
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalemic myrionsumamide A, tetracyclic skeleton alkaloids from Myrioneuron effusum†
Jia-Hui Zhang‡
ac,
Mingming Cao‡a,
Yu Zhanga,
Xiao-Hui Liad,
Yu-Cheng Gu e,
Xiao-Nian Lia,
Ying-Tong Di*a and
Xiao-Jiang Hao*abc
e,
Xiao-Nian Lia,
Ying-Tong Di*a and
Xiao-Jiang Hao*abc
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China. E-mail: haoxj@mail.kib.ac.cn; diyt@mail.kib.ac.cn
bThe Key Laboratory of Chemistry for Natural Products of Guizhou Province and Chinese Academy of Sciences, Guiyang 550014, China
cResearch Unit of Chemical Biology of Natural Anti-Virus Products, Chinese Academy of Medical Sciences, Beijing, 100730, China
dYunnan Institute of Materia Medica, Kunming 650111, China
eJealott's Hill International Research Centre, Syngenta, Bracknell, Berkshire RG42 6EY, UK
First published on 3rd October 2022
Abstract
Investigation of the alkaloids from Myrioneuron effusum leads to the isolation of myrionsumamide A (1), a pair of enantiomeric alkaloids with an unprecedented tetracyclic system skeleton. These two alkaloids were separated by chiral HPLC with a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 from the scalemic mixture. Their structures including absolute configurations were determined by NMR spectroscopy, X-ray diffraction data and ECD calculations. Both (+)-1 and (−)-1 showed antibacterial activity against Staphylococcus aureus with MIC at 7.81 μg ml−1.
:5 from the scalemic mixture. Their structures including absolute configurations were determined by NMR spectroscopy, X-ray diffraction data and ECD calculations. Both (+)-1 and (−)-1 showed antibacterial activity against Staphylococcus aureus with MIC at 7.81 μg ml−1.
Introduction
Myrioneuron alkaloids belong to the category of lysine-based structurally diverse natural products elaborated by plants of the genus Myrioneuron R. Br. (Rubiaceae).1 Some structures of them showed significant biological activities such as antimalarial and anti-hepatitis C virus (HCV),2–4 which along with their complex ring systems (up to 10 hexatomic rings) and amazing multiple chiral centers (up to 12 ones) within one structure have attracted great attention for total synthesis studies.5–7 By July 2022, more than twenty Myrioneuron alkaloids had been obtained with various polycyclic structures (tricyclic-, tetracyclic-, pentacyclic-, hexacyclic-, octacyclic-, and decacyclic-type).1,4,8–10 However, their stereochemistry control and polycyclic skeleton construction logic in nature remain elusive.One basic feature of Myrioneuron alkaloids is their decahydroquinoline (DHQ) core (Fig. 1), and the subsequent attachment of rings C and D onto the DHQ moiety (rings A and B) consistently generated the typical ‘myrionamide carbon skeleton’ with a series of chiral centers (Fig. 1). From the biosynthesis perspective, the five carbon atoms on ring D were assumed to be derived from a lysine after oxidation and decarboxylation reactions.2,8 However, in our continuous investigation of bioactive Myrioneuron alkaloids, (±)-myrionsumamide A (1) was isolated and possessed a different carbon skeleton from myrionamide and schoberine (Fig. 2), whose structures were confirmed by X-ray diffraction experiments.3 This new manner of ring D formation and attachment drew our attention to study its structure and bioactivity.
 | ||
| Fig. 1 Representative Myrioneuron alkaloids with DHQ moiety and ‘myrionamide carbon skeleton’ (highlighted in bold). | ||
 | ||
| Fig. 2 Structures of myrionsumamide A (1) and known typical Myrioneuron alkaloids myrionamide and schoberine. | ||
Here we report the isolation of 1 from Myrioneuron effusum (Pitard) Li and its structural elucidation on the basis of NMR and X-ray diffraction data analysis, and a hypothesized biogenetic pathway of 1 was also discussed. Further we observed that myrionsumamide A is a scalemic mixture, and (+)-1 and (−)-1 were separated by chiral HPLC with a ratio of 5:3. An antibacterial experiment showed both (+)-1 and (−)-1 were active against Staphylococcus aureus with MIC at 7.81 μg ml−1.
Results and discussion
Myrionsumamide A (1) was recrystallized from acetone to give colorless crystal, and its molecular formula C15H24N2O2 was established by positive HR-ESI-MS (m/z 265.1922 [M + H]+, calcd for C15H25N2O2, 265.1911), corresponding to 5 degrees of unsaturation. The 1H and 13C NMR data (Table 1) revealed the presence of two quaternary carbons (δC 174.0 and δC 70.7), four methines, and nine methylenes. The IR absorption bands around 3555, 2932 and 1614 cm−1 showed the presence of hydroxyl, secondary amine, and carbonyl functionality, respectively. Since one carbonyl accounted for one out of the five degrees of unsaturation, the remaining four degrees of unsaturation were assumed for the presence of a tetracyclic system in the structure.| No. | δC | δH-a (mult, J) | δH-b (mult, J) |
|---|---|---|---|
| a Overlapped.b Recorded in DMSO-d6. | |||
| 2 | 42.7 | 4.50 (dd, 13.14, 4.26) | 2.62 (m) |
| 3 | 19.9 | 1.70 (m) | 1.56 (m)a |
| 4 | 27.0 | 1.63 (m) | 1.29 (m) |
| 5 | 42.2 | 1.64 (m) | |
| 6 | 29.7 | 1.61 (m) | 1.53 (m) |
| 7 | 26.5 | 1.54 (m) | 1.24 (m) |
| 8 | 26.4 | 2.12 (m) | 1.79 (dt, 13.14, 3.36) |
| 9 | 36.4 | 1.66 (m) | |
| 10 | 54.6 | 3.86 (t, 5.52) | |
| 11 | 60.9 | 2.80 (d, 2.88) | |
| 13 | 45.0 | 2.91 (dd, 11.52, 3.72) | 2.64 (ddd, 24.42, 11.16, 3.54) |
| 14 | 23.5 | 1.56 (m)a | 1.23 (m) |
| 15 | 36.8 | 2.53 (dd, 12.42, 3.96) | 1.30 (m) |
| 16 | 70.8 | ||
| 17 | 174.2 | ||
| 16-OH | 4.94 (s)b | ||
The gross structure of 1 was constructed by 2D NMR experiment. Based on the 1H–1H COSY and HSQC study, two structural fragments were established as drawn with bold bonds (Fig. 3A). The connectivity of the two structural fragments, via two nitrogen atoms and two quaternary carbons were established by the analysis of HMBC correlations (Fig. 3A). The HMBC correlation from H-2 to C-10 indicated that C-2 and C-10 were connected via N-1, which further confirmed by that methine (C-10, δC 54.5) and methylene (C-2, δC 42.5) were typical carbons bearing one nitrogen atom of Myrioneuron alkaloid. Similarly, the HMBC correlations from H-11 to C-13 and from H-13 to C-11 revealed that C-11 and C-13 were connected through N-12. In addition, the HMBC correlations from H-9, H-11 and H-15 to C-16, from H-2, H-11 and H-15 to C-17, from H-11 to C-15 and from H-15 to C-11 indicated the structural fragments a and b were connected by a quaternary carbon bridge of C-16/C-17. Thus, the 2D structure of 1 was established as shown (Fig. 3A).
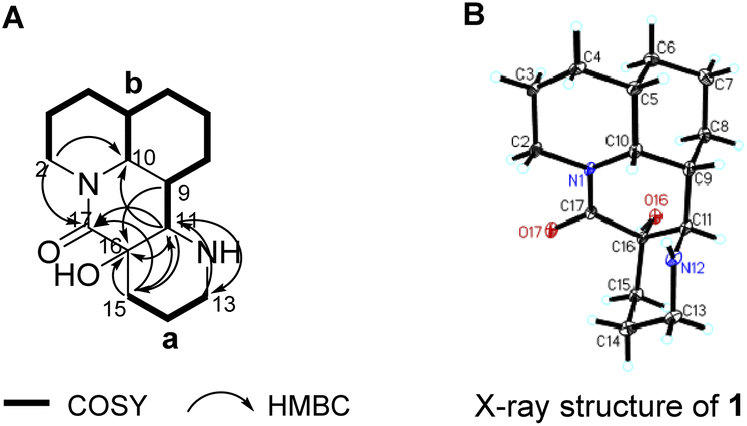 | ||
| Fig. 3 (A) 1H–1H COSY, key HMBC and ROESY correlations of 1; (B) X-ray structure of 1. | ||
The relative configuration of 1 was elucidated by 2J coupling constant analysis and further confirmed by X-ray diffraction data. Since H-10 was split by H-5 and H-9 to give 2J = 5.52 Hz which required cis position of both H-5 and H-9 with H-10, and these three cofacial protons were arbitrarily assigned as β-oriented. The remaining chiral centers at C-11 and C16 were directly confirmed by X-ray diffraction. Compound 1 was carefully recrystallized from acetone. As a result, the planar structure and relative configuration of 1 was confirmed as shown in Fig. 3B. Moreover, analysis of X-ray data suggested 1 to be racemate by the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (data has been deposited to CCDC with no. 2190231). Subsequently, 1 was further analyzed by HPLC with a chiral column and turned out to be an scalemic mixture with a ratio of (+)-1/(−)-1 as 5:3 (Fig. 4), and in agree with its slightly positive rotation value (Fig. S1.10†).
(data has been deposited to CCDC with no. 2190231). Subsequently, 1 was further analyzed by HPLC with a chiral column and turned out to be an scalemic mixture with a ratio of (+)-1/(−)-1 as 5:3 (Fig. 4), and in agree with its slightly positive rotation value (Fig. S1.10†).
 | ||
| Fig. 4 Analysis of a scalemic mixture of (±)-1 by chiral HPLC (Daicel Chiralpak IC 4.6 × 250 mm). | ||
Both racemic and enantiomeric enrichment were observed in alkaloids from Myrioneuron faberi,3 so we suppose that primary polycyclic structures could be formed spontaneously as biomimetic access to C5-lysine-derived nitraramine11 and other scalemic natural products,12,13 while the optical pure species should be resulted from late stage enzymatic modification(s). A proposed biosynthetic pathway for 1 starting from L-lysine was shown in Scheme 1. Three basic building bricks a, b and c could be derived from L-lysine through decarboxylation, deamination, and oxidation, with disappearance of the chiral carbon. First, a and b could transform into intermediates i and ii via Mannich-like reaction. Second, iii could react with c to give imine iv, and the tautomer of iv might undergo intramolecular Manish-like reaction resulting ring D closed vi. Then ring C could be generated to give vii via condensation from vi. Finally, 1 could be obtained by oxidation of vii. Since optical monomer (+)-1 was enriched over (−)-1, it was speculated that the hydroxylation at C-16 was catalyzed by an oxygenase with poor stereoselectivity which favors the formation of (+)-1.
 | ||
| Scheme 1 Plausible biosynthetic pathway for 1. | ||
Based on the relative configurations, the absolute configurations of the two enantiomers were confirmed by comparing the experimental CD with the theoretical calculation ECD (Fig. 5). The ECD spectrum was calculated using the TD-DFT-B3LYP/6-311G+(2d,p) theory of B3LYP/6-31G(d,p)-optimized geometries with the IEFPCM model (in MeOH) in the Gaussian 09 program package.14 The CD of (+)-1 is in agree with the ECD of the model compound (5R, 9S, 10S, 11S, 16R), while the CD of (−)-1 is comparable to the ECD of (5S, 9R, 10R, 11R, 16S) (Fig. 5). Thus, the absolute configuration of (+)-1 and (−)-1 was determined as 5R, 9S, 10S, 11S, 16R and 5S, 9R, 10R, 11R, 16S, respectively.
 | ||
| Fig. 5 Experimental ECD spectrum of (±)-1 (solid) and calculated ECD spectrum of (±)-1 (dash). | ||
Antibacterial activity of (+)-1 and (−)-1 were evaluated against six bacterial strains (Staphylococcus aureus ATCC29213, Bacillus subtilis ATCC 6051, Escherichia coli ATCC25922, Pseudomonas aeruginosa ATCC27853, Acinetobacter baumanii ATCC BAA-1710D and Klebsiella pneumonia ATCC BAA-1705) by MIC method15 after optical density value by spectrophotometer at 490 nm. Both (+)-1 and (−)-1 showed antibacterial activity against Staphylococcus aureus ATCC29213 with MIC value of 7.81 μg ml−1.
Experimental
General procedures
Optical rotations were measured with a Jasco P-1020 polarimeter. UV spectra were obtained using a Shimadzu UV-2401A spectrophotometer. A Tenor 27 spectrophotometer was used for IR spectra as KBr pellets. 1D and 2D NMR spectra were recorded on Bruker spectrometer with TMS as internal standard. HRESIMS was performed on a triple quadrupole mass spectrometer. Semi-preparative HPLC was performed on an Agilent 1100 liquid chromatograph with a Waters X-Bridge Prep Shield RP18 (10 × 150 mm) column. Column chromatography (CC) was performed using silica gel (100–200 mesh and 300–400 mesh, Qingdao Marine Chemical, Inc., Qingdao, People's Republic of China) and Sephadex LH-20 (40–70 μm, Amersham Pharmacia Biotech AB, Uppsala, Sweden). Microplate reader (BioTek ELx800) was used in antibacterial assay. X-ray diffraction was carried out on BRUKER D8 QUEST.Plant material
The twigs and stems of Myrioneuron effusum were collected from Guangxi Province, People's Republic of China, in November 2014. The plant samples were identified by Ligong Lei of Kunming Institute of Botany, Chinese Academy of Science (CAS). A voucher specimen (HXJ20141101) was deposited at the State Key Laboratory of Phytochemistry and Plant Resource in West China, Kunming Institute of Botany, Chinese Academy of Science (CAS).Extraction and isolation
The air-dried, powdered leaves and twigs (57 kg) of M. effusum were extracted three times with MeOH at room temperature. The total extract (4.8 kg) was subjected to normal phase Si gel (100–200 mesh; CHCl3/MeOH, 1:0 to 0:1) to obtain four major fractions (Fr. 1–4). Fraction 2 (311.5 g) was also subjected to Si gel column chromatography with CHCl3/MeOH (20:1) to yield four sub-fractions (2a–2d). Fraction 2c (90.0 g) was chromatographed over a RP-C18 silica gel with step-gradient of MeOH–H2O (v/v, from 10:90 to 90:10) to obtain the crude alkaloids (51.4 g) and then separated to three fractions (2c-1–2c-3) by Si gel column chromatography (300–400 mesh, CHCl3/MeOH, 20:1). Fraction 2c-1 (12.6 g) was separated by preparative HPLC (Waters XSelect CSH prep C18, 5 μm, 19 × 150 mm; MeOH–H2O, from 30:70 to 90:10 in 20 min; velocity of flow 10 ml min−1) to obtained three fractions (2c-1a–2c-1c). Fraction 2c-1b (40.5 mg) was chromatographed over a Sephadex LH-20 gel column to give compound (±)-1 (15.6 mg). Then (+)-1 and (−)-1 were separated by chiral HPLC (Daicel Chiralpak ODH, 4.6 × 250 mm, isopropanol/hexane 13:87, 1 ml min−1).
X-ray crystallographic analysis
The crystals of 1 were obtained from acetone at room temperature. The crystallographic diffraction data were collected with Cu Kα radiation (λ = 1.54178 Å) using a BRUKER D8 QUEST diffractometer at T = 150(2) K. The structure was solved by the SHELXS method and refined based on full-matrix least-squares on F2 using SHELXL-2018/3. Crystal data for 1: C15H24N2O2, M = 264.36, a = 10.0436(5) Å, b = 11.4268(6) Å, c = 12.7535(6) Å, α = 92.7820(10)°, β = 96.8930(10)°, γ = 108.4380(10)°, V = 1372.67(12) Å3, T = 100(2) K, space group P, Z = 4, μ(CuKα) = 0.676 mm−1, 17390 reflections measured, 4629 independent reflections (Rint = 0.0477). The final R1 values were 0.0744 (I > 2σ(I)). The final wR(F2) values were 0.2159 (I > 2σ(I)). The final R1 values were 0.0747 (all data). The final wR(F2) values were 0.2165 (all data). The goodness of fit on F2 was 1.107.
Antibacterial assay
Compounds (+)-1 and (−)-1 were tested for antibacterial activities against Staphylococcus aureus ATCC29213, Bacillus subtilis ATCC 6051, Escherichia coli ATCC25922, Pseudomonas aeruginosa ATCC27853, Acinetobacter baumanii ATCC BAA-1710D and Klebsiella pneumonia ATCC BAA-1705. The minimum inhibitory concentration (MIC) values were determined using a 96-well plate format with LB broth. Briefly, each strain was grown in LB and used as a seed culture when the OD490 reached 2.0. The seed culture (10 μL) was added to 10 ml LB, and 100 μL LB with cells was transferred into the first 6 columns on a 96-well plate ready for assay (LB without cells in column 7 as background, and DMSO instead of compound in column 8 as a negative control). The compounds (+)-1 and (−)-1 and the positive control hygromycin b were dissolved in DMSO to make serial dilutions ready for assay. Then add a series of compound to get final concentrations of 125 μM, 62.5 μM, 31.25 μM, 15.62 μM, 7.81 μM, 3.91 μM in each row. Each strain was also set up in the same way and in triplicate. The 96-well plates were cultured at 37 °C for 8 h and then the OD490 was measured to determine the MIC values.Conclusions
In this work we showed a new ring system construction manner together with stereoselectivity adopted by Myrioneuron alkaloids (±)-Myrionsumamide A. Their structures and absolute configurations were elucidated by NMR data analysis, X-ray diffraction, and ECD calculations. Meanwhile, both (+)-1 and (−)-1 showed antibacterial activity against Staphylococcus aureus.Author contributions
Dr J.-H. Zhang, contributed to data curation, formal analysis, investigation, and validation. Dr M. Cao, contributed to formal analysis, visualization, writing – original draft, and writing – review & editing. Dr Y. Zhang, contributed to methodology and resources. Dr X.-H. Li, contributed to methodology and formal analysis. Dr Y.-C. Gu, contributed to funding acquisition and supervision. Dr X.-N. Li, contributed to data curation and software. Dr Y.-T. Di, contributed to writing – original draft, software, and supervision. Prof. Dr X.-J. Hao, contributed to funding acquisition, project administration, conceptualization and resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported financially by grants from the National Science Foundation of China (U1812403 to X.-J. Hao, 81573323 and 31770392 to Y.-T. Di, 22177050 to M. Cao), Yunnan Provincial Science and Technology Department (No. 202003AD150012 and 202201AS070040), Foundation of Central Asian Drug Discovery and Development Center of Chinese Academy of Sciences (CAM202103, China), and Youth Innovation Promotion Association CAS to M. Cao (2022–2026). The Syngenta postgraduate studentship awarded to J.-H. Zhang (2014–2016) is appreciated.References
- E. Gravel and E. Poupon, Biosynthesis and biomimetic synthesis of alkaloids isolated from plants of the Nitraria and Myrioneuron genera: an unusual lysine-based metabolism, Nat. Prod. Rep., 2010, 27, 32–56 RSC.
- V. C. Pham, A. Jossang, P. Grellier, T. Sévenet, V. H. Nguyen and B. Bodo, Structure and total synthesis of (−)-myrionidine and (−)-schoberine, antimalarial alkaloids from Myrioneuron nutans, J. Org. Chem., 2008, 73, 7565–7573 CrossRef CAS PubMed.
- M. M. Cao, Y. Zhang, S. D. Huang, Y. T. Di, Z. G. Peng, J. D. Jiang, C. M. Yuan, D. Z. Chen, S. L. Li, H. P. He and X. J. Hao, Alkaloids with Different Carbon Units from Myrioneuron faberi, J. Nat. Prod., 2015, 78, 2609–2616 CrossRef CAS.
- M. M. Cao, Y. Zhang, X. H. Li, Z. G. Peng, J. D. Jiang, Y. C. Gu, Y. T. Di, X. N. Li, D. Z. Chen, C. F. Xia, H. P. He, S. L. Li and X. J. Hao, Cyclohexane-fused octahydroquinolizine alkaloids from Myrioneuron faberi with activity against hepatitis C virus, J. Org. Chem., 2014, 79, 7945–7950 CrossRef CAS PubMed.
- J. M. Aquilina and M. W. Smith, Symmetry-Driven Total Synthesis of Myrioneurinol, J. Am. Chem. Soc., 2022, 144, 11088–11093 CrossRef CAS PubMed.
- T. J. Fulton, A. Y. Chen, M. D. Bartberger and B. M. Stoltz, Enantioselective total synthesis of (−)-myrifabral A and B, Chem. Sci., 2020, 11, 10802–10806 RSC.
- A. J. Nocket and S. M. Weinreb, Total synthesis of the tetracyclic antimalarial alkaloid (±)-myrioneurinol, Angew. Chem., Int. Ed., 2014, 53, 14162–14165 CrossRef CAS PubMed.
- M. M. Cao, S. D. Huang, Y. T. Di, C. M. Yuan, G. Y. Zuo, Y. C. Gu, Y. Zhang and X. J. Hao, Myrifabine, the first dimeric Myrioneuron alkaloid from Myrioneuron faberi, Org. Lett., 2014, 16, 528–531 CrossRef CAS PubMed.
- S. D. Huang, Y. Zhang, M. M. Cao, Y. T. Di, G. H. Tang, Z. G. Peng, J. D. Jiang, H. P. He, X. J. Hao and A. Myriberine, A new alkaloid with an unprecedented heteropentacyclic skeleton from Myrioneuron faberi, Org. Lett., 2013, 15, 590–593 CrossRef CAS.
- X. H. Li, Y. Zhang, J. H. Zhang, X. N. Li, M. M. Cao, Y. T. Di, Z. G. Peng, J. D. Jiang, X. J. Hao and A.-C. Myritonines, Alkaloids from Myrioneuron tonkinensis Based on a Novel Hexacyclic Skeleton, J. Nat. Prod., 2016, 79, 1203–1207 CrossRef CAS.
- E. Gravel, E. Poupon and R. Hocquemiller, Biomimetic One-Step Access to Nitraramine from Simple C5 Units. Revision of the Previously Reported Structure of Epinitraramine to Nitraramine, Org. Lett., 2005, 7, 2497–2499 CrossRef CAS PubMed.
- S. W. Haynes, P. K. Sydor, C. Corre, L. Song and G. L. Challis, Stereochemical elucidation of streptorubin B, J. Am. Chem. Soc., 2011, 133, 1793–1798 CrossRef CAS.
- M. A. Schafroth, G. Mazzoccanti, I. Reynoso-Moreno, R. Erni, F. Pollastro, D. Caprioglio, B. Botta, G. Allegrone, G. Grassi, A. Chicca, F. Gasparrini, J. Gertsch, E. M. Carreira and G. Appendino, Δ(9)-cis-Tetrahydrocannabinol: Natural Occurrence, Chirality, and Pharmacology, J. Nat. Prod., 2021, 84, 2502–2510 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji; X. Li; M. Caricato; A. V. Marenich; J. Bloino; B. G. Janesko; R. Gomperts; B. Mennucci; H. P. Hratchian; J. V. Ortiz; A. F. Izmaylov; J. L. Sonnenberg; D. Williams-Young; F. Ding; F. Lipparini; F. Egidi; J. Goings; B. Peng; A. Petrone; T. Henderson; D. Ranasinghe; V. G. Zakrzewski; J. Gao; N. Rega; G. Zheng; W. Liang; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; K. Throssell; J. A. Montgomery Jr; J. E. Peralta; F. Ogliaro; M. J. Bearpark; J. J. Heyd; E. N. Brothers; K. N. Kudin; V. N. Staroverov; T. A. Keith; R. Kobayashi; J. Normand; K. Raghavachari; A. P. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; J. M. Millam; M. Klene; C. Adamo; R. Cammi; J. W. Ochterski; R. L. Martin; K. Morokuma; O. Farkas; J. B. Foresman and D. J. Fox, Gaussian 09 Rev. D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- L. N. Zhao, X. X. Guo, S. Liu, L. Feng, Q. R. Bi, Z. Wang and N. H. Tan, (±)-Zanthonitidine A, a Pair of Enantiomeric Furoquinoline Alkaloids from Zanthoxylum nitidum with Antibacterial Activity, Nat. Prod. Bioprospect., 2018, 8, 361–367 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR, HSQC, HMBC, 1H–1H COSY, NOESY, IR, UV and HR-ESI-MS spectra of 1, CD data for compounds (+)-1 and (−)-1, and X-ray crystallographic data for 1 (CIF). CCDC 2190231. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra05342j |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |