
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization and anticancer activities of Zn2+, Cu2+, Co2+ and Ni2+ complexes involving chiral amino alcohols†
Q. Umar‡
 a,
Y. H. Huang‡a,
A. Nazeera,
H. Yina,
J. C. Zhanga,
M. Luo*a and
X. G. Meng*b
a,
Y. H. Huang‡a,
A. Nazeera,
H. Yina,
J. C. Zhanga,
M. Luo*a and
X. G. Meng*b
aDepartment of Chemistry and Chemical Engineering, Hefei University of Technology, Hefei, 23000, P.R. China. E-mail: luomei@pku.edu.cn; mengxianggao@ccnu.edu.cn
bCollege of Chemistry, Central China Normal University, Wuhan, 430079, P.R. China
First published on 9th November 2022
Abstract
Seven new metal coordination complexes, [NiC15H43N5O11] (I), [Co3C36H98N6O6] (II), [CuC14H32N2O6] (III), [Cu2C32H43Cl2N2O13] (IV), [Zn2C24H32Cl3N3O3] (V), [Co3C48H66Cl6N6O6] (VI), and [Zn (C18H45N3O3] (VII), have been synthesized from some direct reactions of amino-alcoholic ligands with metal salts in anhydrous methanol or ethanol medium. All the crystals of these seven complexes are crystallized in the chiral space groups (P212121 for (I), (IV), (VI) and (VII); P21 for (III) and (V); and C2 for (II), respectively). Their characteristic peaks were analyzed and assigned by FTIR, NMR, and UV–Vis and elemental analysis techniques. The anticancer activities of amino alcohol complexes (I)–(VII) showed cytotoxic effects against the human tumour cell line A549; among them, complex (V) showed the best activity with an IC50 value of 17.8. The higher biological activity should be related to its di-nuclear zinc(II) unit in which one zinc is only four-coordinated by four small chloride anions.
Introduction
Cancer, as one of the worst lethal factors, is affecting human health all over the world.1 As a very cutting-edge subject, it is getting more and more attention from biologists, medical scientists and chemists.2 Especially, designing specific anticancer drug molecules based on a particular targeted cancer location is a major challenge for chemists. Although a large number of anti-cancer drug molecules have been designed so far, there are only a few of them that can be truly highly active during a treatment in clinical applications due to their side effects and drug resistance. Cisplatin, for example, is a molecule that has been recognized as having high therapeutic activity against cancer.3–8 There is no doubt that the discovery of cisplatin, cis-diamminedichloroplatinum(II), represents one of the most significant events for cancer chemotherapy in the 20th century.9–12 Besides cisplatin, several other platinum complexes (carboplatin, oxaliplatin, nedaplatin, and lobaplatin) have also been approved for current tumor therapy.13 Nedaplatin is another platinum derivative developed in Japan which is reported to cause much less nephrotoxicity than cisplatin.13–15 However, this potential drug molecule still needs more further experimental evidence before its real clinical practice application and efficacy recognized by researchers from all walks of life. It is precisely for this reason that chemists are constantly investing a lot of manpower and material resources to design a variety of small chemical molecules including organics, inorganic and metal complexes, hoping to be able to screen out some more artificial molecules with high cancer activities.In more recent years, there have been still some other studies on the therapeutic applications using gold complexes as potential alternatives to the anticancer drug cisplatin in which the ligands are mainly related with some multi-dentate N-donor ligands.16–18 For instance, metal complexes based on ruthenium and iridium have been explored and they also show their respective different characteristics of biological activity in their anticancer activities research.19–21 However, the use of above mentioned nobel metals is still limited by its own shortcomings and limitations just like high preparation cost and poor compatibility of the human body. Recently, amino alcohols involved coordination compounds with the first transitional metal such as Co2+, Cu2+ and Zn2+ and have already been studied because of their good anticancer activities and low toxicity.22–24 What’ more, amino alcohol complexes can also play a key role in catalysis one hand and become a vital aspect of human life on the other hand. Most of the first-transition metal such as cobalt, copper and zinc complexes are all also very essential for living organisms. Aiming to find some new types of functional drug molecules, our research team have successfully synthesized and characterized a series of Cu2+, Zn2+, Co2+, and Ni2+ complexes by using a one-pot synthetic method. Additionally, the synthetic method has some advantages such as high yield, low toxicity, less smoke generation, and low cost, etc. The design strategy of metal coordination complexes are mainly based on the wide range of coordination numbers, inconstant geometries and available redox states.8
Through the one-pot synthetic method, we initially reported herein seven new complexes, [NiC15H43N5O11] (I), [Co3C36H98N6O6] (II), [CuC14H32N2O6] (III), [Cu2C32H43Cl2N2O13] (IV), [Zn2C24H32Cl3N3O3] (V), [Co3C48H66Cl6N6O6] (VI), and [ZnC18H45N3O3] (VII). Interestingly, these complexes demonstrated good cytotoxicity in lung cancer cells but negligible toxicity to normal cells.
Experimental
Materials and methods
D-Valinol, D-phenylglycenol, D-leucinol, Ni (NO3)2·6H2O, Co(OAc)2.4H2O, Cu (OAc) 2.H2O, CuCl2·2H2O, CoCl2·6H2O, ZnCl2, and Cu(ClO4)2·6H2O were purchased from Acros. 1HNMR spectra were obtained using a Bruker AM-300 spectrometer. 1H and 13C NMR spectra were recorded using Bruker AM-500 and Bruker AM-600 spectrometers. Chemical shifts were reported in ppm (δ) with the solvent relative to tetramethylsilane (TMS) used as the internal standard (residual CHCl3, δH 7.26 ppm; CDCl3, C, 77 ppm). The following abbreviations were used to designate multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet. Infrared spectra were recorded on a Mattson Galaxy Series FTIR 3000 spectrometer; peaks are reported in cm−1. Elemental analyses were performed on an Elemental Analyser AE-3000. The crystal structure was determined by a Gemini S Ultra diffractometer.Cytotoxicity assay
The human tumor cell line against A549 (lung cancer) was used in the cytotoxic assay. These cell lines were obtained from ATCC (Manassas, VA, USA). Cells were cultured in RMPI-1640 or DMEM (Biological Industries, Kibbutz Beit Haemek, and Israel) supplemented with 10% foetal bovine serum (Biological Industries) at 37 °C in a humidified atmosphere with 5% CO2. The cytotoxicity assay was evaluated by the MTS (Promega, Madison, WI, USA) assay. The cytotoxicity assay was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) (Promega, Madison, WI, USA) assay. Cells were seeded into each well of a 96-well cell culture plate. After 12 h of incubation at 37 °C, the test compound (100 μM) was added. After incubation for 48 h at 37 °C, the cells were subjected to the MTS assay. Compounds with a growth inhibition rate of 50% were further evaluated at concentrations of compound AH106 is re-screened at concentrations of 100, 50, 25, 12.5, 6.25 μM, and the remaining compounds are re-screened at concentrations of 200, 100, 50, 25, 12.5 μM, in triplicate with cisplatin and paclitaxel (Sigma, St. Louis, MO, USA) as positive controls.The IC50 value of each compound was calculated with Reed and Muench's method,25 and the results are shown in Table 2.
X-ray analyses
X-ray crystal data were collected on a Bruker SMART diffractometer equipped with graphite monochromatic Mo Kα radiation (λ = 0.71073 Å) or Cu Kα radiation (λ = 1.34138 Å). The structure was solved using full-matrix least squares on F2 using the SHELXTL program. All non-H atoms were refined with anisotropic thermal parameters. All hydrogen atoms were located theoretically and refined with riding model position parameters and fixed isotropic thermal parameters. The crystallographic parameters are listed in Table 1.| Complex | I | II | III | IV | V | VI | VII |
|---|---|---|---|---|---|---|---|
| Empirical formula | NiC15H43N5O111 | Co3C36H98N6O17 | CuC14H32N2O6 | Cu2C32H41Cl2N4O13 | Zn2C24H32Cl3N3O3 | Co3C48H66Cl6N6O6 | ZnC18H45Cl2N3O3 |
| Formula mass | 528.25 | 1071.99 | 387.95 | 887.67 | 647.61 | 1212.55 | 487.84 |
| Temp (K) | 293(2) | 293(2) | 200(2) | 100.0(1) | 200(2) | 293(2) | 293(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 1.34138 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P212121 | C2 | P21 | P212121 | P21 | P212121 | P212121 |
| a (Å) | 10.7412(5) | 24.5160(11) | 4.9600(2) | 9.1340(8) | 10.264(3) | 14.3688(11) | 11.3519(12) |
| b (Å) | 12.9279(7) | 13.4214(6) | 22.0851(9) | 15.3961(13) | 9.025(3) | 16.6075(14) | 14.6691(13) |
| c (Å) | 19.3249(12) | 17.9973(6) | 8.5898(4) | 28.660(3) | 15.089(6) | 24.609(2) | 16.6580(16) |
| β (°) | 109.287(1) | 98.14(10) | 91.457(11) | ||||
| Volume (Å3) | 2683.5(3) | 5589.5(4) | 931.45(7) | 4030.4(6) | 1397.3(8) | 5872.4(8) | 2773.9(5) |
| Z | 4 | 4 | 2 | 4 | 2 | 4 | 4 |
| D calcd (g cm−3) | 1.308 | 1.274 | 1.383 | 1.463 | 1.539 | 1.372 | 1.168 |
| μ (mm−1) | 0.779 | 0.944 | 1.201 | 6.872 | 2.033 | 1.158 | 1.097 |
| F (000) | 1136 | 2308 | 414 | 1828 | 664 | 2508 | 1048 |
| θ range (°) | 2.61–26.00 | 2.28–25.50 | 2.40–26.00 | 2.83–48.80 | 1.99–25.50 | 1.64–25.50 | 2.27–25.50 |
| Total reflec. | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 299 299 |
41240 |
9465 | 115086 |
12859 |
28027 |
23297 |
| Unique reflections | 5246 | 10402 |
3608 | 5905 | 5151 | 10885 |
5166 |
| R 1, wR2 [I > 2σ(I)] | 0.0431, 0.0879 | 0.054, 0.1307 | 0.0232, 0.0498 | 0.0909, 0.2565 | 0.0721, 0.1713 | 0.0961, 0.2610 | 0.0514, 0.1076 |
| R 1, wR2 [all data] | 0.0648, 0.0995 | 0.0845, 0.1515 | 0.0255, 0.0512 | 0.1088, 0.2746 | 0.1161, 0.1974 | 0.1363, 0.3040 | 0.0935, 0.1299 |
| Residuals (e.Å3) | 0.346,−0.283 | 0.693,−0.473 | 0.212,−0.224 | 0.957,−0.497 | 1.087,−0.738 | 1.032,−1.218 | 0.304,−0.397 |
| Complex | IC50a (μM) |
|---|---|
| a Cytotoxicity as IC50 values for each cell line, the concentration of complex that caused 50% reduction relative to untreated A549 cells determined by the SRB assay. Cisplatin was used as an experimental control. | |
| I | >200 |
| II | >200 |
| III | 31.58 ± 2.80 |
| IV | 77.53 ± 1.99 |
| V | 17.71 ± 0.45 |
| VI | 97.74 ± 5.30 |
| VII | 67.61 ± 3.50 |
| cis | 24.37 ± 0.13 |
Synthesis of complexes (I)–(VII)
:1) were heated and refluxed for 48 h, filtration was conducted immediately after the reaction, and the filtrate was kept for slow volatilization. The copper-, cobalt-, nickel- and zinc-containing complexes were successfully synthesized by reacting (R)-2-amino-3-methyl butane-1-ol, D-phenyl glycenol and D-leucinol as ligands with Cu(OAc)2·H2O, Co(OAc)2·4H2O, Cu(ClO4)2·6H2O, CoCl2·6H2O and ZnCl2, respectively, and the cultivated crystal was analysed and characterized by X-ray diffraction, IR, 1HNMR, 13CNMR, UV and E.A. The first step is to find the right ligands, and then, the ligands and the corresponding metal salts reaction was performed on the filter residue or during filtrate processing at the end of the reaction to find a suitable solvent for crystal precipitation. This step is the most critical and requires available solvent tetrahydrofuran, anhydrous methanol, ethanol, and chloroform. Crystals can be frozen in a refrigerator if these substances do not precipitate at room temperature.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1387 (–C–C), 1339 (–CH3), 1162 (–C–O), 1129, 1087, 764, 691, 668 (–Ni–O), 614 (–Ni–N). For [NiC15H43 N5O11] anal. calcd., %: C, 34.07; H, 8.14; N, 13.25. Found, %: C, 34.46; H, 7.711; N, 13.32.C), 1403 (–C–C), 1257(–CH3), 1188(–C–O), 1057, 1013, 548 (–Co–O), 650 (–Co–N). For [Co3C36H98N6O6] anal. calcd., %: C, 40.29; H, 9.14; N, 7.83 Found, %: C, 40.44; H, 8.89; N, 8.5%.C), 1228 (–CH3), 1144 (–C–O), 886, 659 (–Cu–O), 618 (Cu–N). For [CuC14H32N2O6] anal. calcd %: C, 43.30; H, 8.24; N, 7.21%. Found, %: C, 43.24; H, 8.682; N, 7.05.C), 1496 (–C–C), 1453, 1270 (–CH3), 1149 (–C–O), 763, 700, 652 (–Cu–O), 618 (Cu–N). anal. calcd., %: C, 43.25; H, 4.84; N, 6.30%. Found, %: C, 42.85; H, 4.86; N, 6.16.C), 1397 (–C0), 1278 (–CH3), 1172 (–C–O), 757, 696, 652 (–Zn–O), 588 (–Zn–N). 1H NMR (600 MHz, DMSO) δ 7.74–7.85 (m, 1H), 7.19–7.30 (m, 4H), 3.98 (s, 1H), 3.68(s, 2H), 3.49 (s, 3H), 13C NMR (151 MHz, DMSO and CDCl3) δ 141.1128.2, 127.3, 127.1, 66.1, 57.2. For [Zn2C24H32Cl3N3O3] anal. calcd., 44.30%; H, 5.56; N, 5.84 Found, %: C, 46.30; H, 5.08; N, 6.38.C), 1533 (–CO), 1495 (–C–C), 1454, 1376(–CH3), 1194, 1156 (–C–O), 696, 649 (–Co–O), 577 (–Co–N). For [Co3C48H66Cl6N6O6] anal. calcd., %: C, 48.61; H, 5.75, N, 6.52. Found, %: C, 48.87; H, 6.126; N, 7.07.C), 1274 (–CO), 1157 (–CH3), 1130 (–C–O), 1022, 688, 648, 592 (–Zn–O), 568 (–Zn–N). 1H NMR (600 MHz, DMSO and CDCl3) δ 3.78(s, 1H), 3.62 (d, J = 8.0 Hz, 1H), 3.29–3.32 (m, 1H), 1.73–1.75 (m, 1H), 1.26–1.36(m, 2H), 0.91, 0.93 (dd, J = 6.6, 6.5 Hz, 6H); 13C NMR (150 MHz, DMSO and CDCl3) 64.6, 61.4, 51.0, 41.8, 29.5, 24.4, 22.9, 22.7. For [ZnC18H45Cl2N3O3] anal. calcd., %: C, 44.30; H, 9.30; N, 8.61%. Found, %: C, 44.45; H, 8.94; N, 8.30.
C), 1387 (–C–C), 1339 (–CH3), 1162 (–C–O), 1129, 1087, 764, 691, 668 (–Ni–O), 614 (–Ni–N). For [NiC15H43 N5O11] anal. calcd., %: C, 34.07; H, 8.14; N, 13.25. Found, %: C, 34.46; H, 7.711; N, 13.32.C), 1403 (–C–C), 1257(–CH3), 1188(–C–O), 1057, 1013, 548 (–Co–O), 650 (–Co–N). For [Co3C36H98N6O6] anal. calcd., %: C, 40.29; H, 9.14; N, 7.83 Found, %: C, 40.44; H, 8.89; N, 8.5%.C), 1228 (–CH3), 1144 (–C–O), 886, 659 (–Cu–O), 618 (Cu–N). For [CuC14H32N2O6] anal. calcd %: C, 43.30; H, 8.24; N, 7.21%. Found, %: C, 43.24; H, 8.682; N, 7.05.C), 1496 (–C–C), 1453, 1270 (–CH3), 1149 (–C–O), 763, 700, 652 (–Cu–O), 618 (Cu–N). anal. calcd., %: C, 43.25; H, 4.84; N, 6.30%. Found, %: C, 42.85; H, 4.86; N, 6.16.C), 1397 (–C0), 1278 (–CH3), 1172 (–C–O), 757, 696, 652 (–Zn–O), 588 (–Zn–N). 1H NMR (600 MHz, DMSO) δ 7.74–7.85 (m, 1H), 7.19–7.30 (m, 4H), 3.98 (s, 1H), 3.68(s, 2H), 3.49 (s, 3H), 13C NMR (151 MHz, DMSO and CDCl3) δ 141.1128.2, 127.3, 127.1, 66.1, 57.2. For [Zn2C24H32Cl3N3O3] anal. calcd., 44.30%; H, 5.56; N, 5.84 Found, %: C, 46.30; H, 5.08; N, 6.38.C), 1533 (–CO), 1495 (–C–C), 1454, 1376(–CH3), 1194, 1156 (–C–O), 696, 649 (–Co–O), 577 (–Co–N). For [Co3C48H66Cl6N6O6] anal. calcd., %: C, 48.61; H, 5.75, N, 6.52. Found, %: C, 48.87; H, 6.126; N, 7.07.C), 1274 (–CO), 1157 (–CH3), 1130 (–C–O), 1022, 688, 648, 592 (–Zn–O), 568 (–Zn–N). 1H NMR (600 MHz, DMSO and CDCl3) δ 3.78(s, 1H), 3.62 (d, J = 8.0 Hz, 1H), 3.29–3.32 (m, 1H), 1.73–1.75 (m, 1H), 1.26–1.36(m, 2H), 0.91, 0.93 (dd, J = 6.6, 6.5 Hz, 6H); 13C NMR (150 MHz, DMSO and CDCl3) 64.6, 61.4, 51.0, 41.8, 29.5, 24.4, 22.9, 22.7. For [ZnC18H45Cl2N3O3] anal. calcd., %: C, 44.30; H, 9.30; N, 8.61%. Found, %: C, 44.45; H, 8.94; N, 8.30.X-ray structure
X-ray diffraction data for complex (I)–(VII) were collected at room temperature using graphite-monochromatic Mo kα radiation (λ = 0.71073 Å) on an Oxford Diffraction GeminiS diffractometer. Structure solution and refinement for complex I–VII were carried out with the programs SHELXT26 and SHELXL-2018/3,27 respectively. MERCURY28 was employed for molecular graphics and OLEX2.29 Non-hydrogen atoms in (I)–(VII) were refined anisotropically while hydrogen atoms were treated by constrained isotropic refinement. Crystal data and refinement parameters for compounds (I)–(VII) are summarized in Table 1. The selected bond lengths and bond angles are listed in Table S1.† Hydrogen bonds of the complex (I)–(VII) is given in Table S2.†Description of the crystal structures for complexes (I)–(VII)
The crystal structure and stereogram of complexes (I)–(III) is shown in Fig. 1 and (IV)–(VII) in Fig. 2. All the complexes from (I) to (VII) are crystallized in the chiral space groups under a certain experimental conditions, i.e., P212121 for (I)/(II)/(VI)/(VII), P21 for (III)/(V), and C2 for (II), respectively (Table 1). For mono-nuclear metal complex (I), (III), (IV) and (VII), there are one (for (I), (III) and (VII)) or two (for IV)) metal ions, two (for (III)), three (for (I), (VII)) or four (for (IV)) ligands, two counter-anions and two water molecules (for (I)) in their asymmetric units. For compound (V), it is a di-nuclear metal coordination complexes, in whose asymmetric units there is two zinc ions, each three D-phenyl glycenol and three chloride anions. For compound (II) and (VI), they are both in a fashion of tri-nuclear cobalt coordination modes except for the main ligands being different. In their asymmetric units, there are three cobalt cations, six main ligands, three (for (II)) or six (for (VI)) counter anions and several solvent molecules.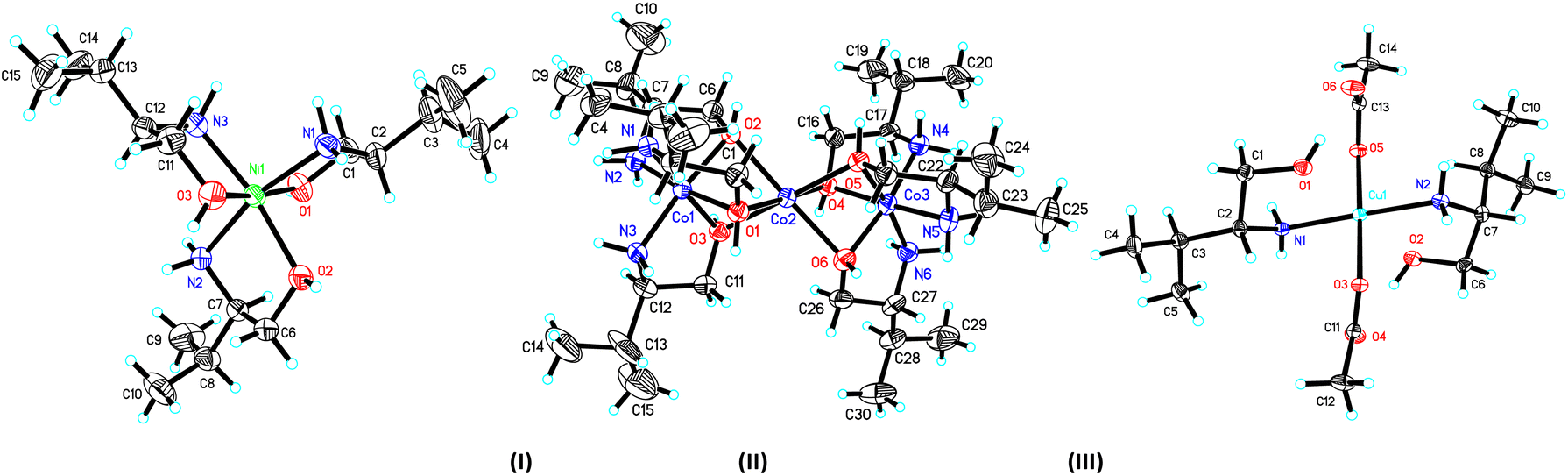 | ||
| Fig. 1 The ORTEP molecular structures of complexes (I) to (III) shown as 30% thermal ellipsoid probabilities. | ||
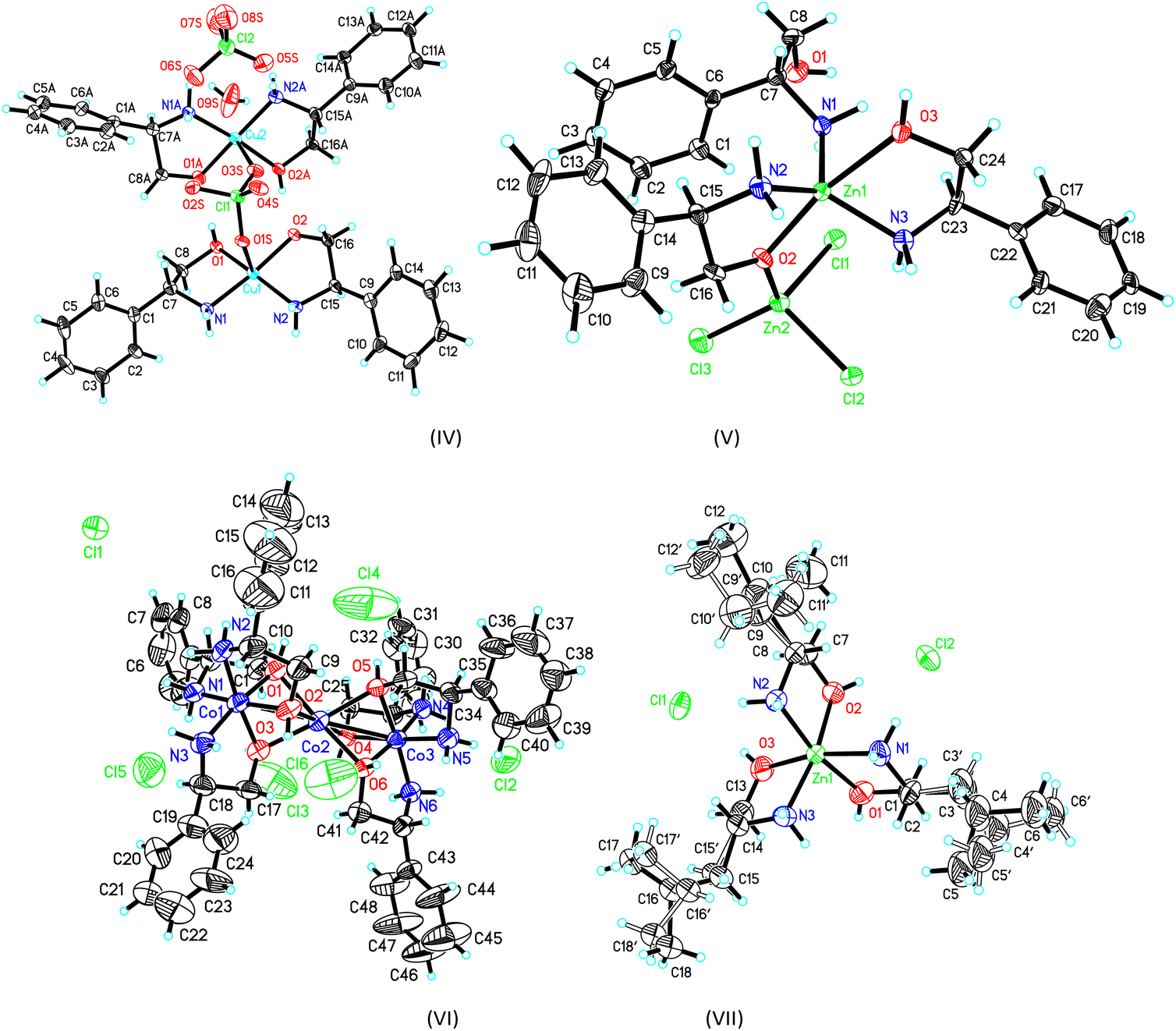 | ||
| Fig. 2 The ORTEP molecular structures of complexes (IV) to (VII) shown as 30% thermal ellipsoid probabilities. | ||
In the crystal of complex (I), the central nickel ion adopts the octahedron coordination sphere by a N3O3 donor set from three separated D-valinol molecules. The bond lengths (dNi–N = 2.071(4) Å/2.085(4) Å, dNi–O = 2.076(3) Å −2.130(3) Å) and angles (79.95(4)° − 98.42(16)° for the cis angles and 164.77(16)° − 171.41(13)° for the trans angles around nickel centre) are comparable to some analogs.30,31 In its crystal packing, the molecules are linked into a three-dimensional hydrogen-bonded network by a combination of N–H⋯O and O–H⋯O interactions (Table S2 and Fig. S1†).
The crystal of complex (II), these three metal centres are linked three μ2-O bridges between each two cobalt ions with the Co1⋯Co2 and Co2⋯Co3 distances of 2.644(2) Å and 2.637(2) Å, respectively. For the two lateral cobalt atoms, they both are in the octahedron coordination environments surrounded by each three nitrogen atoms and three oxygen atoms from three D-valinol ligands. But the middle Co3 is best described as in an inclined trigonal prism coordination by six oxygen atoms. Similar to those in (I), the bond lengths and angles around three cobalt centres are also comparable to its analogs.32 The component ions in (II) are joined together, forming the final three-dimensional network by the extensive hydrogen bonds (Table S2 and Fig. S1†).
In the crystal of complex (III), the central metal copper atom gives a square-planar coordination by each two D-valinol nitrogen atoms and two acetate oxygen atoms when the two valinol oxygen atoms are not considered due to their far distant away from the Cu1 atom [2.493(3) Å/2.502(3) Å]. The four bond lengths are dCu1–N1 = 1.997(3) Å, dCu1–N2 = 2.000(3) Å, dCu1–O1 = 2.003(3) Å, and dCu1–O2 = 2.020(2) Å. And the four cis and two trans angles around copper centre are of 89.08(14)°/88.57(11)° and 177.33(16)°/178.11(12)°, respectively. In the crystal packing, the molecules are linked into a one-dimensional hydrogen-bonded chain along the [100] axis (Table S2 and Fig. S1†).
For the crystal of complex (IV), because the perchlorate O1S, O3S and water O9S atoms are distant more than 2.60(1) Å away from the central metal atoms, the coordination of the both the copper atoms can be described as square-planar configurations. The Cu–N (or O) bond lengths are in a range of 1.935(8) Å to 2.007(10)Å around Cu1, and 1.911(11)Å to 1.986(14) Å around Cu2. The cis and trans angles are of 85.40(1)° to 96.35(1)° and 170.55(1)° to 173.62(4)°, respectively, around Cu1 atom. Those cis and trans angles are of 84.82(4)° to 98.36(4)° and 173.50(4)° to 175.05(4)°,respectively, around Cu2 atom which are comparable with some analogs.33 It should be mentioned that one of the coordinated D-phenyl glycenol is disprotonated when coordinating the copper atom and its Cu–O bond lengths are slightly shorter than the un-disprotonated Cu–O bond. These two discrete coordination cations are joined together into a dimmer by a complementary O–H⋯O hydrogen bonds. Additionally, the molecules are linked into three-dimensional network by a combination of N–H⋯O hydrogen bonds in the crystal packing (Table S2 and Fig. S1†).
The crystal of complex (V) was composed of a di-nuclear zinc complex in which these two metals are joined together by a μ2-O bridge from a D-phenyl glycenol molecule and the Zn1⋯Zn2 distance is 3.444(4) Å. For Zn1 and Zn2, they adopt the bi-pyramidal and tetrahedral coordination sphere, respectively. As for the bi-pyramidal polyhedron around Zn1, two glycenol oxygen O2 and O3 atoms are positioned at the apical sites and the three amine nitrogen atoms locate at the basal plane. As for Zn2 atoms, its tetrahedral coordination is composed of three chloride anions and one glycenol oxygen atom. The coordination bond lengths are ranging from 2.028(11) Å to 2.085(12) Å around Zn1 and from 1.918(11) Å to 2.345(4) Å around Zn2. The cis bond angles are varying from 73.93(1)° to 127.32(4)° and the trans angle is 165.45(4)° around Zn1 atom. For Zn2 atom, the angles are ranging from 99.61(4)° to 119.37(4)°. In the crystal packing, the molecules are linked into a two-dimensional layer structure by the N–H⋯O and O–H⋯O hydrogen bonds running parallel to the (001) plane (Table S2 and Fig. S1†).
In the crystal of complex (VI), the tri-nuclear coordination core is similar to that in complex (II) except for the ligand is different. The Co1⋯Co2 and Co2⋯Co3 distances are both 2.650(4) Å which is slightly longer those in crystal of (II). No more discussions are given due to its structural similarity with crystal (II). The molecules are also linked into the final three-dimensional network by the extensive N–H⋯Cl and O–H⋯Cl hydrogen bonds.
For the crystal of (VII), the mono-nuclear central zinc metal atom adopts the octahedral coordination configuration furnished by a N3O3 donor set. The bond lengths and angles are in a range from 2.094(6) Å to 2.203(5) Å and from 78.70(4) to 100.14(4) which are similar to its analogs.34–36 the component ions are linked into a three-dimensional network by a combination of O–H⋯Cl and N–H⋯Cl hydrogen bonds (Table S2 and Fig. S1†).
Results
NMR spectroscopy for complexes (V) and (VII)
The NMR spectra of complexes (V) and (VII) are shown in Fig. 3 and 4. For the zinc complexes, NMR for complexes (V) and (VII) were also tested further, and from Fig. 3 and 4, we can see that δ 7.76–7.85 and 7.19–7.30 ppm are the characteristic absorption peaks of five protons in the benzene ring. Additionally, δ 3.98, 3.69 and 3.49 ppm are absorption peaks of the CH, CH2 and NH2 group H protons, respectively. In the 13CNMR, δ 140.1, 127.2, 126.3, 126.1, 65.0, and 56.2 ppm are also characteristic absorption peaks, including phenyl carbon protons, tertiary carbon and methylene carbon protons. Similar to complex (VII), in the 1HNMR, δ 3.78, 3.62, 3.30–3.33, and 2.94 each represent the characteristic absorption peaks of the H protons of the CH, CH2 and NH2 and isobutyl groups (δ 1.74, 1.26–1.36, 0.91 and 0.94 ppm). All the NMR results, whether in 1HNMR or 13CNMR, confirmed that the exact structures of complexes (V) and (VII) are consistent with the analytical results of the crystal structures.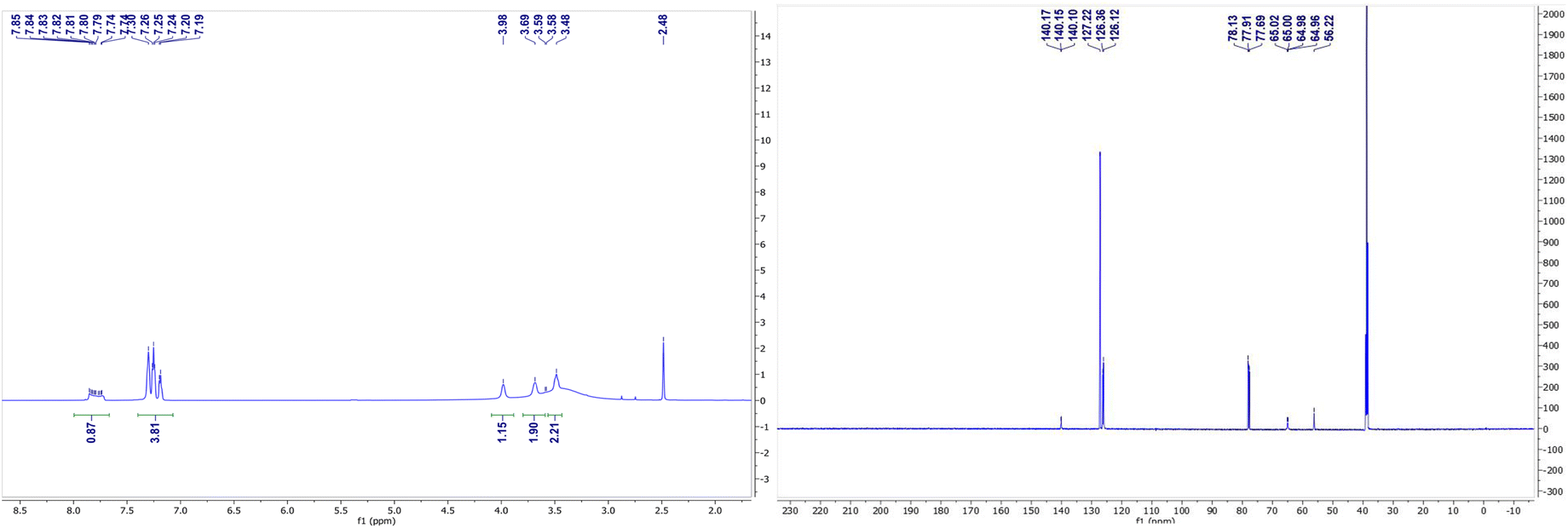 | ||
| Fig. 3 1H NMR (left) and 13C NMR (right) spectrum of complex V. | ||
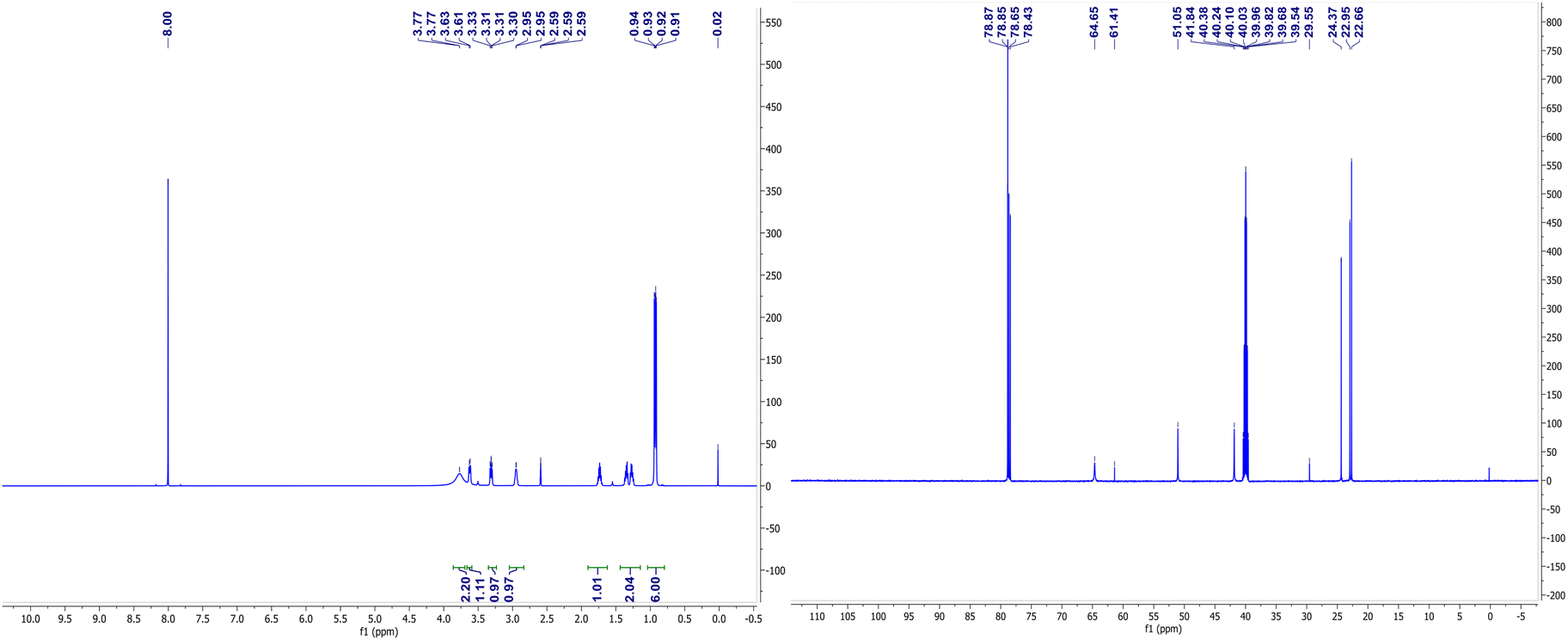 | ||
| Fig. 4 1HNMR and 13CNMR spectrums of complex (VII). | ||
IR spectroscopy of complexes (I)–(VII)
The IR analysis shows a number of peaks that are in all IR spectra (shown in Fig. 5). The IR vibrations of the OH group are usually found to be most sensitive to the environment. As such, they show distinct shifts in the spectra of the hydrogen-bonded species. Generally, the optimum absorption region of non-hydrogen bonded or a free hydroxyl group is in a range of 3550–3700 cm−1. In the case of their presence in a molecule, intra and intermolecular hydrogen bonding reduces the O–H stretching band to the region of 3000–3550 cm−1, signifying the O–H⋯O interactions in the crystal packing. In all our studied complexes, an obvious red shift phenomenon of the hydroxyl groups have been also observed for the infrared absorption vibration peak at 3115–3395 cm−1, which once more demonstrates the weakening of the O–H bond energy due to the delocalization of electrons through hydrogen bonds. The C–C vibrations (1200 cm−1–1400 cm−1) have been shifted toward some regions of very large wavelengths upon complexation, showing that the electron density in C–C bond is increased by transition metal complexation by the stated ligands. Some sharp peaks at 3310–3350 cm−1 representing the existence of NH2 groups. For these functional groups such as all the C–H bonds, their infrared absorption peaks lie between 3000 and 2000 cm−1, CO and CC stretching vibrations are at almost 1720 and 1600 cm−1, and C–O vibrations are at 1280 and 1090 cm−1, respectively.37,38 In more details, there is the 3100 to 3000 cm−1 peak of the aromatic C–H stretching vibration, the stronger absorption at 1600 cm−1, which can be referred to as aromatic CC bonds, and aliphatic C–H stretching vibrations between 3000 and 2800 cm−1. The presence of aromatic structures has been supported by the existence of the spectral region between 1500 and 1630 cm−1. The absorbance bands at 1300 cm−1 to 1400 cm−1 can be assigned to bending vibrations of the CH3 group. These peaks at 1465 cm−1 and 1150 cm−1 are dominated by the C–O stretching vibrations.39–41 There are stretching vibration peaks at 600–650 cm−1 and 500–600 cm−1 for metal-nitrogen and stretching vibration peaks at 594 cm−1, 659 cm−1, 617 cm−1, 649 cm−1, 848 cm−1 and 695 cm−1 for Ni–N, Cu–N, Cu–O, Co–N, Co–O and Zn–N, respectively, that further signifies the interactions between metal and ligands. A peak appeared at 650–670 cm−1 for the C–Cl band and 1500–1600 cm−1 for ClO4 (IR figures are shown in Fig. S2†).
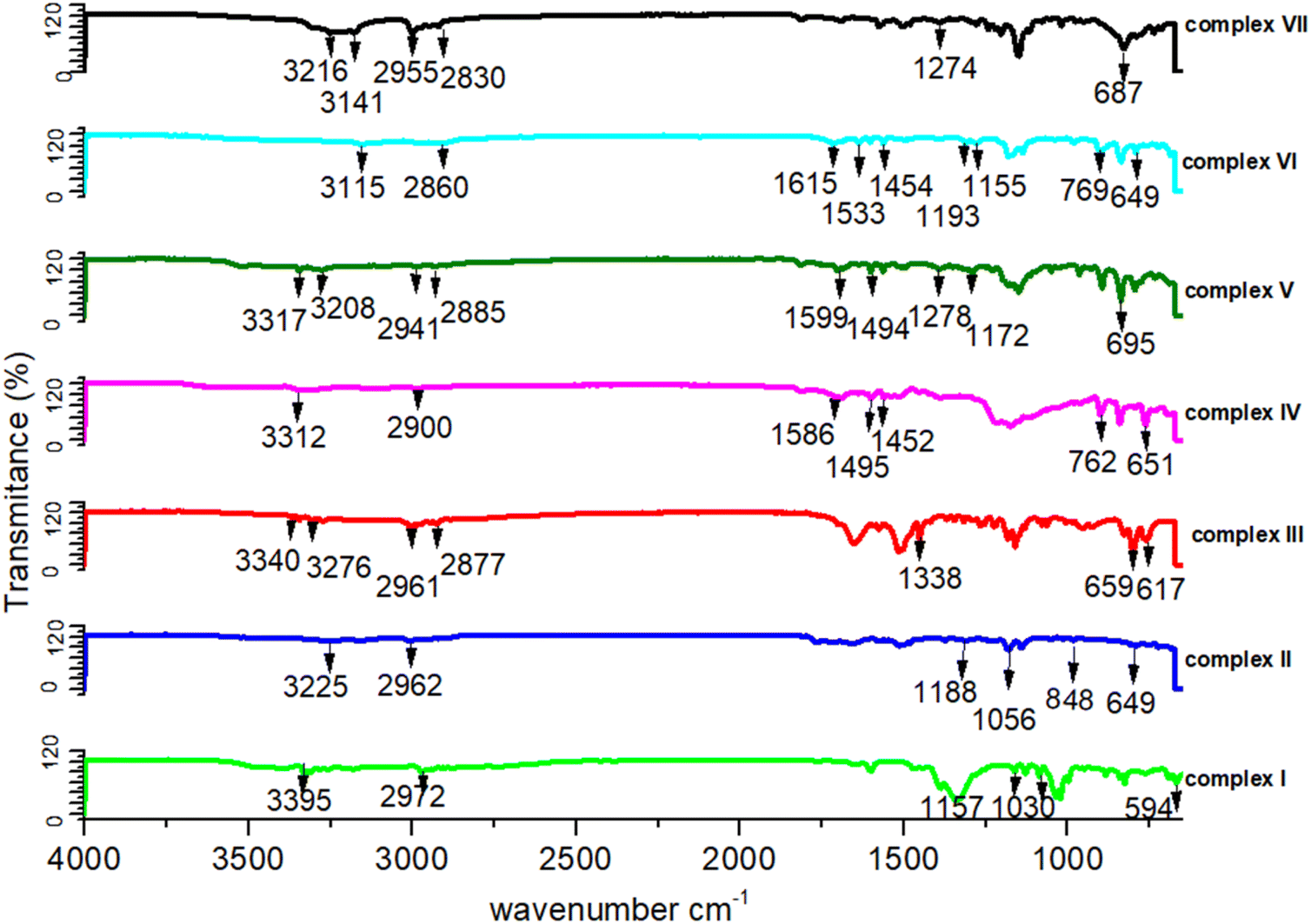 | ||
| Fig. 5 IR spectra of all the complexes (I) to (VII) in range of 4000–500 cm−1 region. | ||
UV-vis spectroscopy for complexes (I)–(VII)
In their methanol solutions, the absorption spectra of the seven complexes and raw materials have been reported.42–45 Transition metal complexes are often coloured owing to the presence of multiple electronic states associated with incompletely filled d orbitals. Evidence of coordination of the metal ion with the corresponding ligands was provided by the d–d transitions of the metal ion observed in the complexes, which were absent in the ligands, the strong band in the ultraviolet region was ascribed to the n → π* transition of the ligands. Interestingly, the complex (I) at 230–234 nm, complex (II) 260–264 nm, 375–379 nm, and 525–527 nm and complex (VII) has broad peaks at 210–213 and 280–282 nm due to σ → σ*,and n → σ* C–O/C–N transition. Whereas complex (III) has a broad band at 278–281 nm due to σ → σ*, and n → σ* C–O/C–N transition while 640–669 nm due to CO n → π* transition. Surprisingly complex (IV), complex (V) and complex (VI) has peaks at 268–271 nm and 360–363 nm, and a broad peak at 250–253 nm. Complex (VI) has a large peak at 255–257 nm, 386–388 nm, and 525–527 nm due to σ → σ*,and n → σ * C–O/C–N transition and due to n → π* CC transition, respectively.27–29 The UV spectra of the complexes are given in Fig. 6. The UV spectra of the complexes (I)–(VII) are given in Fig. S3†.
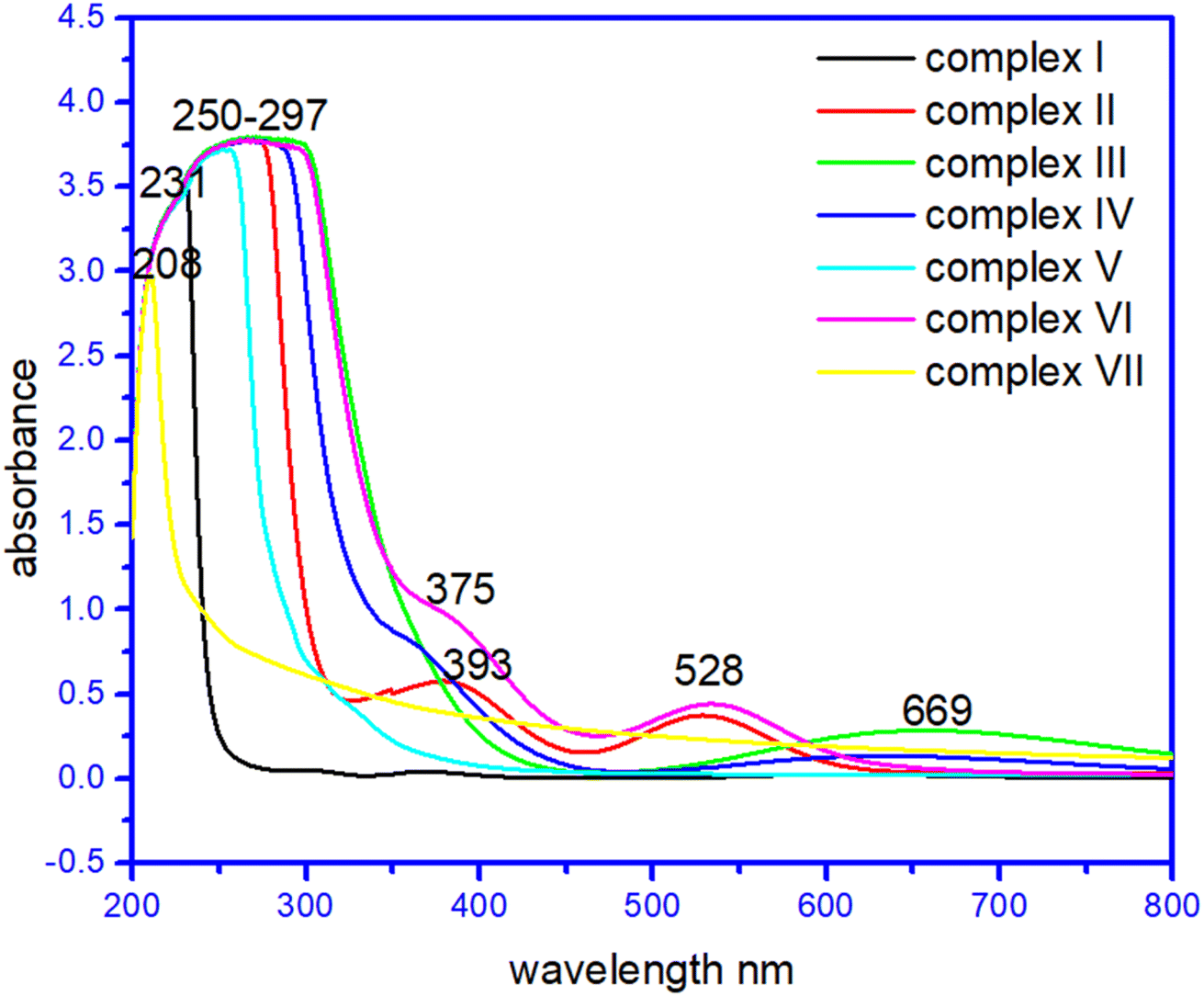 | ||
| Fig. 6 UV visible spectra of complexes (I) to (VII). | ||
In order to ascertain the colour origin of the complexes, it can be deduced from the crystal-field-theory view. As for a first-transition metal atom, its five 3d-orbitals can be splitted into two sets in a certain formed crystal field due to the interactions of the various outside ligands. From Fig. 6, the apparent d–d transitions of compounds (II), (III) and (VI) can be easily observed with their respective maximum absorptions at 528 nm, 528 nm and 669 nm. As is well known that by a combined effect from six ligands in octahedral coordination compounds (II) and (VI), the 3d orbitals for the first-transition metal will be energetically splitting into two groups, three irreducible lower energetic t2g (dxy, dyz and dxz) and two higher energetically e2g (dx2−y2, dz2) ones. Then, the electron-transition in the visible light region should occur between these two groups of obitals. However, the copper atom in compound (III) was four-coordinated which results in its 3d orbitals splitting into four sets in terms of energetic sequence dyz/dxz, dz2, dxy and dx2−y2. These d–d transitions absorb light in visible region, it can make the compounds present its complementary colours such as the red-brown for compound (II) and (VI) and blue for compound (III).
Cytotoxicity assay
The anticancer activity of amino alcohol complexes (I)–(VII) showed cytotoxic effects against the human tumour cell line A549; among them, complex (V) showed the best activity with an IC50 value of 17.8. The IC50 values of the seven compounds along with cisplatin were tested and calculated via Reed and Muench's method.25 In order testify the stabilities of these compounds, we have tested all their UV-vis absorption spectra in a certain solution medium (Fig. S4†) over time process. The results indicate that they didn't undergo apparent change after an overnight standing in air condition, which also clearly proved that the compounds were stable in their cytotoxicity research.From a structural point, we found that the type of metal coordination, the small guest ligands around a metal center and the coordination unsaturation degree are all related to their activities. For example, the activity of zinc compound V is in generally greater than that of copper compound III, which is in turn apparently greater than that of cobalt and nickel compounds II, VI and I. For compound V, its anticancer activity is better than that of compound VII which should be due to the presence of a metal zinc centre coordinated by three chlorine atoms and one oxygen atom. The three coordinated chloride ions may be more easily hydrolyzed during the process of its anticancer activity. For the two copper compounds III and VI, the copper ion in III is more easily exposed to a certain substrate groups due to its planar four-coordinated geometry. Perhaps for this reason, compound III also showed better anticancer activity with the IC50 value of 31.58 ± 2.80 μM, which is kind of like the mechanism of the four-coordinated platinum atom in cisplatin compound. Further relationship between the coordination environment of a metal atom and its activities are ongoing in our lab.
Conclusions
A series of Zn(II), Cu(II), Co(II), and Ni(II) novel complexes containing chiral amino alcohols were synthesized through one-pot method, and characterized spectroscopically using NMR, FTIR, UV-visible, and EA as well as single-crystal X-ray diffraction techniques to confirm the structure of already synthesized chiral amino alcohol complexes. The complexes were then evaluated against anti-lungs cancer cell. They also exhibited cytotoxic activities against A549 cell lines. Compared to the others, complex (V) had an IC50 of 17.8 against A549 lung cancer cells. The observations demonstrated that the anticancer activity of these complexes is dependent on the type of metal ion, cell line, and geometries of the corresponding molecule. The obtained anticancer activities results are mark able and comparable to the available anti-cancer drugs in market. We expect that these complexes could be a remarkable drug in future and replace cis-pt as well as use for other medicinal application. The intriguing results may help in the design and development of new medicinal drug-like compounds. The catalytic activities of complexes (I)–(VII) for different organic reactions are currently ongoing.Author contributions
M. Luo designed the research, analysed the data and revised the article; H. Yin and J. C. Zhang helped with NMR and elemental testing. Q. Umar wrote the draft, performed the research work and contributed equally with Y. H. Huang to this research work. Y. H. Huang and A. Nazeer also performed the research work. X.-G. Meng revised the draft and analysed the data; all authors read and approved the final manuscript.Consent for publication
All authors consent to the publication.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the Hefei University of Technology and State Key Laboratory of Photochemistry and Plant Resources in West China.Notes and references
- A. Jemal, R. L. Siegel, J. Ma, F. Islami, C. DeSantis, A. Goding Sauer, E. P. Simard and E. M. Ward, J. Clin. Oncol., 2015, 33, 829–835 CrossRef PubMed
.
- H. M. Treshalina, V. I. Romanenko, D. N. Kaluzhny, M. I. Treshalin, A. A. Nikitin, A. S. Tikhomirov and A. E. Shchekotikhin, Eur. J. Pharm. Sci., 2017, 109, 631–637 CrossRef CAS PubMed
- W.-Y. Zhang, F. Du, M. He, L. Bai, Y.-Y. Gu, L.-L. Yang and Y.-J. Liu, Eur. J. Med. Chem., 2019, 178, 390–400 CrossRef CAS PubMed
- C. X. Zhang and S. J. Lippard, Curr. Opin. Neurol., 2003, 7, 481–489 CAS
- F. Guarra, A. Pratesi, C. Gabbiani and T. Biver, J. Inorg. Biochem., 2021, 217, 111355 CrossRef CAS PubMed
- Y. Lin, Y. Lin, Z. Lin, C. Chang and C. Cheng, J. Biotechnol., 2019, 305, S31 CrossRef
- L. Conti, E. Macedi, C. Giorgi, B. Valtancoli and V. Fusi, Coord. Chem. Rev., 2022, 469, 214656 CrossRef CAS
- Y. Y. Qi, Q. Gan, Y. X. Liu, Y. H. Xiong, Z. W. Mao and X. Y. Le, Eur. J. Med. Chem., 2018, 154, 220–232 CrossRef CAS PubMed
- C. Hartinger, A. Nazarov, S. Ashraf, P. Dyson and B. Keppler, Curr. Med. Chem., 2008, 15, 2574–2591 CrossRef CAS PubMed
- N. Farrell, L. R. Kelland, J. D. Roberts and M. Van Beusichem, Cancer Res., 1992, 52, 5065–5072 CAS
- J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS PubMed
- M. D. Hall, K. A. Telma, K.-E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Cancer Res., 2014, 74, 3913–3922 CrossRef CAS PubMed
- Z. B. Nojini, F. Yavari and S. Bagherifar, J. Mol. Liq., 2012, 166, 53–61 CrossRef CAS
- H. Matsumoto, Y. Hirabayashi, H. Kubota, H. Murakami, M. Higashida, K. Haruma, J. Hiratsuka, M. Nakamura and T. Hirai, Anticancer Res., 2012, 32, 1827–1832 CAS
- K. Yoshinaga, H. Niikura, Y. Ogawa, K. Nemoto, S. Nagase, T. Takano, K. Ito and N. Yaegashi, Gynecol. Oncol., 2007, 104, 36–40 CrossRef CAS PubMed
- T. Brown, Harv. Bus. Rev., 2008, 86, 84–92 Search PubMed
- C. Nardon, G. Boscutti and D. Fregona, Anticancer Res., 2014, 34, 487–492 CAS
- L. Messori, G. Marcon, M. A. Cinellu, M. Coronnello, E. Mini, C. Gabbiani and P. Orioli, Bioorg. Med. Chem., 2004, 12, 6039–6043 CrossRef CAS PubMed
- A. F. A. Peacock and P. J. Sadler, Chem.–Asian J., 2008, 3, 1890–1899 CrossRef CAS PubMed
- A. Habtemariam, M. Melchart, R. Fernández, S. Parsons, I. D. H. Oswald, A. Parkin, F. P. A. Fabbiani, J. E. Davidson, A. Dawson, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2006, 49, 6858–6868 CrossRef CAS PubMed
- Z. Liu, A. Habtemariam, A. M. Pizarro, S. A. Fletcher, A. Kisova, O. Vrana, L. Salassa, P. C. A. Bruijnincx, G. J. Clarkson, V. Brabec and P. J. Sadler, J. Med. Chem., 2011, 54, 3011–3026 CrossRef CAS PubMed
- Q.-B. Wang, Z.-Y. Jing, X.-M. Hu, W.-X. Lu and P. Wang, CrystEngComm, 2021, 23, 216–220 RSC
- K. J. P. Mahesha, C. S. Karthik, M. K. Hema, P. Mallu and N. K. Lokanath, CrystEngComm, 2021, 23, 4344–4369 RSC
- Z. Mardani, R. Kazemshoar-Duzduzani, K. Moeini, A. Hajabbas-Farshchi, C. Carpenter-Warren, A. M. Z. Slawin and J. D. Woollins, RSC Adv., 2018, 8, 28810–28824 RSC
- M. A. Ramakrishnan, World Journal of Virology, 2016, 5, 85 CrossRef PubMed
- A. A. Amer, J. Heterocycl. Chem., 2017, 55, 297–301 CrossRef
- I. V. Dyachenko, V. D. Dyachenko, P. V. Dorovatovsky, V. N. Khrustalev and V. G. Nenajdenko, Russ. J. Org. Chem., 2020, 56, 974–982 CrossRef CAS
- M. Purushothaman, K. Thanigaimani, S. Arshad, S. Silambarasan, I. A. Razak and K. M. S. Ali, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, o812–o813 CrossRef CAS PubMed
- E. L. Little, W. J. Middleton, D. D. Coffman, V. A. Engelhardt and G. N. Sausen, J. Am. Chem. Soc., 1958, 80, 2832–2838 CrossRef CAS
- B. Zheng, M. Wang, Z. Li, Q. Bian, J. Mao, S. Li, S. Liu, M. Wang, J. Zhong and H. Guo, Tetrahedron: Asymmetry, 2011, 22, 1156–1160 CrossRef CAS
- S. C. Manna, S. Mistri and A. D. Jana, CrystEngComm, 2012, 14, 7415–7422 RSC
- C. Chao, H. Liang, W. Quan, B. Zhang and R. Wang, Chem. Res. Chin. Univ., 2020, 24, 306–311 Search PubMed
- G. Labourdette, D. J. Lee, B. O. Patrick, M. B. Ezhova and P. Mehrkhodavandi, Organometallics, 2009, 28, 1309–1319 CrossRef CAS
- N. Mofaddel, A. A. Adoubel, C. J. Morin, P.-L. Desbène and G. Dupas, J. Mol. Struct., 2010, 975, 220–226 CrossRef CAS
- I. Valencia, H. López-Sandoval, Y. Ávila-Torres, M. Castro and N. Barba-Behrens, J. Mol. Struct., 2020, 1202, 127200 CrossRef CAS
- Y.-H. Ou, R.-K. Du, S.-P. Zhang, Y. Ling, S. Li, C.-J. Zhao, W.-Z. Zhang and L. Zhang, J. Mol. Struct., 2020, 1215, 128229 CrossRef CAS
- K. Ramaswamy and R. Venkatachalapathy, Spectrosc. Lett., 1991, 24, 759–778 CrossRef CAS
- Z. P. Deng, Z. Y. Zhang, L. H. Huo, S. W. Ng, H. Zhao and S. Gao, CrystEngComm, 2012, 14, 6548–6558 RSC
- V. Cepus, M. Borth and M. Seitz, Int. J. Clean Coal Energy, 2016, 5, 13–22 CrossRef CAS
- I. Samfira, S. Rodino, P. Petrache, R. T. Cristina, M. Butu and M. Butnariu, Digest Journal of Nanomaterials and Biostructures, 2015, 10, 557–566 Search PubMed
- M. Butnariu, M. Peana, I. Sarac, S. Chirumbolo, H. Tzoupis, C. T. Chasapis and G. Bjørklund, Chem. Pap., 2021, 75, 5523–5533 CrossRef CAS
- J. Costamagnac and J. Vargasc, J. Mol. Struct., 1996, 367, 97–110 CrossRef
- Q. H. Meng, P. Zhou, F. Song, Y. B. Wang, G. L. Liu and H. Li, CrystEngComm, 2013, 15, 2786–2790 RSC
- J. Han, Y. Wang, J. Wang, C. Wu, X. Zhang and X. Yin, J. Organomet. Chem., 2022, 973–974 Search PubMed
- Z. Li, H. Yan, K. Liu, X. Huang and M. Niu, J. Mol. Struct., 2019, 1195, 470–478 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting information characterization spectra for complexes (I)–(VII), crystallographic information files (CIF). This material is available free of charge via the internet at Crystallographic information for all compounds has been deposited with the Cambridge Crystallographic Data Center (CCDC) as supplementary publications. CCDC 2158086–2158089, 2158091–2158093 and 2158095. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra05576g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |