
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 5-aryl-3,3′-bis-indolyl and bis-7-aza-indolyl methanone derivatives from 5-bromo-7-azaindoles via sequential methylenation using microwave irradiation, CAN oxidation, and Suzuki coupling reactions†
Elavarasan Pavithraa,
Sathananthan Kannadasan *a and
Ponnusamy Shanmugam*b
*a and
Ponnusamy Shanmugam*b
aDepartment of Chemistry, School of Advanced Sciences, VIT, Vellore-632014, India. E-mail: kannadasan.s@vit.ac.in
bOrganic and Bioorganic Chemistry Division, CSIR-Central Leather Research Institute (CLRI), Adyar, Chennai-600020, India. E-mail: shanmu196@rediffmail.com
First published on 27th October 2022
Abstract
A catalyst-free and green chemical method has been developed for the methylenation of indole and N-methyl-7-aza indoles with aqueous formaldehyde afforded respective N,N′-dimethyl-3,3′-bis-7-azaindolylmethanes under microwave irradiation in excellent yield. Subsequent oxidation of the products thus obtained, using one electron chemical oxidant CAN afforded N,N′-dimethyl-3,3′-bis-7-azaindolylmethanone derivatives in excellent yield. This resulted in methanone derivatives with halogen substitution at the aryl ring which when subjected to Suzuki coupling with aryl boronic acids furnished highly functionalized fluorescent biaryl derivatives. Plausible mechanisms, characterization including XRD, and evaluation of photophysical properties of the Suzuki coupled products are described.
Introduction
7-Azaindole and its derivatives exhibit significant biological activities and the framework has contributed to the development of new therapeutic agents.1 For example, natural products, meriolin 1 (1) and meriolin 3 (2) showed improved potency, and several FDA-approved drugs such as vemurafenib (3) are used for the treatment of metastatic melanoma. Pexidartinib (4) is approved for the treatment of giant cell tumours associated with severe morbidity. Moreover, 7-azadindole derivative NVP-QAV680 (5) is known for the treatment of allergies and Zn-azaindole complex (6) has a bright blue emitter property. To demonstrate materials applications, and new functionality to enhance the performance of 7-azaindole derivatives, some of the triaryl boron functionalized compounds such as (7) and (8) have been reported as ‘‘bifunctional materials’’ in OLEDs (Fig. 1).2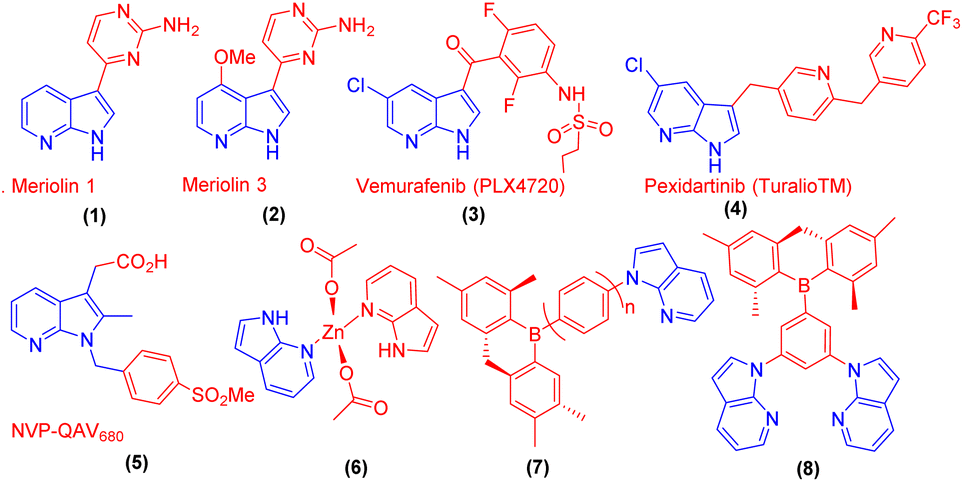 | ||
| Fig. 1 Drugs and materials based on 7-aza indole cores. | ||
Methylenation is a well-known structural modification reaction in organic synthesis.3 Lewis acid-mediated synthesis of 3,3′-bis-7-azaindolylmethane derivatives using zinc- or acid-mediated cross-coupling reaction of 7-azaindoles with a number of diverse aldehydes provides the corresponding C3-linked methylenation products is reported.4 However, the development of the synthesis of 3-methylenation products of indoles and 7-azaindoles by green chemistry protocol using a catalyst-free, aqueous, and microwave irradiation method would be an alternate and efficient method warranted.
Oxidation of benzylic methylene with various oxidizing agents offers functional derivatives such as alcohols, aldehydes and ketones depending upon the substrates and reagents used.5 Development of synthetic methods based on utilizing green chemistry protocols such as green chemistry, clay, microwave irradiation techniques enzymatic method etc., are of current interest in the synthetic organic chemistry.6 Cerium(IV) ammonium nitrate (CAN) has emerged as a versatile reagent for a variety of synthetic transformations which have been well documented.7 Conversion of 3-methylenation products of indoles and 7-azaindoles to corresponding ketones are interesting synthetic modifications and no reports are available using mild and efficient reagents. Thus, we were interested to develop the oxidation of 3-methylenation products of indoles and 7-azaindoles using CAN as a one-electron oxidant into the corresponding ketones. Thus, herein we wish to report the details of the work carried out by initial methylenation, oxidation using CAN, and further synthetic utility of ketone products by using the Suzuki coupling reaction.
To synthesize the title compounds, a retrosynthetic analysis is shown in Scheme 1. Accordingly, the functionalized and fluorescent 3,3′-bis-7-azaindolylmethanone B derivatives were synthesized via steps involving microwave irradiated methylenation of 7-aza indoles D with aqueous formaldehyde would provide the N,N′-dimethyl-3,3′-bis-7-azaindolylmethane C followed by oxidation of compound C using CAN be followed by Suzuki coupling8 of C with aryl boronic acids.
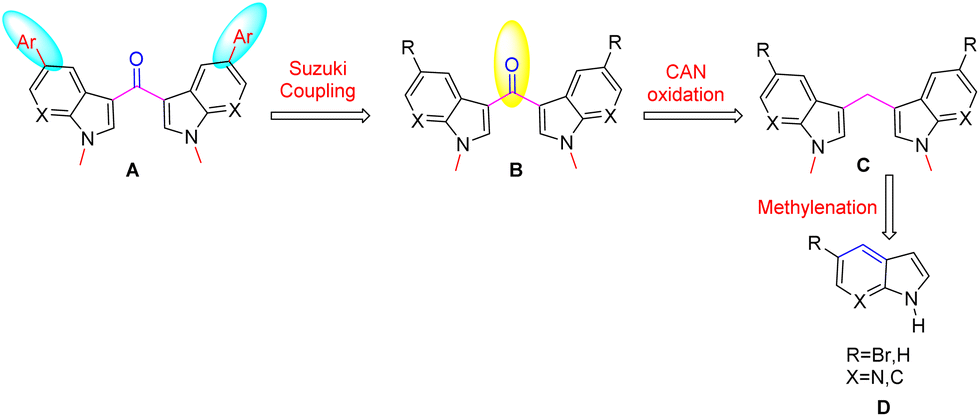 | ||
| Scheme 1 Retrosynthetic route for the title compounds. | ||
As per the retrosynthesis shown in Scheme 1, initially, we followed the reported procedure9 for the synthesis of methylenated compound 2a′ from 7-azaindole 1 under microwave irradiation did not produce compound 2a′ but N-hydroxy methyl product in 3a′ was isolated in 44% yield and the reaction produced multiple spots and are inseparable by column chromatography. However, repeating the above reaction under sealed tube heating conditions produced 95% of a product i.e. methylated and N-hydroxy methyl product exclusively in 3a′ 95% yield (Scheme 2). The reaction suggests that when free N–H is available, under the reaction condition, it produces only N-hydroxy methyl compounds such as 3a′ and exclusive methylenated product is not observed.
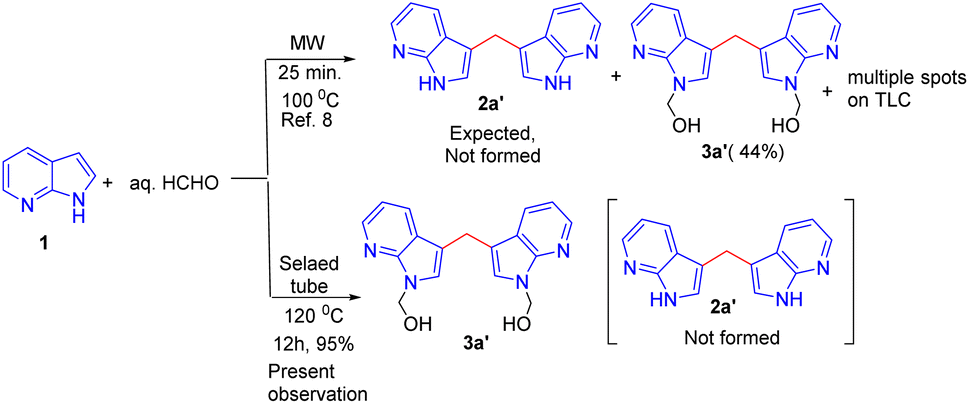 | ||
| Scheme 2 Initial studies. | ||
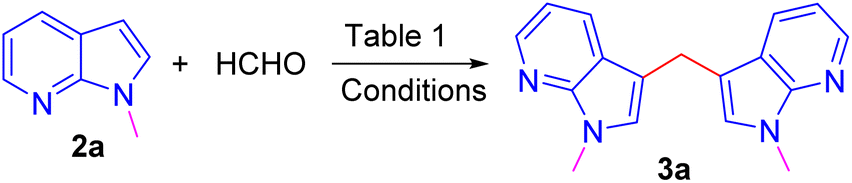
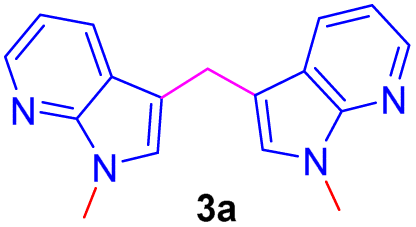
Repeating the reaction of 1.0 equiv. of N-methyl-7-azaindole 2a was treated with 3.0 equiv. of 37% aq. HCHO taken in a sealed tube was heated at a slightly lower temperature of 100 °C for 12 h and gave only a trace amount of bis(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)methane 3a, as evidenced by spectroscopic data (Table 1, entry 1). To improve the yield of compound 3a, reactions were carried out by varying parameters such as temperature, time, and catalyst load (Table 1). Increasing the temperature to 115 °C slightly improved the yield of 3a to 35% and at 120 °C further improved the yield by 55% (Table 1, entries 2 and 3). Extending the reaction time to 15 h further improved the yield to 74% and the addition of 1 mmol amount of water to the reaction did not alter the yield (Table 1, entries 4 and 5). Repeating the reaction with paraformaldehyde at 120 °C for 12 h and 15 h afforded the expected product in 72 and 80% yields, respectively (Table 1, entries 6 and 7). Notably, to reduce the reaction time, and to improve the yield, when the mixture of 2a and aqueous formaldehyde was irradiated in a microwave oven for 15 minutes and furnished product 3a with a 45% yield. However, the best yield (85%) was obtained in a rapid reaction time of 5 min. microwave irradiation (100 °C, 150 psi, power 150) using 50% w/w montmorillonite K 10 clay as a solid acid catalyst, and this was found to be an optimum condition (Table 1, entry 9).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Reagent (3.0 equiv.) | Temp. (°C) | Time (h) | Yield of 3aa (%) |
| a Isolated yield.b Irradiated at 100 W using CEM Discover-300 microwave synthesizer. | |||||
| 1 | 2a | Aq. HCHO | 100 | 12 | Trace |
| 2 | 2a | Aq. HCHO | 115 | 12 | 35 |
| 3 | 2a | Aq. HCHO | 120 | 12 | 55 |
| 4 | 2a | Aq. HCHO | 120 | 15 | 74 |
| 5 | 2a | Aq. HCHO, H2O | 120 | 12 | 74 |
| 6 | 2a | (HCHO)n | 120 | 12 | 72 |
| 7 | 2a | (HCHO)n | 120 | 15 | 80 |
| 8 | 2a | Aq. HCHO | MWb | 15 min | 45 |
| 9 | 2a | Aq. HCHO, 50% w/w Mont. K-10 | MWb | 5 min | 85 |
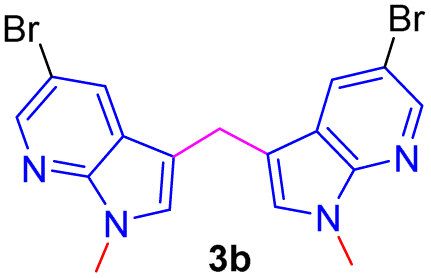
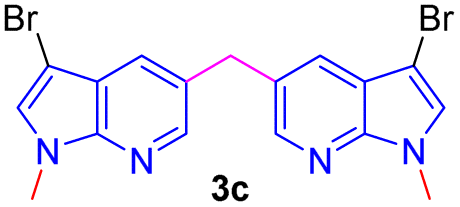
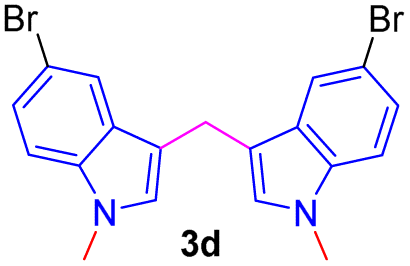
Encouraged by the preliminary results, the scope and diversity of the reaction were explored by selecting a number of N-alkyl-7-azaindole, indole, and carbazole 2b–k (Table 2). Under optimized microwave irradiation conditions, all the reactions underwent smoothly to produce the corresponding methylenation products 3b–k in very good to excellent yields (Table 2, entries 1–7). Further, diversify the methylenation reaction compound 2c is also used as a substrate to produce a novel methylenation at C-5 position to produce compound 3c. The structure of all the products was established by spectroscopic methods such as 1H, 13C NMR, DEPT-135, and HRMS.
| Entry | Substrate 2 | Product 3 | MW timeb,c (min) | Yield (%) |
|---|---|---|---|---|
| a Optimized condition: aq. HCHO, 50% w/w Mont. K-10, MW, 100 W.b CEM Discover-300 microwave synthesizer was used.c Prolonged irradiation results in decomposition or charring. | ||||
| 1 | 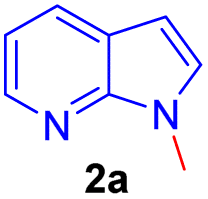 |
 |
5 | 74 |
| 2 | 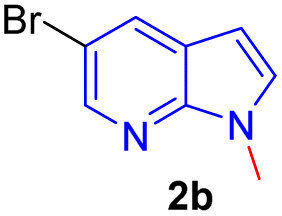 |
 |
8 | 80 |
| 3 | 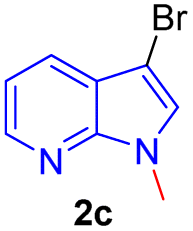 |
 |
8 | 85 |
| 4 | 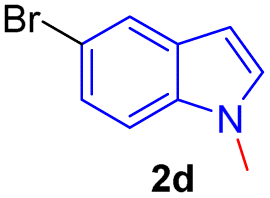 |
 |
5 | 84 |
| 5 | 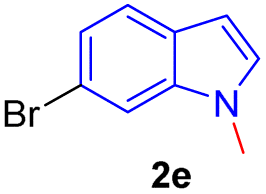 |
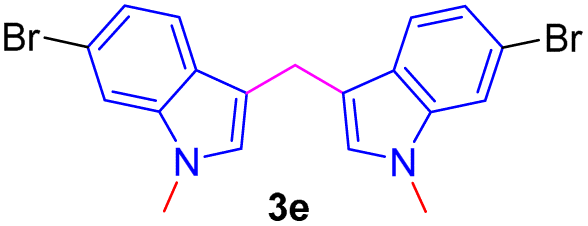 |
5 | 82 |
| 6 | 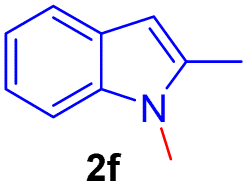 |
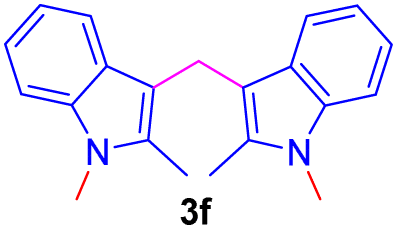 |
6 | 80 |
| 7 | 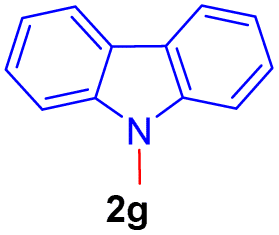 |
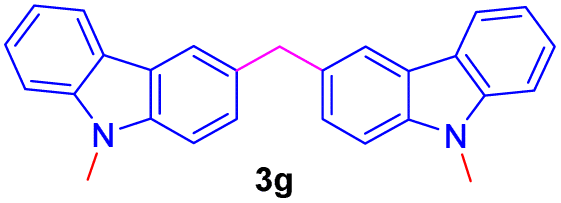 |
12 | 79 |
| 8 | 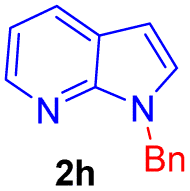 |
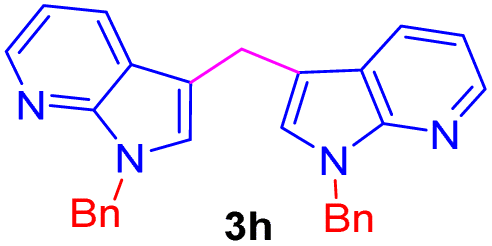 |
9 | 75 |
| 9 | 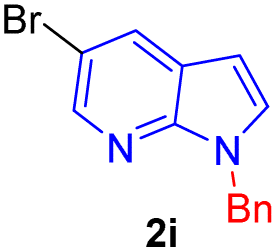 |
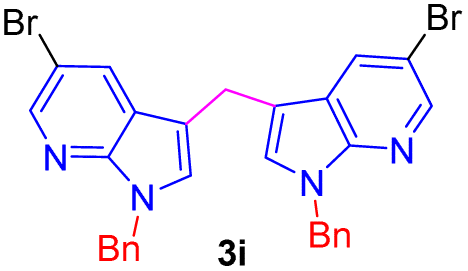 |
7 | 78 |
| 10 | 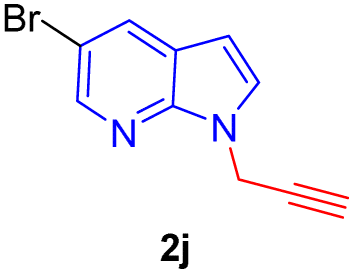 |
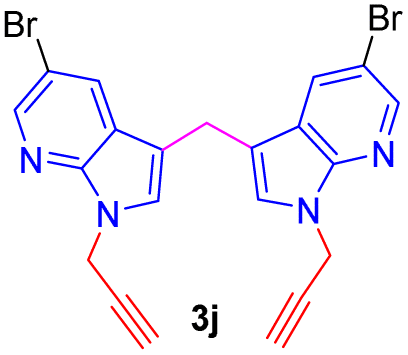 |
10 | 80 |
| 11 | 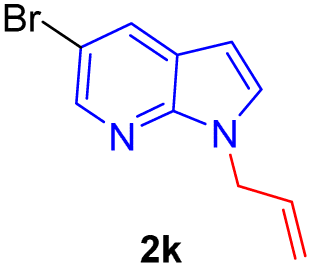 |
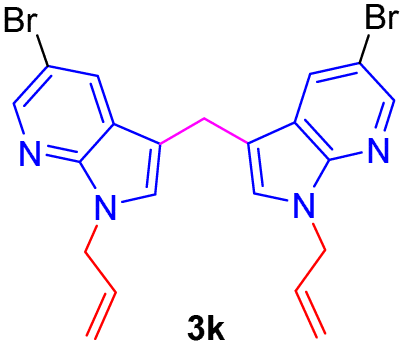 |
10 | 75 |
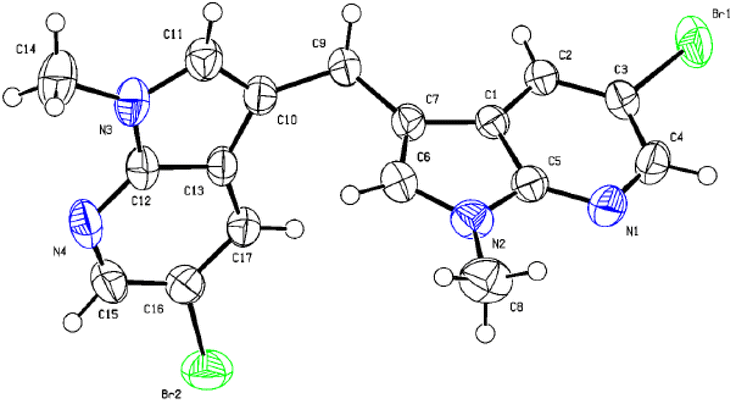
The structure of new compounds was assigned from spectroscopic data and a representative compound 3b structure and relative stereochemistry were assigned based on single-crystal X-ray analysis (Fig. 2).10
A plausible mechanism for the formation of bis(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)methane 3a from N-methyl-7-aza indole 2a with formaldehyde is shown in Scheme 3.11 Under the influence of Mont. K-10 clay, the electrophilicity of the formaldehyde is increased and undergoes a facile conjugate addition of 7-azaindole to provide hydroxy methylated intermediate E. Intermediate E upon elimination of water molecule via intermediate F to provide another intermediate G. Second addition of 7-azaindole to the intermediate G provides the product 3a.
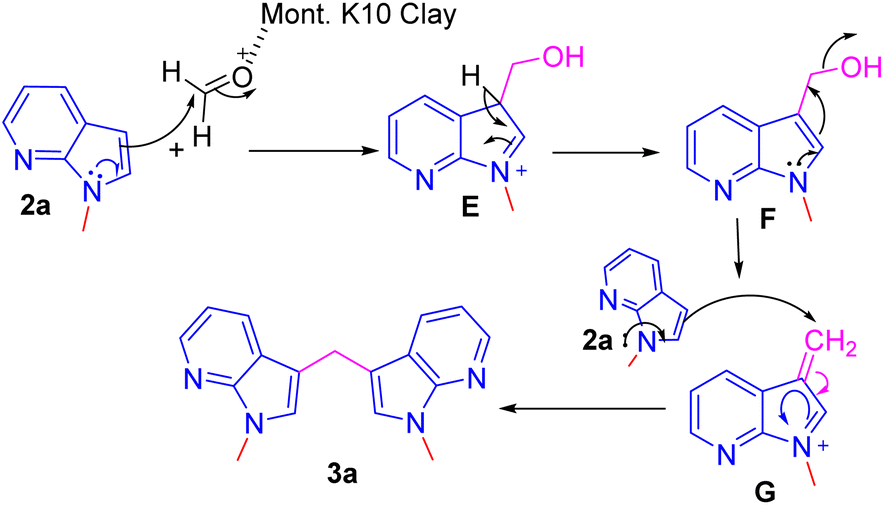 | ||
| Scheme 3 A plausible mechanism for the formation of compounds 3a. | ||
To demonstrate scalability of the reaction a gram scale experiment has been carried out using substrate 2b to afford the expected product in 74% yield (Scheme 4).
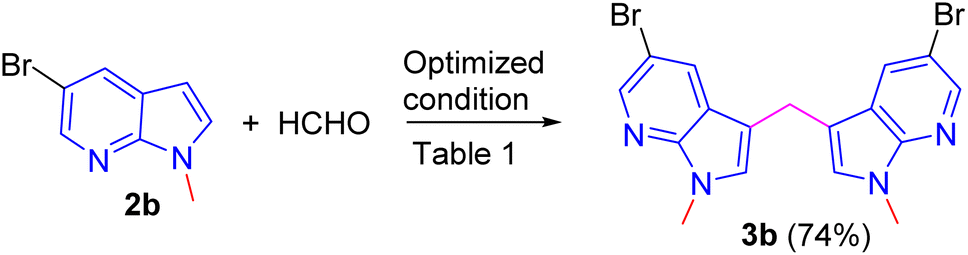 | ||
| Scheme 4 Gram scale synthesis of compound 3b. | ||
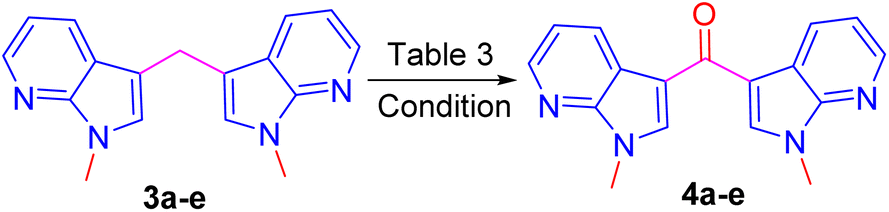
Direct oxidation of bis(1-methyl-1H-pyrrolo[2,3-b] pyridin-3-yl) methane 3a into corresponding ketones 4a are unknown in the literature. However, these ketones were reported by a two-step synthetic protocol starting from respective methylenes.8,12 Hence, we were interested to develop a one-pot, mild and efficient procedure for this direct oxidation of 3a into its ketone 4a. To achieve the oxidation, initially, the benzylic oxidation of methylene carbon in compound 3a was treated with peroxides such as 30% H2O2, and TBHP failed to provide ketone 4a (Table 3, entries 1 and 2). To our dismay, another well-known oxidizing agent SeO2 also failed to provide the ketone (Table 3, entry 3). In addition, Cs2CO3 also failed to provide the expected product (Table 3, entry 4).13 However, a single electron chemical oxidant such as CAN was explored (Table 3, entry 5). Hence ketone 4a was contained in 70% yield by reacting the compound 3a with 3 equivalents of CAN in ACN–MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) solvent system for 30 minutes and was found as a suitable and optimized condition for the oxidation reaction. To diversify the methodology, Further under optimized condition compound 4c was synthesized from the respective 3-bromo-N-methyl-5-methyledine-bis-7-azaindole.
:3) solvent system for 30 minutes and was found as a suitable and optimized condition for the oxidation reaction. To diversify the methodology, Further under optimized condition compound 4c was synthesized from the respective 3-bromo-N-methyl-5-methyledine-bis-7-azaindole.
|
||||
|---|---|---|---|---|
| Entry | Oxidizing agent (equiv.) | Solvent | Condition | Product 4a (yield%) |
| a Decomposed.b SM recovered. | ||||
| 1 | 30% H2O2 (5 equiv.) | MeOH | RT, 12 h | a |
| 2 | TBHP (4 equiv.) | CH3CN | RT, 12 h | Trace |
| 3 | SeO2 (2 equiv.) | 1,4-Dioxane | 120 °C, 12 h | Trace |
| 4 | Cs2CO3 (4 equiv.) | Dry DMSO | 150 °C, 12 h | b |
| 5 | CAN (3 equiv.) | MeOH:CH3CN |
0 °C–RT, 0.5 h | 70 |
Having followed the optimized CAN oxidizing condition, ketones 4a–e were prepared in excellent yield from respective methylated compounds 3a–e (Fig. 3).
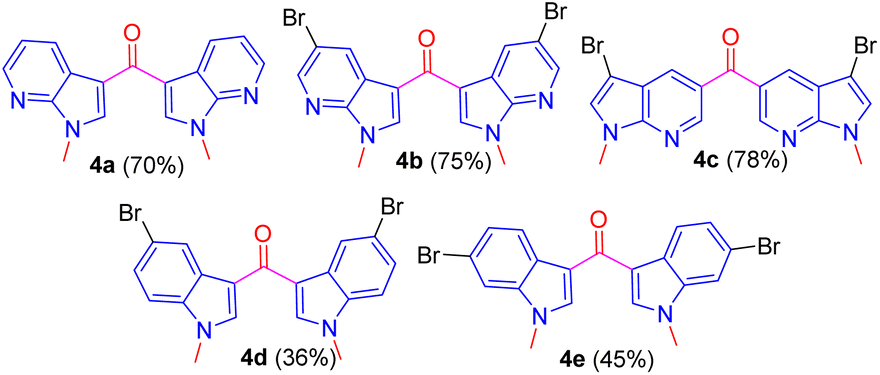 | ||
| Fig. 3 Synthesized ketones 4a–e. | ||
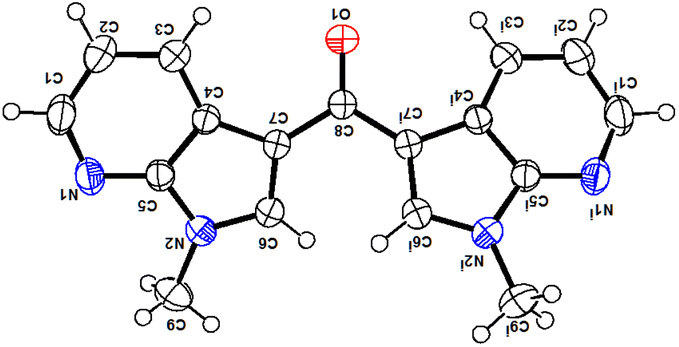
The structure of all the new compounds 4a–e was assigned from spectroscopic data and a representative compound 4a structure and relative stereochemistry were assigned based on single-crystal X-ray analysis (Fig. 4).14
A plausible mechanism for the formation of compound 4 from the bis(1-methyl-1H-pyrrolo[2,3-b] pyridin-3-yl) methane 3a by CAN is shown in Scheme 5.12e Initially, the CAN oxidizes the benzylic methylene group to benzylic radical cation intermediate I, which further undergoes second oxidation by CAN and by liberating H+ ion to form the cation intermediate J. Intermediate J upon reaction with nitrate oxygen of CAN forms C–O bond intermediate K, and subsequent hydride ion elimination results the ketone 4 (Scheme 5).
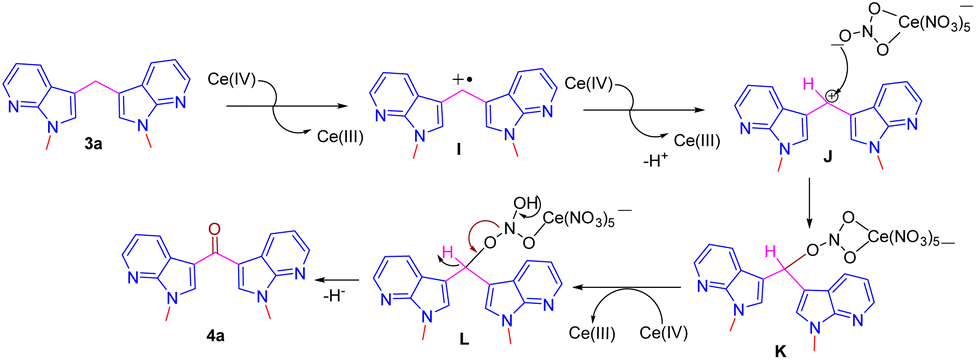 | ||
| Scheme 5 A plausible mechanism for the formation of compounds 4a. | ||
The presence of bromine in product 4b prompted us to derivatize its biphenyl derivatives by exploiting Suzuki coupling. Biphenyls and their derivatives are important structural motifs due to applications both in academic interest and in synthetic use of various carbo- and heterocyclic frameworks.15 The asymmetric structural framework of biaryls has been effectively utilized as asymmetric catalysts, natural product synthesis, supramolecular assemblies, and organic materials.16 Thus, the halogen on the aromatic substitution was successfully utilized for Suzuki coupling with several aryl boronic acids 5a–e to provide aryl-substituted fluorescent 7-aza indole derivatives 6a–e in excellent yield (Scheme 6).17
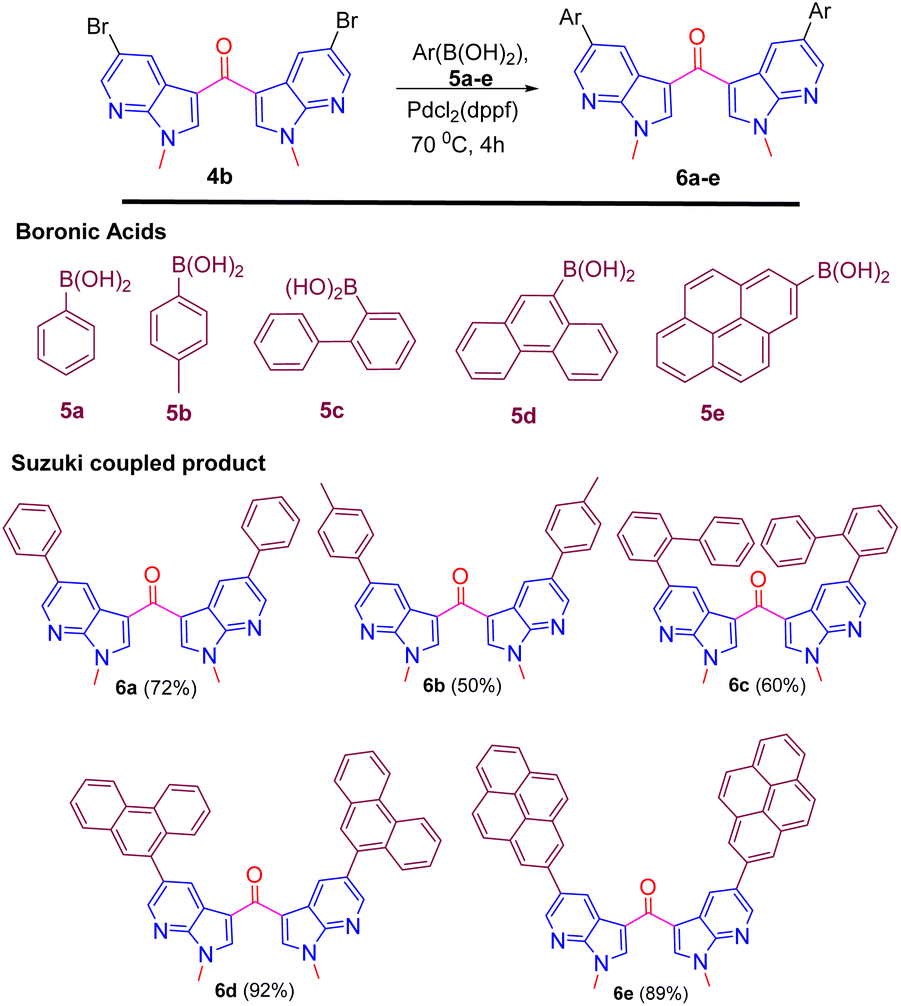 | ||
| Scheme 6 Suzuki coupling of compound 4b with several aryl boronic acids 5a–e. | ||
The nature of biaryl-based derivatives 6a–e prompted us to evaluate the basic photophysical properties. The absorption measured in MeOH showed in the range of λmax.abs. 230–248, emission in the range of λmax.emi. 398–497, and large Stoke's shift (Table 4) has been evaluated. This basic data suggest that they are strong blue emitters (Table 4).
| Entry | Product | Absorptiona λmax.abs. | Emissiona λmax.emi. | Stoke's shift Δῡb (cm−1) |
|---|---|---|---|---|
| a All the spectra were recorded in MeOH at 2 × 10−5 M at 298 K concentration.b Stoke's shift = λmax.abs. − λmax.emi. [cm−1]. | ||||
| 1 | 3b | 232, 299 | 412 | 18831, 9173 |
| 2 | 4b | 282, 316 | 398 | 10335, 6520 |
| 3 | 6a | 244, 326 | 406 | 16353, 6044 |
| 4 | 6b | 249, 328 | 463 | 18562, 8889 |
| 5 | 6c | 230, 324 | 408 | 18969, 6355 |
| 6 | 6d | 252, 325 | 497 | 19562, 10649 |
| 7 | 6e | 274, 343 | 450 | 14274, 6932 |
In conclusion, we have demonstrated a catalyst-free and green chemical method has been developed for the methylenation of indole and N-methyl-7-aza indoles with aqueous formaldehyde afforded respective N,N′-dimethyl-3,3′-bis-7-azaindolylmethane under MW in excellent yield. Subsequent oxidation of product using one electron chemical oxidant CAN afford N,N′-dimethyl-3,3′-bis-7-azaindolylmethanone derivatives in excellent yield. This resulted in methanone derivatives with halogen substitution at the aryl ring subjected to Suzuki coupling with aryl boronic acids furnished with highly functionalized biaryl derivatives. Plausible mechanisms, characterization including XRD, and evaluation of photophysical properties of the Suzuki coupled products are reported.
Experimental section
General remarks
All the reactions were carried out in oven-dried glassware. CEM Discover-300 microwave synthesizer was used for all the microwave irradiation reactions. The progress of the reactions was monitored by thin layer chromatography (TLC) using Merck pre-coated TLC plates (Merck 60 F254) Purification of the products was accomplished by column chromatography packed with silica gel 100–200 mesh. NMR spectra were recorded on a Bruker-400.3 MHz NMR spectrometer (400.3 MHz for 1H NMR and 100.6 MHz for 13C NMR) with CDCl3/TMS as the solvent and TMS as the internal reference. Integrals are in accordance with assignments and coupling constants are reported in Hertz (Hz). All 13C spectra are proton-decoupled. Multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), dt (doublet triplet), td (triplet of doublets), and br s (broad singlet). FTIR spectra were recorded on a PerkinElmer RX-IFT-IR spectrometer and absorbance values are reported in cm−1. ESI HRMS was performed on a Waters(R) Micromass(R) Q-TOF MicroTM mass spectrometer. Yields refer to quantities obtained after chromatography.Experimental procedure
:CH3CN (2:1) was added cerium(IV) ammonium nitrate (3 equiv.) in a portion-wise at 0 °C to RT for 30 min. After the completion of the reaction (monitored by TLC), the reaction mixture was diluted with EtOAc and washed with distilled water. The combined organic layer was dried over anhydrous MgSO4 and the solvent was evaporated under reduced pressure. The crude mixture was purified on a silica gel column chromatography (eluent:hexane/EtOAc:1/1) to afford the corresponding compounds 4a–e in good yields.:H2O mixture (3:1, 2 mL) under N2 atm., was heated at 70 °C for 22 h. After completion of the reaction (monitored by TLC), the mixture was allowed to attain room temperature. The mixture was diluted with EtOAc, filtered through a pad of Celite and concentrated under vacuum. The crude mixture was purified by silica gel column chromatography to afford products 6a–e.Spectroscopic data for synthesized compounds
722, 1024, 764, 705, 602 cm−1. 1H NMR (400.3 MHz, CDCl3/TMS): δ 8.23 (dd, J = 4.7, 1.5 Hz, 2H), 7.68 (dd, J = 7.8, 1.6 Hz, 2H), 7.28–7.12 (m, 6H), 7.07 (dd, J = 7.7, 1.8 Hz, 4H), 6.98–6.78 (m, 4H), 5.36 (s, 4H), 4.07 (s, 2H). 13C NMR (101.6 MHz, CDCl3/TMS): δ 148.1, 143.0, 138.0, 128.6, 127.1, 125.7, 120.1, 115.2, 112.8, 47.5, 21.7. DEPT-135 (101.6 MHz, CDCl3/TMS): δ 148.1, 143.0, 138.0, 128.6, 127.1, 125.7, 120.1, 115.2, 112.8, 47.5, 21.7. HRMS (ESI): calcd for C29H24N4 [M + H]+ m/z: 429.2079; found 429.2072.722, 1024, 764, 705, 602 cm−1. 1H NMR (400.3 MHz, CDCl3/TMS): δ 8.25 (d, J = 2.1 Hz, 2H), 7.77 (d, J = 2.2 Hz, 2H), 7.35–7.12 (m, 6H), 7.13–6.99 (m, 4H), 6.89 (s, 2H), 5.33 (s, 4H), 3.99 (s, 2H). 13C NMR (101.6 MHz, CDCl3/TMS): δ 146.4, 143.6, 137.4, 129.4, 128.8, 127.7, 127.2, 121.4, 111.8, 111.2, 47.8, 21.6. DEPT-135 (101.6 MHz, CDCl3/TMS): δ 143.6, 129.4, 128.8, 127.7, 127.2, 47.8, 21.6. HRMS (ESI): calcd for C29H22Br2N4 [M + H]+ m/z: 584.0289; found 585.0286.595 cm−1. 1H NMR (400.3 MHz, CDCl3/TMS): δ 8.68 (s, 2H), 8.37 (s, 2H), 7.73 (s, 2H), 3.90 (s, 6H). 13C NMR (101.6 MHz, CDCl3/TMS): δ 183.0, 146.7, 145.5, 134.8, 133.1, 121.0, 114.7, 32.3. DEPT-135 (101.6 MHz, CDCl3/TMS): δ 145.5, 134.8, 133.1, 32.3. HRMS (ESI): calcd for C17H12Br2N4O [M + H]+ m/z: 446.9456; found 446.9456.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PE thanks VIT, Vellore for the research fellowship. SK thanks VIT Management for providing infrastructure facilities and VIT SEED GRANT for carrying out this work.Notes and references
- D. R. Motati, R. Amaradhi and T. Ganesh, Org. Chem. Front., 2021, 8, 466–513 RSC.
- S. B. Zhao and S. Wang, Chem. Soc. Rev., 2010, 39, 3142–3156 RSC.
- (a) Y. Chen, Chem.–Eur. J., 2019, 25, 3405–3439 CrossRef CAS PubMed; (b) C. Sun, X. Zou and F. Li, Chem.–Eur. J., 2013, 19, 14030–14033 CrossRef CAS PubMed; (c) C. Qiao, X. F. Liu, H. C. Fu, H. P. Yang, Z. B. Zhang and L. N. He, Chem.–Asian J., 2018, 13, 2664–2670 CrossRef CAS PubMed; (d) P. T. Ha, O. T. Nguyen, K. D. Huynh, T. T. Nguyen and N. T. Phan, Synlett, 2018, 19, 2031–2034 Search PubMed; (e) M. Elagawany, L. Maram and B. Elgendy, RSC Adv., 2021, 11, 7564–7569 RSC.
- (a) S. H. Lee, K. Kim, Y. U. Jeon, A. Kundu, P. Dey, J. Y. Hwang, N. K. Mishra, H. S. Kim and I. S. Kim, Tetrahedron Lett., 2019, 60, 150974 CrossRef; (b) A. Z. Halimehjani and V. Barati, ChemistrySelect, 2018, 3, 3024–3028 CrossRef CAS.
- (a) X. Han, Z. Zhou, C. Wan, Y. Xiao and Z. Qin, Synthesis, 2013, 45, 615 CrossRef CAS; (b) Y. Hu, L. Zhou and W. Lu, Synthesis, 2017, 49, 4007 CrossRef CAS; (c) M. R. Maurya, B. Sarkar, A. Kumar, N. Ribeiro, A. Miliute and J. C. Pessoa, New J. Chem., 2019, 43, 17620 RSC; (d) K. N. Parida, S. Jhulki, S. Mandal and J. N. Moorthy, Tetrahedron, 2012, 68, 9763 CrossRef CAS; (e) S. V. Lieberman and Connor, R., Org. Synth., 1938, 18, 61 CrossRef CAS; (f) T. Nishimura, Org. Synth., 1956, 36, 58 CrossRef CAS; (g) W. H. Hartford and M. Darrin, Chem. Rev., 1958, 58, 1–61 CrossRef CAS; (h) I. Vlattas, I. T. Harrison, L. Tokes, J. H. Fried and A. D. Cross, J. Org. Chem., 1968, 33, 4176–4179 CrossRef CAS; (i) C. S. Marvel, J. S. Saunders and C. G. Overberger, J. Am. Chem. Soc., 1946, 68, 1085–1088 CrossRef CAS PubMed; (j) M. S. Carpenter, W. M. Easter and T. F. Wood, J. Org. Chem., 1951, 16, 586–617 CrossRef CAS; (k) L. Syper, Tetrahedron Lett., 1966, 7, 4493–4498 CrossRef; (l) W. S. Trahanovsky and L. B. Young, J. Org. Chem., 1966, 31, 2033–2035 CrossRef CAS; (m) H. Gilman, C. G. Brannen and R. K. Ingham, J. Am. Chem. Soc., 1956, 78, 1689–1692 CrossRef CAS; (n) E. Ganin and I. Amer, Synth. Commun., 1995, 25, 3149–3154 CrossRef CAS; (o) R. Hosseinzadeh, M. Tajbakhsh and H. Vahedi, Synlett, 2005, 18, 2769–2770 CrossRef; (p) M. Ghaffarzadeh, M. Bolourtchian, M. Gholamhosseni and F. Mohsenzadeh, Appl. Catal., A, 2007, 333, 131–135 CrossRef CAS.
- (a) M. Tobiszewski, M. Marć, A. Gałuszka and J. Namieśnik, Molecules, 2015, 20, 10928–10946 CrossRef CAS PubMed; (b) I. T. Horvath and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef CAS PubMed; (c) H. E. D. M. Saleh, and M. Koller, in Green Chemistry, IntechOpen, 2018 Search PubMed; (d) Z. Zhang, J. Wen, M. Wang, C. G. Yan and Z. Shi, Green Synthesis and Catalysis, 2021, 2, 275–285 CrossRef; (e) Y. Kim and C. J. Li, Green Synthesis and Catalysis, 2020, 1, 1–11 CrossRef; (f) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- (a) V. Sridharan and J. C. Menéndez, Chem. Rev., 2010, 110, 3805–3849 CrossRef CAS PubMed; (b) V. Nair and A. Deepthi, Chem. Rev., 2007, 107, 1861–1891 CrossRef; (c) V. Nair, L. Balagopal, R. Rajan and J. Mathew, Acc. Chem. Res., 2004, 37, 21–30 CrossRef CAS PubMed and references cited therein. (d) P. Shanmugam, V. Vaithiyanathan and V. Baby, Tetrahedron Lett., 2006, 47, 6851–6855 CrossRef CAS; (e) P. Shanmugam and V. Vaithiyanathan, Can. J. Chem., 2009, 87, 591 CrossRef CAS.
- (a) S. Darses, Tetrahedron Lett., 1996, 37, 3857–3860 CrossRef CAS; (b) L. Chen, D. R. Sanchez, B. Zhang and B. P. Carrow, J. Am. Chem. Soc., 2017, 139, 12418–12421 CrossRef CAS; (c) L. Chen, H. Francis and B. P. Carrow, ACS Catal., 2018, 8, 2989–2994 CrossRef CAS; (d) I. A. Sanhueza, Angew. Chem., Int. Ed., 2021, 60, 7007–7012 CrossRef CAS.
- T. Pillaiyar, M. Köse, K. Sylvester, H. Weighardt, D. Thimm, G. Borges, I. Förster, I. von Kügelgen and C. E. Müller, J. Med. Chem., 2017, 60, 3636–3655 CrossRef CAS PubMed.
- CCDC-2191474 (3b) contains the supplementary crystallographic data for this paper†..
- F. Pu, Y. Li, Y. H. Song, J. Xiao, Z. W. Liu, C. Wang, Z. T. Liu, J. G. Chen and J. Lu, Adv. Synth. Catal., 2016, 358, 539–542 CrossRef CAS.
- (a) Y. Ma, J. You and F. Song, Chem.–Eur. J., 2013, 19, 1189–1193 CrossRef CAS PubMed; (b) R. Shen, T. Kusakabe, K. Takahashi and K. Kato, Org. Biomol. Chem., 2014, 12, 4602–4609 RSC; (c) X. Zheng, Z. Huang, Q. Zheng, L. Wang, C. Zhang and G. Gao, Org. Lett., 2022, 24, 4197–4201 CrossRef CAS; (d) S. Kotha, R. Ali, V. Srinivas and N. G. Krishna, Tetrahedron, 2015, 71, 129–138 CrossRef CAS; (e) T. Pillaiyar, M. Köse, K. Sylvester, H. Weighardt, D. Thimm, G. Borges, I. Förster, I. von Kügelgen and C. E. Müller, J. Med. Chem., 2017, 60, 3636–3655 CrossRef CAS PubMed; (f) S. K. Guchhait, M. Kashyap and H. Kamble, J. Org. Chem., 2011, 76, 4753–4758 CrossRef CAS PubMed; (g) P. Neupane, A. A. Salim and R. J. Capon, Tetrahedron Lett., 2020, 61, 151651 CrossRef CAS; (h) T. Das, A. Chakraborty and A. Sarkar, Tetrahedron Lett., 2014, 55, 7198–7202 CrossRef CAS.
- (a) A. T. Khan, T. Khan, L. H. Choudhury and S. Ghosh, Tetrahedron Lett., 2007, 48, 2271–2274 CrossRef CAS; (b) D. Shi, Y. Ren, H. Jiang, J. Lu and X. Cheng, Dalton Trans., 2013, 42, 484–491 RSC; (c) J. Młochowski and H. Wójtowicz-Młochowska, Molecules, 2015, 20, 10205–10243 CrossRef PubMed; (d) R. Chebolu, A. Bahuguna, R. Sharma, V. K. Mishra and P. C. Ravikumar, Chem. Commun., 2015, 51, 15438–15441 RSC; (e) P. Shanmugam, V. Vaithiyanathan and K. Selvakumar, Tetrahedron Lett., 2008, 49, 2119–2123 CrossRef CAS.
- CCDC-2102023 (4a) contains the supplementary crystallographic data for this paper†..
- (a) J. A. Lopez and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766–6798 RSC; (b) C. Ni, D. Zha, H. Ye, Y. Hai, Y. Zhou, E. V. Anslyn and L. You, Angew. Chem., Int. Ed., 2018, 57, 1300–1305 (Angew. Chem., 2018, 130, 1314–1319) CrossRef CAS PubMed; (c) Z. J. Jain, P. S. Gaide and R. S. Kankte, Arabian J. Chem., 2017, 10, S2051–S2066 CrossRef CAS; (d) J. Wang, G. Cooper, D. Tulumello and A. P. Hitchcock, J. Phys. Chem. A, 2005, 109, 10886–10896 CrossRef CAS PubMed; (e) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed; (f) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed; (g) Y. Tuanli, A. C. Marino and C. L. Richard, J. Org. Chem., 2005, 70, 3511–3517 CrossRef PubMed; (h) H. He, C. Wang, T. Wang, N. Zhou, Z. Wen, S. Wang and L. He, Dyes Pigm., 2015, 113, 174–180 CrossRef CAS; (i) J. Zhang, J. Su, Y. Ma and H. H. Guo, J. Phys. Chem. B, 2012, 116, 2075–2089 CrossRef CAS PubMed.
- (a) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 (Angew. Chem., 2005, 117, 5518–5563) CrossRef CAS PubMed; (b) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed; (c) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (d) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed; (e) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (f) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed; (g) S. Yuan, Y. Chen, J. Qin, W. Lu, L. Zou, Q. Zhang, X. Wang, X. Sun and H. Zhou, J. Am. Chem. Soc., 2016, 138, 8912–8919 CrossRef CAS PubMed; (h) T. Qin, S. L. Skraba-Joiner, Z. G. Khalil, R. P. Johnson, R. J. Capon Jr and J. A. Porco, Nat. Chem., 2015, 7, 234–240 CrossRef CAS PubMed; (i) V. Bhat, E. R. Welin, X. Guo and B. M. Stoltz, Chem. Rev., 2017, 117, 4528–4561 CrossRef CAS PubMed; (j) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed.
- P. Kannaboina, K. A. Kumar and P. Das, Org. Lett., 2016, 18, 900–903 CrossRef CAS PubMed.
- P. Dudhe, M. A. Krishnan, K. Yadav, D. Roy, K. Venkatasubbaiah, B. Pathak and V. Chelvam, Beilstein J. Org. Chem., 2021, 17, 1453–1463 CrossRef CAS PubMed.
- G. Shi, T. Yoon, S. Cha, S. Kim, M. Yousuf, N. Ahmed, D. Kim, H. W. Kang and K. S. Kim, ACS Sens., 2018, 3, 1102–1108 CrossRef CAS PubMed.
- S. H. Lee, K. Kim, Y. U. Jeon, A. Kundu, P. Dey, J. Y. Hwang, N. K. Mishra, H. S. Kim and I. S. Kim, Tetrahedron Lett., 2019, 60, 150974 CrossRef.
- K. Takaishi, H. Kosugi, R. Nishimura, Y. Yamada and T. Ema, Chem. Commun., 2021, 57, 8083–8086 RSC.
- C. Qiao, X. F. Liu, H. C. Fu, H. P. Yang, Z. B. Zhang and L. N. He, Chem.–Asian J., 2018, 13, 2664–2670 CrossRef CAS PubMed.
- P. K. Pradhan, S. Dey, V. S. Giri and P. Jaisankar, Synthesis, 2005, 2005, 1779–1782 CrossRef.
- Y. C. Zheng, M. L. Zheng, S. Chen, Z. S. Zhao and X. M. Duan, J. Mater. Chem. B, 2014, 2, 2301–2310 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. Copies of 1H NMR, 13C NMR, DEPT-135, HRMS spectra for all the new compounds, and single-crystal XRD data for compounds 3b and 4a are provided. CCDC 2191474 and 2102023. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra05849a |
| This journal is © The Royal Society of Chemistry 2022 |