
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, and biological evaluation of novel bioactive thalidomide analogs as anticancer immunomodulatory agents†
Anas Ramadan Kotba,
Dina A. Bakhotmah*b,
Abdallah E. Abdallah a,
Hazem Elkadya,
Mohammed S. Taghoura,
Ibrahim. H. Eissa*a and
Mohamed Ayman El-Zahabi*a
a,
Hazem Elkadya,
Mohammed S. Taghoura,
Ibrahim. H. Eissa*a and
Mohamed Ayman El-Zahabi*a
aPharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo, 11884, Egypt. E-mail: malzahaby@azhar.edu.eg; Ibrahimeissa@azhar.edu.eg
bDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia. E-mail: dbakhotmah@kau.edu.sa
First published on 22nd November 2022
Abstract
Cancer is still a dangerous disease with a high mortality rate all over the world. In our attempt to develop potential anticancer candidates, new quinazoline and phthalazine based compounds were designed and synthesized. The new derivatives were built in line with the pharmacophoric features of thalidomide. The new derivatives as well as thalidomide were examined against three cancer cell lines, namely: hepatocellular carcinoma (HepG-2), breast cancer (MCF-7) and prostate cancer (PC3). Then the effects on the expression levels of caspase-8, VEGF, NF-κB P65, and TNF-α in HepG-2 cells were evaluated. The biological data revealed the high importance of phthalazine based compounds (24a–c), which were far better than thalidomide with regard to the antiproliferative activity. 24b showed IC50 of 2.51, 5.80 and 4.11 μg mL−1 compared to 11.26, 14.58, and 16.87 μg mL−1 for thalidomide against the three cell lines respectively. 24b raised caspase-8 level by about 7 folds, compared to 8 folds reported for thalidomide. Also, VEGF level in HepG-2 cells treated with 24b was 185.3 pg mL−1, compared to 432.5 pg mL−1 in control cells. Furthermore, the immunomodulatory properties were proven to 24b, which reduced TNF-α level by approximately half. At the same time, NF-κB P65 level in HepG-2 cells treated with 24b was 76.5 pg mL−1 compared to 278.1 and 110.5 pg mL−1 measured for control cells and thalidomide treated HepG-2 cells respectively. Moreover, an in vitro viability study against Vero non-cancerous cell line was investigated and the results reflected a high safety profile of all tested compounds. This work suggests 24b as a promising lead compound for development of new immunomodulatory anticancer agents.
1. Introduction
In the last few decades, there has been a tremendous amount of effort and progress in cancer treatment. However, cancer is still a disease with an increasingly high mortality rate all over the world.1 Many challenges have been reported clinically, including resistance of cancer to the used drugs, which leads to failure of the treatment.2–4 In addition to that, the toxicity of many anticancer drugs is not limited to cancer cells.5 As a result, severe and serious toxic effects were reported6,7 as well as difficulties in optimizing the drug regimen.8 These two main problems were taken into account in this study which aimed to develop anticancer candidates with potential low resistance rate and few side effects.9In our journey to find a suitable lead, thalidomide emerged as a lead with many significant criteria. Aside from teratogenicity, thalidomide was found to exhibit few side effects.10 It was modified to give what is known as immunomodulatory drugs (IMiDs), such as lenalidomide and pomalidomide, that were approved for cancer treatment.11–13 The significant anticancer effects of thalidomide and the IMiDs have been proven to involve different targets and several mechanisms, which include immunomodulatory action. Vascular endothelial growth factor (VEGF) and hence angiogenesis were reported to be inhibited by these drugs.14 Also, the apoptosis rate and cell cycle arrest were increased along with the elevation of caspase-8 levels.14 Furthermore, the immunomodulatory effects that include co-stimulation of T-cells, inhibition of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), IL-1, IL-12. In addition to nuclear factor kappa B (NFκB) inhibition.14 On the other hand, the levels of the anti-inflammatory cytokines were elevated. The elevated cytokines include interferon-gamma (IFN-γ) and IL-10, IL-2.11,15–19 Thalidomide's immunomodulatory properties account for its efficacy in various autoimmune and inflammatory diseases.20,21 Based on these characteristics, we suggest that thalidomide modification may afford new effective and safe candidates with a low resistance rate.
Thalidomide structural modifications have resulted in many effective anticancer analogs such as lenalidomide, pomalidomide,22 and avadomide (CC-122).23 Thalidomide was approved for treatment of acute ENL in 1998 and for treatment of multiple myeloma (MM) in 2006.24 Lenalidomide was found to be a more potent TNF-α inhibitor than thalidomide.25 In 2006, it received FDA approval for treatment of MM.26 Pomalidomide showed more significant results than lenalidomide in both inhibition of TNF-α and stimulation of IL-2.27 In 2013, it was approved by FDA for treatment of MM.28 Avadomide exhibited potent anti-proliferative, anti-angiogenic and immunomodulatory activities.29 It was proven to be effective in treatment of MM, diffuse large B-cell lymphoma (DLBCL) and solid tumors.23
The structural modification of thalidomide that involved the replacement of the phthalimido moiety by quinazoline nucleus afforded avadomide.30 Quinazoline containing candidates revealed potent inhibition to NF-κB,31 TNF-α, IL-6, and IL1β,32–34 antiproliferative35 and immunomodulatory activities36 along with induction of apoptosis and elevation of caspase levels.37 The compounds based on phthalazine, a bio-isostere of quinazoline, showed similar properties which included TNF-α and NF-κB inhibition,38 antiproliferative,39 apoptosis induction40 and antiangiogenic effect.41
1.1. Rationale of molecular design
It is clear that thalidomide has many targets involved in the expression of its high efficacy in cancer, autoimmune and inflammatory diseases. As a result, analyzing the binding interactions to one or two targets does not provide an explanation for all the biological effects reported for thalidomide. But analyzing the chemical structure of thalidomide may be a better way to design new candidates with similar properties. Hence, the ligand-based design approach has been followed in this study. The molecular structure of thalidomide as a lead compound can be analyzed into four main regions as illustrated in Fig. 1. Four pharmacophoric features are identified as follows: (1) a flat hetero aromatic ring, (2) a spacer, (3) a hydrogen bond forming region, (4) a hydrophobic moiety. | ||
| Fig. 1 Illustration for the rationale of the design. | ||
Herein, we carried out modifications on the analyzed four regions. Firstly, the phthalimide moiety was replaced with two other bio-isostere heteroaromatic nuclei; quinazoline and phthalazine. It was reported that quinazoline was a significant alternative to phthalimide and afforded the highly effective avadomide molecule. Secondly, it is noticed that the rotation of the sigma bond between glutarimide moiety and phthalimide nucleus of thalidomide is almost restricted. We aimed to increase the flexibility by having one or two NH groups which will allow more flexibility of the attached planar benzenesulfonyl, the clororfluroranilide moieties or the puckered glutarimide ring or ring open modification of the semicarbazide moiety. Consequently, such possible variations may result in biological variations due to possible variations of the way of binding to the target binding sites as shown in Fig. 1. Thirdly, the hydrogen bond forming group was designed to include imide, urea, sulfamoyl, and even hydrogen bond forming halogens e.g., fluoro and chloro. Finally, aliphatic and aromatic groups were constructed to examine which would be the most effective as a hydrophobic moiety in terms of thalidomide activities.
2. Results and discussion
2.1. Chemistry
For synthesis of the target compounds, reactions sequence is clarified in Schemes 1–3. Initially, quinazoline-2,4(1H,3H)-dione 3 was prepared by fusion of anthranilic acid 1 with urea 2 according to the reported methods.42 Next, heating of 3 with phosphorus oxychloride in the presence of Et3N for 7 h afforded the dichloro derivative 4.43 The produced 2,4-dichloroquinazoline 4 was refluxed with 3-chloro-4-fluoroaniline 5 in isopropyl alcohol in the presence of two mole equivalents of Et3N to produce the final target compound 6. Refluxing two-mole equivalents of methyl and ethyl amine with compound 6 in acetonitrile gave compounds 7a,b respectively (Scheme 1). The structures of compounds 6 and 7a,b were verified by elemental analyses and spectral data. The IR spectra of these compounds revealed the presence of NH absorption bands at a range of 3380–3277 cm−1. Moreover, the 1H NMR spectra of compound 6 showed peak at 10.26 ppm due to NH. On the other hand, the 1H NMR spectra of compounds 7a,b showed deshielded peaks at a range of 8.30 to 9.5 ppm corresponding to the two NHs. Additionally, the aliphatic protons appeared at a range of 1.02 to 3.48 ppm.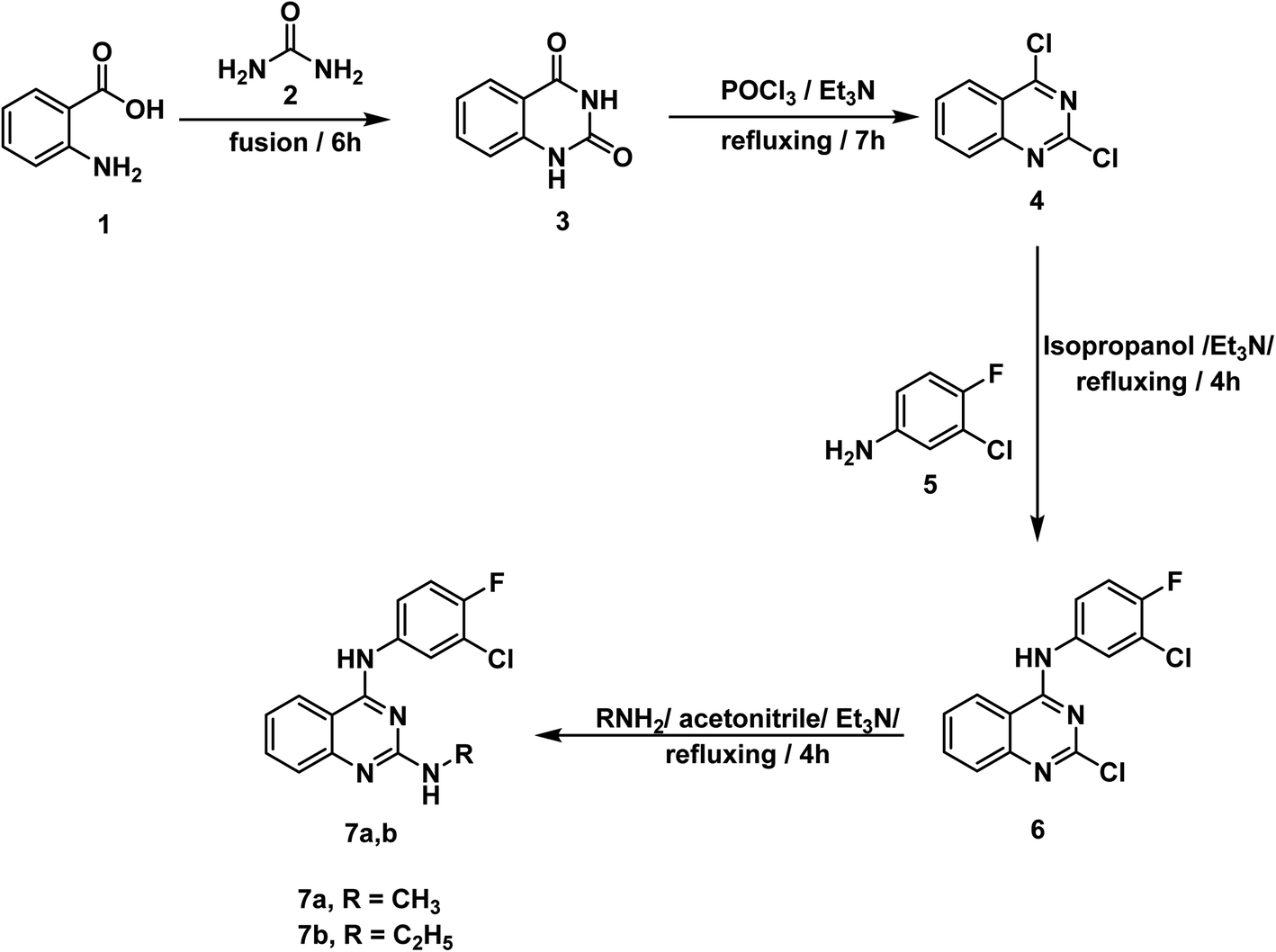 | ||
| Scheme 1 General procedure for preparation of target compounds 6 and 7a,b. | ||
In Scheme 2, the starting materials 9a,b were reported to be prepared by fusion of urea with 5-chloroanthranilic acid 8a or 6-chloroanthranilic acid 8b, respectively. Chlorination of the produced quinazoline-2,4-dione derivatives 9a,b was achieved by refluxing in POCl3 in the presence of N,N-dimethyl aniline (DMA) following the reported procedure.44 The target compounds 11a,b were finalized by refluxing 2,4,5-trichloro-1,2,3,4-tetrahydroquinazoline 10a and/or 2,4,6-trichloro-1,2,3,4-tetrahydroquinazoline 10b with 3-chloro-4-fluoroaniline 5 in isopropyl alcohol in the presence of Et3N. IR spectra revealed the presence of NH absorption bands at 3419 and 3381 cm−1. On the other hand, the 1H NMR spectra of compounds 11a,b showed deshielded peaks for the NH protons at a range of 10.31 to 9.87 ppm. In the meantime, compounds 13a,b were furnished by refluxing the appropriate 2,4-dichloroquinazoline derivatives 10a,b with 3-aminopiperidine-2,6-dione HCl 12 under basic conditions. Also, under the same condition reactions, compounds 15a,b was produced by reaction of 10a,b with semicarbazide HCl 14. Treatment of 2,4,5-trichloroquinazoline 10a with excess hydrazine hydrate 16 in absolute ethanol afforded 2,5-dichloro-4-hydrazinylquinazoline 17.45 Finally, synthesis of compounds 19a,b was attained in good yields by dropwise addition of the appropriate benzenesulfonyl chloride 18a,b to a mixture of 2,5-dichloro-4-hydrazinylquinazoline 17 and Et3N in DMF in ice salt bath. The IR spectra of these compounds were characterized by appearance of characteristic bands of the two NH groups at 3397–3306 cm−1 and very characteristic bands of SO2 group at a range of 1166–1163 cm−1. On the other hand, the 1H NMR spectrum of 19a showed peaks at 10.04 and 7.82 ppm due to sulfonhydrazide moiety while compound 19b showed peaks at 10.24, 7.97 ppm.
 | ||
| Scheme 2 General procedure for preparation of target compounds 11a,b, 13a,b, 15a,b, and 19a,b. | ||
2,3-Dihydrophthalazine-1,4-dione 21 was prepared by condensation of equimolar amounts of phthalic anhydride 20 and hydrazine hydrate 16 in absolute ethanol, according to a reported procedure46 as illustrated in Scheme 3. Refluxing of 21 in POCl3 afforded 1,4-dichlorophthalazine 22, which was then treated with an excess of hydrazine hydrate 16 to give 1-chloro-4-hydrazinylphthalazine 23 as shown in Scheme 3. Finally, the target compounds 24a–c were obtained by addition of an appropriate benzenesulfonyl chloride 18a–c dropwise to a cooled solution of 23 in DMF. The 1H NMR spectra of 24a–c showed peaks at about 11.75 and 9.44 ppm due to sulfonhydrazide moiety.
 | ||
| Scheme 3 General methods for preparation of the new derivatives 24a–c. | ||
2.2. Biological testing
| Comp | In vitro cytotoxicity IC50a (μg mL−1) | ||
|---|---|---|---|
| HepG-2 | MCF-7 | PC3 | |
| a Three independent experiments were performed for each concentration. | |||
| 6 | 74.81 ± 4.1 | 81.52 ± 4.2 | 84.96 ± 4.7 |
| 7a | 40.50 ± 2.8 | 28.13 ± 2.1 | 47.20 ± 2.8 |
| 7b | 11.68 ± 0.8 | 9.45 ± 0.9 | 15.48 ± 1.3 |
| 11a | 77.24 ± 4.2 | 86.14 ± 4.4 | 91.26 ± 4.9 |
| 11b | 38.74 ± 2.8 | 45.91 ± 3.0 | 51.20 ± 3.5 |
| 13a | 46.32 ± 3.1 | 35.29 ± 2.4 | 54.31 ± 3.0 |
| 13b | 59.01 ± 3.3 | 29.33 ± 2.3 | 38.36 ± 3.1 |
| 15a | 24.51 ± 2.0 | 17.33 ± 1.3 | 26.08 ± 2.0 |
| 15b | 16.18 ± 1.4 | 9.87 ± 0.9 | 13.79 ± 1.2 |
| 19a | 19.07 ± 1.5 | 12.76 ± 1.0 | 21.48 ± 1.6 |
| 19b | 28.92 ± 2.2 | 20.48 ± 1.5 | 32.71 ± 2.3 |
| 24a | 3.87 ± 0.2 | 6.07 ± 0.7 | 8.63 ± 0.6 |
| 24b | 2.51 ± 0.1 | 5.80 ± 0.4 | 4.11 ± 0.3 |
| 24c | 7.96 ± 0.5 | 8.12 ± 0.7 | 14.92 ± 1.1 |
| Thalidomide | 11.26 ± 0.54 | 14.58 ± 0.57 | 16.87 ± 0.7 |
It is noticeable that the 24a–c compounds, which are based on the phthalazine core carrying the sulfonhydrazide moiety, showed the best antiproliferative results. Such compounds are, in fact, significantly better than thalidomide versus the selected cancer cell lines. The most significant candidate was 24b, which showed IC50 of 2.51, 5.80 and 4.11 μg mL−1 compared to 11.26, 14.58, and 16.87 μg mL−1 for thalidomide against the three cell lines respectively. Compound 24a came in second with IC50 of 3.87, 6.07, and 8.63 μg mL−1 respectively. The third was compound 24c, which showed an IC50 of 7.96, 8.12, and 14.92 μg mL−1 respectively.
The best quinazoline derivative was 7b, in which the quinazoline nucleus was attached to chlorofluoroanilide group at position 4 and ethyl amino group at position 2. It showed results better than thalidomide against both MCF-7 and PC3, and comparable to that of thalidomide against HepG-2. It is also noticed that compound 15b was stronger than thalidomide against MCF-7 and PC3, but weaker than it against HepG-2. Compounds 19a, 15a, and 19b revealed moderate activities. It was found clear that HepG-2 was the most sensitive cancer cell line for the most promising candidates (24a–c). Accordingly, HepG-2 cell line was selected for further evaluation. Two compounds were selected for such evaluation; phthalazine based 24b (the most effective candidate) and 7b (the most promising quinazoline based derivative).
Regarding quinazoline containing derivatives, comparing the cytotoxic activity of compounds 11b, 13b, and 15b (incorporating 2,6-dichloroquinazoline moiety) with compounds 11a, 13a, and 15a (incorporating 2,5-dichloroquinazoline moiety) indicated that 2,6-dichloroquinazoline moiety is more advantageous than 2,5-dichloroquinazoline moiety for cytotoxic activity against all cell lines. Moreover, comparing the activity of compounds 15a and 15b having semicarbazide moiety with compounds 13a and 13b having glutarimide moiety indicated that semicarbazide moieties are more beneficial for activity. Regarding 2-chloroquinazoline derivatives 6, 7a,b, it was noticed that replacement of chlorine atom at position-2 with methylamine increased the cytotoxic activity against all cell lines. However, replacement of methylamine with ethylamine increased the cytotoxic activity against all cell lines as well. With regard to phthalazine containing derivatives, comparing the activity of compound 24a (in which 4-methylbenzenesulfonohydrazide moiety is incorporated) and compound 24b (in which 4-fluorobenzenesulfonohydrazide moiety is incorporated) with compound 24a (in which benzenesulfonohydrazide moiety is incorporated), revealed that substituted moieties are more potent than unsubstituted one. In the meantime, comparing the activity of compound 24c (incorporating 4-fluorobenzenesulfonohydrazide moiety) with compound 24b (incorporating 4-methylbenzenesulfonohydrazide moiety), exhibited that the electron withdrawing group is more preferred biologically than electron donating one (Fig. 2).
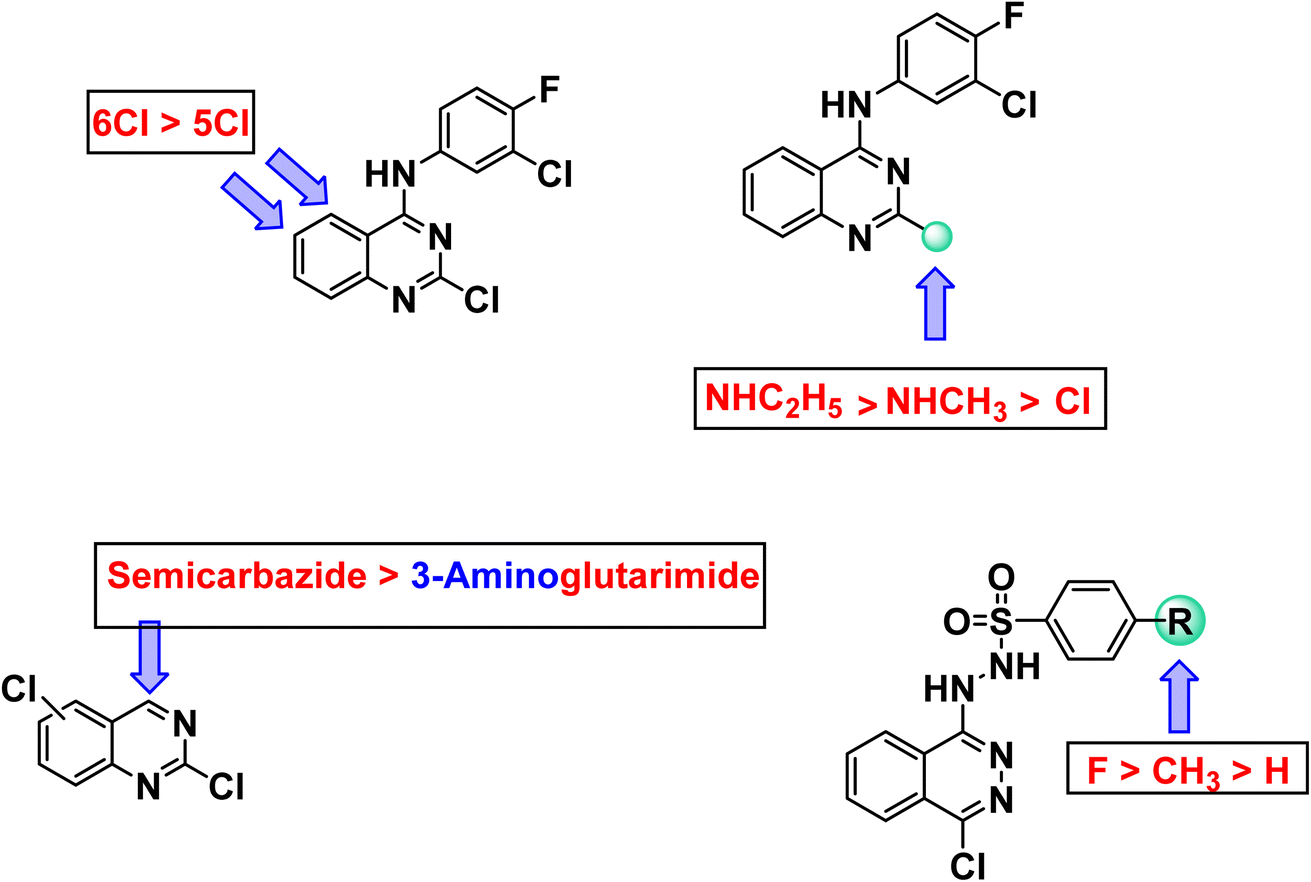 | ||
| Fig. 2 Structure–activity relationship based on in vitro anti-proliferative activity. | ||
2.2.3.1. The effect on the expression level of caspase-8. It was found that compound 24b was able to increase caspase-8 level in HepG-2 cells by about 7 folds, compared to 8 folds reported for thalidomide. While 7b revealed a threefold increase in caspase-8 level as shown in Table 2.
| Comp. no. | CASPASE-8 (ng mL−1) | VEGF (pg mL−1) | NFκB P65 (pg mL−1) | TNF-α (pg mL−1) |
|---|---|---|---|---|
| 24b | 6.9 | 185.3 | 76.5 | 82.5 |
| 7b | 3.2 | 218.5 | 113.7 | 79.3 |
| Control | 1.08 | 432.5 | 278.1 | 162.5 |
| Thalidomide | 8.3 | 153.2 | 110.5 | 53.1 |
2.2.3.2. The effect on the expression level of vascular endothelial growth factor (VEGF). The data presented in Table 2 show that compound 24b reduced the level of VEGF to less than half of that of control cells. HepG-2 cells treated with 24b revealed VEGF level of 185.3 pg mL−1, compared to 432.5 pg mL−1 calculated for control cells. Additionally, 7b reduced VEGF level in HepG-2 cells by about half (218.5 pg mL−1). On the other hand, VEGF level in HepG-2 cells treated with thalidomide was found to be 153.2 pg mL−1.
2.2.4.1. The effect on NF-κB P65 expression level. The obtained data indicated that the impact of 24b on NF-κB P65 was much better than thalidomide. It can be seen that NF-κB P65 level in HepG-2 cells treated with 24b was 76.5 pg mL−1 compared to 278.1 and 110.5 pg mL−1 measured for control HepG-2 cells and thalidomide treated HepG-2 cells respectively. At the same time compound 7b showed a result that was almost comparable to that of thalidomide (Table 2).
2.2.4.2. The effect on TNF-α expression level. The data presented in Table 2 shows that both 24b and 7b reduced TNF-α levels in HepG-2 cells by about half. TNF-α levels were reduced from 162.5 pg mL−1 in control cells to 82.5 and 79.3 pg mL−1 under the effects of 24b and 7b respectively. On the other hand, the level in cells treated with thalidomide was 53.1 pg mL−1.
| Compounds | Cytotoxicity against Vero (IC50 μg mL−1) | Selectivity index (SI) | ||
|---|---|---|---|---|
| HepG-2a | MCF-7b | PC3c | ||
| a SI = cytotoxicity against Vero cells/cytotoxicity against HepG-2 cell line.b SI = cytotoxicity against Vero cells/cytotoxicity against MCF-7 cell line.c SI = cytotoxicity against Vero cells/cytotoxicity against PC3 cell line. | ||||
| 7b | 163.61 ± 1.12 | 13.78 | 17.04 | 10.40 |
| 24b | 161.12 ± 1.42 | 65.18 | 8.59 | 39.80 |
The selectivity index (SI) of a particular compound is the ratio of its toxic concentration against the effective concentration (SI = IC50 on non-cancer cell/IC50 on cancer cell).47 From the results presented in Table 3, the tested compounds showed very high SI against the cancer cell lines. Interestingly, compound 24b showed the highest SI values of 65.18 and 39.80 against the HepG2, and PC3 cell lines, respectively. Compound 7b showed SI values of 13.78, 17.04, and 10.40 against the HepG2, MCF-7, and PC3 cell lines, respectively.
2.3. In silico investigation
The results presented in Table 4 showed that the tested compounds showed no contravention of Lipinski's and Veber's rules and hence display a drug-like molecular nature. In details, the partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) values were less than 5 except compounds 6, 7b, and 11a, and 13b. Molecular weight (Mwt) values of the tested members were less than 500. In addition, number of H-bond donors (HBD) and number of H-bond acceptors (HBA) of these compounds are within the accepted values of less than 5 and 10, respectively. Regarding Veber's rule, the number of rotatable bonds and topological polar surface area (TPSA) of such candidates are within the acceptable values of less than 10 and 140 Å2, respectively. These findings conclude that all the tested compounds meet the criteria of Lipinski and Veber's rules and displayed acceptable physicochemical properties.
P) values were less than 5 except compounds 6, 7b, and 11a, and 13b. Molecular weight (Mwt) values of the tested members were less than 500. In addition, number of H-bond donors (HBD) and number of H-bond acceptors (HBA) of these compounds are within the accepted values of less than 5 and 10, respectively. Regarding Veber's rule, the number of rotatable bonds and topological polar surface area (TPSA) of such candidates are within the acceptable values of less than 10 and 140 Å2, respectively. These findings conclude that all the tested compounds meet the criteria of Lipinski and Veber's rules and displayed acceptable physicochemical properties.
| Comp | Lipinski rules | Veber's rules | ||||
|---|---|---|---|---|---|---|
| HBD | HBA | Mwt | logP |
Rotatable bonds | TPSA | |
| 6 | 1 | 3 | 308.138 | 5.202 | 2 | 37.81 |
| 7a | 2 | 4 | 302.734 | 4.677 | 3 | 49.84 |
| 7b | 2 | 4 | 316.761 | 5.025 | 4 | 49.84 |
| 11a | 1 | 3 | 342.583 | 5.867 | 2 | 37.81 |
| 11b | 2 | 5 | 325.15 | 2.49 | 2 | 83.97 |
| 13a | 3 | 4 | 272.091 | 2.379 | 2 | 92.93 |
| 13b | 1 | 3 | 342.583 | 5.867 | 2 | 37.81 |
| 15a | 2 | 5 | 325.15 | 2.49 | 2 | 83.97 |
| 15b | 3 | 4 | 272.091 | 2.379 | 2 | 92.93 |
| 19a | 2 | 5 | 383.252 | 4.443 | 4 | 92.36 |
| 19b | 2 | 5 | 387.216 | 4.162 | 4 | 92.36 |
| 24a | 2 | 5 | 348.807 | 3.529 | 4 | 92.36 |
| 24b | 2 | 5 | 352.771 | 3.248 | 4 | 92.36 |
| 24c | 2 | 5 | 334.781 | 3.043 | 4 | 92.36 |
| Thalidomide | 1 | 4 | 258.229 | 0.097 | 1 | 83.55 |
| Comp | BBB level | Solubility | Absorption | CYP2D6 inhibition | Plasma protein binding |
|---|---|---|---|---|---|
| 6 | 0 | 1 | 0 | True | True |
| 7a | 1 | 1 | 0 | True | True |
| 7b | 1 | 1 | 0 | True | True |
| 11a | 0 | 1 | 1 | True | True |
| 11b | 3 | 1 | 1 | True | True |
| 13a | 3 | 1 | 0 | False | True |
| 13b | 0 | 1 | 0 | False | True |
| 15a | 3 | 2 | 0 | False | False |
| 15b | 3 | 2 | 0 | False | True |
| 19a | 2 | 2 | 0 | False | False |
| 19b | 2 | 2 | 0 | False | False |
| 24a | 2 | 2 | 0 | False | True |
| 24b | 2 | 2 | 0 | False | True |
| 24c | 3 | 2 | 0 | False | True |
| Thalidomide | 3 | 3 | 0 | False | False |
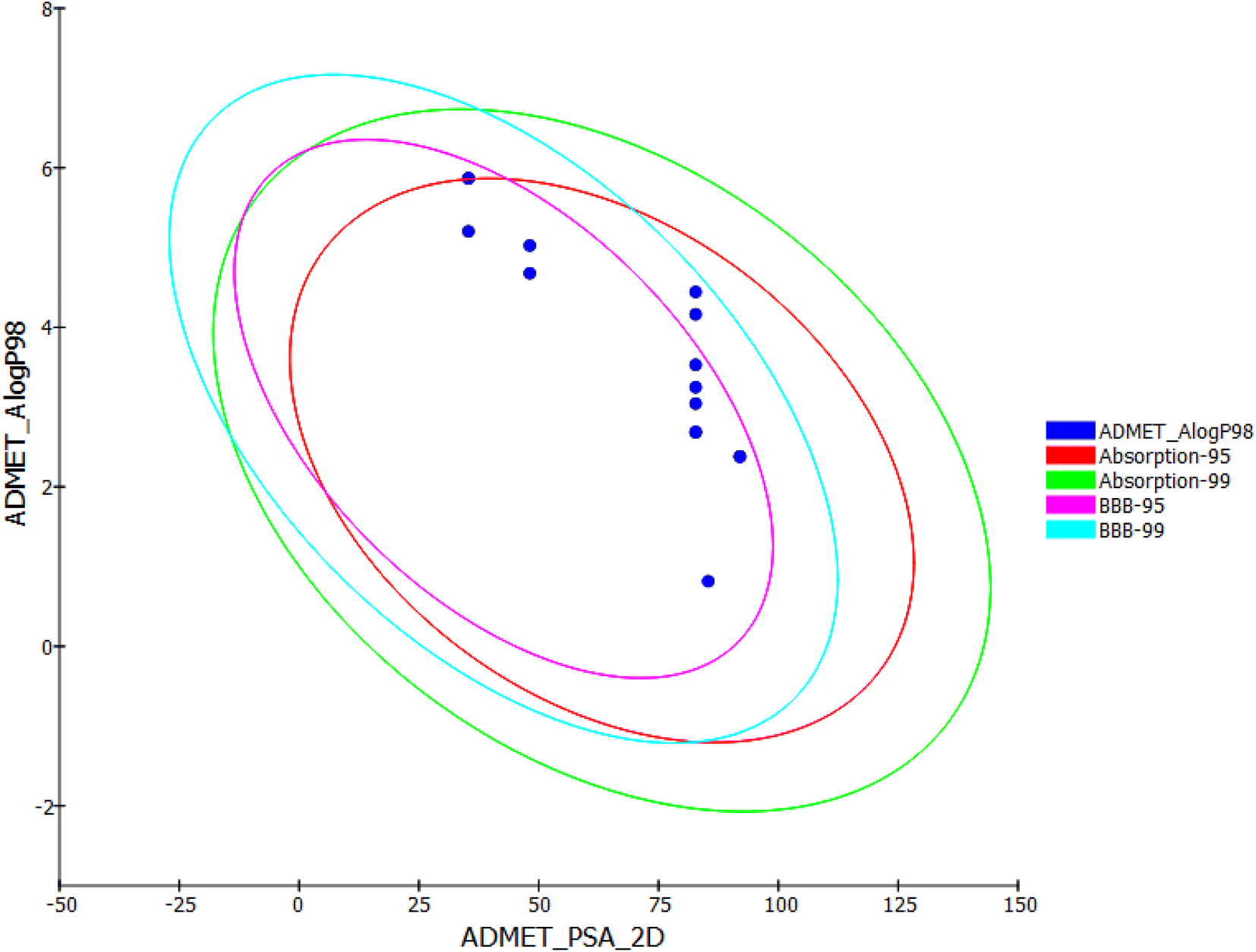 | ||
| Fig. 3 Computational prediction of ADMET parameters. | ||
Among the tested compounds, 11b, 13a, 15a, 15b, and 24c are exhibited low blood–brain barrier (BBB) penetration levels. As a result, it was anticipated that these compounds would not have CNS adverse effects. Regarding aqueous solubility parameters, all compounds showed low to very low levels of aqueous solubility. Next, the intestinal absorption levels of all compounds look like in the good and moderate range. It is interesting to note that compounds 13a,b, 15a,b, 19a,b, and 24a–c were predicted to not inhibit CYP2D6. As a result, it might be successfully metabolized and eliminated. In terms of plasma protein binding, compounds 15a, 19a,b behave similarly to thalidomide in binding in a range of less than 90%. The remainder of compounds, however, was anticipated to bind plasma protein over 90%.
| Comp | FDA rodent carcinogenicity (rat-female) | Carcinogenic potency TD50 (rat)a | Ames mutagenicity | Rat maximum tolerated dose (feed)b | Rat Oral LD50 b | Rat chronic LOAELb | Skin irritancy | Ocular irritancy | DTP |
|---|---|---|---|---|---|---|---|---|---|
| a Unit: mg per kg body weight per day.b Unit: g per kg body weight. | |||||||||
| 6 | Non-carcinogen | 18.598 | Mutagen | 0.106 | 0.023 | 0.021 | Non-irritant | Mild | Non-toxic |
| 7a | Non-carcinogen | 12.040 | Mutagen | 0.240 | 0.156 | 0.022 | Non-irritant | Mild | Non-toxic |
| 7b | Non-carcinogen | 35.737 | Non-mutagen | 0.299 | 0.115 | 0.020 | Non-irritant | Mild | Non-toxic |
| 11a | Non-carcinogen | 15.970 | Non-mutagen | 0.088 | 0.019 | 0.013 | Non-irritant | Mild | Non-toxic |
| 11b | Non-carcinogen | 0.679 | Non-mutagen | 0.146 | 0.169 | 0.037 | Non-irritant | Moderate | Non-toxic |
| 13a | Non-carcinogen | 11.211 | Non-mutagen | 0.282 | 0.064 | 0.038 | Non-irritant | Mild | Non-toxic |
| 13b | Non-carcinogen | 6.050 | Non-mutagen | 0.088 | 0.008 | 0.010 | Non-irritant | Moderate | Non-toxic |
| 15a | Non-carcinogen | 0.257 | Non-mutagen | 0.146 | 0.119 | 0.034 | Non-irritant | Moderate | Non-toxic |
| 15b | Non-carcinogen | 29.594 | Mutagen | 0.282 | 0.091 | 0.041 | Non-irritant | Mild | Non-toxic |
| 19a | Non-carcinogen | 10.189 | Non-mutagen | 0.174 | 0.924 | 0.041 | Non-irritant | Mild | Non-toxic |
| 19b | Non-carcinogen | 12.243 | Non-mutagen | 0.185 | 0.139 | 0.038 | Non-irritant | Mild | Toxic |
| 24a | Non-carcinogen | 12.655 | Non-mutagen | 0.204 | 3.675 | 0.097 | Non-irritant | Mild | Non-toxic |
| 24b | Non-carcinogen | 15.153 | Non-mutagen | 0.217 | 0.566 | 0.087 | Non-irritant | Mild | Non-toxic |
| 24c | Non-carcinogen | 38.125 | Non-mutagen | 0.246 | 1.043 | 0.105 | Non-irritant | Mild | Non-toxic |
| Thalidomide | Carcinogen | 26.375 | Non-mutagen | 0.047 | 0.835 | 0.133 | Non-irritant | None | Non-toxic |
3. Conclusion
Fourteen new compounds, based on quinazoline and phthalazine, were designed and synthesized according to the pharmacophoric features of thalidomide in order to get potential immunomodulatory anticancer agents. N-phthalazinylbenzenesulfonhydrazide (24a–c) emerged as the most potent derivatives in terms of antiproliferative activity. Among which, 24b was the most significant, showing IC50 of 2.51, 5.80 and 4.11 μg mL−1 compared to 11.26, 14.58, and 16.87 μg mL−1 for thalidomide against HepG-2, MCF-7, and PC3 cell lines respectively. Furthermore, it exhibited immunomodulatory properties, including considerable reduction of the expression levels of TNF-α and NF-κB P65 in HepG-2 cells. Additionally, other biological activities of thalidomide were proven to 24b such as inhibition of VEGF and caspase-8. Consequently, this work suggests that compound 24b showed significant biological activity on many targets in a manner similar to thalidomide, and hence it should be considered for further evaluation and modification in order to obtain new potential immunomodulatory anticancer agents.4. Experimental
4.1. Chemistry
All the reagents, chemicals, and apparatus were described in ESI.† The targeted compounds were furnished following the reported methods.4.1.1.1. 2-Chloro-N-(3-chloro-4-fluorophenyl)quinazolin-4-amine 6.
Solid (yield, 77.51%); m.p. = 286–288 °C. IR (KBr, cm−1): 3380 (1NH), 3059 (CH aromatic); 1H NMR (DMSO-d6) δ ppm: 10.26 (s, 1H, N

![[H with combining low line]](https://www.rsc.org/images/entities/b_char_0048_0332.gif) ), 8.52 (s, 1H, Ar–H), 8.08 (dd, J = 7.2, 2.6 Hz, 1H, Ar–H), 7.90 (dd, J = 8.0, 2.4 Hz, 1H, Ar–H), 7.79 (m, 1H, Ar–H), 7.73 (d, J = 8.4 Hz, 1H, Ar–H), 7.67 (dd, J = 7.8, 2.6 Hz, 1H, Ar–H), 7.49 (d, J = 8.0, 4.2 Hz, 1H, Ar–H); anal. (calcd) for C14H8Cl2FN3 (308.14): C, 54.57 (54.73); H, 2.62 (2.85); N, 13.64 (13.81).
), 8.52 (s, 1H, Ar–H), 8.08 (dd, J = 7.2, 2.6 Hz, 1H, Ar–H), 7.90 (dd, J = 8.0, 2.4 Hz, 1H, Ar–H), 7.79 (m, 1H, Ar–H), 7.73 (d, J = 8.4 Hz, 1H, Ar–H), 7.67 (dd, J = 7.8, 2.6 Hz, 1H, Ar–H), 7.49 (d, J = 8.0, 4.2 Hz, 1H, Ar–H); anal. (calcd) for C14H8Cl2FN3 (308.14): C, 54.57 (54.73); H, 2.62 (2.85); N, 13.64 (13.81).
4.1.2.1. N4-(3-Chloro-4-fluorophenyl)-N2-methylquinazoline-2,4-diamine 7a.
Solid (yield, 85.50%); m.p. = 264–266 °C. IR (KBr, cm−1): 3371, 3277 (2NH), 3125 (CH aromatic), 2954 (CH aliphatic); 1H NMR (DMSO-d6) δ ppm: 9.48 (s, 1H, N
 Ph), 8.30 (s, 1H, NCH3), 8.25 (s, 1H, Ar–H), 7.93 (dd, J = 8.6, 4.2 Hz, 1H, Ar–H), 7.58 (dd, J = 8.2, 2.6 Hz, 1H, Ar–H), 7.38 (m, 2H, Ar–H), 7.15 (d, J = 7.6 Hz, 1H, Ar–H), 6.83 (m, 1H, Ar–H), 2.87 (d, J = 8.2 Hz, 3H,C
Ph), 8.30 (s, 1H, NCH3), 8.25 (s, 1H, Ar–H), 7.93 (dd, J = 8.6, 4.2 Hz, 1H, Ar–H), 7.58 (dd, J = 8.2, 2.6 Hz, 1H, Ar–H), 7.38 (m, 2H, Ar–H), 7.15 (d, J = 7.6 Hz, 1H, Ar–H), 6.83 (m, 1H, Ar–H), 2.87 (d, J = 8.2 Hz, 3H,C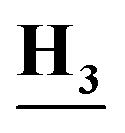 ); anal. calcd for C15H12ClFN4 (302.74): C, 59.51 (59.38); H, 4.00 (4.12); N, 18.51 (18.32).
); anal. calcd for C15H12ClFN4 (302.74): C, 59.51 (59.38); H, 4.00 (4.12); N, 18.51 (18.32).
4.1.2.2. N4-(3-Chloro-4-fluorophenyl)-N2-ethylquinazoline-2,4-diamine 7b.
Solid (yield, 85.60%); m.p. = 270–272 °C. IR (KBr, cm−1): 3374, 3217 (2NH), 2979 (CH aliphatic); 1H NMR (DMSO-d6) δ ppm: 9.66 (s, 1H, N
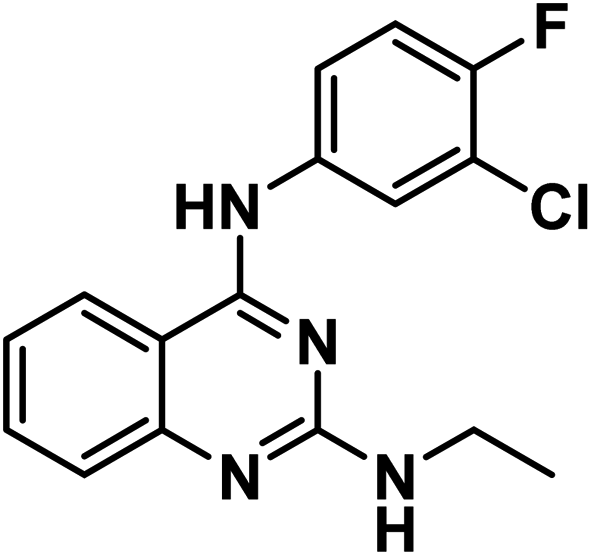 Ph), 9.20 (s, 1H, NCH3), 8.36 (s, 1H, Ar–H), 8.13 (d, J = 8.3 Hz, 1H, Ar–H), 8.03 (m, 1H, Ar–H), 7.68 (dd, J = 7.5, 4.6 Hz, 1H, Ar–H), 7.46 (d, J = 8.4 Hz, 1H, Ar–H), 7.33 (m, 2H, Ar–H), 3.48 (m, 2H, CH3C
Ph), 9.20 (s, 1H, NCH3), 8.36 (s, 1H, Ar–H), 8.13 (d, J = 8.3 Hz, 1H, Ar–H), 8.03 (m, 1H, Ar–H), 7.68 (dd, J = 7.5, 4.6 Hz, 1H, Ar–H), 7.46 (d, J = 8.4 Hz, 1H, Ar–H), 7.33 (m, 2H, Ar–H), 3.48 (m, 2H, CH3C ), 1.02 (t, J = 7.1 Hz, 3H,C
), 1.02 (t, J = 7.1 Hz, 3H,C CH2); 13C NMR (101 MHz, DMSO-d6) δ 158.59, 152.19, 137.34, 133.68, 123.43, 122.69, 122.38, 119.35, 119.16, 117.02, 116.81, 38.73, 15.08; mass (m/z): 318 (M++2), 316 (M+), and 78 (100%, base peak); anal. calcd for C16H14ClFN4 (316.76): C, 60.67 (60.91); H, 4.46 (4.42); N, 17.69 (17.87).
CH2); 13C NMR (101 MHz, DMSO-d6) δ 158.59, 152.19, 137.34, 133.68, 123.43, 122.69, 122.38, 119.35, 119.16, 117.02, 116.81, 38.73, 15.08; mass (m/z): 318 (M++2), 316 (M+), and 78 (100%, base peak); anal. calcd for C16H14ClFN4 (316.76): C, 60.67 (60.91); H, 4.46 (4.42); N, 17.69 (17.87).
4.1.3.1. 2,5-Dichloro-N-(3-chloro-4-fluorophenyl)quinazolin-4-amine 11a.
Solid (yield, 81.78%); m.p. = 132–135 °C. IR (KBr, cm−1): 3381 (1NH), 3059 (CH aromatic); 1H NMR (DMSO-d6) δ ppm: 9.87 (s, 1H, N
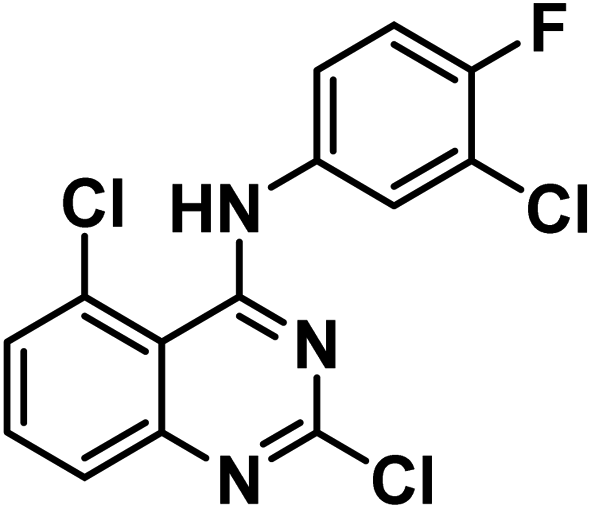 ), 8.00 (s, 1H, Ar–H), 7.86 (d, J = 8.0 Hz, 1H, Ar–H), 7.73 (m, 3H, Ar–H), 7.52 (dd, J = 6.8, 2.6 Hz, 1H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 158.73, 156.19, 153.85, 134.56, 129.74, 129.08, 127.23, 125.62, 124.50, 124.43, 117.35, 117.13, 111.97; mass (m/z): 343 (M+ + 1), 342 (M+), and 157 (100%, base peak). Anal. calcd for C14H7Cl3FN3 (342.58): C, 49.08 (49.29); H, 2.06 (2.18); N, 12.27 (12.49).
), 8.00 (s, 1H, Ar–H), 7.86 (d, J = 8.0 Hz, 1H, Ar–H), 7.73 (m, 3H, Ar–H), 7.52 (dd, J = 6.8, 2.6 Hz, 1H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 158.73, 156.19, 153.85, 134.56, 129.74, 129.08, 127.23, 125.62, 124.50, 124.43, 117.35, 117.13, 111.97; mass (m/z): 343 (M+ + 1), 342 (M+), and 157 (100%, base peak). Anal. calcd for C14H7Cl3FN3 (342.58): C, 49.08 (49.29); H, 2.06 (2.18); N, 12.27 (12.49).
4.1.3.2. 2,6-Dichloro-N-(3-chloro-4-fluorophenyl)quinazolin-4-amine 11b.
Solid (yield, 79.06%); m.p. = 145–146 °C. IR (KBr, cm−1): 3419 (1NH), 3065 (CH aromatic); 1H NMR (DMSO-d6) δ ppm: 10.31 (s, 1H, N
 ), 8.70 (s, 1H, Ar–H), 8.09 (s, 1H, Ar–H), 7.94 (d, J = 8.1 Hz, 1H, Ar–H), 7.79 (m, 2H, Ar–H), 7.52 (dd, J = 6.8, 2.6 Hz, 1H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 158.77, 156.68, 149.91, 134.92, 131.40, 129.49, 124.62, 123.41, 123.34, 123.13, 117.37, 117.15, 115.05; mass (m/z): 343 (M+ + 1), 342 (M+), and 100 (100%, base peak). Anal. calcd for C14H7Cl3FN3 (342.58): C, 49.08 (49.24); H, 2.06 (2.18); N, 12.27 (12.44).
), 8.70 (s, 1H, Ar–H), 8.09 (s, 1H, Ar–H), 7.94 (d, J = 8.1 Hz, 1H, Ar–H), 7.79 (m, 2H, Ar–H), 7.52 (dd, J = 6.8, 2.6 Hz, 1H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 158.77, 156.68, 149.91, 134.92, 131.40, 129.49, 124.62, 123.41, 123.34, 123.13, 117.37, 117.15, 115.05; mass (m/z): 343 (M+ + 1), 342 (M+), and 100 (100%, base peak). Anal. calcd for C14H7Cl3FN3 (342.58): C, 49.08 (49.24); H, 2.06 (2.18); N, 12.27 (12.44).
4.1.4.1. 3-((2,5-Dichloroquinazolin-4-yl)amino)piperidine-2,6-dione 13a.
Solid (yield, 51.70%); m.p. = 218–220 °C. IR (KBr, cm−1): 3344, 3195 (2NH), 3088 (CH aromatic), 2855 (CH aliphatic), 1698 (C

![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O imide); 1H NMR (DMSO-d6) δ ppm: 11.04 (s, 1H, CONCO), 8.72 (d, J = 8.1 Hz, 1H, CNCH), 7.76 (m, 1H, Ar–H), 7.63 (m, 2H, Ar–H), 5.07 (m, 1H, C-piperidine), 3.13 (m, 1H, CH2C2CONH-piperidine), 2.81 (m, 1H, CH2C2CONH-piperidine), 2.56 (m, 1H, C2CH2CONH-piperidine), 2.27 (m, 1H, C2CH2CONH-piperidine); anal. calcd for C13H10Cl2N4O2 (325.15): C, 48.02 (48.28); H, 3.10 (3.34); N, 17.23 (17.41).
O imide); 1H NMR (DMSO-d6) δ ppm: 11.04 (s, 1H, CONCO), 8.72 (d, J = 8.1 Hz, 1H, CNCH), 7.76 (m, 1H, Ar–H), 7.63 (m, 2H, Ar–H), 5.07 (m, 1H, C-piperidine), 3.13 (m, 1H, CH2C2CONH-piperidine), 2.81 (m, 1H, CH2C2CONH-piperidine), 2.56 (m, 1H, C2CH2CONH-piperidine), 2.27 (m, 1H, C2CH2CONH-piperidine); anal. calcd for C13H10Cl2N4O2 (325.15): C, 48.02 (48.28); H, 3.10 (3.34); N, 17.23 (17.41).
4.1.4.2. 3-((2,6-Dichloroquinazolin-4-yl)amino)piperidine-2,6-dione 13b.
Solid (yield, 45.96%); m.p. = 198–200 °C. IR (KBr, cm−1): 3316, 3186 (2NH), 3093 (CH aromatic), 2875 (CH aliphatic), 1725 (C
 O imide); 10.99 (s, 1H, CONCO), 9.02 (d, J = 8.2 Hz, 1H, CNCH), 8.47 (s, 1H, Ar–H), 7.89 (d, J = 8.9 Hz, 1H, Ar–H), 7.71 (d, J = 8.9 Hz, 1H, Ar–H), 5.19 (m, 1H, C-piperidine), 2.90 (m, 1H, CH2C2CONH-piperidine), 2.62 (m, 1H, CH2C2CONH-piperidine), 2.25 (m, 1H, C2CH2CONH-piperidine), 2.11 (m, 1H, C2CH2CONH-piperidine); 13C NMR (101 MHz, DMSO-d6) δ 173.38, 171.97, 160.98, 157.32, 149.60, 134.72, 130.91, 129.39, 123.00, 114.76, 51.18, 31.36, 23.95; mass (m/z): 327 (M+ + 2), 326 (M+ + 1), 325 (M+), and 304 (100%, base peak). Anal. calcd for C13H10Cl2N4O2 (325.15): C, 48.02 (48.25); H, 3.10 (3.31); N, 17.23 (17.59).
O imide); 10.99 (s, 1H, CONCO), 9.02 (d, J = 8.2 Hz, 1H, CNCH), 8.47 (s, 1H, Ar–H), 7.89 (d, J = 8.9 Hz, 1H, Ar–H), 7.71 (d, J = 8.9 Hz, 1H, Ar–H), 5.19 (m, 1H, C-piperidine), 2.90 (m, 1H, CH2C2CONH-piperidine), 2.62 (m, 1H, CH2C2CONH-piperidine), 2.25 (m, 1H, C2CH2CONH-piperidine), 2.11 (m, 1H, C2CH2CONH-piperidine); 13C NMR (101 MHz, DMSO-d6) δ 173.38, 171.97, 160.98, 157.32, 149.60, 134.72, 130.91, 129.39, 123.00, 114.76, 51.18, 31.36, 23.95; mass (m/z): 327 (M+ + 2), 326 (M+ + 1), 325 (M+), and 304 (100%, base peak). Anal. calcd for C13H10Cl2N4O2 (325.15): C, 48.02 (48.25); H, 3.10 (3.31); N, 17.23 (17.59).
4.1.5.1. 2-(2,5-Dichloroquinazolin-4-yl)hydrazine-1-carboxamide 15a.
Solid (yield, 75.51%); m.p. = 189–191 °C. IR (KBr, cm−1): 3457, 3349, 3274, 3194 (br, 4NH), 3050 (CH aromatic); 1H NMR (DMSO-d6) δ ppm: 9.87 (s, 1H, NHN
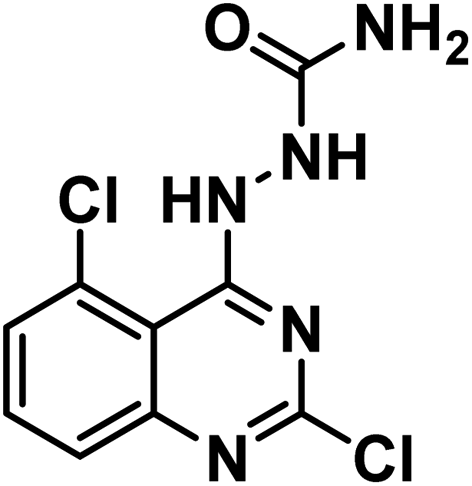 CO), 8.53 (s, 1H, NNHCO), 7.70 (d, J = 8.2 Hz, 1H, Ar–H), 7.56 (d, J = 8.00 Hz, 1H, Ar–H), 7.45 (m, 1H, Ar–H), 6.25 (s, 2H, N
CO), 8.53 (s, 1H, NNHCO), 7.70 (d, J = 8.2 Hz, 1H, Ar–H), 7.56 (d, J = 8.00 Hz, 1H, Ar–H), 7.45 (m, 1H, Ar–H), 6.25 (s, 2H, N ); anal. calcd for C9H7Cl2N5O (272.09): C, 39.73 (39.88); H, 2.59 (2.75); N, 25.74 (25.53).
); anal. calcd for C9H7Cl2N5O (272.09): C, 39.73 (39.88); H, 2.59 (2.75); N, 25.74 (25.53).
4.1.5.2. 2-(2,6-Dichloroquinazolin-4-yl)hydrazine-1-carboxamide 15b.
Solid (yield, 68.65%); m.p. = 171–173 °C. IR (KBr, cm−1): 3471, 3426, 3267, 3190 (br, 4NH), 3027 (CH aromatic); 1H NMR (DMSO-d6) δ ppm: 10.43 (s, 1H, NHN
 CO), 8.45 (s, 1H, NNHCO),8.24 (d, J = 8.2 Hz, 1H, Ar–H), 7.88 (d, J = 8.4 Hz, 1H, Ar–H), 7.72 (m, 1H, Ar–H), 6.20 (s, 2H, N
CO), 8.45 (s, 1H, NNHCO),8.24 (d, J = 8.2 Hz, 1H, Ar–H), 7.88 (d, J = 8.4 Hz, 1H, Ar–H), 7.72 (m, 1H, Ar–H), 6.20 (s, 2H, N ); 13C NMR (101 MHz, DMSO-d6) δ 161.90, 157.71, 134.74, 130.83, 129.18, 123.26, 113.78; mass (m/z): 274 (M+ + 2), 272 (M+), and 127 (100%, base peak); anal. calcd for C9H7Cl2N5O (272.09): C, 39.73 (39.90); H, 2.59 (2.78); N, 25.74 (25.91).
); 13C NMR (101 MHz, DMSO-d6) δ 161.90, 157.71, 134.74, 130.83, 129.18, 123.26, 113.78; mass (m/z): 274 (M+ + 2), 272 (M+), and 127 (100%, base peak); anal. calcd for C9H7Cl2N5O (272.09): C, 39.73 (39.90); H, 2.59 (2.78); N, 25.74 (25.91).
4.1.6.1. N′-(2,5-Dichloroquinazolin-4-yl)-4-methylbenzenesulfonohydrazide 19a.
Solid (yield, 83.68%); m.p. = 155–157 °C. IR (KBr, cm−1): 3397, 3306 (2NH), 3086 (CH aromatic), 1163 (SO2 group); 1H NMR (DMSO-d6) δ ppm: 10.04 (s, 1H, NHN
 SO2), 7.82 (s, 1H, NNHSO2), 7.78 (s, 1H, Ar–H), 7.70 (d, J = 8.4 Hz, 1H, Ar–H), 7.64 (d, J = 8.3 Hz, 1H, Ar–H), 7.53 (d, J = 8.2 Hz, 1H, Ar–H), 7.46 (m, 1H, Ar–H), 7.33 (d, J = 8.0 Hz, 2H, Ar–H), 2.31 (s, 3H, CH3); anal. calcd for C15H12Cl2N4O2S (383.25): C, 47.01 (47.27); H, 3.16 (3.38); N, 14.62 (14.89).
SO2), 7.82 (s, 1H, NNHSO2), 7.78 (s, 1H, Ar–H), 7.70 (d, J = 8.4 Hz, 1H, Ar–H), 7.64 (d, J = 8.3 Hz, 1H, Ar–H), 7.53 (d, J = 8.2 Hz, 1H, Ar–H), 7.46 (m, 1H, Ar–H), 7.33 (d, J = 8.0 Hz, 2H, Ar–H), 2.31 (s, 3H, CH3); anal. calcd for C15H12Cl2N4O2S (383.25): C, 47.01 (47.27); H, 3.16 (3.38); N, 14.62 (14.89).
4.1.6.2. N′-(2,5-Dichloroquinazolin-4-yl)-4-fluorobenzenesulfonohydrazide 19b.
Solid (yield, 66.26%); m.p. = 168–170 °C. IR (KBr, cm−1): 3396, 3306 (2NH), 3065 (CH aromatic), 1166 (SO2 group); 1H NMR (DMSO-d6) δ ppm: 10.24 (s, 1H, NHN
 SO2), 7.97 (s, 1H, NNHSO2), 7.95 (s, 1H, Ar–H), 7.69 (m, 2H, Ar–H), 7.64 (d, J = 8.3 Hz, 1H, Ar–H), 7.53 (d, J = 8.2 Hz, 1H, Ar–H), 7.39 (m, 2H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 163.69, 146.43, 144.82, 135.62, 133.76, 133.42, 131.41, 129.66, 129.57, 126.21, 116.52, 116.30, 115.42, 103.97; mass (m/z): 389 (M++2), 387 (M+), and 193 (100%, base peak); anal. calcd for C14H9Cl2FN4O2S (387.21): C, 43.43 (43.54); H, 2.34 (2.50); N, 14.47 (14.63).
SO2), 7.97 (s, 1H, NNHSO2), 7.95 (s, 1H, Ar–H), 7.69 (m, 2H, Ar–H), 7.64 (d, J = 8.3 Hz, 1H, Ar–H), 7.53 (d, J = 8.2 Hz, 1H, Ar–H), 7.39 (m, 2H, Ar–H); 13C NMR (101 MHz, DMSO-d6) δ 163.69, 146.43, 144.82, 135.62, 133.76, 133.42, 131.41, 129.66, 129.57, 126.21, 116.52, 116.30, 115.42, 103.97; mass (m/z): 389 (M++2), 387 (M+), and 193 (100%, base peak); anal. calcd for C14H9Cl2FN4O2S (387.21): C, 43.43 (43.54); H, 2.34 (2.50); N, 14.47 (14.63).
4.1.7.1. N′-(4-Chlorophthalazin-1-yl)-4-methylbenzenesulfonohydrazide 24a.
Solid (yield, 80.35%); m.p. = 235–237 °C. IR (KBr, cm−1): 3330, 3188 (2NH), 3062 (CH aromatic), 2921 (CH aliphatic), 1166 (SO2 group); 1H NMR (DMSO-d6) δ ppm: 11.72 (s, 1H, N
 SO2), 9.24 (s, 1H, CNNH), 7.98 (d, J = 7.8 Hz, 1H, Ar–H), 7.83 (s, 1H, Ar–H), 7.79 (m, 4H, Ar–H), 7.42 (d, J = 7.9 Hz, 2H, Ar–H), 2.40 (s, 3H, C
SO2), 9.24 (s, 1H, CNNH), 7.98 (d, J = 7.8 Hz, 1H, Ar–H), 7.83 (s, 1H, Ar–H), 7.79 (m, 4H, Ar–H), 7.42 (d, J = 7.9 Hz, 2H, Ar–H), 2.40 (s, 3H, C ); anal. calcd for C15H13ClN4O2S (348.81): C, 51.65 (51.89); H, 3.76 (3.85); N, 16.06 (16.32).
); anal. calcd for C15H13ClN4O2S (348.81): C, 51.65 (51.89); H, 3.76 (3.85); N, 16.06 (16.32).
4.1.7.2. N′-(4-Chlorophthalazin-1-yl)-4-fluorobenzenesulfonohydrazide 24b.
Solid (yield, 83.71%); m.p. = 220–222 °C. IR (KBr, cm−1): 3334, 3189 (2H), 3067 (CH aromatic); 1H NMR (DMSO-d6) δ ppm: 11.83 (s, 1H, N
 SO2), 9.33 (s, 1H, CNNH), 8.00 (dd, J = 8.00, 4.4 Hz, 3H, Ar–H), 7.77 (m, 3H, Ar–H), 7.47 (m, 2H, Ar–H); anal. calcd for C14H10ClFN4O2S (352.77): C, 47.67 (47.85); H, 2.86 (2.97); N, 15.88 (15.62).
SO2), 9.33 (s, 1H, CNNH), 8.00 (dd, J = 8.00, 4.4 Hz, 3H, Ar–H), 7.77 (m, 3H, Ar–H), 7.47 (m, 2H, Ar–H); anal. calcd for C14H10ClFN4O2S (352.77): C, 47.67 (47.85); H, 2.86 (2.97); N, 15.88 (15.62).
4.1.7.3. N′-(4-Chlorophthalazin-1-yl)benzenesulfonohydrazide 24c.
Solid (yield, 77.24%); m.p. = 192–194 °C. IR (KBr, cm−1): 3331, 3199 (2NH), 3063 (CH aromatic); 1H NMR (DMSO-d6) δ ppm: 11.75 (s, 1H, N
 SO2), 9.44 (s, 1H, CNNH), 8.04 (s, 1H, Ar–H), 7.92 (d, J = 7.6 Hz, 2H, Ar–H), 7.85 (s, 3H, Ar–H), 7.65 (d, J = 7.2 Hz, 1H, Ar–H), 7.61 (d, J = 7.5 Hz, 2H, Ar–H); anal. calcd for C14H11ClN4O2S (334.78): C, 50.23 (50.34); H, 3.31 (3.45); N, 16.74 (16.90).
SO2), 9.44 (s, 1H, CNNH), 8.04 (s, 1H, Ar–H), 7.92 (d, J = 7.6 Hz, 2H, Ar–H), 7.85 (s, 3H, Ar–H), 7.65 (d, J = 7.2 Hz, 1H, Ar–H), 7.61 (d, J = 7.5 Hz, 2H, Ar–H); anal. calcd for C14H11ClN4O2S (334.78): C, 50.23 (50.34); H, 3.31 (3.45); N, 16.74 (16.90).
4.2. Biological testing
4.2.5.1. Pharmacokinetic profiling study. This study was applied using Discover studio 4 (ref. 57) in accordance with the comprehensive description in ESI.†
4.2.5.2. ADMET studies. ADMET descriptors were determined using Discovery studio 4.0 as according to the reported method58–61 (ESI†).
4.2.5.3. Toxicity studies. The toxicity parameters of the synthesized compounds were calculated using Discovery studio 4.0 as described62–65 in ESI.†
Conflicts of interest
There is no conflict of interest.Acknowledgements
This paper is based upon work supported by Science, Technology & Innovation Funding Authority (STIFA) under grant number 43040.References
- A. E. Abdallah, R. R. Mabrouk, M. R. Elnagar, A. M. Farrag, M. H. Kalaba, M. H. Sharaf, E. M. El-Fakharany, D. A. Bakhotmah, E. B. Elkaeed and M. M. S. Al Ward, New Series of VEGFR-2 Inhibitors and Apoptosis Enhancers: Design, Synthesis and Biological Evaluation, Drug Des., Dev. Ther., 2022, 16, 587 CrossRef PubMed.
- A.-M. Florea and D. Büsselberg, Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects, Cancers, 2011, 3(1), 1351–1371 CrossRef CAS PubMed.
- M. Nikolaou, A. Pavlopoulou, A. G. Georgakilas and E. Kyrodimos, The challenge of drug resistance in cancer treatment: a current overview, Clin. Exp. Metastasis, 2018, 35(4), 309–318 CrossRef CAS PubMed.
- N. Chatterjee and T. G. Bivona, Polytherapy and targeted cancer drug resistance, Trends Cancer, 2019, 5(3), 170–182 CrossRef CAS PubMed.
- J. A. Simon, P. Szankasi, D. K. Nguyen, C. Ludlow, H. M. Dunstan, C. J. Roberts, E. L. Jensen, L. H. Hartwell and S. H. Friend, Differential toxicities of anticancer agents among DNA repair and checkpoint mutants of Saccharomyces cerevisiae, Cancer Res., 2000, 60(2), 328–333 CAS.
- S. Eckhardt, Recent progress in the development of anticancer agents, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2(3), 419–439 CrossRef CAS PubMed.
- A. Colombo, C. Cipolla, M. Beggiato and D. Cardinale, Cardiac toxicity of anticancer agents, Curr. Cardiol. Rep., 2013, 15(5), 1–11 Search PubMed.
- A. Iliadis and D. Barbolosi, Optimizing drug regimens in cancer chemotherapy by an efficacy–toxicity mathematical model, Comput. Biomed. Res., 2000, 33(3), 211–226 CrossRef CAS PubMed.
- H. Elkady, A. Elwan, H. A. El-Mahdy, A. S. Doghish, A. Ismail, M. S. Taghour, E. B. Elkaeed, I. H. Eissa, M. A. Dahab and H. A. Mahdy, New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 403–416 CrossRef CAS PubMed.
- M. B. Steins, T. Padró, R. Bieker, S. Ruiz, M. Kropff, J. Kienast, T. Kessler, T. Buechner, W. E. Berdel and R. M. Mesters, Efficacy and safety of thalidomide in patients with acute myeloid leukemia, Blood, 2002, 99(3), 834–839 CrossRef CAS PubMed.
- Y. X. Zhu, K. M. Kortuem and A. K. Stewart, Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma, Leuk. Lymphoma, 2013, 54(4), 683–687 CrossRef CAS PubMed.
- C. Galustian, B. Meyer, M.-C. Labarthe, K. Dredge, D. Klaschka, J. Henry, S. Todryk, R. Chen, G. Muller and D. Stirling, The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells, Cancer Immunol. Immunother., 2009, 58(7), 1033–1045 CrossRef CAS PubMed.
- G. Görgün, E. Calabrese, E. Soydan, T. Hideshima, G. Perrone, M. Bandi, D. Cirstea, L. Santo, Y. Hu and Y.-T. Tai, Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma, Blood, 2010, 116(17), 3227–3237 CrossRef PubMed.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem, S. I. Eissa, H. Elkady and I. H. Eissa, Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents, Bioorg. Chem., 2020, 104, 104218 CrossRef CAS PubMed.
- K. C. Anderson, Lenalidomide and thalidomide: mechanisms of action—similarities and differences, in Seminars in Hematology, Elsevier, 2005, pp. S3–S8 Search PubMed.
- S. Kumar and S. V. Rajkumar, Thalidomide and lenalidomide in the treatment of multiple myeloma, Eur. J. Cancer, 2006, 42(11), 1612–1622 CrossRef CAS PubMed.
- S. Lindner and J. Krönke, The molecular mechanism of thalidomide analogs in hematologic malignancies, J. Mol. Med., 2016, 94(12), 1327–1334 CrossRef CAS PubMed.
- T. Ito and H. Handa, Molecular mechanisms of thalidomide and its derivatives, Proc. Jpn Acad., Ser. B, 2020, 96(6), 189–203 CrossRef CAS PubMed.
- T. Reske, M. Fulciniti and N. C. Munshi, Mechanism of action of immunomodulatory agents in multiple myeloma, Med. Oncol., 2010, 27(1), 7–13 CrossRef PubMed.
- D. Millrine and T. Kishimoto, A brighter side to thalidomide: its potential use in immunological disorders, Trends Mol. Med., 2017, 23(4), 348–361 CrossRef CAS PubMed.
- J. Cortés-Hernández, M. Torres-Salido, J. Castro-Marrero, M. Vilardell-Tarres and J. Ordi-Ros, Thalidomide in the treatment of refractory cutaneous lupus erythematosus: prognostic factors of clinical outcome, Br. J. Dermatol., 2012, 166(3), 616–623 CrossRef PubMed.
- A. Lopez-Girona, D. Mendy, T. Ito, K. Miller, A. Gandhi, J. Kang, S. Karasawa, G. Carmel, P. Jackson and M. Abbasian, Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide, Leukemia, 2012, 26(11), 2326–2335 CrossRef CAS PubMed.
- P. R. Hagner, H.-W. Man, C. Fontanillo, M. Wang, S. Couto, M. Breider, C. Bjorklund, C. G. Havens, G. Lu and E. Rychak, CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL, Blood, 2015, 126(6), 779–789 CrossRef CAS PubMed.
- G. E. Diggle, Thalidomide: 40 years on, Int. J. Clin. Pract., 2001, 55(9), 627–631 CAS.
- A. K. Akobeng and P. C. Stokkers, Thalidomide and thalidomide analogues for maintenance of remission in Crohn’s disease, Cochrane Database Syst. Rev., 2009,(2), CD007350 Search PubMed.
- M. Attal, V. Lauwers-Cances, G. Marit, D. Caillot, P. Moreau, T. Facon, A. M. Stoppa, C. Hulin, L. Benboubker and L. Garderet, Lenalidomide maintenance after stem-cell transplantation for multiple myeloma, N. Engl. J. Med., 2012, 366(19), 1782–1791 CrossRef CAS PubMed.
- C. Galustian and A. Dalgleish, Google-2 is logged in Drugs of the Future, 2011, vol. 36, 10, p. 741, ISSN 0377-8282 Copyright 2011 Prous Science CCC: 0377-8282 Search PubMed.
- M. Offidani, L. Corvatta, P. Caraffa, P. Leoni, C. Pautasso, A. Larocca and A. Palumbo, Pomalidomide for the treatment of relapsed–refractory multiple myeloma: a review of biological and clinical data, Expert Rev. Anticancer Ther., 2014, 14(5), 499–510 CrossRef CAS PubMed.
- P. Hagner, M. Wang, S. Couto, M. Breider, C. Fontanillo, M. Trotter, C. C. Bjorklund, C. G. Havens, H. K. Raymon and R. K. Narla, CC-122 has potent anti-lymphoma activity through destruction of the Aiolos and Ikaros transcription factors and induction of interferon response pathways, Blood, 2014, 124(21), 3035 CrossRef.
- A. Renneville, J. A. Gasser, D. E. Grinshpun, P. M. Jean Beltran, N. D. Udeshi, M. E. Matyskiela, T. Clayton, M. McConkey, K. Viswanathan and A. Tepper, Avadomide Induces Degradation of ZMYM2 Fusion Oncoproteins in Hematologic MalignanciesAvadomide Induces ZMYM2 Fusion Oncoprotein Degradation, Blood Cancer Discovery, 2021, 2(3), 250–265 CrossRef CAS PubMed.
- L. Xu and W. A. Russu, Molecular docking and synthesis of novel quinazoline analogues as inhibitors of transcription factors NF-κB activation and their anti-cancer activities, Bioorg. Med. Chem., 2013, 21(2), 540–546 CrossRef CAS PubMed.
- N.-C. Cho, J. H. Cha, H. Kim, J. Kwak, D. Kim, S.-H. Seo, J.-S. Shin, T. Kim, K. D. Park and J. Lee, Discovery of 2-aryloxy-4-amino-quinazoline derivatives as novel protease-activated receptor 2 (PAR2) antagonists, Bioorg. Med. Chem., 2015, 23(24), 7717–7727 CrossRef CAS PubMed.
- J. Hu, Y. Zhang, L. Dong, Z. Wang, L. Chen, D. Liang, D. Shi, X. Shan and G. Liang, Design, synthesis, and biological evaluation of novel quinazoline derivatives as anti-inflammatory agents against lipopolysaccharide-induced acute lung injury in rats, Chem. Biol. Drug Des., 2015, 85(6), 672–684 CrossRef CAS PubMed.
- Y. Pu, D. Cao, C. Xie, H. Pei, D. Li, M. Tang and L. Chen, Anti-arthritis effect of a novel quinazoline derivative through inhibiting production of TNF-α mediated by TNF-α converting enzyme in murine collagen-induced arthritis model, Biochem. Biophys. Res. Commun., 2015, 462(4), 288–293 CrossRef CAS PubMed.
- A. E. Abdallah, S. I. Eissa, M. M. S. Al Ward, R. R. Mabrouk, A. B. Mehany and M. A. El-Zahabi, Design, synthesis and molecular modeling of new quinazolin-4 (3H)-one based VEGFR-2 kinase inhibitors for potential anticancer evaluation, Bioorg. Chem., 2021, 109, 104695 CrossRef CAS PubMed.
- G. A. Bennett, L. A. Radov, L. A. Trusso and V. S. Georgiev, Synthesis and immunomodulating activity of 5H-thiazolo [2,3-b] quinazolin-3 (2H)-one, J. Pharm. Sci., 1987, 76(8), 633–634 CrossRef CAS PubMed.
- A. E. Abdallah, R. R. Mabrouk, M. M. S. Al Ward, S. I. Eissa, E. B. Elkaeed, A. B. Mehany, M. A. Abo-Saif, O. A. El-Feky, M. S. Alesawy and M. A. El-Zahabi, Synthesis, biological evaluation, and molecular docking of new series of antitumor and apoptosis inducers designed as VEGFR-2 inhibitors, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 573–591 CrossRef CAS PubMed.
- D.-C. Liu, G.-H. Gong, C.-X. Wei, X.-J. Jin and Z.-S. Quan, Synthesis and anti-inflammatory activity evaluation of a novel series of 6-phenoxy-[1, 2, 4] triazolo [3, 4-a] phthalazine-3-carboxamide derivatives, Bioorg. Med. Chem. Lett., 2016, 26(6), 1576–1579 CrossRef CAS PubMed.
- K. K. To, D. C. Poon, Y. Wei, F. Wang, G. Lin and L.-w. Fu, Data showing the circumvention of oxaliplatin resistance by vatalanib in colon cancer, Data Brief, 2016, 7, 437–444 CrossRef PubMed.
- D.-Q. Xue, X.-Y. Zhang, C.-J. Wang, L.-Y. Ma, N. Zhu, P. He, K.-P. Shao, P.-J. Chen, Y.-F. Gu and X.-S. Zhang, Synthesis and anticancer activities of novel 1, 2, 4-triazolo [3, 4-a] phthalazine derivatives, Eur. J. Med. Chem., 2014, 85, 235–244 CrossRef CAS PubMed.
- W. M. Eldehna, S. M. Abou-Seri, A. M. El Kerdawy, R. R. Ayyad, A. M. Hamdy, H. A. Ghabbour, M. M. Ali and D. A. Abou El Ella, Increasing the binding affinity of VEGFR-2 inhibitors by extending their hydrophobic interaction with the active site: Design, synthesis and biological evaluation of 1-substituted-4-(4-methoxybenzyl) phthalazine derivatives, Eur. J. Med. Chem., 2016, 113, 50–62 CrossRef CAS PubMed.
- N. Jiang, X. Zhai, Y. Zhao, Y. Liu, B. Qi, H. Tao and P. Gong, Synthesis and biological evaluation of novel 2-(2-arylmethylene) hydrazinyl-4-aminoquinazoline derivatives as potent antitumor agents, Eur. J. Med. Chem., 2012, 54, 534–541 CrossRef CAS PubMed.
- D. Shah, H. Lakum and K. Chikhalia, Synthesis and in vitro antimicrobial evaluation of piperazine substituted quinazoline-based thiourea/thiazolidinone/chalcone hybrids, Russ. J. Bioorg. Chem., 2015, 41(2), 209–222 CrossRef CAS.
- K. S. Van Horn, X. Zhu, T. Pandharkar, S. Yang, B. Vesely, M. Vanaerschot, J.-C. Dujardin, S. Rijal, D. E. Kyle and M. Z. Wang, Antileishmanial activity of a series of N 2, N 4-disubstituted quinazoline-2, 4-diamines, J. Med. Chem., 2014, 57(12), 5141–5156 CrossRef CAS PubMed.
- M. M. Gineinah, M. A. El-Sherbeny, M. N. Nasr and A. R. Maarouf, Synthesis and Antiinflammatory Screening of Some Quinazoline and Quinazolyl-4-oxoquinazoline Derivatives, Arch. Pharm., 2002, 335(11–12), 556–562 CrossRef CAS PubMed.
- H. Drew and R. Garwood, 177. Chemiluminescent organic compounds. Part VII. Substituted phthalaz-1: 4-diones. Effect of substituents on the luminescent power, J. Chem. Soc., 1939, 836–837 RSC.
- J. C. Pritchett, L. Naesens and J. Montoya, Treating HHV-6 infections: The laboratory efficacy and clinical use of ati-HHV-6 agents, 2014 Search PubMed.
- N. A. Alsaif, M. A. Dahab, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, H. A. Mahdy and H. Elkady, New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: design, molecular modeling, and synthesis, Bioorg. Chem., 2021, 110, 104807 CrossRef CAS PubMed.
- S. A. El-Metwally, M. M. Abou-El-Regal, I. H. Eissa, A. B. Mehany, H. A. Mahdy, H. Elkady, A. Elwan and E. B. Elkaeed, Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents, Bioorg. Chem., 2021, 112, 104947 CrossRef CAS PubMed.
- N. A. Alsaif, M. S. Taghour, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, A. Elwan and H. Elkady, Discovery of new VEGFR-2 inhibitors based on bis ([1, 2, 4] triazolo)[4, 3-a: 3', 4'-c] quinoxaline derivatives as anticancer agents and apoptosis inducers, J. Enzyme Inhib. Med. Chem., 2021, 36(1), 1093–1114 CrossRef CAS PubMed.
- M. M. Alanazi, I. H. Eissa, N. A. Alsaif, A. J. Obaidullah, W. A. Alanazi, A. F. Alasmari, H. Albassam, H. Elkady and A. Elwan, Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers, J. Enzyme Inhib. Med. Chem., 2021, 36(1), 1760–1782 CrossRef CAS PubMed.
- M. S. Taghour, H. A. Mahdy, M. H. Gomaa, A. Aglan, M. G. Eldeib, A. Elwan, M. A. Dahab, E. B. Elkaeed, A. A. Alsfouk and M. M. Khalifa, Benzoxazole derivatives as new VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, in silico studies, and antiproliferative evaluation, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 2063–2077 CrossRef CAS PubMed.
- D. Gerlier and N. Thomasset, Use of MTT colorimetric assay to measure cell activation, J. Immunol. Methods, 1986, 94(1–2), 57–63 CrossRef CAS PubMed.
- R. M. Talaat, Soluble angiogenesis factors in sera of Egyptian patients with hepatitis C virus infection: correlation with disease severity, Viral Immunol., 2010, 23(2), 151–157 CrossRef CAS PubMed.
- MyBioSource ELISA Test Kits, https://www.mybiosource.com/human-elisa-kits/casp8/2513072#QLAPP_MBS2513072_TD Search PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. El-Deeb, A. M. Kenawy, E. B. Elkaeed, B. A. Alsfouk, M. S. Alesawy, D. Z. Husein and A. M. Metwaly, Design, synthesis, anti-proliferative evaluation, docking, and MD simulations studies of new thiazolidine-2, 4-diones targeting VEGFR-2 and apoptosis pathway, PLoS One, 2022, 17(9), e0272362 CrossRef CAS PubMed.
- A. Belal, H. Elkady, A. A. Al-Karmalawy, A. H. Amin, M. M. Ghoneim, M. El-Sherbiny, R. H. Al-Serwi, M. A. Abdou, M. H. Ibrahim and A. Mehany, Discovery of Some Heterocyclic Molecules as Bone Morphogenetic Protein 2 (BMP-2)-Inducible Kinase Inhibitors: Virtual Screening, ADME Properties, and Molecular Docking Simulations, Molecules, 2022, 27(17), 5571 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. Gobaara, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, The Assessment of Anticancer and VEGFR-2 Inhibitory Activities of a New 1H-Indole Derivative: In Silico and In Vitro Approaches, Processes, 2022, 10(7), 1391 CrossRef CAS.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. Gobaara, B. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, Design, synthesis, docking, DFT, MD simulation studies of a new nicotinamide-based derivative: In vitro anticancer and VEGFR-2 inhibitory effects, Molecules, 2022, 27(14), 4606 CrossRef CAS PubMed.
- E. B. Elkaeed, I. H. Eissa, H. Elkady, A. Abdelalim, A. M. Alqaisi, A. A. Alsfouk, A. Elwan and A. M. Metwaly, A Multistage In Silico Study of Natural Potential Inhibitors Targeting SARS-CoV-2 Main Protease, Int. J. Mol. Sci., 2022, 23(15), 8407 CrossRef CAS PubMed.
- E. B. Elkaeed, F. S. Youssef, I. H. Eissa, H. Elkady, A. A. Alsfouk, M. L. Ashour, M. A. El Hassab and S. M. Abou-Seri, Metwaly AM: Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease, Int. J. Mol. Sci., 2022, 23(13), 6912 CrossRef CAS PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. M. El-Deeb, A. M. Kenawy, E. B. Elkaeed, A. A. Alsfouk, M. S. Alesawy, A. M. Metwaly and I. H. Eissa, Design and synthesis of thiazolidine-2, 4-diones hybrids with 1, 2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: in-vitro anticancer evaluation and in-silico studies, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 1903–1917 CrossRef CAS PubMed.
- A. Belal, N. M. Abdel Gawad, A. B. Mehany, M. A. Abourehab, H. Elkady, A. A. Al-Karmalawy and A. S. Ismael, Design, synthesis and molecular docking of new fused 1 H-pyrroles, pyrrolo [3, 2-d] pyrimidines and pyrrolo [3, 2-e][1, 4] diazepine derivatives as potent EGFR/CDK2 inhibitors, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 1884–1902 CrossRef CAS PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani, M. M. Alanazi, M. A. Alharbi, I. H. Eissa and M. A. Dahab, New quinoxaline-based VEGFR-2 inhibitors: design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies, RSC Adv., 2021, 11(48), 30315–30328 RSC.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, W. A. Alanazi, A. M. Al-Hossaini, M. A. Alharbi, I. H. Eissa and M. A. Dahab, Discovery of new quinoxaline-based derivatives as anticancer agents and potent VEGFR-2 inhibitors: Design, synthesis, and in silico study, J. Mol. Struct., 2021, 132220 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra06188k |
| This journal is © The Royal Society of Chemistry 2022 |