9, 143.7, 142.4, 142.0, 138.8, 138.5, 136.3, 131.4 (2C), 129.8 (2C), 129.7 (2C), 128.2 (2C), 128.0 (2C), 126.7, 126.3, 118.8 (2C), 79.8, 39.6, 38.6, 34.9, 33.7, 29.2, 28.6 (3C), 26.8, 26.7, 13.7.
HRMS (ESI), m/z: calcd for C35H43N3O4 569.3254, found 592.3165 (M + Na+).
:1 v/v n-hex/AcOEt), sat. NaHCO3 was added (50 mL) and the aqueous phase was extracted with CH2Cl2 (50 mL) and EtOAc (2 × 50 mL). The collected organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography (silicagel, eluent mixture 8:2 v/v CH2Cl2/MeOH) to obtain pure target 12 (1.23 g, 2.63 mmol, 90% yield) as a beige solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 7.33 (m, 2H), 7.28–6.92 (m, 10H), 6.84 (d, J = 8.0 Hz, 2H), 4.45 (bs, 3H), 3.27–2.95 (m, 2H), 2.84–2.76 (m, 2H), 2.62–2.42 (m, 2H), 2.29–1.99 (m, 4H), 1.95–1.75 (m, 2H), 1.75–1.53 (m, 2H), 0.96 (t, J = 7.2 Hz, 3H).
13C-NMR (100 MHz, CDCl3): δ = 173.9, 171.3, 143.6, 142.4, 141.6, 138.8, 138.4, 136.4, 131.5 (2C), 130.0 (2C), 129.3 (2C), 128.2 (2C), 127.9 (2C), 126.7, 126.2, 118.7 (2C), 40.2, 38.6, 34.3, 33.4, 29.2, 27.5, 26.8, 13.7.
HRMS (ESI), m/z: calcd for C32H36F3N3O3 567.2709, found 492.2634 (M-TFA + Na+).
:1 v/v n-hex/AcOEt). Then it was washed with water (15 mL), and the collected organic phases were dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography (silicagel, eluent mixture 3:1 v/v n-hex/EtOAc) to obtain pure target 21 (1.42 g, 3.28 mmol, 54% yield) as a white solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 7.41–7.28 (m, 5H), 5.12 (s, 2H), 5.09 (bs, 1H), 4.34–4.26 (m, 1H), 4.24–4.18 (m, 2H), 2.56–2.36 (m, 2H), 2.29–2.12 (m, 1H), 2.02–1.89 (m, 1H), 1.43 (s, 9H), 1.02–0.98 (m, 2H), 0.04 (s, 9H).
13C-NMR (100 MHz, CDCl3): δ = 172.5, 172.1, 156.0, 135.8, 128.5 (2C), 128.3 (2C), 128.0, 79.4, 66.0, 63.4, 52.9, 30.6, 28.3 (3C), 27.3, 17.8, −1.6 (3C).
HRMS (ESI), m/z: calcd for C22H35NO6Si 437.2234, found 460.2139 (M + Na+).
:1 v/v n-hex/AcOEt). The suspension was then filtered on celite and then concentrated under reduced pressure. The crude was purified by flash chromatography (silicagel, eluent mixture 9:1 v/v n-hex/EtOAc) to obtain pure target 22 (0.705 g, 2.01 mmol, 91% yield) as a white solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 5.17 (m, 1H), 4.42–4.27 (m, 1H), 4.24–4.20 (m, 2H), 2.49–2.40 (m, 2H), 2.23–2.17 (m, 1H), 1.96–1.89 (m, 1H), 1.46 (s, 9H), 1.04–0.99 (m, 2H), 0.06 (s, 9H).
13C-NMR (100 MHz, CDCl3): δ = 177.9, 172.1, 155.4, 80.1, 64.0, 52.8, 30.1, 28.3 (3C), 27.8, 17.4, −1.5 (3C).
HRMS (ESI), m/z: calcd for C15H29NO6Si 347.1764, found 370.1670 (M + Na+).
:3 water–dioxane mixture (4.6 mL). The reaction mixture was left stirring at 60 °C for 3 h until reaction completion (TLC monitoring, eluent mixture 8:2 v/v CH2Cl2/MeOH). The reaction was then quenched with a NaOH solution (2 mL), the solvent was evaporated under reduced pressure and the crude was purified by flash chromatography (silicagel, eluent mixture 8:2 v/v CH2Cl2/MeOH) to obtain pure target 18 (0.447 g, 1.90 mmol, quant. yield) as a white solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 4.27–4.22 (m, 3H), 2.65–2.61 (m, 2H), 2.33–2.26 (m, 2H), 1.07–0.94 (m, 2H), 0.05 (s, 9H).
13C-NMR (100 MHz, CDCl3): δ = 174.6, 174.1, 62.3, 53.2, 30.8, 29.1, 16.9, −1.4 (3C).
HRMS (ESI), m/z: calcd for C10H21NO4Si 247.1240, found 270.1143 (M + Na+).
:1 v/v CHCl3/MeOH). The mixture was poured in a stirred suspension of water (44 mL), AcOH (1.28 mL) and Et2O (11.6 mL). The resulting precipitate was filtered, dried and purified by flash chromatography (silicagel, eluent mixture 9:1 v/v CHCl3/MeOH) to obtain pure target 17 (0.165 g, 0.326 mmol, 51% yield) as a yellow solid.
Analytical characterization. 1H-NMR (400 MHz, DMSO-d6): δ = 11.76 (bs, 1H), 11.68 (bs, 1H), 8.88 (s, 1H), 8.14 (s, 1H), 7.66 (t, J = 6.1 Hz, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.61–7.58 (m, 1H), 7.09 (s, 1H), 6.77 (d, J = 8.8 Hz, 2H), 4.66 (d, J = 6.1 Hz, 2H), 4.34–4.25 (m, 2H), 1.09–1.00 (m, 2H), 0.05 (s, 9H).
13C-NMR (100 MHz, DMSO-d6): δ = 164.9, 159.4, 159.2, 154.9, 154.6, 153.0, 151.4, 149.2, 138.0, 132.7 (2C), 130.1, 129.7, 118.7, 117.6, 111.7 (2C), 64.6, 45.6, 17.0, −1.5 (3C).
HRMS (ESI), m/z: calcd for C23H26N8O4Si 506.1846, found 529.1760 (M + Na+).
:1 v/v mixture of 1 M AcOH/MeOH (36 mL) and with 2:1 v/v water/MeOH (2 × 54 mL); then it was dried over Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography (silicagel, eluent mixture 17:1:2:0.08 v/v CHCl3/MeOH/EtOAc/AcOH) to obtain pure 16 (46.2 mg, 0.0667 mmol, 33% yield) as a yellow solid.
Analytical characterization. 1H-NMR (400 MHz, CDCl3): δ = 11.91 (bs, 1H), 8.75 (s, 1H), 7.55 (d, J = 7.9 Hz, 2H), 7.12 (s, 1H), 6.48 (s, 2H), 4.78–4.72 (m, 1H), 4.55 (s, 1H), 4.37 (t, J = 8.7 Hz, 3H), 4.23 (t, J = 8.4 Hz, 3H), 2.53–2.48 (m, 2H), 2.40–2.27 (m, 1H), 2.18–2.14 (m, 1H), 1.12 (t, J = 8.8 Hz, 3H), 1.02 (t, J = 8.7 Hz, 3H), 0.08 (s, 9H), 0.03 (s, 9H).
13C-NMR (100 MHz, CDCl3): δ = 173.6, 172.1, 166.2, 159.3, 154.8, 154.4, 151.9, 150.5, 149.0, 148.9, 129.8 (2C), 128.9, 121.2, 111.1 (2C), 64.6, 62.4, 52.0, 46.0, 30.2, 25.8, 17.1, 16.8, −1.43 (3C), −1.45 (3C).
HRMS (ESI), m/z: calcd for C30H43N7O8Si2 685.2712, found 708.2618 (M + Na+).
:1 v/v CH2Cl2/MeOH). The reaction mixture was then poured in water (5 mL) and the formed precipitate was filtered and dried to obtain pure 15 (49.4 mg, 0.0630 mmol, 94% yield) as a yellow solid.
Analytical characterization. 1H-NMR (400 MHz, DMSO-d6): δ = 11.72 (bs, 1H), 8.84 (s, 1H), 8.31 (d, J = 7.4 Hz, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.04 (t, J = 6.2 Hz, 1H), 6.66 (d, J = 8.3 Hz, 2H), 4.59 (d, J = 6.1 Hz, 2H), 4.47–4.37 (m, 1H), 4.29 (t, J = 8.5 Hz, 2H), 4.13 (t, J = 8.3 Hz, 2H), 2.87–2.74 (m, 6H), 2.14–2.05 (m, 2H), 1.05 (t, J = 8.5 Hz, 2H), 0.93 (t, J = 8.4 Hz, 2H), 0.06 (s, 9H), 0.00 (s, 9H).
13C-NMR (100 MHz, DMSO-d6): δ = 171.1, 170.0, 169.2 (2C), 169.0, 166.3, 162.4, 154.8, 154.4, 151.8, 150.5, 149.1, 148.9, 129.8 (2C), 128.8, 121.1, 111.3 (2C), 64.7, 62.4, 51.9, 45.9, 30.4, 25.8, 17.6 (2C), 16.8, −1.43 (3C), −1.40 (3C).
HRMS (ESI), m/z: calcd for C34H46N8O10Si2 782.2875, found 805.2788 (M + Na+).
:1 v/v CH2Cl2/MeOH). Water (4.5 mL) was added until formation of a precipitate, which was centrifuged, washed with water (3 × 5 mL) and dried. The crude was purified by flash chromatography (silicagel, eluent mixture 96:4 v/v CH2Cl2/MeOH) to obtain pure target 14 (70.8 mg, 0.0616 mmol, 40% yield) as a light brown solid.
Analytical characterization. 1H-NMR (400 MHz, DMSO-d6): δ = 9.77 (s, 1H), 8.70 (s, 1H), 8.29 (d, J = 7.3 Hz, 1H), 7.81 (dt, J = 11.9, 5.5 Hz, 2H), 7.65–7.57 (m, 3H), 7.39–7.35 (m, 2H), 7.32–7.17 (m, 7H), 7.14–7.08 (m, 3H), 7.03–6.92 (m, 2H), 6.73–6.71 (m, 2H), 6.65 (d, J = 8.4 Hz, 2H), 4.52 (d, J = 5.9 Hz, 2H), 4.31–4.26 (m, 1H), 4.19–4.08 (m, 4H), 3.09–2.95 (m, 4H), 2.42–2.32 (m, 2H), 2.23–1.99 (m, 6H), 1.92–1.86 (m, 1H), 1.67–1.54 (m, 4H), 1.00–0.88 (m, 4), 0.87–0.82 (m, 3H), 0.05 (s, 9H), 0.02 (s, 9H).
13C-NMR (100 MHz, DMSO-d6): δ = 174.0, 173.2, 172.9, 171.6, 167.2, 162.4, 153.3, 151.9, 151.2, 149.7, 149.6, 145.6, 142.2, 140.7, 138.6, 137.4, 136.1, 132.4, 131.6, 130.8, 130.2 (2C), 128.6 (2C), 128.5 (2C), 128.4 (2C), 127.6 (2C), 127.5, 123.6, 123.1 (2C), 117.5 (2C), 112.6 (2C), 63.7, 63.6, 52.7, 42.1, 39.9, 39.8, 34.3, 33.9, 32.2, 28.7, 27.3, 24.4, 24.3, 17.9, 17.8, 14.4, −1.4 (3C), −1.4 (3C).
HRMS (ESI), m/z: calcd for C60H76N10O9Si2 1136.5335, found 1159.5248 (M + Na+).
:1 v/v mixture of CHCl3/EtOAc (14.05 mL) in a failed attempt to precipitate the product. Solvents were then evaporated under reduced pressure and the solid residue was dissolved in a 1:1 v/v mixture of EtOH/MeOH (3.93 mL). 0.2 M NaOAc in MeOH (0.90 mL) was then added, observing the formation of a precipitate, which was then centrifuged, washed with a 1:1 v/v mixture of MeOH/EtOH (3 × 5 mL) and dried to obtain pure target 2 (34.2 mg, 0.0371 mmol, 66% yield) as a yellow solid.
Analytical characterization. 1H-NMR (400 MHz, DMSO-d6): δ = 9.94 (s, 1H), 8.98 (s, 1H), 8.64 (s, 1H), 7.88 (bs, 2H), 7.64–7.58 (m, 3H), 7.38 (t, J = 7.4 Hz, 3H), 7.28 (d, J = 8.4 Hz, 2H), 7.22–7.16 (m, 4H), 7.12 (d, J = 7.8 Hz, 3H), 6.86 (d, J = 5.8 Hz, 2H), 6.73 (d, J = 7.9 Hz, 2H), 6.67 (d, J = 8.1 Hz, 1H), 4.49 (d, J = 5.9 Hz, 2H), 4.12 (bs, 2H), 3.04–3.00 (m, 4H), 2.41–2.35 (m, 2H), 2.27–2.02 (m, 6H), 1.76–1.69 (m, 1H), 1.68–1.56 (m, 4H), 0.86 (t, J = 7.0 Hz, 3H).
13C-NMR (100 MHz, DMSO-d6): δ = 178.8, 178.7, 174.7, 173.4, 171.7, 167.4, 162.9, 159.6, 152.1, 149.9, 145.8, 142.4, 141.2, 139.5, 137.6, 136.1, 133.0, 132.3 (2C), 131.2 (2C), 130.3 (2C), 129.0 (2C), 128.8 (2C), 128.6, 127.9, 127.7 (2C), 122.7, 122.4, 117.6 (2C), 112.7 (2C), 53.0, 41.8, 40.2, 40.0, 34.3, 34.0, 32.0, 28.8, 27.1, 23.5 (2C), 14.0.
HRMS (ESI), m/z: calcd for C49H52N10O7 892.402, found 915.392 (M + Na+).
Nanoparticles assembly
The two conjugates 1 and 2 are mixed with the desired ratio (0%, 5%, 10% and 15% mol, for a total of 4.0 mg). To achieve better results, while compound 1 was weighted and added as a powder, compound 2 was used as a 1 mg mL−1 solution 2% DMSO in THF. THF is added to the mixture, to reach the total volume of 1 mL. The resulting solution was added dropwise to a round bottom flask containing MilliQ grade distilled water (2 mL) under magnetic stirring (500 rpm). The resulting suspension was stirred for 5 min, then THF was thoroughly evaporated under reduced pressure, obtaining an opalescent suspension of hetero-NPs (2 mL, 2 mg mL−1).Nanoparticles characterization
:2 dilutions. As control, cells were incubated with self-assembly inducer 5 and with free PTX 6 at concentrations ranging from 5 μM to 312 nM, obtained once again by serial 1:2 dilutions. After 72 h of incubation, 20 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) stock solution (Promega, Milano, Italy) were added to each well, and cells were incubated for additional 3 h at 37 °C. Afterwards, the absorbances at 490 nm were measured with EnSight™ Multimode Plate Reader (PerkinElmer, Waltham, MA, USA) and cell viability was calculated normalizing the detected Abs against the one recorded in the untreated sample. Results were expressed as mean ± standard deviation (n = 3). Each IC50 was calculated as the concentration that reduced cell viability by 50% after setting both a shared bottom and top constrains for all samples.Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Shen, Y. Wu, Y. Liu and D. Wu, Int. J. Nanomed., 2017, 12, 4085–4109 CrossRef CAS.
- Y. Wang, A. Santos, A. Evdokiou and D. Losic, J. Mater. Chem. B, 2015, 3, 7153–7172 RSC.
- Y. Zhang, F. Fang, L. Li and J. Zhang, ACS Biomater. Sci. Eng., 2020, 6, 4816–4833 CrossRef CAS.
- S. Borrelli, M. S. Christodoulou, I. Ficarra, A. Silvani, G. Cappelletti, D. Cartelli, G. Damia, F. Ricci, M. Zucchetti, F. Dosio and D. Passarella, Eur. J. Med. Chem., 2014, 85, 179–190 CrossRef CAS PubMed.
- E. Ntungwe, E. M. Domínguez-Martín, G. Bangay, C. Garcia, I. Guerreiro, E. Colombo, L. Saraiva, A. M. Díaz-Lanza, A. Rosatella, M. M. Alves, C. P. Reis, D. Passarella and P. Rijo, Int. J. Mol. Sci., 2021, 22, 10210 CrossRef CAS PubMed.
- E. Colombo, L. Polito, M. Biocotino, P. Marzullo, M. Hyeraci, L. D. Via and D. Passarella, ACS Med. Chem. Lett., 2020, 11, 895–898 CrossRef CAS.
- E. Colombo, M. Biocotino, G. Frapporti, P. Randazzo, M. S. Christodoulou, G. Piccoli, L. Polito, P. Seneci and D. Passarella, Pharmaceutics, 2019, 11, 422 CrossRef CAS.
- G. Frapporti, E. Colombo, H. Ahmed, G. Assoni, L. Polito, P. Randazzo, D. Arosio, P. Seneci and G. Piccoli, Pharmaceutics, 2022, 14, 862 CrossRef CAS PubMed.
- G. Fumagalli, B. Stella, I. Pastushenko, F. Ricci, M. S. Christodoulou, G. Damia, D. Mazza, S. Arpicco, C. Giannini, L. Morosi, F. Dosio, P. A. Sotiropoulou and D. Passarella, ACS Med. Chem. Lett., 2017, 8, 953–957 CrossRef CAS PubMed.
- G. Fumagalli, G. Giorgi, M. Vágvölgyi, E. Colombo, M. S. Christodoulou, V. Collico, D. Prosperi, F. Dosio, A. Hunyadi, M. Montopoli, M. Hyeraci, A. Silvani, G. Lesma, L. D. Via and D. Passarella, ACS Med. Chem. Lett., 2018, 9, 468–471 CrossRef CAS PubMed.
- S. Borrelli, D. Cartelli, F. Secundo, G. Fumagalli, M. S. Christodoulou, A. Borroni, D. Perdicchia, F. Dosio, P. Milla, G. Cappelletti and D. Passarella, Chempluschem, 2015, 80, 47–49 CrossRef CAS.
- G. Fumagalli, D. Mazza, M. S. Christodoulou, G. Damia, F. Ricci, D. Perdicchia, B. Stella, F. Dosio, P. A. Sotiropoulou and D. Passarella, Chempluschem, 2015, 80, 1380–1383 CrossRef CAS.
- G. Fumagalli, M. S. Christodoulou, B. Riva, I. Revuelta, C. Marucci, V. Collico, D. Prosperi, S. Riva, D. Perdicchia, I. Bassanini, A. García-Argáez, L. D. Via and D. Passarella, Org. Biomol. Chem., 2017, 15, 1106–1109 RSC.
- G. Fumagalli, L. Polito, E. Colombo, F. Foschi, M. S. Christodoulou, F. Galeotti, D. Perdicchia, I. Bassanini, S. Riva, P. Seneci, A. García-Argáez, L. Dalla Via and D. Passarella, ACS Med. Chem. Lett., 2019, 10, 611–614 CrossRef CAS PubMed.
- R. K. Huang, N. F. Steinmetz, C. Y. Fu, M. Manchester and J. E. Johnson, Nanomedicine, 2011, 6, 55–68 CrossRef CAS.
- B. Ding, X. Wu, W. Fan, Z. Wu, J. Gao, W. Zhang, L. Ma, W. Xiang, Q. Zhu, J. Liu, X. Ding and S. Gao, Int. J. Nanomed., 2011, 6, 1991–2005 CrossRef CAS.
- L. Yang, H. Mao, Y. A. Wang, Z. Cao, X. Peng, X. Wang, H. Duan, C. Ni, Q. Yuan, G. Adams, M. Q. Smith, W. C. Wood, X. Gao and S. Nie, Small, 2008, 5, 235–243 CrossRef.
- S. A. Kularatne and P. S. Low, Methods Mol. Biol., 2010, 624, 249–265 CrossRef CAS PubMed.
- N. Parker, M. J. Turk, E. Westrick, J. D. Lewis, P. S. Low and C. P. Leamon, Anal. Biochem., 2005, 338, 284–293 CrossRef CAS.
- D. O'Shannessy, G. Yu, R. Smale and Y. Fu, Oncotarget, 2012, 3, 414–425 CrossRef.
- L. S. F. Boogerd, M. C. Boonstra, A. J. Beck, A. Charehbili, C. E. S. Hoogstins, H. A. J. M. Prevoo, S. Singhal, P. S. Low, C. J. H. van de Velde and A. L. Vahrmeijer, Oncotarget, 2016, 7, 17442–17454 CrossRef.
- S. Markert, S. Lassmann, B. Gabriel, M. Klar, M. Werner, G. Gitsch, F. Kratz and A. Hasenburg, Anticancer Res., 2008, 28, 3567–3572 Search PubMed.
- N. Hanna, E. Juhász, C. Cainap, O. Gladkov, R. Ramlau, O. Juan-Vidal, R. Lal, J. Symanowski, W. Perez, B. Nguyen and W. Harb, Abstract Book of the 39th ESMO Congress (ESMO 2014) Madrid, Spain 26–30 September 2014, 2014, vol. 25, p. v1 Search PubMed.
- K. N. Moore, D. M. O'Malley, I. Vergote, L. P. Martin, A. Gonzalez-Martin, K. Malek and M. J. Birrer, Gynecol. Oncol., 2018, 151, 46–52 CrossRef CAS PubMed.
- Q. R. J. G. Tummers, C. E. S. Hoogstins, K. N. Gaarenstroom, C. D. de Kroon, M. I. E. van Poelgeest, J. Vuyk, T. Bosse, V. T. H. B. M. Smit, C. J. H. van de Velde, A. F. Cohen, P. S. Low, J. Burggraaf and A. L. Vahrmeijer, Oncotarget, 2016, 7, 32144–32155 CrossRef PubMed.
- S. Wang and P. S. Low, J. Controlled Release, 1998, 53, 39–48 CrossRef CAS PubMed.
- D. Luong, P. Kesharwani, B. A. Killinger, A. Moszczynska, F. H. Sarkar, S. Padhye, A. K. Rishi and A. K. Iyer, J. Colloid Interface Sci., 2016, 484, 33–43 CrossRef CAS.
- M. Ahir, S. Bhattacharya, S. Karmakar, A. Mukhopadhyay, S. Mukherjee, S. Ghosh, S. Chattopadhyay, P. Patra and A. Adhikary, Biomaterials, 2016, 76, 115–132 CrossRef CAS PubMed.
- C. Figliola, E. Marchal, B. R. Groves and A. Thompson, RSC Adv., 2019, 9, 14078–14092 RSC.
- L. Battaglia and M. Gallarate, Expert Opin. Drug Delivery, 2012, 9, 497–508 CrossRef CAS.
- A. K. Pearce and R. K. O'Reilly, Bioconjugate Chem., 2019, 30, 2300–2311 CrossRef CAS.
- A. M. Alkilany, L. Zhu, H. Weller, A. Mews, W. J. Parak, M. Barz and N. Feliu, Adv. Drug Delivery Rev., 2019, 143, 22–36 CrossRef CAS PubMed.
- M. S. Christodoulou, M. Zarate, F. Ricci, G. Damia, S. Pieraccini, F. Dapiaggi, M. Sironi, L. lo Presti, A. N. García-Argáez, L. Dalla Via and D. Passarella, Eur. J. Med. Chem., 2016, 118, 79–89 CrossRef CAS PubMed.
- Q. Liu, W. Lu, M. Ma, J. Liao, A. Ganesan, Y. Hu, S. Wen and P. Huang, RSC Adv., 2014, 5, 1109–1112 RSC.
- G. A. Roth, J. Org. Chem., 1995, 60, 8105–8109 CrossRef CAS.
- M. Nomura, S. Shuto and A. Matsuda, J. Org. Chem., 2000, 65, 5016–5021 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06306a |
| This journal is © The Royal Society of Chemistry 2022 |