
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradation of ciprofloxacin by persulfate activated with pyrite: mechanism, acidification and tailwater reuse†
Hui Liu,
Peng Fu,
Fenwu Liu,
Qingjie Hou,
Zhenye Tong and
Wenlong Bi *
*
College of Resources and Environment, Shanxi Agricultural University, Shanxi 030801, China. E-mail: jiayangbihu@126.com
First published on 20th October 2022
Abstract
Residues of ciprofloxacin (CIP) in the environment pose a threat to human health and ecosystems. This study investigated the degradation of CIP by persulfate (PS) activated with pyrite (FeS2). Results showed that when [CIP] = 30 μM, [FeS2] = 2.0 g L−1, and [PS] = 1 mM, the CIP removal rate could reach 94.4% after 60 min, and CIP mineralization rate reached 34.9%. The main free radicals that degrade CIP were SO4˙− and HO˙, with contributions of 34.4% and 35.7%, respectively. Additionally, compared to the control (ultrapure water), CIP in both tap water and river water was not degraded. However, acidification could eliminate the inhibition of CIP degradation in tap water and river water. Furthermore, acidic tailwater from CIP degradation could be utilized to adjust the pH of untreated CIP, which could greatly promote the degradation of CIP and further reduce disposal costs. The reaction solution was not significantly biotoxic and three degradation pathways of CIP were investigated. Based on the above results and the characterization of FeS2, the mechanism of CIP degradation in the FeS2/PS system was that FeS2 activated PS to generate Fe(III) and SO4˙−. The sulfide in FeS2 reduced Fe(III) to Fe(II), thus achieving an Fe(III)/Fe(II) cycle for CIP degradation.
1. Introduction
With the development of technology and medical advances, antibiotics are increasingly used in a wide range of medical and livestock farming processes.1 Fluoroquinolones are a large group of antibiotics used in clinical treatment.2 Among these, ciprofloxacin (CIP) is a broad-spectrum antibiotic that belongs to third-generation fluoroquinolones.3,4 Due to its large molecular weight and complex structure, CIP cannot be completely absorbed by living organisms, and only a small proportion will be used.5 The remainder will be discharged from organisms as metabolites in the form of hydrolysates, oxidation products, conjugated states, and so on.6 Then, they enter the natural environment through different pathways and with other pollutants, forming municipal wastewater, medical wastewater, farming and waste, manure, and leachate from landfills.7,8 Human medical wastewater is the main source of CIP in wastewater, with livestock farming and factory discharges also accounting for a significant proportion, and most wastewater is discharged directly into municipal domestic wastewater without treatment.9 Antibiotics remain in various water environments, forming a drug concentration antibiotic wastewater with high drug concentration, relatively complex substance composition, and high biological toxicity.10 Herein, antibiotic wastewater without proper treatment can generate antibiotic-resistant bacteria and antibiotic-resistance genes,11 which not only causes serious environmental pollution but poses threats to human health.12,13 Therefore, it is crucial to deal effectively with the environmental pollution caused by antibiotic wastewater discharge.At present, there are various methods for treating wastewater containing antibiotics, which can be roughly divided into three categories: physical, biological, and chemical methods.14 Nickel oxide nanoparticles could remove ciprofloxacin by adsorption,15 but the adsorption capacity was limited and saturation tends to occur during use. Although the activated sludge process could effectively treat pharmaceutical wastewater contaminated with antibiotics,16 this method would cause a potentially harmful situation by creating a large number of resistant bacteria and genes in the activated sludge. In recent years, advanced oxidation processes (AOPs) have received increasing attention owing to their different advantages, such as high oxidation, non-selectivity, and fast reaction rates.17,18 Sulfate radical-based advanced oxidation processes (SR-AOPs) are processes that degrade large organic substances into small molecules such as CO2 and H2O by activating persulfate (PS) or peroxymonosulfate (PMS) to generate SO4˙−.19,20 Usually, PS can be activated to generate SO4˙− under the conditions of heat, ultraviolet, ultrasound, alkali, and transition metal ions.21–25 Nevertheless, the most common Fe(II) activated PS was limited by acid conditions and can generate iron sludge.26,27 Therefore, it is necessary to find an environmentally friendly activator for SR-AOPs to treat wastewater containing antibiotics.
As reported, metal sulfides are used as co-catalysts in catalytic reactions, reducing Fe(III) to Fe(II) and having the ability to promote the regeneration of Fe(II).28–30 Pyrite (FeS2) was a sulfide mineral with high iron ion content and could activate PS to degrade organic pollutants.31 Pyrite-activated PS system could degrade p-chloroaniline, and both SO4˙− and HO˙ played a significant role in the degradation.32 Pyrite-activated PS could degrade tris(2-chloroethyl) phosphate, and the acidic environment favors the generation of free radicals.33 Pyrite could activate PS to degrade 2,4-dichlorophenol (2,4-DCP), and reductive sulfur species were important electron donors in Fe(II)/Fe(III) cycle.34 Moreover, FeS2 was a low-cost and readily available mineral, and it was a high potential and promising activator.35 However, there are few studies on the FeS2 activated PS for the degradation of antibiotics in different water substrates. In this study, FeS2 was selected as the activator to investigate the effect of FeS2-activated PS to degrade CIP. Considering that in practical applications, livestock farming processes mostly use tap water, and livestock wastewater is directly discharged into river water, we further simulated the degradation of CIP in different water substrates in FeS2/PS system. Furthermore, post-reaction biotoxicity, mineralization and intermediates were investigated to explore the mechanism of antibiotic degradation by FeS2/PS system.
2. Materials and methods
2.1. Chemicals and materials
Ciprofloxacin (CIP, >99%) was purchased from Sigma-Aldrich, and its physicochemical properties are shown in Table S1.† Pyrite (FeS2) was purchased from Hebei, China. Sodium persulfate, methanol (MeOH), tert-butanol (TBA), ferrous sulfate (FeSO4·7H2O), sulfuric acid, and sodium hydroxide were all analytically pure reagents. All aqueous solutions were prepared using ultrapure water, except for river water (Uma River in China) and tap water (laboratory) (Table S2† showed physicochemical properties of river water and tap water).2.2. Experimental procedures
Degradation experiments were performed in a photoreactor (XPA-7(G8)). The reaction was initiated by adding 50 mL of CIP (30 μM) reaction solution to a quartz tube and adding the desired FeS2 and PS in a photoreactor. At designed intervals, 1 mL of the reaction solution was passed through a 0.45 μm filter membrane and mixed with a rapid addition of 150 μL of methanol to quench the reaction, and then the post-reaction CIP concentrations were measured.Free radical contribution experiment was probed by adding 1 mL MeOH or 1 mL TBA to the CIP solution. In pH effect experiments, the pH of 30 μM CIP solutions were adjusted to 2, 4, 6, 8 and 10 with 0.5 mol L−1 sulfuric acid and 0.5 mol L−1 sodium hydroxide, and FeS2 and PS were added for subsequent experiments. CIP solutions were prepared using ultrapure water, tap water, and river water for degradation experiments with different water substrates, and the other experimental procedures were the same as above.
2.3. Analytical methods
CIP concentrations were measured by High Performance Liquid Chromatography (HPLC, LC-20AD, Japan) equipped with a C18 column (150 mm × 4.6 mm, 5 μm). The mixture of 0.1% formic acid and acetonitrile (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was employed as the mobile phase at a flow rate of 1 mL min−1 with a detection wavelength of 279 nm. The minerals before and after the reaction were qualitatively analyzed by X-ray diffractometer (XRD, MiniFlex II, Germany), and the functional groups of minerals before and after the reaction were determined by Fourier Transform Infrared Spectrometer (FTIR, Tensor 27, Germany). Scanning electronic microscope observed the mineral characteristics and performed the spectrum analysis (SEM & EDS, JSM-7001F, Japan), minerals surface elements were analyzed by Al-Kα monochromatic X-ray photoelectron spectroscopy (XPS, AXIS ULTRA DLD, Britain). The mineralization of the solution after the reaction was determined by a total organic carbon analyzer (TOC, Multi N/C-3100, Germany). The intermediates of CIP were analyzed by Liquid Chromatograph Mass Spectrometer (LC-MS, Agilent 1290 II-6470, USA).
:1, v/v) was employed as the mobile phase at a flow rate of 1 mL min−1 with a detection wavelength of 279 nm. The minerals before and after the reaction were qualitatively analyzed by X-ray diffractometer (XRD, MiniFlex II, Germany), and the functional groups of minerals before and after the reaction were determined by Fourier Transform Infrared Spectrometer (FTIR, Tensor 27, Germany). Scanning electronic microscope observed the mineral characteristics and performed the spectrum analysis (SEM & EDS, JSM-7001F, Japan), minerals surface elements were analyzed by Al-Kα monochromatic X-ray photoelectron spectroscopy (XPS, AXIS ULTRA DLD, Britain). The mineralization of the solution after the reaction was determined by a total organic carbon analyzer (TOC, Multi N/C-3100, Germany). The intermediates of CIP were analyzed by Liquid Chromatograph Mass Spectrometer (LC-MS, Agilent 1290 II-6470, USA).
3. Results and discussion
3.1. Optimal concentration of FeS2 and PS for CIP degradation
 | ||
| Fig. 1 Analytical results of CIP with FeS2/PS after 60 min of response surface methodology (a) and degradation of CIP in different reaction systems (b) ([CIP] = 30 μM, [FeS2] = 2.0 g L−1, [PS] = 1 mM). | ||
| 2FeS2 + 7O2 + 2H2O → 2Fe(II) + 4SO42− + 4H+ | (1) |
| Fe(II) + S2O82− → Fe(III) + SO42− + SO4˙− | (2) |
| SO4˙− + H2O → H+ + SO42− + HO˙ | (3) |
3.2. Determination of reactive oxygen species and system concentration
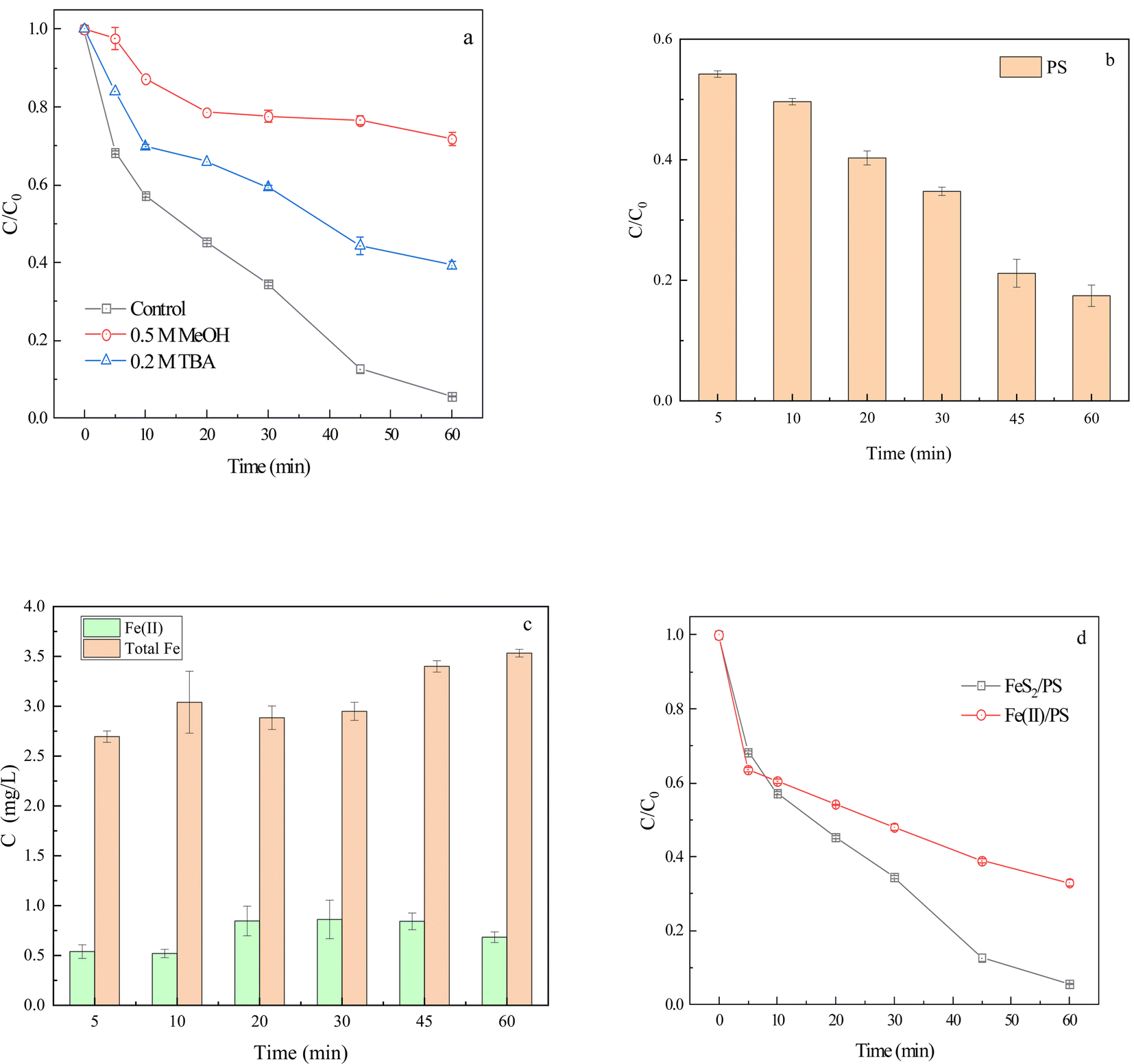 | ||
| Fig. 2 Determination of reactive oxygen species (a), PS concentration (b), Fe(II) and total Fe concentration (c) and validation of FeS2 interface effect (d) in degradation of CIP by FeS2/PS system ([CIP] = 30 μM, [FeS2] = 2.0 g L−1, [Fe(II)] = 4 mg L−1, [PS] = 1 mM). | ||
To further investigate the interface effect of FeS2, 1 mM PS and 4 mg L−1 Fe(II) (FeSO4·7H2O) were added to CIP solution to form the Fe(II)/PS homogeneous system. The degradation rate of CIP at 60 min in Fe(II)/PS system was 67.0%, and it was presumed that the sulfide in FeS2 promoted the degradation of CIP (Fig. 2d). FeS2 might react with Fe(III) (eqn (4)),45 leading to dynamic changes in Fe(III) and Fe(II) concentrations. Therefore, FeS2 could reduce Fe(III) to Fe(II), and further react with PS to degrade CIP.
| FeS2 + 14Fe(III) + 8H2O → 15Fe(II) + 2SO42− + 16H+ | (4) |
3.3. Effect of pH on the degradation of CIP by FeS2/PS system
The effect of different pH on the removal of CIP by FeS2 activated PS was shown in Fig. 3. When the pH was 10, the degradation of CIP in FeS2/PS system decreased slowly with time, and the removal of CIP after 60 min of reaction was only 62.4%. The dissolution of Fe(II) was inhibited under alkaline conditions, allowing only small amounts of SO4˙− to be generated at low Fe(II) concentrations. When pH were 8 and 6, the degradation rate of CIP was first fast and then slow, but the degradation rate of CIP was slightly lower than that of control, which might be due to the addition of H2SO4 and NaOH when adjusting the pH. When the pH was 4, the CIP was rapidly degraded within the first 45 min, and the removal rate reached 95.7% after 60 min. This result might be due to acidic conditions prompting the dissolution of Fe(II) from FeS2, PS in the presence of high concentrations of activator to rapidly generate large amounts of SO4˙− and HO˙.46 The degradation trend of CIP was slow at pH was 2, with 83.8% of CIP removed at 60 min, which was lower than the removal of CIP at pH = 4. Although large amounts of Fe(II) were generated under acidic conditions, the excess Fe(II) could consume SO4˙− and HO˙, which affected the degradation of the CIP. | ||
| Fig. 3 Effect of pH on the degradation of CIP by FeS2/PS system ([CIP] = 30 μM, [FeS2] = 2.0 g L−1, [PS] = 1 mM). | ||
As can be seen in Table S3,† there was a varying degree of pH decrease after 60 min of reaction. Except for pH = 2 (pH remained at 1.98 after 60 min of reaction), the other pH systems had a pH between 2.94 and 3.07 after 60 min of reaction. Fig. S3† shows the pH of the control at different reaction times. The results showed that pH degraded rapidly and then slowly, with a pH of 2.93 after 60 min. Weak acidic environment further promoted FeS2/PS degradation of CIP. In addition, the reaction of FeS2 with PS to generate H+, Fe(III), and SO42− caused the solution pH to be low.47
3.4. Characterization of pyrite before and after reaction
XPS spectrometer was used to determine the binding energy of pyrite before and after the reaction to identify the chemical properties and composition of the sample surface. As shown in Fig. 4a, FeS2 before and after the reaction consists mainly of S, C, O, Fe, and Na. Fig. S4† shows the EDS energy spectrum of FeS2 before and after the reaction whose main elements were in accordance with the above results. C, O, and Na elements may be FeS2 in contact with non-metals and metals in air.48 The FeS2 surface elements were significantly lower after use, especially Fe and S, probably due to leaching from FeS2 into the water. XRD (Fig. 4b) and FTIR (Fig. S5†) results showed that the diffraction intensities and functional groups of fresh and used FeS2 had changed slightly accordingly. | ||
| Fig. 4 Full spectrum of XPS measurements of fresh and used pyrite (a), XRD spectrum (b), high-resolution Fe 2p (c), high-resolution S 2p (d). | ||
Fig. 4c shows the high-resolution Fe 2p spectra of FeS2 before and after the reaction. The fit revealed three peaks in the Fe 2p3/2 orbitals at 707.2, 709.8, and 712.1 eV for Fe(II)–S, Fe(II)–O, and Fe(III)–O.49 The pre-reaction ratios were 35.6%, 26.5%, and 37.9%. After the reaction, the Fe(III)–O peak increased by 17.2 percentage points, and the Fe(II)–S peak reduced by 19.5 percentage points. Fig. 4d shows the high-resolution S 2p spectra of FeS2 before and after the reaction. The fitted peaks of S 2p3/2 and S 2p1/2 orbitals of S22− were 162.6 and 163.9 eV, respectively,50,51 with the pre-reaction ratio of 64.9% and post-reaction ratio of 32.5%. Besides, the S 2p3/2 and S 2p1/2 orbitals for SO42− peaks were 168.5 and 169.8 eV with a post-reaction ratio of 31.5% and a post-reaction ratio of 67.5%. This result indicated that the S22− in FeS2 was oxidized to SO42− during the oxidation process. Moreover, as the proportion of elemental S decreased much more than elemental Fe after the reaction, S22− could promote the Fe(III)/Fe(II) cycle.52
3.5. Effect of different water substrates on the degradation of CIP by FeS2/PS system
In practical applications, livestock farming processes mostly use tap water, and livestock wastewater is directly discharged into river water. Hence, this study investigated the degradation of CIP by FeS2/PS in tap water and river water, and the results were compared with that in ultrapure water.As shown in Fig. 5a, the CIP was not effectively degraded in either tap or river water. At 60 min, the degradation rates of CIP in tap water and river water were 85.6 and 82.3 percentage points lower than that in ultrapure water. It was possible that the anions in tap water and river water reacted with SO4˙− and HO˙ to generate low reactive radicals,53 thus slowing down the degradation rate of CIP. Furthermore, the presence of organic matter in river water and tap water competed with CIP for the reaction with SO4˙− and HO˙, resulting in ineffective degradation of CIP. In addition, the greater degree of inhibition in river water than that in tap water may be due to the high concentration and more complex composition of anions in river water.
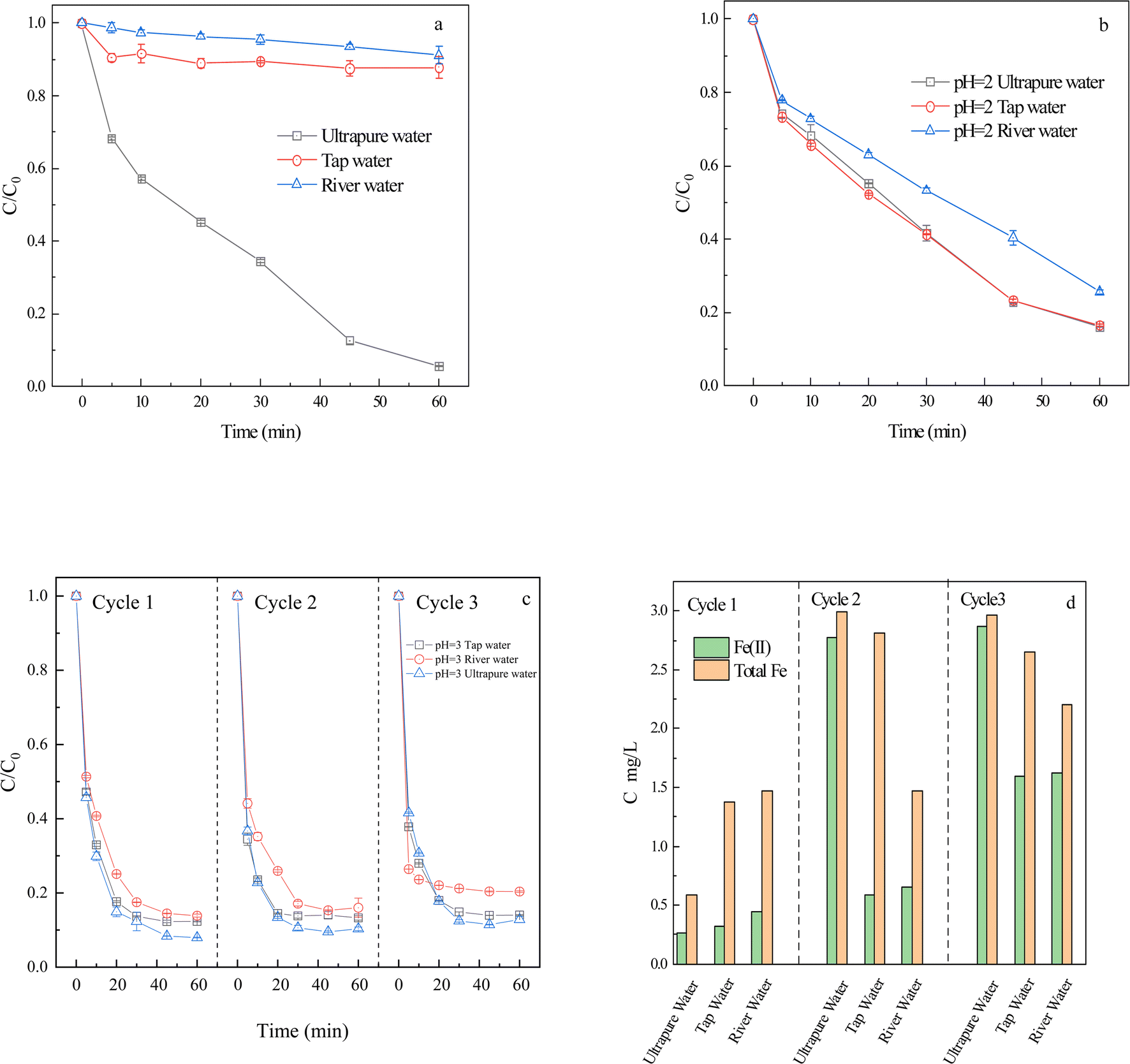 | ||
| Fig. 5 Effect of different water substrates without pH adjustment (a) and pH = 2 (b); acidic tailwater after reaction to the adjustment of pH to 3 of different water substrates (c) and the Fe(II) and total Fe concentration of solution before reaction (d) on the degradation of CIP by FeS2/PS system ([CIP] = 30 μM, [FeS2] = 2.0 g L−1, [PS] = 1 mM). | ||
The physicochemical properties of tap water and river water (Table S2†) indicated that tap water and river water contain high concentrations of HCO3− and Cl−. Higher HCO3− and Cl− could react with SO4˙− and HO˙ to form low-activity free radicals and affect the degradation of CIP. According to the latest distribution diagram of CO2, CO32−, and HCO3− at different pH values, only free CO2 is found in the water when pH < 4. Furthermore, the pH experiment (Fig. 3) shows that the weak acid nature promoted the degradation of CIP, thereby accelerating the dissolution of Fe(II) and speeding up the reaction process. Therefore, acidification could greatly weaken the inhibition of water substrates to CIP. The pH of different water substrates was adjusted to 4 for the experiment (Fig. S6†), but no effective treatment effect was achieved. Therefore, the pH of different water substrates was adjusted to 2 for further experiments (Fig. 5b). The degradation rates of CIP in ultrapure water, tap water, and river water at 60 min were 83.8%, 83.4%, and 74.4%, respectively. The degradation rate of CIP in river water was 8.4 times that in unadjusted pH, and that in tap water was 6.9 times that in unadjusted pH. It was possible that the inhibitory effect of the anions was weakened in the more acidic environment, so we chose pH 2 for CIP degradation by different water substrates. Therefore, acidification (pH = 2) of FeS2/PS system could promote degradation of CIP in different water substrates.
To progress a reduction in disposal cost, the pH of the untreated CIP was adjusted by replacing the acid with degraded tailwater. As shown in Fig. 5c, after 60 min of reaction, the CIP degradation rates of river water, tap water, and ultrapure water were 86.1%, 87.7%, and 92.1%, respectively. The CIP removal rate was higher than that in Fig. 5b. In addition, the pH of the reaction solution was approximately 2.6, and this tailwater was adjusted to pH 3 for the untreated CIP for the second cycle. After 60 min of reaction, the CIP degradation rates of river water, tap water, and ultrapure water were 84.0%, 86.8%, and 89.7%, respectively. The pH of the solution after reaction was about 2.5. The tailwater was adjusted to pH 3 of the untreated CIP solution for the third cycle. After 60 min of reaction, the CIP degradation rates were 80.0%, 86.0%, and 87.2% for river water, tap water, and ultrapure water, respectively. The reason may be due to the weak anion interference in the system at pH < 4 and the tailwater still contained a small amount of ferrous iron to promote the FeS2/PS system. In summary, the reuse of tailwater three times still has a good degradation effect on CIP.
To investigate the mechanism of tailwater reuse, Fe(II) and total Fe concentrations of the reaction solutions were measured before cycling in different water substrates (Fig. 5d). The concentrations of Fe(II) and total Fe in the solution generally increased with increasing number of cycles. The total Fe concentration before the first cycle was less than 1.5 mg L−1 and the Fe(II) concentration was less than 0.5 mg L−1. Before the second cycle, the total Fe and Fe(II) concentrations were generally higher than those in cycle 1, but did not exceed 3 mg L−1. The total Fe and Fe(II) concentrations in cycle 3 were slightly higher than those in cycle 2. Therefore, the increase in the concentration of Fe(II) in solution was also responsible for promoting the rapid degradation of CIP (Fig. 5c).
3.6. Effect of biotoxicity and mineralization of CIP degradation by FeS2/PS system
To investigate the changes in biotoxicity before and after treatment, the solution before and after the reaction was added to the bacterial culture medium and the absorbance value at 600 nm (OD600) was measured to characterize the growth of soil microorganisms, so as to determine the biotoxicity of each treatment system.54 Fig. 6a shows that microbial growth was slow from 3 to 6 h and probably in the acclimatization phase, and became rapid from 6 to 24 h. At 24 h, the OD600 values for the H2O, CIP, FeS2/PS/H2O, and FeS2/PS/CIP treatments were all around 2.0, with no significant differences between each treatment compared to the blank (H2O). Although chemicals were added to generate SO4˙− to degrade CIP, no significant toxicity was present compared to H2O. Therefore, the FeS2/PS system could effectively remove CIP without increasing toxicity and has good potential for application.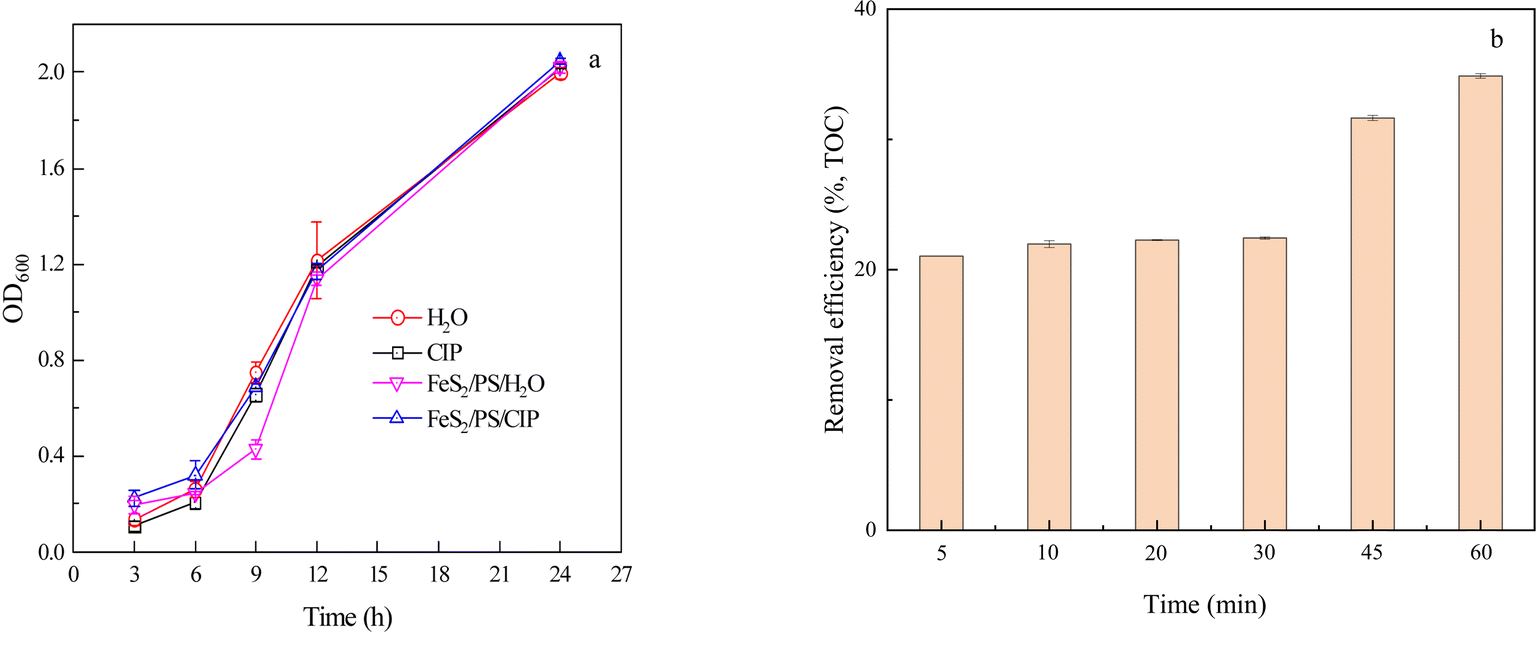 | ||
| Fig. 6 Effect of biotoxicity (a) and mineralization (b) of CIP degradation in FeS2/PS system ([CIP] = 30 μM, [FeS2] = 2.0 g L−1, [PS] = 1 mM). | ||
Fig. 6b shows the mineralization of CIP in FeS2/PS system. The removal of TOC increased slowly in the first 30 min and increased rapidly after 30 min. The TOC removal was 34.9% after 60 min. This result may be due to the slow release of Fe(II) from FeS2 in the early stages and the start of a large amount of Fe(II) dissolution after 30 min, producing more SO4˙− to degrade CIP and increase the TOC removal.
3.7. Proposed degradation of CIP pathways and mechanism in FeS2/PS system
The intermediates of CIP degradation were analyzed by LC-MS (Fig. S7 and Table S4†). The result shows that quinolones may be degraded at the cyclopropyl, piperazine ring, quinolone ring, and F atomic structures.55,56 Fig. 7a shows three proposed degradation pathways according to the structure of intermediates. As shown in pathway-I, P1 (m/z = 330.1) was the product of substitution reaction, decarboxylation (–COOH) of quinolone ring further formed P2 (m/z = 285.2), and P3 (m/z = 260.1) was the product of piperazine ring destruction, and then degraded to CO2, H2O, and other small molecular products. In pathway-II, P4 (m/z = 292.1) was formed by SO4˙− and HO˙ oxidizing the cyclopropyl group of CIP, then the quinolone ring was decarboxylated and the piperazine ring was broken to form P5 (m/z = 178.1). P6 (m/z = 170.1) was the product of the continued breakage of the quinolone ring, followed by decarboxylation and deamination to form P7 (m/z = 127.1) and P8 (m/z = 112.1). Finally, it was degraded to form small molecular products such as CO2 and H2O. As shown in pathway-III, the piperazine ring of CIP was de-ethylation to obtain P9 (m/z = 306.1), then the piperazine ring continued to be broken to form P10 (m/z = 263.1), then decarboxylation of the quinolone ring to form P11 (m/z = 219.1), and further decyclopropyl to give P5. The rest of the degradation process was as in pathway-II. Based on these results, Fig. 7b shows the presumed degradation mechanism of CIP in the FeS2/PS system. | ||
| Fig. 7 Proposed degradation of CIP pathways (a) and mechanism (b) in FeS2/PS system. | ||
4. Conclusion
This study showed that CIP could be removed effectively in FeS2/PS system. Both SO4˙− and HO˙ were the main contributors to the degradation of CIP. Furthermore, FeS2/PS system could effectively degrade CIP in a wide pH range, and acidification (pH = 2) could eliminate the inhibition of CIP degradation in tap water and river water. Besides, reaction tailwater could be used to adjust the pH of untreated CIP in different water substrates to 3, which could promote the degradation of CIP in FeS2/PS system. After 3 cycles of pH adjustment of the acidic tailwater after the reaction, the degradation of CIP was accelerated and significant. The mechanism of the degradation of CIP in FeS2/PS system was that FeS2 activated PS to generate Fe(III) and SO4˙− to degrade CIP. The sulfide in FeS2 reduced Fe(III), thus achieving the Fe(III)/Fe(II) cycle and continuously and effectively activating PS to degrade CIP. This work might bring valuable insights into the treatment of antibiotic wastewater by FeS2/PS system.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shanxi Province, China (No. 202103021224130, No. 202103021224139); the Youth Foundation of Shanxi Province, China (Grant No. 201901D211353). We thank the staff of the School of Resources and Environment, Shanxi Agricultural University, and the State Key Laboratory of Coal Conversion, Institute of coal chemistry, Chinese Academy of Sciences, Taiyuan. We thank the anonymous reviewers and editors for their valuable suggestions and comments on the revision and improvement of this paper.References
- Y. Hu, N. Habibul, Y.-Y. Hu, F.-L. Meng and G.-P. Sheng, Sci. Total Environ., 2021, 771, 144787 CrossRef CAS PubMed.
- J. Feng, L. Wang, X. Ran, B. Xiao, L. Lei, J. Zhu, R. Li, X. Xi and G. Feng, Environ. Technol. Innov., 2022, 28, 102785 CrossRef CAS.
- Y. Huang, L.-c. Nengzi, X. Zhang, J. Gou, Y. Gao, G. Zhu, Q. Cheng and X. Cheng, Chem. Eng. J., 2020, 388, 124274 CrossRef CAS.
- Z. Liu, J. Wan, Y. Ma and Y. Wang, Chemosphere, 2021, 273, 129747 CrossRef CAS PubMed.
- P. V. F. d. Sousa, A. F. d. Oliveira, A. A. d. Silva, B. G. Vaz and R. P. Lopes, Chem. Pap., 2018, 73, 249–260 CrossRef.
- R. Yu, J. Zhao, Z. Zhao and F. Cui, J. Hazard. Mater., 2020, 390, 121998 CrossRef CAS PubMed.
- Z. L. Li, R. Cheng, F. Chen, X. Q. Lin, X. J. Yao, B. Liang, C. Huang, K. Sun and A. J. Wang, J. Hazard. Mater., 2021, 405, 124366 CrossRef CAS PubMed.
- J. Ma, Y. Xiong, X. Dai and F. Yu, Chem. Eng. J., 2020, 380, 122387 CrossRef CAS.
- M. Salari, M. R. Nikoo, A. Al-Mamun, G. R. Rakhshandehroo and M. G. Mooselu, J. Environ. Manage., 2022, 317, 115469 CrossRef CAS PubMed.
- D. Cheng, H. Liu, Y. E, F. Liu, H. Lin and X. Liu, Sci. Total Environ., 2021, 773, 145102 CrossRef CAS PubMed.
- D. Cheng, Y. Feng, Y. Liu, J. Li, J. Xue and Z. Li, Sci. Total Environ., 2018, 634, 1148–1156 CrossRef CAS PubMed.
- S. K. Kuila, D. K. Gorai, B. Gupta, A. K. Gupta, C. S. Tiwary and T. K. Kundu, Chemosphere, 2021, 268, 128780 CrossRef CAS PubMed.
- H. Tang, Z. Dai, X. Xie, Z. Wen and R. Chen, Chem. Eng. J., 2019, 356, 472–482 CrossRef CAS.
- J. Wang and R. Zhuan, Sci. Total Environ., 2020, 701, 135023 CrossRef CAS PubMed.
- S. Rahdar, A. Rahdar, C. A. Igwegbe, F. Moghaddam and S. Ahmadi, Desalin. Water Treat., 2019, 141, 386–393 CrossRef CAS.
- S. I. Abou-Elela and M. El-Khateeb, J. Ind. Pollut. Control, 2015, 1, 1–5 Search PubMed.
- J. Du, Y. Wang, F. Faheem, T. Xu, H. Zheng and J. Bao, RSC Adv., 2019, 9, 20323–20331 RSC.
- J. Tang and J. Wang, RSC Adv., 2017, 7, 50829–50837 RSC.
- P. Shao, J. Tian, F. Yang, X. Duan, S. Gao, W. Shi, X. Luo, F. Cui, S. Luo and S. Wang, Adv. Funct. Mater., 2018, 28, 1705295 CrossRef.
- S. Al Hakim, A. Baalbaki, O. Tantawi and A. Ghauch, RSC Adv., 2019, 9, 33472–33485 RSC.
- H. Shi, G. Zhou, Y. Liu, Y. Fu, H. Wang and P. Wu, RSC Adv., 2019, 9, 31370–31377 RSC.
- Y.-R. Huang, Y. Kong, H.-Z. Li and X.-M. Wei, Environ. Technol. Innov., 2020, 18, 100780 CrossRef.
- L. Yang, J. Xue, L. He, L. Wu, Y. Ma, H. Chen, H. Li, P. Peng and Z. Zhang, Chem. Eng. J., 2019, 378, 122146 CrossRef CAS.
- R. Garcia-Cervilla, A. Santos, A. Romero and D. Lorenzo, Sci. Total Environ., 2021, 751, 141782 CrossRef CAS PubMed.
- Z.-H. Diao, J.-C. Jin, M.-Y. Zou, H. Liu, J.-Q. Qin, X.-H. Zhou, W. Qian, P.-R. Guo, L.-J. Kong and W. Chu, Sep. Purif. Technol., 2021, 278, 119620 CrossRef.
- E. Aseman-Bashiz and H. Sayyaf, Chemosphere, 2021, 274, 129772 CrossRef CAS PubMed.
- T. Li, P. Zhu, D. Wang, Z. Zhang and L. Zhou, Environ. Res., 2022, 209, 112830 CrossRef CAS PubMed.
- G. Gou, S. Kang, H. Zhao, C. Liu, N. Li, B. Lai and J. Li, Sep. Purif. Technol., 2022, 290, 120828 CrossRef CAS.
- C. Lai, X. Shi, L. Li, M. Cheng, X. Liu, S. Liu, B. Li, H. Yi, L. Qin, M. Zhang and N. An, Sci. Total Environ., 2021, 775, 145850 CrossRef CAS PubMed.
- H. Dong, B. Wang, L. Li, Y. Wang, Q. Ning, R. Tian, R. Li, J. Chen and Q. Xie, Sep. Purif. Technol., 2019, 218, 113–119 CrossRef CAS.
- J. Ye, Y. Zang, Q. Wang, Y. Zhang, D. Sun, L. Zhang, G. Wang, X. Zheng and J. Zhu, J. Energy Chem., 2021, 56, 283–289 CrossRef CAS.
- Y. Zhang, H. P. Tran, X. Du, I. Hussain, S. Huang, S. Zhou and W. Wen, Chem. Eng. J., 2017, 308, 1112–1119 CrossRef CAS.
- W. Lian, X. Yi, K. Huang, T. Tang, R. Wang, X. Tao, Z. Zheng, Z. Dang, H. Yin and G. Lu, Ecotoxicol. Environ. Saf., 2019, 174, 667–674 CrossRef CAS PubMed.
- P. He, J. Zhu, Y. Chen, F. Chen, J. Zhu, M. Liu, K. Zhang and M. Gan, Chem. Eng. J., 2021, 406, 126758 CrossRef CAS.
- Z. H. Diao and W. Chu, Sci. Total Environ., 2021, 754, 142155 CrossRef CAS PubMed.
- W. Song, J. Li, Z. Wang, C. Fu, X. Zhang, J. Feng, Z. Xu and Q. Song, Sci. Total Environ., 2020, 699, 134258 CrossRef CAS PubMed.
- X. Wang, Y. Wang, N. Chen, Y. Shi and L. Zhang, Chemosphere, 2020, 244, 125568 CrossRef CAS PubMed.
- Z.-H. Diao, Q. Wei, P.-R. Guo, L.-J. Kong and S.-Y. Pu, Chem. Eng. J., 2018, 349, 683–693 CrossRef CAS.
- Y. Chen, Y. Liu, L. Zhang, P. Xie, Z. Wang, A. Zhou, Z. Fang and J. Ma, J. Hazard. Mater., 2018, 353, 18–25 CrossRef CAS PubMed.
- X. Chang, T. Lin, W. Chen, H. Xu, H. Tao, Y. Wu, Q. Zhang and S. Yao, Sci. Total Environ., 2020, 737, 139711 CrossRef CAS PubMed.
- S. Guo, Q. Wang, C. Luo, J. Yao, Z. Qiu and Q. Li, Chemosphere, 2020, 243, 125390 CrossRef CAS PubMed.
- S. Xing, W. Li, B. Liu, Y. Wu and Y. Gao, Chem. Eng. J., 2020, 382, 122837 CrossRef CAS.
- R. Yin, L. Hu, D. Xia, J. Yang, C. He, Y. Liao, Q. Zhang and J. He, Catal. Today, 2020, 358, 294–302 CrossRef CAS.
- X. Shi, Y. Li, Z. Zhang, L. Sun and Y. Peng, Chem. Eng. J., 2019, 372, 1113–1121 CrossRef CAS.
- Y. Zhou, X. Wang, C. Zhu, D. D. Dionysiou, G. Zhao, G. Fang and D. Zhou, Water Res., 2018, 142, 208–216 CrossRef CAS PubMed.
- H. Luo, C. Ni, C. Zhang, W. Wang, Y. Yang, W. Xiong, M. Cheng, C. Zhou, Y. Zhou, S. Tian, Q. Lin, G. Fang, Z. Zeng and G. Zeng, J. Colloid Interface Sci., 2022, 610, 221–233 CrossRef CAS PubMed.
- H. Liu, F. Liu, J. Zhang, J. Zhou, W. Bi, J. Qin, Q. Hou, Y. Ni, S. Xu and C. Yang, Water Sci. Technol., 2022, 85, 2912–2927 CrossRef CAS PubMed.
- S. Gao, Y. Liu, J. Zhu, Y. Wang, X. Han, X. Xia and X. Zhao, Environ. Sci.: Water Res. Technol., 2021, 7, 1430–1442 RSC.
- Y. Gao, Z. Li, Z. Fu, H. Zhang, G. Wang and H. Zhou, Sep. Purif. Technol., 2021, 262, 118336 CrossRef CAS.
- H. Xu and Y. Sheng, Chem. Eng. J., 2021, 414, 128823 CrossRef CAS.
- Y. Chen, S. Xu, S. Zhu, R. J. Jacob, G. Pastel, Y. Wang, Y. Li, J. Dai, F. Chen, H. Xie, B. Liu, Y. Yao, L. G. Salamanca-Riba, M. R. Zachariah, T. Li and L. Hu, Nano Res., 2019, 12, 2259–2267 CrossRef CAS.
- W. Liu, Y. Wang, Z. Ai and L. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28534–28544 CrossRef CAS PubMed.
- H. Alamgholiloo, N. N. Pesyan, R. Mohammadi, S. Rostamnia and M. Shokouhimehr, J. Environ. Chem. Eng., 2021, 9, 105486 CrossRef CAS.
- Y. Li, Y. Shi, D. Huang, Y. Wu and W. Dong, J. Hazard. Mater., 2021, 413, 125420 CrossRef CAS PubMed.
- Z. Chen, W. Lai, Y. Xu, G. Xie, W. Hou, P. Zhanchang, C. Kuang and Y. Li, J. Hazard. Mater., 2021, 405, 124262 CrossRef CAS PubMed.
- N. S. Shah, J. A. Khan, M. Sayed, Z. Ul Haq Khan, H. S. Ali, B. Murtaza, H. M. Khan, M. Imran and N. Muhammad, Chem. Eng. J., 2019, 356, 199–209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05412d |
| This journal is © The Royal Society of Chemistry 2022 |