
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanostructured IrOx supported on N-doped TiO2 as an efficient electrocatalyst towards acidic oxygen evolution reaction†
Guoqiang Li *a,
Hongrui Jiaa,
Huan Liub,
Xin Yanga and
Meng-Chang Lina
*a,
Hongrui Jiaa,
Huan Liub,
Xin Yanga and
Meng-Chang Lina
aCollege of Energy Storage Technology, Shandong University of Science and Technology, Qingdao 266590, China. E-mail: ligq@sdust.edu.cn
bQingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
First published on 11th October 2022
Abstract
Reducing the Ir consumption without compromising the catalytic performance for the oxygen evolution reaction (OER) is highly paramount to promote the extensive development of the environmentally-friendly solid polymer electrolyte water electrolysis (SPEWE) system. Herein, TiO2 is doped with N through facile NH3 gas treatment and innovatively employed to support IrOx nanoparticles towards acidic OER. N-doping action not only dramatically boosts the electrical conductivity and dispersing/anchoring effects of TiO2, but also effectively improves the electron-transfer procedure. As a result, the IrOx/N–TiO2 electrocatalyst exhibits prominent catalyst utilization, catalytic activity and stability. Specifically, the overpotential required to deliver 10 mA cm−2 is only 270 mV, and the mass activity climbs to 278.7 A gIr−1 @ 1.55 VRHE. Moreover, the single cell voltage is only 1.761 V @ 2.0 A cm−2 when adopting IrOx/N–TiO2 as the anode catalyst, which is 44 mV lower than that of the commercial IrO2 counterpart.
Introduction
Energy shortage and environmental issues urgently call for clean and sustainable fuels; due to the high energy density, extensive sources and environmental friendliness, hydrogen (H2) is regarded as a promising alternative to traditional fossil fuels.1–3 At present, H2 is mainly generated from coal gasification and methane-steam reforming; irreconcilable conflict between the hydrogen production and environmental destruction is bound to exist.3,4 Alternatively, photocatalytic hydrogen production is also very popular, due to the endless solar energy and environmental friendliness.5–8 More notably, water electrolysis powered by renewable electricity is regarded as an extremely promising technique, especially the solid polymer electrolyte water electrolysis (SPEWE) with prominent advantages of high H2 purity, high current density, low ohmic loss, compact system design and rapid system response, has attracted widespread research focus.9–13 The oxygen evolution reaction (OER) occurring on the anode side of a water electrolyzer dominates the overall efficiency because of the sluggish kinetics.12,14–16 At present, Ir and Ru oxides are still the most representative OER electrocatalysts, especially the Ir oxide with more prominent practical potentiality, regrettably, extremely low reserve of Ir element in the earth's crust (0.001 ppm) impedes its widespread application.17–21 Hence, reducing the Ir consumption without compromising the OER catalytic performance is highly paramount.Benefiting from the comprehensive support effect including effective dispersing, anchoring, electronic structure modulation and coordination, support materials have achieved widespread application in several typical electrocatalytic systems, such as the hydrogen oxidation/evolution reactions (HOR/HER),22–26 oxygen reduction reaction/OER (ORR/OER),27–29 CO2 reduction reaction (CO2RR),30,31 N2 reduction reaction (N2RR),32,33 etc. Among them, high anodic potential and harsh acidic circumstances make the widely used carbon-based materials unstable as the support towards acidic OER,34,35 seeking for suitable corrosion-resistant support candidate is urgently yet highly challenging.
Up to now, acid-resistant metal compounds based on Ti and Sn elements have been widely researched and confirmed the effectiveness as support materials on catalyzing OER. It is well-known that TiO2 and SnO2 are semiconductors with poor electrical conductivity, active species supported on them are strictly limited to participate the reaction. There are two mainstream strategies to overcome this issue, nonstoichiometric and heteroatom-doping treatments. The electrical conductivity of TiO2 that generally below 10−4 S cm−1 can dramatically increase to ultrahigh 103 S cm−1 of nonstoichiometric Ti4O7.36–38 Gago et al. synthesized Ir/Ti4O7 catalyst with Ti4O7 as the support, the mass activity was as high as 4.2 A gIr−1 @ 1.48 V, remarkably larger than that of Ir-black (1.6 A gIr−1), indicating the improved Ir metal utilization.38 Hu et al. applied Nb-doped TiO2 to support IrO2 nanoparticles, the mass activity significantly increased from 198 A gIrO2−1 of unsupported IrO2 to 471 A gIrO2−1 of supported IrO2/Nb–TiO2 at 1.6 V, accompanied with enhanced stability.39 For SnO2, Sb-doped SnO2 (ATO) is the most common improver. Böhm et al. prepared macroporous ATO with the electrical conductivity of 3.6 S cm−1, obviously higher than that of SnO2 (3.8 × 10−4 S cm−1), the corresponding IrOx/ATO catalyst exhibited the mass activity of 63 A gIr−1 @ 1.53 V and enhanced stability.40 Similarly, Strasser et al. also confirmed the promoting effect of ATO support on the Ir nanodendrites.41 Expect for the metal oxide, metal nitride such as TiN also displays promising support effect. Xing et al. revealed the high electrical conductivity (28.18 S cm−1) of TiN, and its effectiveness of dispersing, anchoring and electronic structure modulation on IrO2@Ir nanoparticles.28 Afterwards, Hu et al. verified that TiN significantly improved the IrO2 utilization, the mass activity climbed to 874 A gIrO2−1 @ 1.6 V.42
As another style of heteroatom-doping treatment, non-metal such as N, B, F, P can also be doped into TiO2 and generally applied in various photocatalytic systems. Saini et al. synthesized N–TiO2 and B–TiO2 through a sol–gel route and verified the effectiveness on the photocatalytic degradation of emerging micro-pollutants.43 Xu et al. successfully prepared F–TiO2, which showed enhanced photocatalytic activity for the phenol degradation.44 Chevalier et al. also prepared F–TiO2 by an emulsions method and exhibited remarkably higher efficiency for the photocatalytic degradation of nitrobenzene.45 O'Shea et al. co-doped TiO2 with N, F and P, the photocatalytic activity of this modified TiO2 to produce hydroxyl radicals under ultra violet (UV) and visible light irradiation can be dramatically improved.46 These cases mentioned-above mainly benefit from the decreased band-gap and the red-shift of light absorption profile to the visible region of TiO2 through non-metal element doping.
Herein, we successfully doped TiO2 with N element and synthesized IrOx/N–TiO2 electrocatalyst to catalyze OER. N-doping procedure dramatically increases the electrical conductivity of TiO2, and subsequently improves the dispersing/stabilizing effects on IrOx, weakens the oxidative dissolution of Ir species. Satisfactory, enhanced catalyst utilization and catalytic performance are simultaneously realized.
Results and discussion
Fig. 1a illustrates the procedure to synthesize N–TiO2 support through NH3 treatment and IrOx/N–TiO2 catalyst through ethylene glycol (EG) refluxing reduction method, respectively. Interestingly, the as-obtained N–TiO2 appears light-blue color, apparently different from TiO2 with pure-white, implying the band gap change due to the N-doping treatment. As shown in Fig. 1b, the common X-ray diffraction (XRD) peaks of N–TiO2 and TiO2 ascribe to the anatase phase of TiO2.47 The apparently sharper diffraction peaks of N–TiO2 indicate the enlarged crystal size through the heating treatment. Notably in the inset of Fig. 1b, the N–TiO2 (101) crystal diffraction facet slightly moves to a lower diffraction angle compared with TiO2, demonstrating the successful incorporation of N into the TiO2 lattice. Furthermore, IrOx/N–TiO2, IrOx/TiO2 and IrOx show common crystal diffraction facets of (111), (200), (220) and (311) (Fig. 1c), ascribing to the cubic phase of Ir.41,48 Meanwhile, diffraction peaks of anatase phase TiO2 are also apparently recognized for the supported catalysts.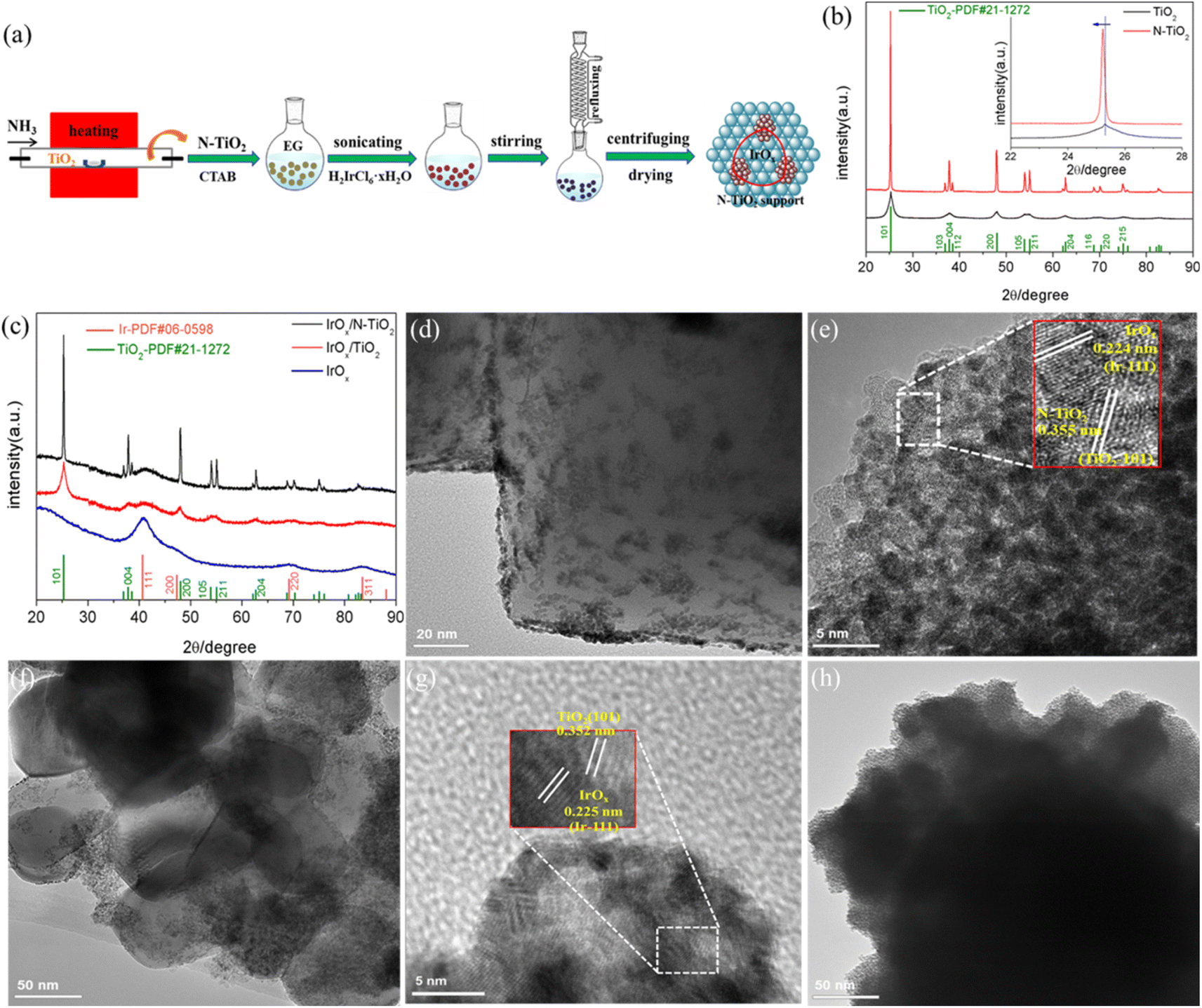 | ||
| Fig. 1 (a) Synthesis route of the N–TiO2 support and IrOx/N–TiO2 catalyst. XRD patterns of (b) TiO2 and N–TiO2 (inset: XRD patterns within 22–28°), and (c) IrOx/N–TiO2, IrOx/TiO2 and IrOx. TEM images of (d) IrOx/N–TiO2, (f) IrOx/TiO2 and (h) IrOx. HRTEM images of (e) IrOx/N–TiO2 and (g) IrOx/TiO2. | ||
The morphology characteristics of these as-prepared samples were revealed by transmission electron microscopy (TEM). As shown in Fig. S1 (ESI†), N peak appears distinctly in the energy-dispersive X-ray spectroscopy (EDX), and the atomic/weight contents of N element are 3.60 at%/1.89 wt%, respectively, again verifies the effective doping procedure. TEM image in Fig. 1d shows the uniform dispersion of IrOx nanoparticles on the N–TiO2 support, while the relatively nonuniform dispersion appears when using TiO2 support (Fig. 1f). The keys lie in the formation of Ti3+ species and the oxygen vacancies due to the N-doping treatment.49,50 Ti3+ with reductibility can easily adsorb the IrCl62− precursor ion with high-valent Ir4+ species during the synthesis process, and oxygen vacancies can act as the anchoring sites for effectively adsorbing IrCl62−. Therefore, improved dispersion of IrOx on N–TiO2 is acquired. Furthermore, unsupported IrOx catalyst displays more noticeable particles aggregation (Fig. 1h). IrOx/N–TiO2 and IrOx/TiO2 were further characterized by the high-resolution TEM (HRTEM) (Fig. 1e and g), for the supported IrOx nanoparticles, the interplanar spacings of 0.224 and 0.225 nm are all slightly larger than that of the standard Ir (111) crystal facet with 0.221 nm (JCPDS no. 06-0598), implying that the formation of surface oxidation state can enlarge the interplanar spacing of metallic Ir to some extent. Similarly, Luo et al. has reported that the interplanar spacing of Ir (111) crystal facet increased to 0.225 nm when alloyed with W metal.51 For N–TiO2, the interplanar spacing of 0.355 nm is slightly larger than that of the standard TiO2 (101) crystal facet with 0.352 nm (JCPDS No. 21-1272), mainly resulted from the partly substitution of O2− ion by N3− ion with larger radius. By contrast, IrOx/TiO2 displays standard TiO2 (101) crystal facet. Moreover, the adjacent IrOx and N–TiO2 (TiO2) nanoparticles confirm the supported structure. The HAADF-STEM and corresponding elemental mapping images (Fig. S2, ESI†) of IrOx/N–TiO2 reveal that Ir, N, O and Ti element are uniformly distributed throughout the IrOx/N–TiO2 nanoparticles.
X-ray photoelectron spectroscopy (XPS) was applied to investigate the surface structure of the samples. The binding energy of all peaks has been calibrated with the C 1s orbit of 284.6 eV. XPS survey spectrum for N–TiO2 (Fig. S3a, ESI†) depicts the typical Ti 2p, O 1s/KLL and N 1s orbits, and the atomic content of N is 3.79 at%, almost in accordance with the EDX result. After supporting IrOx, Ir 4p/4d/4f orbits can also be clearly recognized (Fig. S3b, ESI†). The N 1s spectrum for IrOx/N–TiO2 was deconvoluted and obtained only one prominent peak (Fig. 2a). This peak at 396.70 eV is assigned to the substitutional N in Ti–N bonds, indicating that the N atoms enter into the TiO2 lattice through replacing the O atoms.52,53 Compared with IrOx, the Ir 4f spectra for IrOx/N–TiO2 and IrOx/TiO2 all shifted negatively to lower electronic binding energy direction (Fig. 2b and S3c, ESI†), indicating the electron-transfer process from N–TiO2 and TiO2 to IrOx. Especially the IrOx/N–TiO2 with the Ir 4f7/2 orbit locates at 61.10 eV, lower by 0.16 and 0.24 eV than those of IrOx/TiO2 and IrOx (Table S1, ESI†), respectively. Next, the relative contents of Ir0 and Ir4+ species were obtained through deconvoluting the Ir 4f spectra, and shown in Table S1 (ESI†). For IrOx/N–TiO2, the content of Ir0 species is the highest one with 66.1% (64.1% for IrOx/TiO2 and 63.2% for IrOx), mainly due to the more significant electron-transfer effect. In addition, the contents of Ir4+ species all surpass 30%, confirming the formation of IrOx oxide component on the catalyst surface.
 | ||
| Fig. 2 (a) High-resolution XPS spectrum of the deconvoluted N 1s for IrOx/N–TiO2. (b) The comparison of the XPS spectra of Ir 4f for IrOx/N–TiO2, IrOx/TiO2 and IrOx. High-resolution XPS spectra of the deconvoluted Ir 4f for (c) IrOx/N–TiO2 and (d) IrOx. | ||
The electrical conductivity of N–TiO2 was measured to reach 0.13 S cm−1, apparently higher than that of TiO2 (7.9 × 10−6 S cm−1). The significant effect of the N-doping treatment on improving the electrical conductivity of TiO2 is undoubtable, which is beneficial for driving the supported active sites to participate the reaction. The main reason should be that strongly correlated interaction emerges between N and Ti due to the N-doping treatment, which will lead the electrons in the 3d orbit of Ti move to the 2p orbit of N. Consequently, conduction band becomes lower, band gap becomes narrower and the electrical conductivity is bound to improve.50,54 Cyclic voltammogram (CV) tests at high scanning rate of 300 mV s−1 were firstly performed in N2-saturated 0.5 M H2SO4 solution (Fig. 3a), subsequently, the outer charge (Qouter) that assesses the accessible active surface and directly correlates with the OER activity was obtained through integrating the CV curve between 0.70 and 1.40 V.28,55,56 As shown in the inset of Fig. 3a, the Qouter of IrOx/N–TiO2 is 8.93 mC, about 2.83 times of IrOx/TiO2 (3.16 mC) and 1.28 times of IrOx (6.97 mC), respectively. The improvement mainly resulted from the enhanced dispersing effect and electrical conductivity of the N–TiO2 support. Furthermore, although the better dispersion of IrOx nanoparticles on TiO2 than the unsupported IrOx, the ultralow electrical conductivity of TiO2 can strictly limit the effective participation of the supported IrOx in OER, hence, the Qouter is apparently lower than that of the IrOx catalyst. During the linear sweep voltammetry (LSV) tests (Fig. 3b), the overpotentials of the catalysts to deliver current density of 10 mA cm−2 follow this order: IrOx/N–TiO2 (270 mV) < IrOx (286 mV) < IrO2(CM) (307 mV) < IrOx/TiO2 (313 mV). Furthermore, IrOx/N–TiO2 shows competitively catalytic effect compared with those reported supported Ir-based catalysts (Table S2, ESI†). As for the pure supports, N–TiO2 and TiO2 all perform nearly negligible catalytic activity for OER, although N–TiO2 performs slightly better. The corresponding mass activity curves are shown in Fig. 3c, IrOx/N–TiO2 (278.7 A gIr−1) exhibits 2.70 and 2.50 times higher mass activity than those of IrOx/TiO2 (75.4 A gIr−1) and IrOx (79.7 A gIr−1) at 1.55 V, respectively. Meanwhile, it outperforms the majority of the ever reported catalysts with similar types (Table S3, ESI†). Therefore, N–TiO2 significantly improves the catalyst utilization of IrOx for OER.
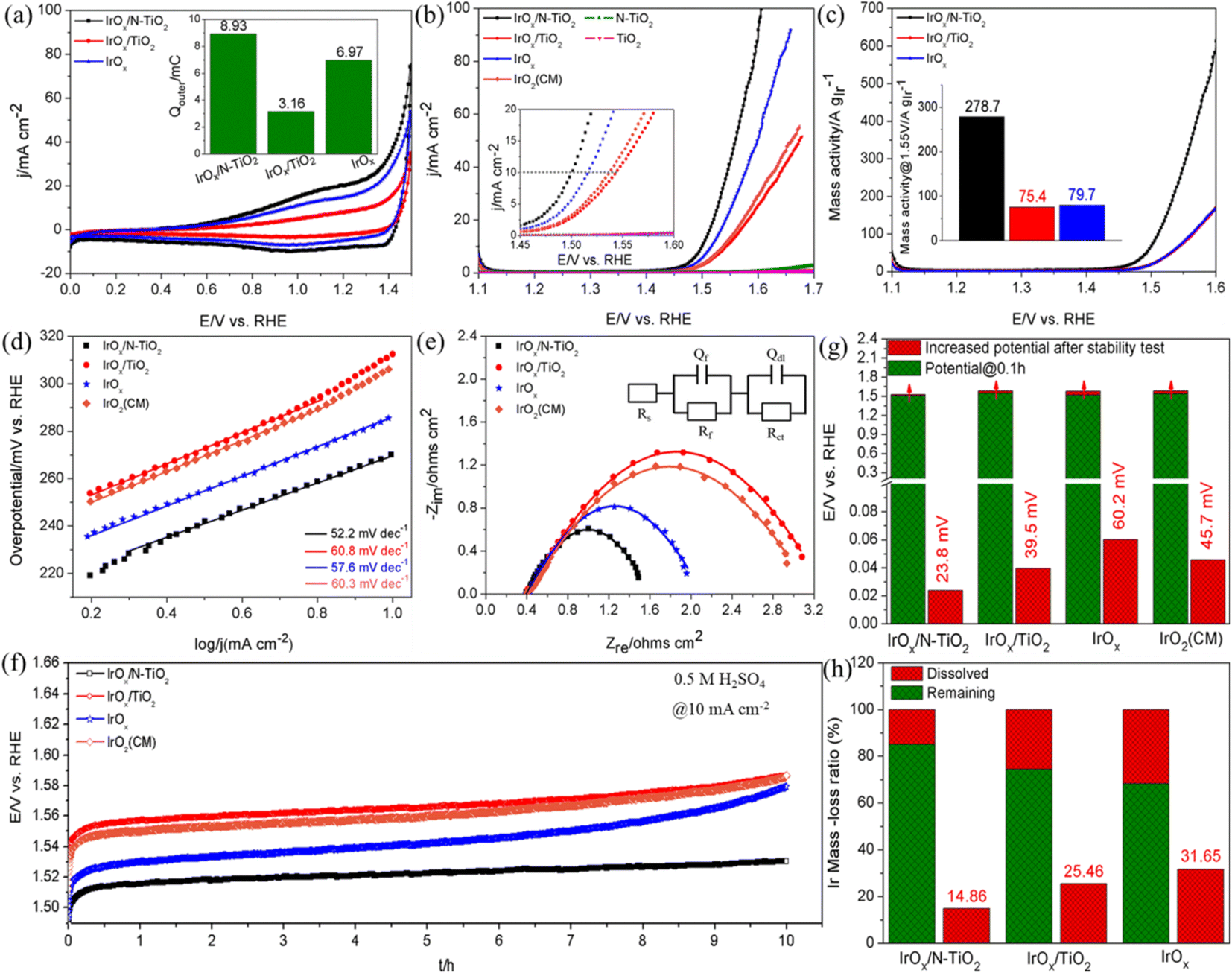 | ||
| Fig. 3 (a) CV curves at the scanning rate of 300 mV s−1 in N2-saturated 0.5 M H2SO4 solution (inset: histograms of outer charge). (b) LSV curves at the scanning rate of 5 mV s−1 (inset: LSV curves of the current density within 0 and 20 mA cm−2). (c) Mass activity obtained from the LSV tests. (d) Tafel plots obtained from the LSV curves in (b). (e) Nyquist plots for OER at 1.55 V in a sweeping frequency range from 0.1 Hz to 10 kHz (inset: equivalent circuit). (f) Potential-time curves at the constant current density of 10 mA cm−2 for 10 h in N2-saturated 0.5 M H2SO4 solution. (g) Histograms of the potential change. (h) Histograms of the Ir mass-loss ratio after the stability tests. | ||
To evaluate the reaction kinetics, Tafel slopes extracted from the LSV curves were firstly investigated (Fig. 3d). IrOx/N–TiO2 possesses the smallest value of 52.2 mV dec−1, indicating the most favorable OER kinetics. Subsequently, electrochemical impedance spectroscopy (EIS) tests were performed, the corresponding Nyquist plots are depicted in Fig. 3e and an equivalent circuit (EC) was adopted to fit them. As a key component, Rct represents the charge transfer resistance.57 The Rct of IrOx/N–TiO2 is only 14.5 Ω, apparently smaller than those of IrOx/N–TiO2 (35.3 Ω), IrOx (20.6 Ω) and IrO2(CM) (35.3 Ω) (Table S4, ESI†), indicating the fastest charge transfer rate during the reaction.
Reaction stability was evaluated through galvanostatic mode, as shown in Fig. 3f, the potentials increase to varying degrees, indicating the degradation of catalytic activity. Even so, IrOx/N–TiO2 maintains favorable stability, the potential only increased by 23.8 mV after 10 h, outperforms other counterparts (39.5 mV for IrOx/TiO2, 60.2 mV for IrOx, 45.7 mV for IrO2(CM)) (Fig. 3g). Subsequently, LSV tests were applied again to investigate the maintained catalytic activity after the stability tests (Fig. S4, ESI†). IrOx/N–TiO2 still shows outstanding performance, with the potential only increased by 21.7 mV at 10 mA cm−2. Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was also carried to investigate the electrochemically dissolved Ir species (Fig. 3h). Thanks to the transferred electrons from N–TiO2, the Ir mass-loss ratio of supported IrOx is only 14.86%, by contrast, the values of IrOx/TiO2 and IrOx reach as high as 25.46 and 31.65%, respectively. Hence, N–TiO2 can weaken the oxidative dissolution of Ir species.
A two-electrode cell driving overall water splitting was assembled by employing IrOx/N–TiO2, IrOx and IrO2(CM) as the anode catalysts, Pt/C(CM) as the cathode catalyst. Polarization curves are shown in Fig. 4a, the IrOx/N–TiO2||Pt/C(CM)-based cell performs the lowest voltage of 1.785 V to deliver 100 mA cm−2, meaning the total overpotential of only 555 mV. Compared with IrOx||Pt/C(CM) and IrO2(CM)||Pt/C(CM), 46 and 118 mV can be saved at 100 mA cm−2, respectively. Furthermore, IrOx/N–TiO2||Pt/C(CM) exhibits favorable stability during the 20 h galvanostatic tests at constant 10 mA cm−2 (Fig. 4b). The structural stability was confirmed by TEM characterization and shown in Fig. 4c, well dispersion of IrOx nanoparticles on the whole verifies the enhanced anchoring effect of N–TiO2.
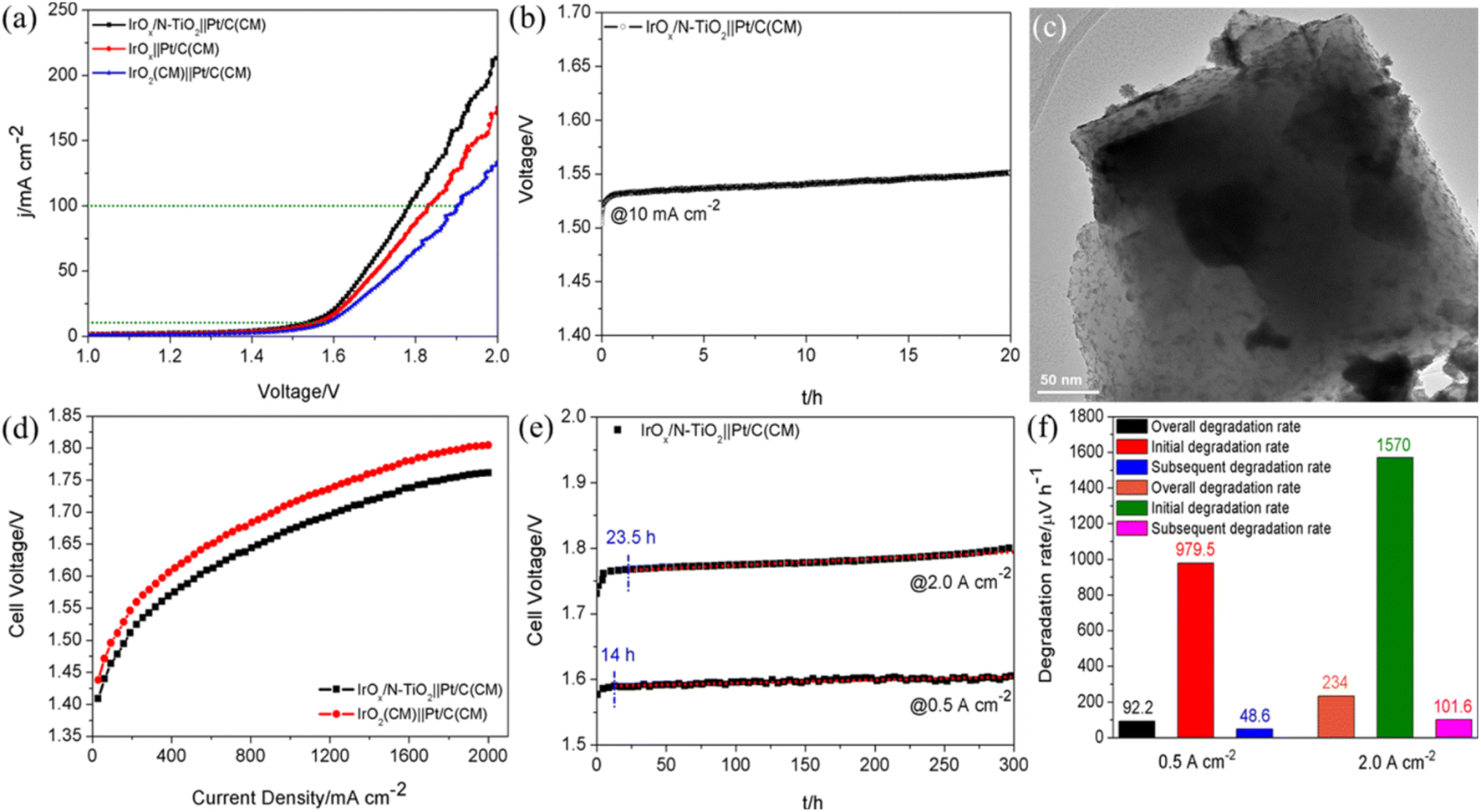 | ||
| Fig. 4 (a) Polarization curves for two-electrode overall water splitting with IrOx/N–TiO2, IrOx or IrO2(CM) as the anode catalyst (loading of 0.3 mg cm−2), and Pt/C(CM) as the cathode catalyst (loading of 0.1 mg cm−2) at the scanning rate of 5 mV s−1. (b) Voltage–time curve of IrOx/N–TiO2||Pt/C(CM) at the constant current density of 10 mA cm−2 for 20 h. (c) TEM image of IrOx/N–TiO2 after the stability test. (d) Steady-state polarization curves of the SPEWE single cells with IrOx/N–TiO2 or IrO2(CM) as the anode catalyst (loading of 2.0 mg cm−2), and Pt/C(CM) as the cathode catalyst (loading of 1.0 mg cm−2). (e) Stability tests of IrOx/N–TiO2||Pt/C(CM)-based cell operated at the constant current densities of 0.5 and 2.0 A cm−2 for 300 h. The subsequent degradation rates are evaluated by means of a linear regression fit (dashed black lines). (f) Histograms of the degradation rate within the stability tests at 0.5 and 2.0 A cm−2. | ||
To scrutinize the practical application potential of IrOx/N–TiO2, proton exchange membrane water electrolysis (PEMWE) single cell was assembled with Nafion® 115 proton exchange membrane as the SPE, IrOx/N–TiO2 as the anode catalyst and Pt/C(CM) as the cathode catalyst. By contrast, the IrO2(CM)||Pt/C(CM)-based cell was also assembled. For IrOx/N–TiO2||Pt/C(CM), the required voltages to deliver 1 and 2 A cm−2 are only 1.672 and 1.761 V, respectively, lower by 41|44 mV than those of IrO2(CM)||Pt/C(CM) (Fig. 4d). Finally, long-term stability tests of IrOx/N–TiO2||Pt/C(CM)-based cell were performed at constant 0.5 and 2.0 A cm−2 for 300 h, respectively (Fig. 4e). Significant degradations at the beginning of the stability tests (0–14 h for 0.5 A cm−2; 0–23.5 h for 2.0 A cm−2) are observed, which could be ascribed to two main reasons, the mass transfer polarization and the modification of the oxidation state at the anode catalyst surface.58 The overall, initial and subsequent degradation rates are calculated and shown in Fig. 4f. Apparently, the stability performance at 2.0 A cm−2 (overall/initial/subsequent degradation rates of 234/1570/101.6 μV h−1) is inferior to that at 0.5 A cm−2 (overall/initial/subsequent degradation rates of 92.2/979.5/48.6 μV h−1), mainly resulted from the more seriously oxidative dissolution of Ir species and the damage of other cell components under harsher environment.
However, compared with these reported PEMWE cells employing Ir-based anode catalysts,59–65 this IrOx/N–TiO2||Pt/C(CM)-based cell displays relatively mediocre level for the stability performance. Therefore, systematic optimization of the cell fabrication should be the further research emphasis for enhancing the practical cell performance, especially the operating stability.
Conclusions
In summary, supported IrOx/N–TiO2 catalyst was synthesized to effectively catalyze OER. N–TiO2 exhibits comprehensive support effect, firstly, the remarkably high electrical conductivity of 0.13 S cm−1 is beneficial for driving the active sites to adequately participate the reaction. Secondly, N–TiO2 can effectively disperse and anchor the IrOx nanoparticles. Thirdly, prominent electron-transfer from N–TiO2 to IrOx significantly weakens the electrochemically oxidative dissolution of Ir species. Consequently, IrOx/N–TiO2 performs enhanced catalytic performance, the required overpotential to drive 10 mA cm−2 is only 270 mV, accompanied with improved catalyst utilization, favorable reaction kinetics and enhanced stability. Moreover, the single cell employing IrOx/N–TiO2 performs the voltages of only 1.672 V @ 1 A cm−2 and 1.761 V @ 2 A cm−2.Author contributions
G. L. conceived and coordinated the research. G. L. and H. J. conducted the catalyst preparation, physical characterization and electrochemical measurement. G. L., X. Y. and M-C. L. contributed to the analysis of experiment results. The manuscript was primarily written and modified by G. L. and H. L. All authors contributed to the discussion and manuscript review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Elite Program of Shandong University of Science and Technology (0104060540812), and the Qingdao Scientific and Technological Innovation High-Level Talents Project: Aluminum-Ion Power and Energy Storage Battery (17-2-1-1-zhc).Notes and references
- X. J. Zeng, D. R. Duan, X. F. Zhang, X. H. Li, K. Li, R. H. Yu and M. Moskovits, J. Mater. Chem. C, 2022, 10, 4140–4147 RSC.
- L. G. Li, P. T. Wang, Q. Shao and X. Q. Huang, Adv. Mater., 2022, 33, 2004243 CrossRef PubMed.
- S. Roy, Z. H. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. D. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS.
- X. X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- G. F. Liao, C. X. Li, X. Z. Li and B. Z. Fang, Cell Rep. Phys. Sci., 2021, 2, 100355 CrossRef CAS.
- G. F. Liao, C. X. Li, S.-Y. Liu, B. Z. Fang and H. M. Yang, Phys. Rep., 2022, 983, 1–41 CrossRef CAS.
- G. F. Liao, C. X. Li, S.-Y. Liu, B. Z. Fang and H. M. Yang, Trends Chem., 2022, 4, 111–127 CrossRef CAS.
- G. F. Liao, X. Y. Tao and B. Z. Fang, Matter, 2022, 5, 377–379 CrossRef CAS.
- Z. W. Lei, T. Y. Wang, B. T. Zhao, W. B. Cai, Y. Liu, S. H. Jiao, Q. Li, R. G. Cao and M. L. Liu, Adv. Energy Mater., 2020, 10, 2000478 CrossRef CAS.
- B. H. Zhou, R. J. Gao, J.-J. Zou and H. M. Yang, Small, 2022, 18, 2202336 CrossRef CAS PubMed.
- S. D. Ghadge, P. P. Patel, M. K. Datta, O. I. Velikohatnyi, R. Kuruba, P. M. Shanthi and P. N. Kumta, RSC Adv., 2017, 10, 17311–17324 RSC.
- Y. Z. Wen, P. N. Chen, L. Wang, S. Y. Li, Z. Y. Wang, J. Abed, X. N. Mao, Y. M. Min, C. D. Dinh, P. D. Luna, R. Huang, L. S. Zhang, J. Wang, L. P. Wang, R. J. Nielsen, H. H. Li, T. T. Zhuang, C. C. Ke, O. Voznyy, Y. F. Hu, Y. Y. Li, W. A. Goddard III, B. Zhang, H. S. Peng and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 6482–6490 CrossRef CAS PubMed.
- L. An, C. Wei, M. Lu, H. W. Liu, Y. B. Chen, G. G. Scherer, A. C. Fisher, P. X. Xi, Z. J. Xu and C. H. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS PubMed.
- Y. H. Zhao, M. H. Xi, Y. B. Qi, X. D. Sheng, P. F. Tian, Y. H. Zhu, X. L. Yang, C. Z. Li and H. L. Jiang, J. Energy Chem., 2022, 69, 330–337 CrossRef CAS.
- C. J. Liu, B. B. Sheng, Q. Zhou, D. F. Cao, H. H. Ding, S. M. Chen, P. J. Zhang, Y. J. Xia, X. J. Wu and L. Song, Nano Res., 2022, 15, 7008–7015 CrossRef CAS.
- Y. P. Liu, X. Liang, H. Chen, R. Q. Gao, L. Shi, L. Yang and X. X. Zou, Chin. J. Catal., 2021, 42, 1054–1077 CrossRef CAS.
- Z. J. Chen, X. G. Duan, W. Wei, S. B. Wang and B.-J. Ni, Nano Energy, 2020, 78, 105392 CrossRef CAS.
- Z. L. Fan, Y. J. Ji, Q. Shao, S. Z. Geng, W. X. Zhu, Y. Liu, F. Liao, Z. W. Hu, Y.-C. Chang, C.-W. Pao, Y. Y. Li, Z. H. Kang and M. W. Shao, Joule, 2021, 5, 3221–3234 CrossRef CAS.
- Y. B. Wang, S. Hou, R. P. Ma, J. D. Jiang, Z. P. Shi, C. P. Liu, J. J. Ge and W. Xing, ACS Sustainable Chem. Eng., 2021, 9, 10710–10716 CrossRef CAS.
- H. Chen, L. Shi, X. Liang, L. N. Wang, T. Asefa and X. X. Zou, Angew. Chem., Int. Ed., 2020, 59, 19654–19658 CrossRef CAS PubMed.
- L. F. Lu, H. Zheng, Y. X. Li, Y. H. Zhou and B. Z. Fang, Chem. Eng. J., 2023, 451, 138668 CrossRef CAS.
- G. H. Bae, D. H. Youn, S. Han and J. S. Lee, Carbon, 2013, 51, 274–281 CrossRef CAS.
- M. K. Kundu, T. Bhowmik, R. Mishra and S. Barman, ChemSusChem, 2018, 11, 2388–2401 CrossRef CAS PubMed.
- Y. H. Liu, Q. L. Wang, J. C. Zhang, J. Ding, Y. Q. Cheng, T. Wang, J. Li, F. X. Hu, H. B. Yang and B. Liu, Adv. Energy Mater., 2022, 12, 2200928 CrossRef CAS.
- Q. Yang, H. X. Liu, P. Yuan, Y. Jia, L. Z. Zhuang, H. W. Zhang, X. C. Yan, G. H. Liu, Y. F. Zhao, J. Z. Liu, S. Q. Wei, L. Song, Q. L. Wu, B. Q. Ge, L. Z. Zhang, K. Wang, X. Wang, C.-R. Chang and X. D. Yao, J. Am. Chem. Soc., 2022, 144, 2171–2178 CrossRef CAS PubMed.
- L. F. Lu, S. H. Zou and B. Z. Fang, ACS Catal., 2021, 11, 6020–6058 CrossRef CAS.
- J. Liu, M. G. Jiao, B. B. Mei, Y. X. Tong, Y. P. Li, M. B. Ruan, P. Song, G. Q. Sun, L. H. Jiang, Y. Wang, Z. Jiang, L. Gu, Z. Zhou and W. L. Xu, Angew. Chem., Int. Ed., 2019, 58, 1163–1167 CrossRef CAS.
- G. Q. Li, K. Li, L. Yang, J. F. Chang, R. P. Ma, Z. J. Wu, J. J. Ge, C. P. Liu and W. Xing, ACS Appl. Mater. Interfaces, 2018, 10, 38117–38124 CrossRef CAS PubMed.
- X. Z. Zheng, M. K. Qin, S. X. Ma, Y. Z. Chen, H. H. Ning, R. Yang, S. J. Mao and Y. Wang, Adv. Sci., 2022, 9, 2104636 CrossRef CAS PubMed.
- C. Lim, W. H. Lee, J. H. Won, Y.-J. Ko, S. Kim, B. K. Min, K.-Y. Lee, W. S. Jung and H.-S. Oh, Adv. Sustainable Syst., 2022, 6, 2200019 CrossRef.
- B. H. Zhang, Y. Z. Jiang, M. X. Gao, T. Y. Ma, W. P. Sun and H. G. Pan, Nano Energy, 2021, 80, 105504 CrossRef CAS.
- M. L. Yuan, H. H. Zhang, D. L. Gao, H. Y. He, Y. Sun, P. L. Hu, S. Dipazir, Q. G. Li, L. Zhou, S. W. Li, Z. J. Liu, J. H. Yang, Y. B. Xie, H. Zhao and G. J. Zhang, J. Mater. Chem. A, 2020, 8, 2691–2700 RSC.
- L. Shi, S. N. Bi, Y. Qi, R. F. He, K. Ren, L. R. Zheng, J. O. Wang, G. L. Ning and J. W. Ye, ACS Catal., 2022, 12, 7655–7663 CrossRef CAS.
- G. C. da Silva, S. I. Venturini, S. Y. Zhang, M. Löffler, C. Scheu, K. J. J. Mayrhofer, E. A. Ticianelli and S. Cherevko, ChemElectroChem, 2020, 7, 2330–2339 CrossRef CAS.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef PubMed.
- S. H. Yang, Y. Li, J. Sun and B. Q. Cao, J. Power Sources, 2019, 431, 220–225 CrossRef CAS.
- F. C. Walsh and R. G. A. Wills, Electrochim. Acta, 2010, 55, 6342–6351 CrossRef CAS.
- L. Wang, P. Lettenmeier, U. Golla-Schindler, P. Gazdzicki, N. A. Cañas, T. Morawietz, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Phys. Chem. Chem. Phys., 2016, 18, 4487–4495 RSC.
- W. Hu, S. L. Chen and Q. H. Xia, Int. J. Hydrogen Energy, 2014, 39, 6967–6976 CrossRef CAS.
- D. Böhm, M. Beetz, M. Schuster, K. Peters, A. G. Hufnagel, M. Döblinger, B. Böller, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2019, 30, 1906670 CrossRef.
- H.-S. Oh, H. N. Nong, T. Reier, M. Gliech and P. Strasser, Chem. Sci., 2015, 6, 3321–3328 RSC.
- K. K. Zhang, W. S. Mai, J. Li, H. Wang, G. Q. Li and W. Hu, J. Mater. Sci., 2020, 55, 3507–3520 CrossRef CAS.
- V. Yadav, H. Sharma, A. Rana and V. K. Saini, J. Ind. Eng. Chem., 2022, 107, 126–136 CrossRef CAS.
- Y. J. Gao and Y. M. Xu, Acta Phys.-Chim. Sin., 2012, 28, 641–646 Search PubMed.
- N. Fessi, M. F. Nsib, Y. Chevalier, C. Guillard, F. Dappozze, A. Houas, L. Palmisano and F. Parrino, Langmuir, 2020, 36, 13545–13554 CrossRef CAS PubMed.
- A. M. Abdullah, M. Á. Gracia-Pinilla, S. C. Pillai and K. O'Shea, Molecules, 2019, 24, 2147 CrossRef CAS PubMed.
- H. J. Chen, G. R. Deng, Z. S. Feng, Z. Q. Xu, M. Y. Yang, Y. Huang, Q. L. Peng, T. S. Li and Y. Wang, Chem. Commun., 2022, 58, 3214–3217 RSC.
- L. H. Zu, X. Y. Qian, S. L. Zhao, Q. H. Liang, Y. E. Chem, M. Liu, B.-J. Su, K.-H. Wu, L. B. Qu, L. L. Duan, H. L. Zhan, J.-Y. Zhang, C. Li, W. Li, J. Y. Juang, J. W. Zhu, D. Li, A. B. Yu and D. Y. Zhao, J. Am. Chem. Soc., 2022, 144, 2208–2217 CrossRef CAS PubMed.
- S. Khan, T. L. Ruwer, N. Khan, A. Köche, R. W. Lodge, H. Coelho-Júnior, R. L. Sommer, M. J. L. Santos, C. F. Malfatti, C. P. Bergmann and J. A. Fernandes, J. Mater. Chem. A, 2021, 9, 12214–12224 RSC.
- M. N. Fan, Z. H. Lin, P. Zhang, X. D. Ma, K. P. Wu, M. L. Liu and X. H. Xiong, Adv. Energy Mater., 2021, 11, 2003037 CrossRef CAS.
- L. H. Fu, X. Hu, Y. B. Li, G. Z. Cheng and W. Luo, Nanoscale, 2019, 11, 8898–8905 RSC.
- Q. Q. Chen, A. Ozkan, B. Chattopadhyay, K. Baert, C. Poleunis, A. Tromont, R. Snyders, A. Delcorte, H. Terryn, M. P. Delplancke-Ogletree, Y. H. Geerts and F. Reniers, Langmuir, 2019, 35, 7161–7168 CrossRef CAS PubMed.
- Y. Wang, C. X. Feng, M. Zhang, J. J. Yang and Z. J. Zhang, Appl. Catal., B, 2010, 100, 84–90 CrossRef CAS.
- L. Xu, C. Q. Tang, L. Dai, D. H. Tang and X. G. Ma, Acta Phys. Sin., 2007, 56, 1048–1053 CrossRef.
- G. F. Li, H. M. Yu, X. Y. Wang, D. L. Yang, Y. K. Li, Z. G. Shao and B. L. Yi, J. Power Sources, 2014, 249, 175–184 CrossRef CAS.
- W. Hu, Y. Q. Wang, X. H. Hu, Y. Q. Zhou and S. L. Chen, J. Mater. Chem., 2012, 22, 6010–6016 RSC.
- R. P. Ma, Y. Wang, G. Q. Li, L. Yang, S. W. Liu, Z. Jin, X. Zhao, J. J. Ge and W. Xing, Nano Res., 2021, 14, 4321–4327 CrossRef CAS.
- S. Siracusano, S. Trocino, N. Briguglio, F. Pantò and A. S. Aricò, J. Power Sources, 2020, 468, 228390 CrossRef CAS.
- G. F. Li, H. M. Yu, W. Song, X. Y. Wang, Y. K. Li, Z. G. Shao and B. L. Yi, Int. J. Hydrogen Energy, 2012, 37, 16786–16794 CrossRef CAS.
- J. B. Cheng, H. M. Zhang, G. B. Chen and Y. N. Zhang, Electrochim. Acta, 2009, 54, 6250–6256 CrossRef CAS.
- K. A. Lewinski, D. F. van der Vliet and S. M. Luopa, ECS Trans., 2015, 69, 893–917 CrossRef CAS.
- M. Möckl, M. F. Ernst, M. Kornherr, F. Allebrod, M. Bernt, J. Byrknes, C. Eickes, C. Gebauer, A. Moskovtseva and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 064505 CrossRef.
- S. C. Sun, Z. G. Shao, H. M. Yu, G. F. Li and B. L. Yi, J. Power Sources, 2014, 267, 515–520 CrossRef CAS.
- G. F. Li, H. M. Yu, X. Y. Wang, S. C. Sun, Y. K. Li, Z. G. Shao and B. L. Yi, Phys. Chem. Chem. Phys., 2013, 15, 2858–2866 RSC.
- C. Rakousky, U. Reimer, K. Wippermann, S. Kuhri, M. Carmo, W. Lueke and D. Stolten, J. Power Sources, 2017, 342, 38–47 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, detailed experimental section, additional experimental Figures and Tables. See https://doi.org/10.1039/d2ra05374h |
| This journal is © The Royal Society of Chemistry 2022 |