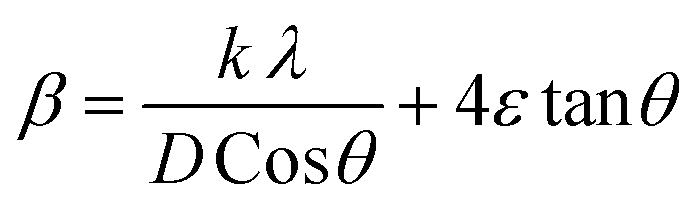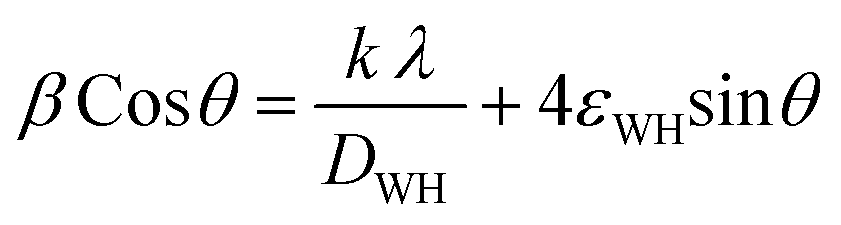DOI:
10.1039/D2RA05304G
(Paper)
RSC Adv., 2022,
12, 34584-34600
Structural, optical, magnetic, and enhanced antibacterial properties of hydrothermally synthesized Sm-incorporating α-MoO3 2D-layered nanoplates
Received
24th August 2022
, Accepted 24th November 2022
First published on 2nd December 2022
Abstract
In this study, we have synthesized pristine and [0.5,1.5, and 2.5] M% samarium (Sm)-incorporating α-MoO3 2D-layered nanoplates utilizing a facile hydrothermal process, and investigated the physical properties along with antibacterial effectiveness. X-ray diffraction (XRD) patterns confirmed the single-phase, stable orthorhombic polycrystalline structure of the as-prepared samples. The crystallite size, lattice strain, and dislocation density were measured using both Debye–Scherrer (D–S) and Williamson-Hall (W-H) techniques. Both pristine and Sm-incorporating α-MoO3 samples showed two-dimensional (2D) layered nanoplate-type surface morphology, revealed by field emission scanning electron microscopy (FE-SEM) images. Energy dispersive X-ray spectroscopy (EDS) confirmed the presence of Sm contents in the α-MoO3 matrix. After Sm incorporation in α-MoO3, the different functional groups as well as vibrational groups were observed by Fourier-transform infrared (FTIR) spectroscopy and Raman spectroscopy analyses, respectively. The optical band gaps were measured from UV-vis diffuse reflectance spectroscopy (DRS) by employing the Kubelka–Munk formula and interestingly it is found that the bandgap energy (Eg) gradually decreased from 2.96 to 2.83 eV with the increment of Sm content. When compared to pristine α-MoO3, the Sm-incorporating samples experienced a steady improvement in room temperature ferromagnetic (RTFM) behavior as Sm content increased, as measured by hysteresis loops. The antibacterial activities of both samples were assessed against Gram-positive: Staphylococcus aureus (S. aureus), and Gram-negative: Escherichia coli (E. coli) and Salmonella enteritidis (S. enteritidis) bacteria by the agar well diffusion method and enhanced antibacterial activity was observed as the Sm concentration increased, compared to pristine nanoplates. The obtained results suggest that the synthesized Sm-incorporating α-MoO3 2D-layered nanoplate could be a potential antibacterial agent.
1 Introduction
Because of the large influence on patient wellbeing and environmental safety issues, healthcare-associated (nosocomial) infections (HAIs) have been a serious worry among scientists in recent years due to rapidly rising death rates and increasing annual costs for HAI prevention.1,2 The ubiquitous nature and ability of bacteria in the environment to grow, transfer, and develop multidrug resistance are significant challenges in this sector. A variety of detailed studies are required to produce new and effective antibacterial drugs to eliminate the ever-increasing concerns about bacterial infections. Nanotechnology, a new multidisciplinary discipline combining physics, chemistry, biology, and materials science, has enabled the most promising new insights to overcome the drawbacks of current antimicrobial treatments and the ongoing growth of bacterial resistance.
The increased interest in nanomaterials, particularly inorganic nanomaterials, for bioscience applications is due to their distinct features such as the high surface area to volume ratio, uniform size, shape, size distribution, morphology, surface functionalization, improved stability, and safety compared to organic antimicrobial agents.3–7 Antimicrobial nanostructures offer a wide range of uses, including antibacterial textiles, catheter devices, biofilm suppression, and additions in paints for antimicrobial surfaces, among others.8 In the last few decades, antimicrobial characteristics of nanoparticles such as Ag, Au, Cu, ZnO, CuO, Fe2O3, TiO2, CaO, Fe@Co3O4, MoS2, and MgO have become an interesting area of investigation.9–13 However, these oxide materials' limited active surface area, low durability, significant cytotoxicity in nature, and high costs limit their applicability as antibacterial devices. As a result, the scientific community faces a challenge in producing a new antibacterial agent using a low-cost nano-oxide material. Among several reported semiconductor nanomaterials, detailed studies of ZnO, CuO, Fe2O3, and TiO2 have been reviewed and reported by several groups as compared to other materials as antimicrobial agents.14–18
Transition metal oxides (TMO) have recently received a lot of study attention due to their wide range of prospective applications in the emerging fields of photodetectors,19 thin-film transistors,20 photo-catalytic, optoelectronic, magneto-optic, photonic, gas sensors, transparent conductors, and photovoltaic solar cells, among others.21–27 These materials can be used by incorporating transition metal (TM) ions into cation sites in host conventional semiconductors like MoO3, ZnO, and TiO2, and then introducing ferromagnetic properties into nonmagnetic semiconductors, allowing for simultaneous utilization of charge and spin carriers.28–30 Moreover tuning the properties of various nanostructured materials like CdS, boron nitride (BN), PbS, and PbO have received attention for various applications.31–33
Molybdenum trioxide (MoO3), in particular, among intrinsic n-type II–VI semiconductors, has a wide direct band gap of 2.8–3.2 eV at room temperature.34 One of the most important and extensively studied TMO is MoO3 because of its rich chemistry associated with multiple states, high thermal-chemical stability, suitable electronic, electrical, and optical properties, and numerous potential technological applications in the fields of spintronic devices, optoelectronic devices, magneto-optic devices, photonic devices, organic light-emitting diodes (OLEDs), charge injection or extraction layer in organic photovoltaic (OPVs), gas sensors, transparent conductors, photo-catalytic, and photovoltaic solar cells.35,36
There are three basic crystal forms in MoO3 material. The orthorhombic MoO3 phase (α-MoO3) is based on an octahedral MoO6 basis, with all of the MoO3 components sharing edges and corners, resulting in a zigzag chain and layered structure.37 Whereas, there are two metastable MoO3 phases namely monoclinic MoO3 (β-MoO3) and hexagonal MoO3 (h-MoO3) exist. It is necessary to modify the physical properties of MoO3 nanoparticles to expand their applications in a variety of nanotechnology sectors, and this tuning may be accomplished via a variety of methods, including doping, annealing, and modifying production techniques.38,39
Doping, or the inclusion of a particular extrinsic element, is another straightforward, practical, and very effective method for altering and/or enhancing many properties of fundamental materials, such as electrical, magnetic, and optical capabilities.40 Thus, as previously reported, the characteristics of MoO3 may be altered by alloying it with a variety of elements such as cobalt (Co),29 europium (Eu),41 and dysprosium (Dy).42 It is important to consider the radius of the dopant element since dopant atoms with comparable radii are recommended for producing compounds with minimum lattice imperfections.42 Previously many research groups had replaced the small ionic radius Mo6+ (0.65 Å) with various larger ionic (0.74–0.97 Å) elements like Eu3+ (0.96 Å),43 Cd2+ (0.97 Å),44 Ce3+ (0.87 Å),45 Dy3+ (0.91 Å),46 Zn2+ (0.74 Å),47 and Tb3+ (0.92 Å)48 in order to get the desired properties for various applications. The benefit of doping metal oxides with rare earth metals such as Dy, Sm Ce, etc. is that the 4f empty orbitals make them easily mix with functional groups. Additionally, doping rare earth metals leads in an absorbance shift to the visible portion of the electromagnetic spectrum by manipulating structural parameters. As the rare-earth samarium ion (Sm3+) has a radius of 1.08 Å, therefore, it would also be a good dopant for replacing the Mo ion (Mo6+) in the MoO3 lattice. Some recent reviews on rare-earth oxides and rare-earth elements as doping agents in various applications are also notable.49–54 Another significant parameter affecting the quality of nanostructured materials is the annealing temperature. Several studies have been conducted on the influence of annealing temperature on unincorporated MoO3 nanomaterials.46,55
The properties of generated nanostructures vary according to their size, shape, and many aspects such as synthesis processes. Thus far, unincorporated and incorporated nanostructured α-MoO3 may be manufactured utilizing a variety of approaches, including hydrothermal synthesis, solution combustion synthesis, and probe ultrasonic synthesis, among others, to enhance the performance of these nanomaterials.40,56 Each synthesis approach offers several distinct benefits over others. According to a review of the literature, the hydrothermal process is the most practical and effective method for modifying the size, shape, and other features of nanomaterials. Furthermore, the hydrothermal process is an enthralling, simple, affordable, and successful strategy.43,57 However, the effects of Sm-incorporated in nanostructured α-MoO3 synthesized by the hydrothermal method have not been explored so far.
In this work, nanostructured pristine α-MoO3 and [0.5, 1.5, and 2.5 M%] Sm-incorporated α-MoO3 were synthesized by a facile environment-friendly hydrothermal approach. The effect of Sm incorporation on the microstructural (lattice parameters, average crystallite size, crystalline nature, etc.), morphological, functional, Raman, optical and magnetic properties of the α-MoO3 nanoplates are then investigated comprehensively using a variety of characterization techniques such as FE-SEM, XRD, EDX, FTIR, Raman spectroscopy, UV-vis spectroscopy, and a vibrating sample magnetometer (VSM) of a Quantum Design Physical Property Measurement System (PPMS). In addition, the antimicrobial activity of both samples was investigated against one Gram-positive (S. aureus) and two Gram-negative (E. coli and S. enteritidis) bacteria by the agar well diffusion method.
2 Experimental details
2.1 Materials
The ammonium heptamolybdate tetrahydrate (AHM), (NH4)6Mo7O24·4H2O, was employed as the precursor material of Mo for the hydrothermal synthesis of pristine and [0.5, 1.5, 2.5] M% Sm-incorporated α-MoO3 samples. Samarium(III) oxide (Sm2O3) was used as the source of the samarium. Other chemicals including concentrated nitric acid (37% HNO3), de-ionized (DI) water, and ethanol were utilized in this process. All chemicals (Sigma Aldrich Ltd, USA) were analytical grades used without further purification.
2.2 Nanoplates preparation
To obtain a pure MoO3 sample, 6 g of AHM was dissolved in 250 mL de-ionized (DI) water. The aqueous solution was magnetically stirred for 1 h at 75 rpm at room temperature. After that to achieve yellow precipitation, 24.5 mL of 37% HNO3 was added drop by drop at 100 °C for 1 h using a hotplate with magnetic stirring. The precipitated MoO3 was cleaned several times with ethanol and DI water. Following that, the washed MoO3 was desiccated for 12 h at 90 °C in an electric oven to obtain MoO3 nanopowders. Finally, the resulting MoO3 nanopowder was annealed at 450 °C for 3 h in an electric furnace to obtain the final product of α-MoO3 nanoplates. The above procedures were followed to obtain 0.5, 1.5, or 2.5 M% Sm-incorporated α-MoO3 nanoplates. For this, the required amount of samarium(III) oxide (Sm2O3) was added with AHM. The possible chemical reaction to synthesize α-MoO3 is given by eqn (1). In Fig. 1, the creation of α-MoO3 nanoplates is depicted as well.| | |
(NH4)6Mo7O24·4H2O(aq) + 6HNO3 → 7MoO3(s) + 6NH4NO3(aq) + 7H2O(l)
| (1) |
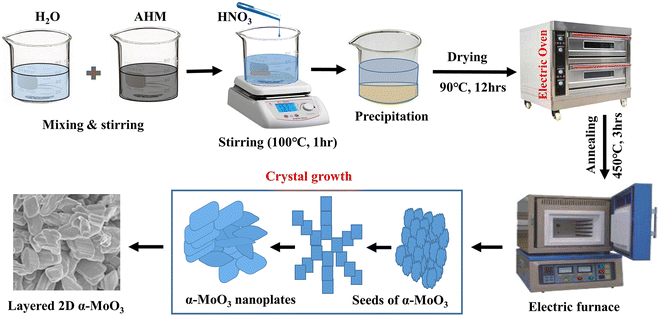 |
| | Fig. 1 The schematic diagram for the synthesis of pristine and Sm-incorporated α-MoO3 nanoplates. | |
2.3 Characterization
An X-ray diffractometer (3040-X'Pert PRO, Philips) was used to collect the X-ray diffraction (XRD) patterns of the pure and Sm-incorporated MoO3 powder samples to evaluate phase purity and structural features. XRD analysis was performed using CuKα radiation of wavelength 1.54056 Å, where 2θ = 10° to 70°, operating voltage = 40 kV, current = 30 mA, and scanning speed = 1° min−1. An FE-SEM integrated with EDS (JEOL JSM-7600F) was utilized to investigate the surface morphologies along with elemental composition58,59 of the synthesized samples, where the accelerating electric field was 5 kV. An FTIR (FTIR-ATR, Model: PerkinElmer Spectrum Two) with a scanning wavenumber range of 4000–400 cm−1 was used to identify the existing functional groups. A confocal Raman spectrometer (Monovista CRS+500) was used to obtain the Raman spectra with laser excitation at 532 nm. The optical investigation was carried out using a UV-vis DRS (Lambda 1050, PerkinElmer, USA) in the wavelength range of 250 to 800 nm. A vibrating sample magnetometer (VSM) of a Quantum Design PPMS was used to measure magnetic characteristics at room temperature.
2.4 Antibacterial testing of pristine and Sm-incorporated α-MoO3 nanoplates
The antimicrobial effects of pristine and Sm-incorporated α-MoO3 nanoplates against a Gram-positive bacterium, Staphylococcus aureus (ATCC 6538), and two Gram-negative bacteria, Escherichia coli (ATCC 8739) and Salmonella enterica (ATCC 14028) were analyzed using the agar well diffusion method with Mueller–Hilton Agar (MHA). Bacterial strains were cultured on Nutrient Agar media overnight at 37 °C. The bacterial colonies were suspended in 10 mL of sterile normal saline. Using UV-vis spectroscopy (Hitachi, Model: U-2910, Japan), the optical density (OD) was recorded at 580 nm and adjusted to achieve a cell density of 106–107 CFU mL−1 The good diffusion experiment was performed by pouring 25 mL of sterile MHA into Petri plates and allowing them to set. MHA plates were uniformly infected after solidification by swabbing test microorganisms with sterile cotton buds. Wells were bored into MHA plates after swabbing with a sterile borer (6 mm in diameter). Then, 100 μl of each concentration of 20 k, 40 k, 50 k, and 80 k ppm pristine and Sm-incorporated α-MoO3 were added to each well. The plates were then incubated for 24 hours at 37 °C. To examine the antibacterial properties of both pristine and Sm-incorporated α-MoO3, the zone of inhibition (ZOI) was measured in millimeters after incubation with a slide caliper.
3 Results and discussion
3.1 Structural properties
Fig. 2 demonstrates the XRD patterns of as-prepared pristine and [0.5, 1.5, 2.5] M% Sm-incorporated MoO3 powder samples. The diffracted peaks at (020), (110), (040), (021), (111), (060), (002), and (081) planes are clearly distinguishable which are well-matched with the orthorhombic phase of MoO3 (JCPDS # 05-0508) with space group Pbnm having a = 3.962 Å, b = 13.858 Å, c = 3.697 Å.60 All samples show polycrystalline in nature. The sharp and well-defined peaks indicate a good crystalline structure and the absence of unwanted peaks confirms the phase purity of all samples. The most intense peak (021) at 27.28° of pristine α-MoO3 is defined as the preferred orientation and no remarkable shifting was observed after the incorporation of Sm into the MoO3 lattice. The lattice parameters determined from equation61 are a = 3.962 Å, b = 13.858 Å, and c = 3.697 Å, which are in good agreement with the literature values and presented in Table 1. Using Debye Scherrer (D–S) equation (eqn (2)),42 the crystallite size (DD–S) is determined from the full width at half maximum (FWHM) values of the XRD peaks.| |
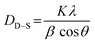 | (2) |
Where, DD−S, λ, K, β, and θ signify the crystallite size, X-ray wavelength (λ = 1.54056 Å for CuKα), shape factor (K = 0.90), FWHM of the corresponding diffraction peak (in radian) positioned at 2θ, and Bragg angle (in degree), respectively. With the inclusion of Sm3+ atoms, the lattice strain (ε) and dislocation density (δ) develop in the α-MoO3 lattice which can be determined from the Stokes–Wilson formula eqn (3) and eqn (4), respectively.62| |
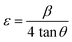 | (3) |
| |
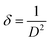 | (4) |
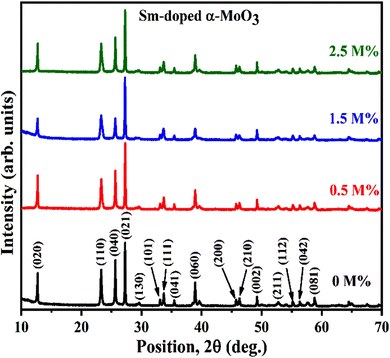 |
| | Fig. 2 XRD patterns of pristine and Sm-incorporated α-MoO3 nanoplates. | |
Table 1 Lattice parameters (a, b, c) of pristine and Sm-incorporated α-MoO3
| Sample (M%) |
Lattice parameters |
| a (Å) |
b (Å) |
c (Å) |
| 0% |
3.9675 |
13.9223 |
3.6981 |
| 0.5% |
3.9633 |
13.9351 |
3.6981 |
| 1.5% |
3.9638 |
13.9351 |
3.6983 |
| 2.5% |
3.9642 |
13.9128 |
3.6975 |
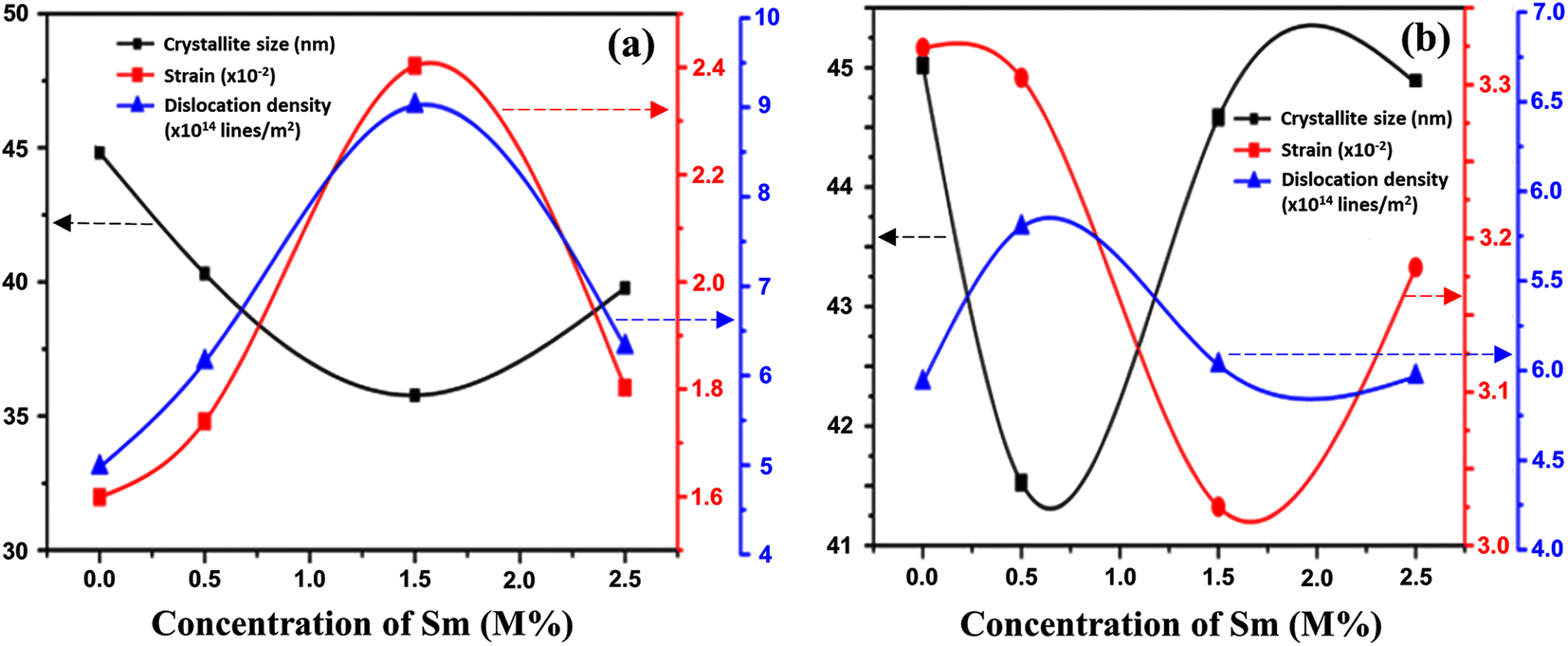
Table 2 summarizes the estimated crystallite sizes, dislocation density, lattice strain, and other parameters. According to Table 2, the average crystallite sizes (DD−S) considering all major peaks estimated from Debye–Scherrer formula is 44.8, 40.6, 35.8, and 39.8 nm for 0, 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3, respectively. The DD−S value declined steadily from 44.8 to 35.8 nm up to 1.5 M% Sm, then increased slightly for 2.5 M% but was still lower than the pristine sample. As a result, the incorporated samples exhibited reduced crystallites. It's important to note that the corresponding dislocation density and strain values follow the reverse way of DD−S assessed from Debye–Scherrer formula. To get more precise crystallographic properties, the Williamson-Hall (W-H) method is used.63 The W-H approach is the most often employed method that takes into account size (Scherer) broadening (βD), and strain broadening (βS). Using this technique, eqn (5) represents the individual contribution to the broadening of reflection.
Table 2 Structural parameters for pristine and Sm-incorporated α-MoO3
| Sample (M%) |
Debye–Scherer method |
W-H method |
|
|
| Strain, ε × 10−2 |
δ(lines per m2) × 1014 |
D (nm) |
Strain, εWH × 10−4 |
δ(lines per m2) × 1014 |
DWH (nm) |
SF |
Crystallinity (%) |
| 0% |
1.60 |
4.98 |
44.8 |
3.32 |
4.93 |
45.0 |
0.10 |
72.1% |
| 0.5% |
1.74 |
6.15 |
40.6 |
3.30 |
5.80 |
41.5 |
0.11 |
56.6% |
| 1.5% |
2.40 |
9.02 |
35.8 |
3.02 |
5.03 |
44.6 |
0.15 |
66.7% |
| 2.5% |
1.80 |
6.32 |
39.8 |
3.18 |
4.97 |
44.9 |
0.11 |
70.7% |
Applying the formula βD = 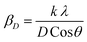 and βS = 4εtanθ, one gets eqn (6).
and βS = 4εtanθ, one gets eqn (6).
| |
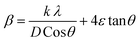 | (6) |
The crystallite size, DWH and lattice strain, εWH, are calculated using the following format (eqn (7)) of the above equation (eqn (6)):
| |
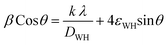 | (7) |
The linear equation (eqn (7)) is known as the W-H equation. As illustrated in Fig. 3, the lattice strain (εWH) and crystallite size (DWH) of pristine and Sm-incorporated α-MoO3 can be estimated from the slope and y-intercept of linear fitted eqn (7), respectively. Using Fig. 3, the measured values of crystallite size (DWH) are found to be 45.0, 41.5, 44.6, and 44.9 nm for 0, 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3, respectively. It can be noted that the reduction of DWH as well as the increment of εWH and δ resembles the Debye–Scherer trend. The varied nature of structural characteristics calculated using both methods are presented in Table 2 and the graphical representation is depicted in Fig. 4(a) and (b). According to calculated crystallite size values in both Debye–Scherrer and Williamson-Hall (W-H) formulae, a deterioration of crystallinity is observed as the crystallite size decreases from pristine to 1.5 M% Sm incorporated samples. The crystallite size is decreasing which might be the segregation of crystallites into smaller ones. In another way, the crystallite size decreases with increasing Sm amount, which may be attributed to the Sm cation interfering with the α-MoO3 crystalline lattice. This interference effect could be attributable to the deformation of the crystal lattice caused by the larger ionic radius of Sm3+ (1.04 Å) compared to Mo6+ (0.74–0.97 Å). The lattice expands as the Sm concentration rises, which is also caused by the higher ion radius of Sm than that of Mo resulting in the declination of crystallinity.64 Lattice strain is a measurement of the distribution of lattice constants brought on by crystal defects, particle size, dislocations, and oxygen vacancies, and more information regarding the number of crystal defects in the materials is revealed by the dislocation density. But for 2.5 M% Sm-incorporated α-MoO3, the strain and dislocation density showed lower values and inversely crystallite size increased slightly. Here, it was expected to the movement of defects and atoms for a larger amount (2.5 M%) of Sm, and thus consequently, a larger cluster was formed due to the agglomeration of minute crystallites by crumbling the grain boundary that enhances the average crystallite size.63 Some research groups reported a similar trend of change in crystallite size after doping. For example, Boukhachem et al.7 investigated the Sn-doped MoO3, and observed a considerable increase in crystallite size for 1 and 2 M% and then decreased for 3 and 4 M% Sn-doped MoO3. Similarly, Al-Otaibi et al.8 studied Zn-doped MoO3 nanobelts and found that the crystallite size increased from 42.63 to 46.56 nm for 2 wt% Zn doping, decreased to 45.30 nm for 2.5 wt% Zn, increased to 51.98 nm for 3.3 wt% Zn, and finally decreased to 43.07 nm for 5 wt% Zn. The stacking fault indicates the crystal imperfections follow the decrement trend of εWH and δ. The crystallinity trended in the same manner as the crystallite size. The intensity and full width at half maximum (FWHM) of the XRD peaks are widely considered indicators of crystallinity and crystallite size.
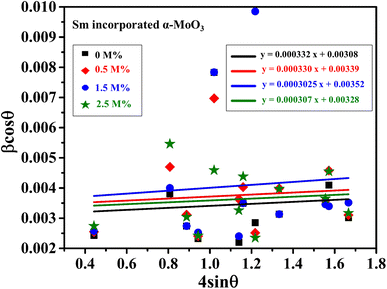 |
| | Fig. 3 W-H plot assuming UDM of 0.0, 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3. | |
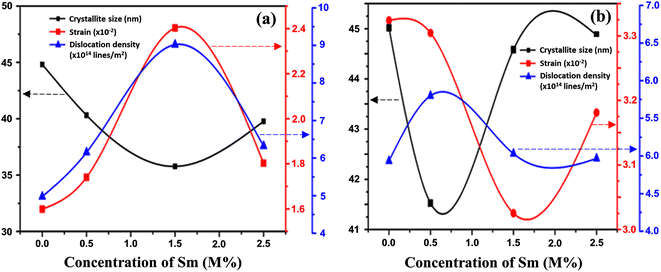 |
| | Fig. 4 Changes in strain, dislocation density, and crystallite size with Sm concentration variation in α-MoO3 using (a) Debye–Scherer, and (b) Williamson-Hall methods. | |
3.2 Surface morphology and elemental analysis
Fig. 5(a)–(d) show the FE-SEM images of surface morphology of pristine and Sm-incorporated α-MoO3 nanoplates. The multilayered heap structure was clearly constructed from a two-dimensional (2D) nanoplate-like structure of α-MoO3. To build these 2D layered α-MoO3 structures, these layers are stacked in a staggered pattern and possibly held together by weak van der Waals forces.65 In the FE-SEM micrographs shown in Fig. 5(a)–(d), the number of convolution layers ranges from three to forty-four. The thickness of a stack constructed with 44 layers is about 1 μm. A similar nanoplate morphology has been reported by various research groups.66,67 The FE-SEM images of the doped and pristine α-MoO3 samples show almost identical structures.
 |
| | Fig. 5 2D layered nanoplate type micrographs of (a) pristine (b) 0.5 M%; (c) 1.5 M% and (d) 2.5 M% Sm-incorporated α-MoO3. | |
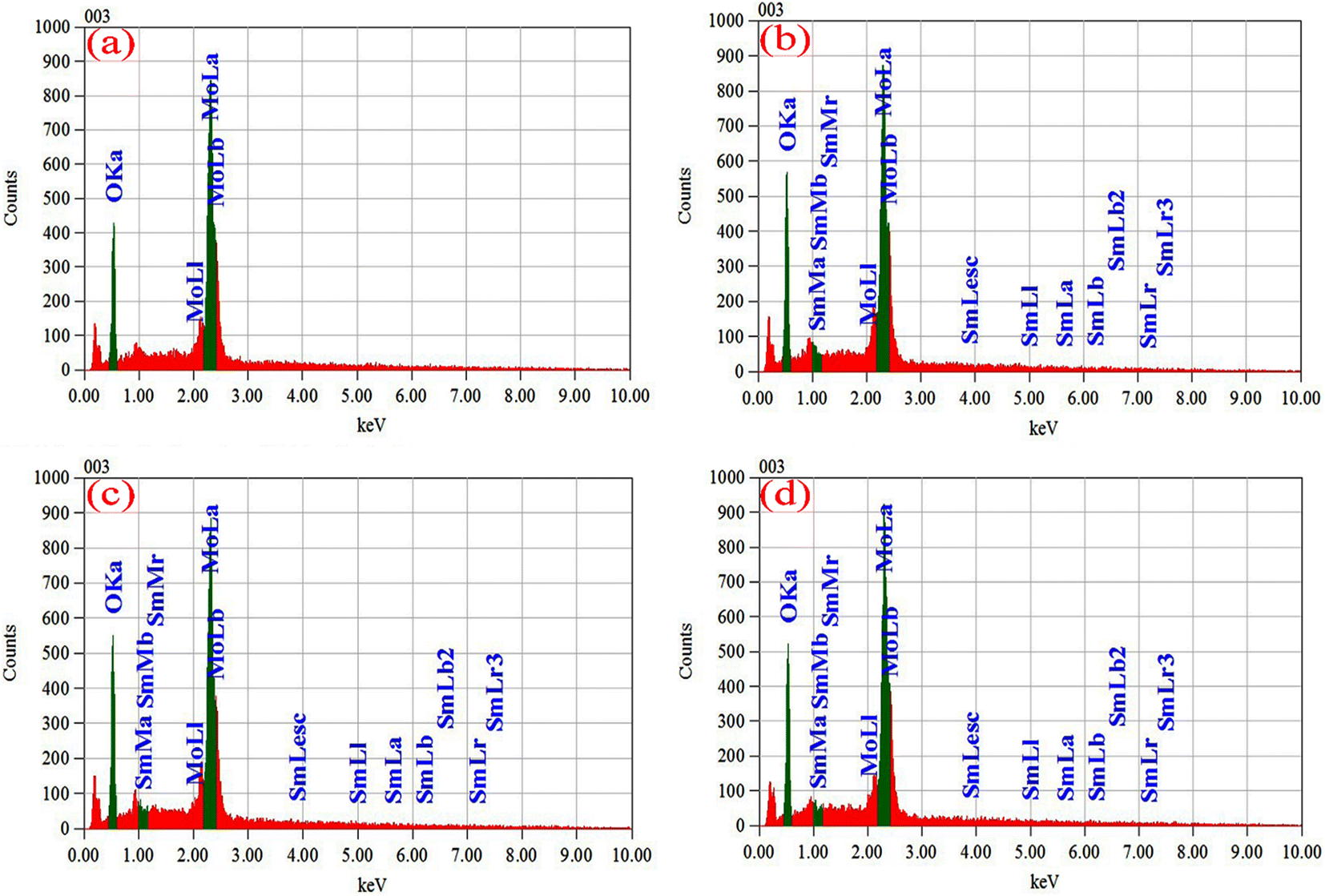
EDS has been used to determine the percentages of basic compositions present in the experimental samples. Fig. 6(a)–(d) show the EDS profile of pristine and doped MoO3 nanoplates respectively. The elemental mass and atoms' presence is shown in Table 3. In Fig. 6(a) the EDS spectra peak at 0.525 keV and 2.36 keV confirm oxygen (O) and molybdenum (Mo), respectively. The atomic percentage shows the molar ratio of Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O is around 1:3 confirming the stoichiometry of MoO3. The EDS peak at 2.36 keV in Fig. 6(b)–(d) shows the Sm-incorporation in α-MoO3 nanoplates. As the atomic percentages of Sm increase with doping percentage variation are clearly in Table 3 confirmed that Sm was successfully incorporated into the α-MoO3 matrix.
:O is around 1:3 confirming the stoichiometry of MoO3. The EDS peak at 2.36 keV in Fig. 6(b)–(d) shows the Sm-incorporation in α-MoO3 nanoplates. As the atomic percentages of Sm increase with doping percentage variation are clearly in Table 3 confirmed that Sm was successfully incorporated into the α-MoO3 matrix.
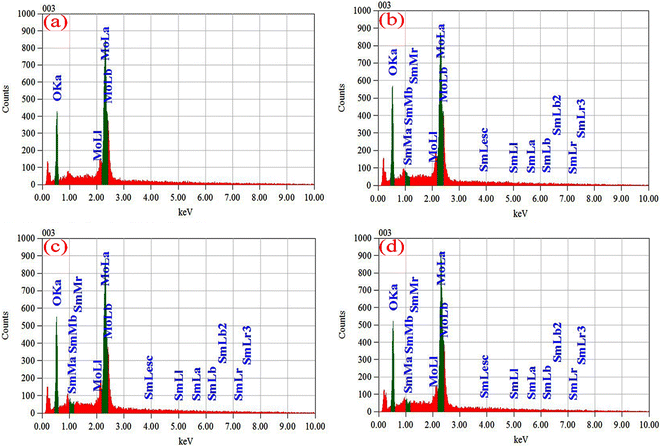 |
| | Fig. 6 EDX spectra of (a) pristine, (b) 0.5 M%, (c) 1.5 M%, (d) 2.5 M% Sm-incorporated α-MoO3 nanoplate. | |
Table 3 The mass% and atom% of pristine and Sm-incorporated α-MoO3
| Sample |
Elements |
Mass% |
Atom% |
| Pure α-MoO3 |
O |
37.40 |
78.18 |
| Mo |
62.60 |
21.82 |
| 0.5% Sm-incorporated α-MoO3 |
O |
30.48 |
72.45 |
| Mo |
68.67 |
27.26 |
| Sm |
0.85 |
0.29 |
| 1.5% Sm-incorporated α-MoO3 |
O |
38.73 |
78.66 |
| Mo |
60.15 |
20.89 |
| Sm |
1.12 |
0.45 |
| 2.5% Sm-incorporated α-MoO3 |
O |
29.03 |
70.31 |
| Mo |
69.27 |
28.96 |
| Sm |
1.70 |
0.73 |
3.3 Functional properties
Functional groups have been identified at room temperature using Fourier transform inferred (FTIR) spectroscopy. Fig. 7 reveals the FTIR spectra of all samples in the wavenumber range of 400 to 4000 cm−1 (desired range 1000 to 450 cm−1). All (pristine and Sm-incorporated) α-MoO3 samples show three main characteristic vibrational bands of orthorhombic crystal structure (α-MoO3) and no unwanted vibrational bands, confirming the chemical stability of the orthorhombic phase of as-synthesized materials after Sm incorporation and which results support the XRD examination. The two major vibrational zones (i.e., 1000–600 cm−1 and 600–400 cm−1) were observed in the spectra of all the samples. Four significant vibrational peaks are seen in the IR spectra of pristine α-MoO3 nanoplates, located at 570.45, 812.88, 861.06, and 982.58 cm−1, respectively; where a weaker peak is observed at 486.26 cm−1. For pristine α-MoO3, the sharp and intense peak at 982.58 cm−1 corresponds to the stretching vibration between oxygen and molybdenum atoms (Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), which confirms the possible layered orthorhombic phase (α phase) or structure of MoO3.67–69 This band is found at 981.82, 981.82, and 983.48 cm−1 for 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3, respectively.
O), which confirms the possible layered orthorhombic phase (α phase) or structure of MoO3.67–69 This band is found at 981.82, 981.82, and 983.48 cm−1 for 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3, respectively.
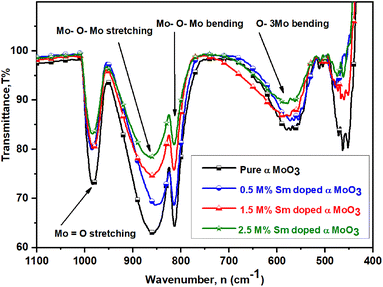 |
| | Fig. 7 FTIR spectra of pristine and Sm-incorporated α-MoO3 nanoplates. | |
The pristine sample has a highly-intense signal at 861.06 cm−1, which is attributed due to stretching vibrations of the bridge oxygen bond (O–Mo–O)56 and is seen at 860.15, 861.82, 861.82 cm−1 for 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3, accordingly. Interestingly, it is clearly seen that the peak intensity of stretching vibration of O(3) atoms in the Mo–O–Mo units gradually decreased with rising Sm concentration in α-MoO3 whereas almost no change in peak location is observed. A strongly sharp band is identified ascribed for Mo–O–Mo bending vibration located at the same position (812.88 cm−1) of pristine and all Sm-incorporated α-MoO3 samples although peak intensity sharply decreased with increasing Sm contents. The peak at 570.45 cm−1 is related to the bending vibrations of an oxygen atom (O(2)) coupled to three metal atoms ν(O–3Mo) in pristine α-MoO3 nanoplates70,71 and this bending vibration can also be detected at 572.27, 573.18, and 576.67 cm−1 in 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3, respectively. A faint peak at 461.97 cm−1 is attributed to Mo(2)–O(4) interaction with the water molecule and its strength declined as the Sm content increased.72 Table 4 indicates the comparison of functional groups of α-MoO3 nanoplates as a function of wavenumber values after Sm incorporation. From the above discussion, it is clearly concluded that Sm contents affect the peak position and intensity of FTIR spectra of α-MoO3 nanoplates without destroying the orthorhombic crystal structure.
Table 4 Functional groups in pristine and (0.5, 1.5, and 2.5 M%) Sm-incorporated α-MoO3
| Functional groups |
Wavenumber, n (cm−1) |
| 0 M% |
0.5 M% |
1.5 M% |
2.5 M% |
| MoO stretching vibration |
982.58 |
981.82 |
981.82 |
983.48 |
| Mo–O–Mo stretching vibration |
861.06 |
860.15 |
861.82 |
861.82 |
| Mo–O–Mo bending vibration |
812.88 |
812.88 |
812.88 |
812.88 |
| O–3Mo bending vibration |
570.45 |
572.27 |
573.18 |
576.67 |
3.4 Raman spectroscopy
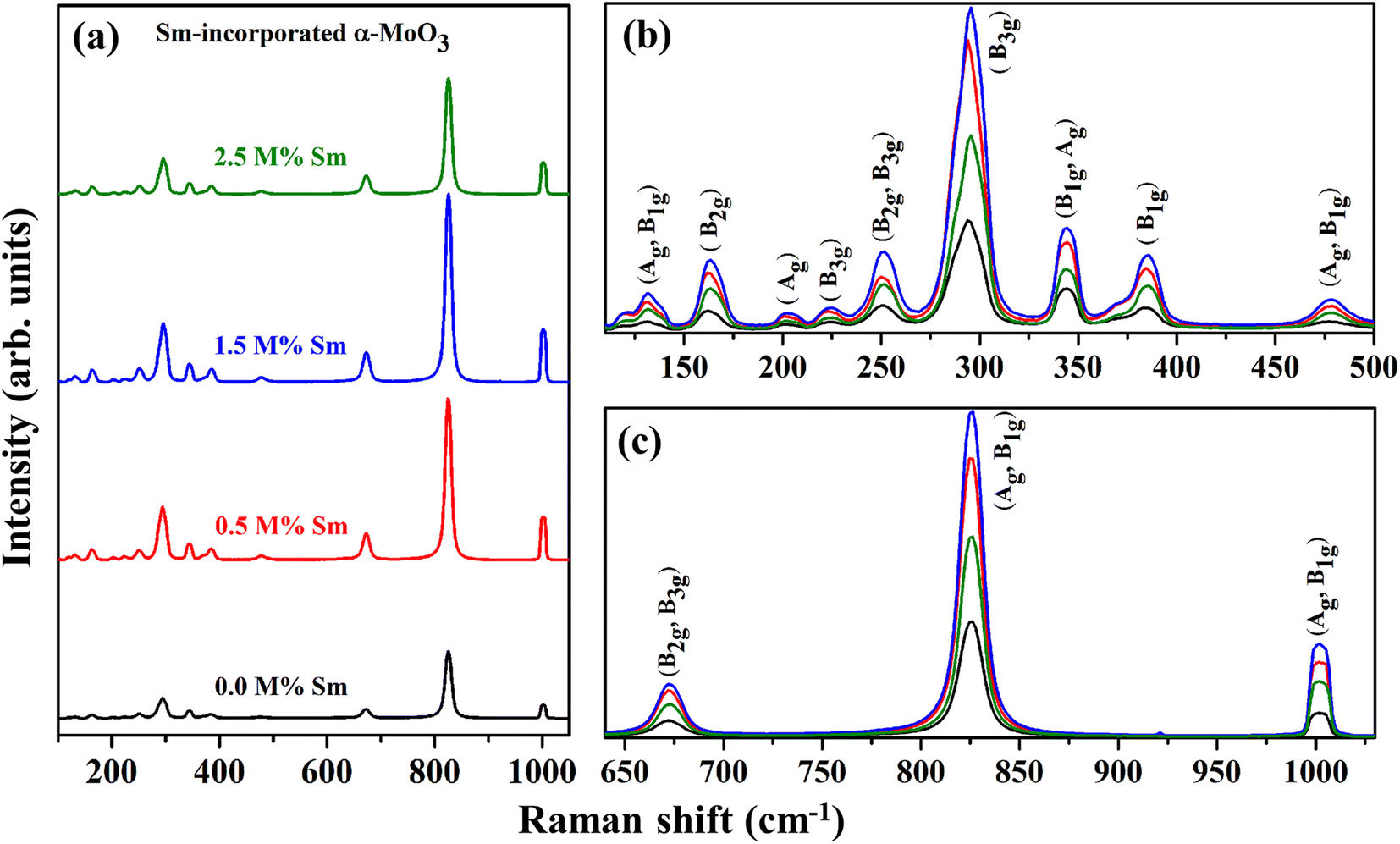
In addition to the information provided by XRD patterns, Raman spectroscopy is an effective technique that can be used to investigate further changes in the phase formation, defect generation (oxygen vacancy), and microstructural parameters of the prepared nanocrystalline pristine and Sm-incorporated α-MoO3 nanoplates due to Sm incorporation. Py and Maschke reported the Raman active phonon modes/characteristic Raman bands of the orthorhombic structure of molybdenum trioxide (space group: Pbnm) at 157 (Ag, B1g), 195 (B2g), 217 (Ag), 244 (B3g), 287 (B2g, B3g), 295 (B3g), 337 (B1g, Ag), 375 (B1g), 470 (Ag, B1g), 666 (B2g, B3g), 819 (Ag, B1g) and 995 (Ag, B1g).73 The room-temperature typical Raman spectra within the wavenumber range of 50–1100 cm−1 of pristine and Sm-incorporated α-MoO3 nanoplates were collected using 632.8 nm excitation and are shown in Fig. 8. The positions (Raman shift) of the Raman vibrational bands and their corresponding active phonon modes are presented in Table 5. All of the detected Raman bands exhibit inappropriate positions that are very similar to those described in the nature of the orthorhombic layered structure of molybdenum trioxide (α-MoO3) in the literature.74 The characteristic bands of molybdenum trioxide in the 1050–600 and 500–100 cm−1 ranges are predominantly due to Mo–O stretching (νas) and bending (δ) modes, according to the vibrational analysis of Raman spectra as depicted in Fig. 8(b) and (c), respectively. The distinct, sharply intense peaks corresponding to molybdenum trioxide suggest that all α-MoO3 have good crystallization.
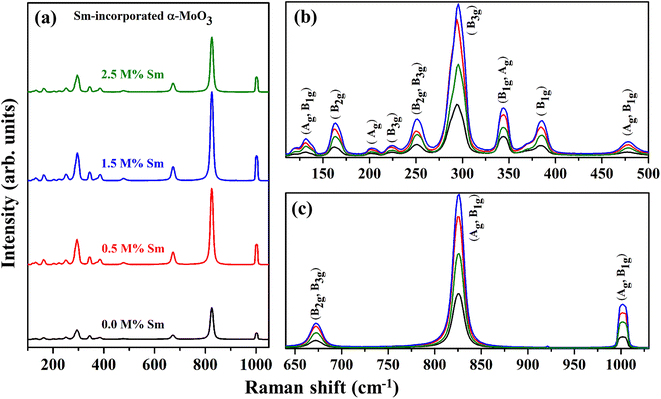 |
| | Fig. 8 Raman spectra (in the wavenumber range of (a) 100–1050 cm−1, (b) 75–500 cm−1, and (c) 640–1030 cm−1) of (0.0, 0.5, 1.5, and 2.5 M%) Sm-incorporated α-MoO3 nanoplates. | |
Table 5 Raman shift of active phonon modes in pristine and (0.5, 1.5, and 2.5 M%) Sm-incorporated α-MoO3
| Raman active phonon modes |
Assignment |
Raman shift (cm−1) |
| Standard (ref. 73,75) |
0% |
0.5% |
1.5% |
2.5% |
| (Ag, B1g) |
Translational rigid MoO4-chain mode, (Tb) |
157 |
132 |
131 |
131 |
132 |
| (B2g) |
Twisting mode of the terminal oxygen, (τ OMO twist) |
195 |
161 |
162 |
164 |
163 |
| (Ag) |
Rotational rigid MoO4-chain mode, (Rc) |
217 |
201 |
200 |
201 |
201 |
| (B3g) |
Twisting mode of the terminal oxygen, (τ OMO twist) |
244 |
225 |
223 |
225 |
224 |
| (B2g, B3g) |
Wagging modes of the terminal oxygen, (δ OMO waging) |
287 |
251 |
250 |
251 |
251 |
| (B3g) |
δ OMO waging |
295 |
294 |
293 |
295 |
294 |
| (B1g, Ag) |
δ O–M–O bend |
337 |
343 |
343 |
343 |
344 |
| (B1g) |
δ OMO scissor |
375 |
384 |
383 |
385 |
385 |
| (Ag, B1g) |
νas O–M–O stretch and bend |
470 |
477 |
477 |
479 |
477 |
| (B2g, B3g) |
Asymmetric stretching of Mo–O–Mo bridge along the c axis, (νas OMO stretch) |
666 |
673 |
682 |
673 |
677 |
| (Ag, B1g) |
Symmetric stretch of the terminal oxygen atoms, (νs OMO stretch) |
819 |
825 |
822 |
831 |
826 |
| (Ag, B1g) |
Asymmetric stretch of the terminal oxygen atoms, (νas MO) stretch |
995 |
1001 |
1002 |
1002 |
1001 |
The pristine samples exhibit Raman peaks at 132, 161, 201, 225, 251, 294, 343, 384, 477, 673, 825, and 1001 cm−1, all of which are indicative of the α-MoO3 phase.74 The most intense peak positioned at 1001 cm−1 (Ag, B1g) is ascribed due to the asymmetric stretch of the terminal oxygen atoms (νas Mo6+O) along the a- and b-axis of pristine α-MoO3. This is the main characteristic vibrational mode of the α-phase.41 The formation of oxygen vacancies should result in anion vacancies along the c-axis. As a result of the loss of bridging oxygen, the Mo atom is expected to shift towards the terminal oxygen in the b-direction, weakening the terminal bond along the a-axis.41,76 The phonon mode observed at 825 cm−1 (Ag, B1g) is assigned to the doubly coordinated oxygen (Mo–O–Mo) in the stretching mode of α-MoO3.77 Mo–O–Mo bridge is stretched asymmetrically along the c axis at 673 cm−1 (B2g, B3g). 343 and 384 cm−1 vibrational bands are attributed to δ (O–Mo–O) bending (B1g, Ag) mode and δ (O–Mo–O) scissoring (B1g) mode, respectively for pristine sample.48 The peak at 294 and 251 cm−1 are due to the wagging mode of the terminal oxygen atoms, denoted as δ (OMO). Some low intense Raman peaks located at the lower Raman shift as a function of wavenumber value (Fig. 8(b)) at 132, 161, 201, 225, and 477 cm−1 originated due to the translational rigid MoO4-chain mode (Tb) of (Ag, B1g) symmetry, twisting mode of the terminal oxygen τ (OMO twist) of (B2g), rotational rigid MoO4-chain mode (Rc) of (Ag), twisting mode of the terminal oxygen τ (O = MO twist) of (B3g) and symmetric νas O–M–O stretching and bending vibration of (Ag, B1g) mode, respectively.78 There is no extra Raman peak due to the incorporation of Sm3+, although the intensity of the phonon modes fluctuates with Sm admixing, indicating that Sm might only cause crystallographic defects and oxygen vacancies, not structural transformation.
3.5 Optical properties
Fig. 9(a) and (b) show the optical absorbance and reflectance spectra of pristine and Sm-incorporated α-MoO3 nanoplates in the wavelength range 200–800 nm at room temperature utilizing UV-vis-NIR diffuse reflectance spectroscopy (DRS). Pristine and Sm-incorporated α-MoO3 nanoplates exhibit high bandgap absorption in the UV spectra, as illustrated in Fig. 9(a) and (b). Absorption levels climb to peak values and then progressively decline over time in the visible range of the reflected light spectrum. An absorption band edge ranging from 375 to 425 nm was found in all samples, which may be due to the electronic bandgap transition. An explanation for the electronic bandgap transition is provided by the absorption of visible light by hybridized O-2p orbital with Mo 4d in the valence and conduction bands, respectively (which is mainly composed of Mo 4d orbital hybridized with O 2p).79,80 Here, for the α-MoO3 powder sample, Kubelka–Munk (K–M) function was utilized to estimate the optical bandgap (Eg) as follows (Eq. (8)).63
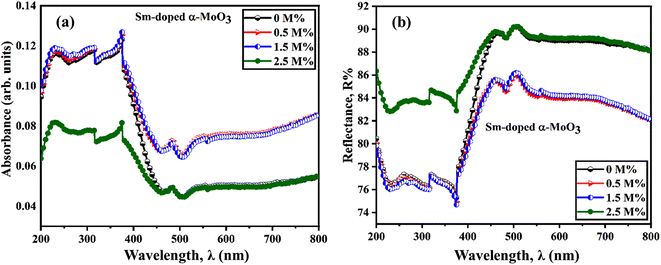 |
| | Fig. 9 (a) UV-vis absorbance spectra, and (b) UV-vis reflectance spectra derived from diffused reflectance spectroscopy of pristine and Sm-incorporated α-MoO3 nanoplates, respectively. | |
As it is known to us, absorption co-efficient (α) is proportional relation to the K–M function, it can be written as eqn (9).
| | |
F(R) ∝ α = (hν − Eg)n/hν
| (9) |
Where
F(
R) is the Kubelka–Munk (K–M) function,
R is the diffuse reflectance,
α is the absorption coefficient,
h is the plank's constant (6.626 × 10
−34 J s),
ν is light frequency,
hν is incident light energy, and
Eg is the optical bandgap of the nanoplates. The presence of a straight line in the spectra indicates the optical bandgap of the as-synthesized nanoplates is of direct type. The characterization of the optical transition process is indicated by the factor
n, and the value of
n depends on the transition modes.
n = 1/2, 2, 3/2, and 3 for direct allowed, indirect allowed, direct forbidden, and indirect forbidden modes, respectively.
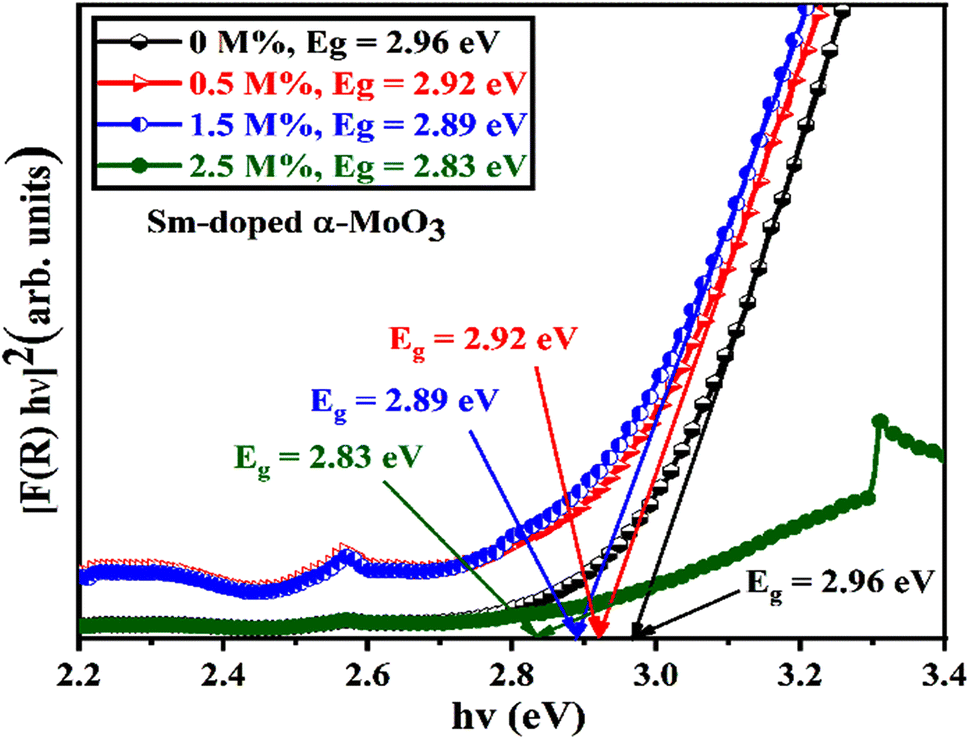
The optical band gap energy of the corresponding materials is calculated by extrapolating the linear portion of the plot of [F(R) × hν]2 versus hν, and the tangent line with [F(R) × hν]2 = 0.81 Fig. 10 shows that the direct bandgap energies (Eg) of pristine and 0.5, 1.5, and 2.5 M% Sm-incorporated α-MoO3 nanoplates are found to be 2.96, 2.92, 2.89, and 2.83, respectively (Table 6). The bandgap energy is gradually reduced from 2.96 to 2.83 eV after incrementing of samarium contents up to 2.5 M%. Sm incorporation changes the optical characteristics vis-a-vis pristine α-MoO3 nanoplates, as seen by the considerable shift of optical band gap energy into the visible region. It is generally known that the value of Eg in bulk semiconductors is constant, but in nanostructured materials it can fluctuate according to the size of crystallites or crystal defects. The red shift trend of Eg values with increased Sm contents could be explained by the overall reduction of crystallite size caused by an increase in defects, disorders, and lattice strain, which is consistent with the findings of X-ray diffraction as shown in Fig. 11. When the Mo6+ ion in the MoO3 matrix is replaced by Sm3+ ion, strain, defects, and charge imbalance might occur in the nanoplates. This may cause the valence band of incorporated nanoplates to broaden or the bandgap to narrow. In addition, the formation of sub-stoichiometric product,82,83 as well as oxygen vacancies capable of capturing one or two electrons may contribute to the lowering of optical bandgap; the oxygen vacancies occupied by electrons act as donor centers. As a consequence, the impurity band is pushed farther into the forbidden gap region. Since the optical bandgap of the α-MoO3 nanoplates has been reduced by substituting Sm3+ ions for Mo6+, this may be the reason for this reduction. In the forbidden gap area, Sm3+ ions may create an impurity band.84,85 Using Sm incorporation in α-MoO3 to induce impurity levels suggests that a novel antibonding orbital has been discovered. There were two types of molecular orbitals: bonding and antibonding. The new antibonding orbital with lower energy provided unoccupied orbitals between the valence and conduction bands. There were changes in the valence and conduction bands due to oxygen vacancies as well as lattice instability, which reduced the bandgap.78
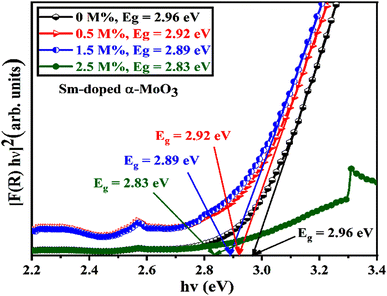 |
| | Fig. 10 [F(R) × hν]2 vs. incident photon energy, hν plots to calculate bandgap energy of pristine and Sm incorporated α-MoO3 nanoplates. | |
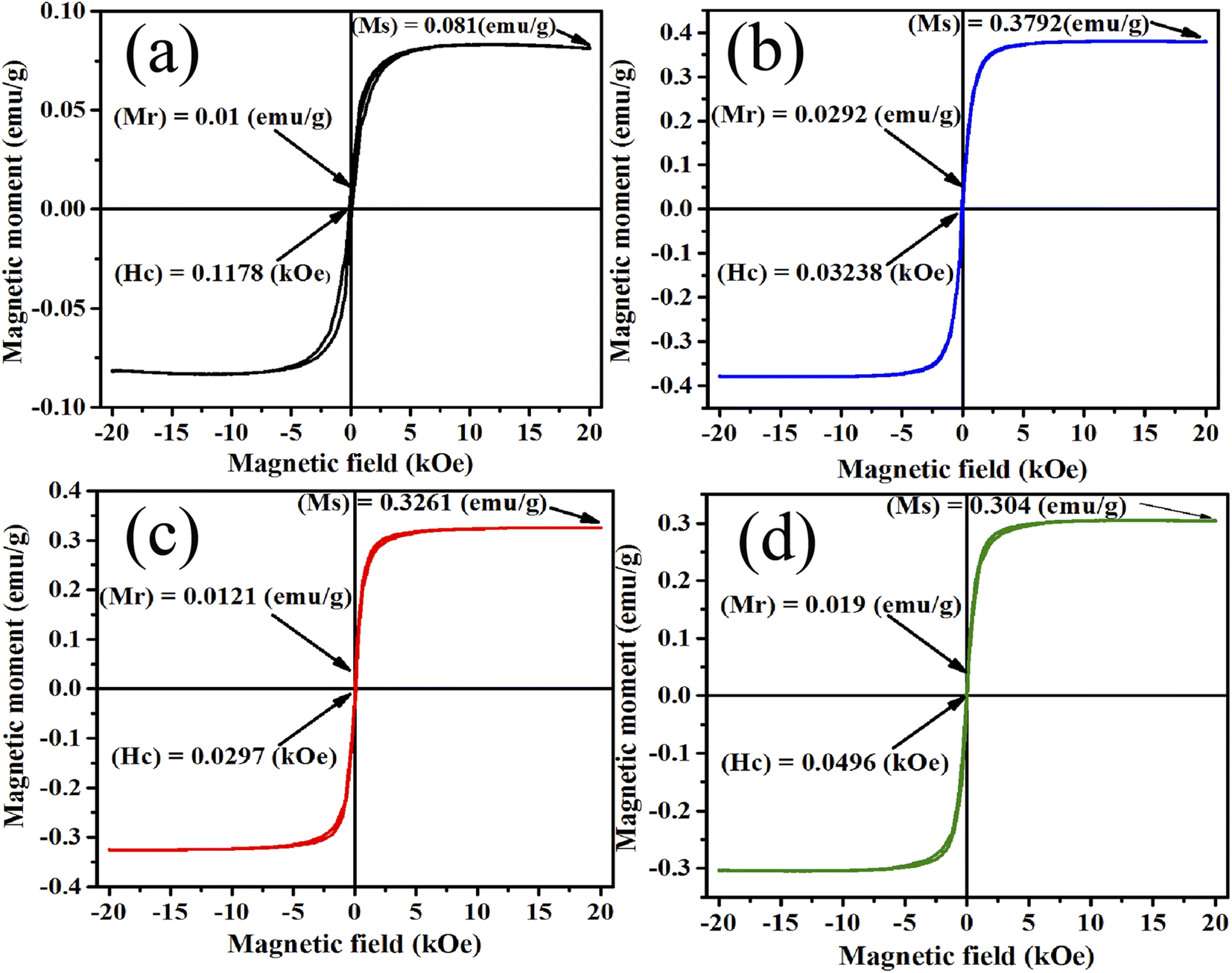
Table 6 Summary of the band gap energy, saturation magnetization (Ms), and coercivity (Hc) of the pristine and Sm-incorporated α-MoO3
| Sample (M%) |
Band gap energy (eV) |
Magnetic properties |
| Saturation magnetization, Ms(emu g−1) |
Remanence, Mr (emu g−1) |
Coercivity, Hc (kOe) |
| 0% |
2.96 |
0.0810 |
0.0100 |
0.1178 |
| 0.5% |
2.92 |
0.3792 |
0.0292 |
0.0323 |
| 1.5% |
2.89 |
0.3261 |
0.0121 |
0.0297 |
| 2.5% |
2.83 |
0.3040 |
0.0190 |
0.0496 |
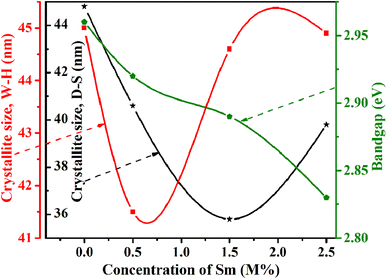 |
| | Fig. 11 Relation of crystallite size and bandgap with Sm concentration. | |
3.6 Magnetic properties
The PPMS technique was used to study the magnetic properties of pristine and samarium-incorporated 2D layered α-MoO3 nanoplates. The behavior of magnetism of all samples was observed from the typical magnetization hysteresis (M–H) loops depicted in Fig. 12 by plotting the magnetic moment (M) against the applied magnetic field (H).86 Magnetic properties such as saturation magnetization (Ms), remanence (Mr), and coercivity (Hc) of samples are listed in Table 6. All doped samples, including the pristine α-MoO3 nanoplates, exhibited ferromagnetic hysteresis behaviors at room temperature. Similarly, the ferromagnetic nature of pristine α-MoO3 was observed in several studies.30,87 However, some previous reports also mentioned that the stoichiometric MoO3 (bulk) is a paramagnetic semiconductor with no unpaired electron spins.88,89
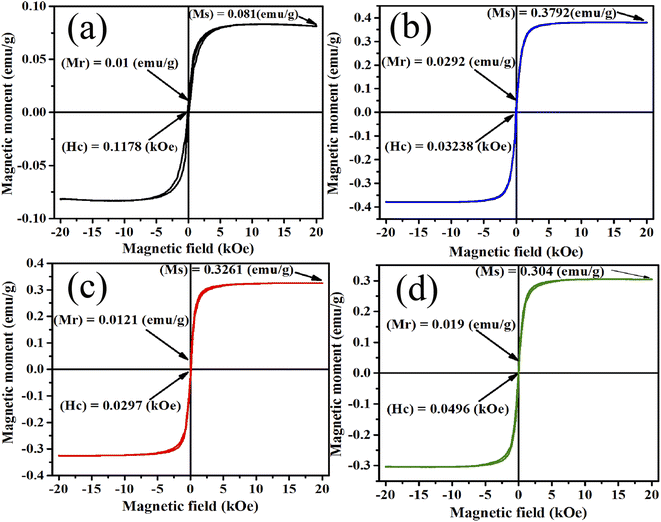 |
| | Fig. 12 M–H curve (a) 0% (b) 0.5% (c) 1.5% and (d) 2.5% Sm-incorporated α-MoO3. | |
In this experiment, the saturation magnetization (Ms) remarkably increased from 0.081 to 0.3792 emu g−1 after only 0.5% Sm incorporation in α-MoO3 and remains high with increasing doping concentration. However, the coercivity (Hc) decreased with Sm incorporation. Because of oxygen vacancies, the ferromagnetic behavior observed in the pristine sample could be related to the non-stoichiometry composition found in MoO3 nanoplates.29 Ferromagnetic property enhanced after Sm incorporation may be due to (i) defect formation after doping in MoO3 matrix, and (ii) generation of micro-cluster of Sm.90 The relatively large radius of Sm increased strain in the MoO3 matrix may lead to the segregation of Sm clusters in the doped sample. Raman and XRD studies show that the crystallinity has decreased; and the defects, oxygen vacancies, and faults increased with Sm doping concentration which clearly indicates the ferromagnetism enhancement at room temperature.
3.7 Antibacterial activity
Pristine and Sm-incorporated α-MoO3 nanoplates were examined to determine their efficacy as a potential material for various biological uses, primarily as an antibacterial agent due to their lower toxicity. Fig. 13 shows the antimicrobial properties as a function of the zone of inhibition of pristine and Sm-incorporated α-MoO3 nanoplates with varying concentrations against three bacteria. The images as a function of the inhibition zone seen in this figure are indicative of the results obtained after three repetitions of antimicrobial treatment of both nanoplate samples. According to the results, both nanomaterials are successful in producing a zone of inhibition against Gram-positive and Gram-negative bacterial strains, as depicted in Table 7. Nevertheless, Sm-incorporated α-MoO3 nanoplates were found to be more active against the examined bacteria than pristine nanoplates.
 |
| | Fig. 13 Antibacterial activity of pristine and Sm-incorporated α-MoO3 nanoplates against Gram-positive (S. aureus) and Gram-negative (E. coli and S. enteritidis) bacteria as a function of diameter (mm) of the zone of inhibition. | |
Table 7 Zone of inhibition (diameter in mm) caused by pristine and Sm-incorporated α-MoO3 nanoplates
| Organisms |
Conc. (ppm) |
Zone of inhibition (mm) |
| 0.0% Sm |
0.5% Sm |
1.5% Sm |
2.5% Sm |
| Staphylococcus aureus |
20000 |
12.56 |
13.47 |
11.25 |
14.74 |
| 40000 |
15.09 |
13.80 |
15.03 |
15.90 |
| 60000 |
15.61 |
17.12 |
16.15 |
16.21 |
| 80000 |
16.68 |
16.70 |
16.25 |
18.27 |
| Escherichia coli |
20000 |
N/A |
N/A |
10.01 |
9.31 |
| 40000 |
10.51 |
N/A |
10.96 |
10.21 |
| 60000 |
8.50 |
10.81 |
11.50 |
10.25 |
| 80000 |
11.45 |
10.87 |
11.52 |
10.29 |
| Salmonella enteritidis |
20000 |
N/A |
N/A |
N/A |
8.31 |
| 40000 |
8.34 |
9.01 |
9.71 |
8.76 |
| 60000 |
8.44 |
9.20 |
9.86 |
9.03 |
| 80000 |
9.03 |
9.33 |
9.91 |
10.35 |
In the context of pristine α-MoO3 nanoplates, the largest zone of inhibition (16.68 mm in diameter) was recorded against S. aureus for the concentration of 80000 ppm, which is good agreement with our previously reported largest zone of inhibition for orthorhombic molybdenum trioxide nanoplates.61 At a lower concentration (20000 ppm), the pristine sample was inactive against both Gram-negative bacterial strains such as E. coli and S. enteritidis, and no zone was identified in Petri plates. Therefore, the ZOI of bared α-MoO3 linearly depends on concentration and after increasing the concentration of molybdenum trioxide, its ZOI increased up to 11.45 and 9.03 mm for E. coli and S. enteritidis, respectively.
The antibacterial activity of α-MoO3 nanoplates showed its increased value of ZOI due to 0.5% Sm admixing against all examined bacteria and the observed highest ZOI is (17.12 mm) against S. aureus at the concentration of 80000 ppm. Although, no ZOI was found at comparatively lower concentrations against E. coli (20000 and 40000 ppm) and S. enteritidis (20000 ppm) for 0.5% Sm. Similar enhanced inhibiting bacterial growth or killing bacterial cells (highest 17.25 mm, against S. aureus at 80000 ppm) was identified for 1.5% Sm-incorporated α-MoO3 nanoplates. From Fig. 13 and Table 7, it is clearly seen that the highest ZOI (18.27 mm) was determined against S. aureus at the concentration of 80000 ppm for 2.5% Sm compared to other samples. As a result, these findings suggest the enhanced antibacterial effectiveness of α-MoO3 nanoplates due to samarium incorporation in the α-MoO3 matrix. It has been revealed that Sm-doped α-MoO3 nanoplates are highly effective and that the growth of bacteria is inhibited when concentrations of Sm-doped α-MoO3 nanoplates rise.
Understanding the interaction between the pure orthorhombic phase of MoO3 nanomaterials and bacterial cell walls, which results in cell wall degradation, is a crucial one.91 Several studies have suggested that the antibacterial properties of various nanomaterials are highly influenced by their morphologies (crystallite size and shape), crystallographic structure, surface defects, and composition of nanomaterials.2,9,92–94
In the case of metal oxide semiconductor nanomaterials, suitable doping element also plays a vital role in antimicrobial applications.95,96 Chandar et al. reported that the incorporation of the Ni element in the MoO3 matrix enhanced the antibacterial activity of MoO3 nanomaterials compared to the undoped sample.97 Ag also increased the effectiveness of MoO3 against bacterial strains, explored by Raj et al.95 Among various dopant elements, samarium is considered a less toxic and good one for enhancing the physical properties of transition metal oxides in the case of biological and other relevant applications.96 As a result, samarium doping may improve the antimicrobial activity of α-MoO3 nanoplates. The experimental results of the antibacterial activity of doped MoO3 nanomaterials compared with previous reports are shown in Table 8.
Table 8 Comparative assessment of antibacterial activity (highest ZOI) of doped MoO3 with earlier reports
| Sl. no. |
Materials |
Dopant |
Bacteria |
Zone of inhibition (mm) |
Ref. |
| Pure |
Doped |
| 1 |
h-MoO3 nanorods |
Ag |
B. cereus |
12 ± 0.4 |
07 ± 0.9 |
95 |
| C. koseri |
16 ± 0.7 |
22 ± 0.5 |
| P. aeruginosa |
17 ± 0.3 |
23 ± 0.4 |
| 2 |
α-MoO3 nanorods |
Ni |
E.coli |
16 ± 0.5 |
18 ± 0.5 |
97 |
| S. aureus |
16 ± 1 |
19 ± 0.5 |
| 3 |
α-MoO3 nanoplates |
Sm |
S. aureus |
16.68 |
18.27 |
Present |
| E. coli |
11.45 |
10.29 |
| S. enteritidis |
9.03 |
10.35 |
The antimicrobial activity of the Sm doped α-MoO3 nanoplates can be explained by three main mechanisms namely: (a) production of enhanced levels of reactive oxygen species (ROS)98 on the surface of α-MoO3 nanoplates, (b) release of Mo6+ ions from the α-MoO3 nanoplates, and (c) size of the nanomaterials.96,97,99 Fig. 13 clearly reveals inhibitory zones against bacterial strains, indicating that the Sm-doped α-MoO3 nanoplates have damaged the bacteria's membrane wall. Bacteria are destroyed when oxygen species are released from the surface of Sm-incorporated α-MoO3 nanoplates. Tables 2 and 7 show that the reduction in crystallite size of the Sm-incorporated α-MoO3 nanoplates increased the size of the inhibitory zone resulting in increased surface area to volume ratio and formation of more H2O2.95 The interstitial occupancy of the Sm-incorporated α-MoO3 lattice structure increased as the percentage of Sm increased, resulting in free carriers in the α-MoO3 lattice. The free carriers play a crucial role in antibacterial effectiveness. The antibacterial efficiency as a function of ZOI of examined Sm-incorporated α-MoO3 nanoplate samples is inversely propositional to crystallite size and linearly proportional to the surface area to volume ratio. Thus, the crystallite size of Sm-incorporated α-MoO3 2D layered type nanoplate orthorhombic structure has a significant impact on activity most likely due to a large number of nanomaterials with oxygen species accumulating in the cell membrane.
The substitution of Sm ions (Sm3+) into the Mo positions leads to a larger amount of Mo interstitials, causing more molybdenum ions to be released into the system. Interstitial Mo6+ ions are expected to have a higher chance of breaking out from the α-MoO3 lattice. The liberated Mo6+ ions may have a strong electrostatic interaction with bacterial cell membranes, causing significant bacterial cell destruction. Therefore, the formation of Mo6+ ions is also one of the causes of the enhancement of antimicrobial activities of Sm-incorporated α-MoO3 nanoplates. Furthermore, Sm ions (Sm3+) contribute to damage to the bacterial cell membranes.
Basically, the antibacterial activity of Sm-incorporated α-MoO3 2D layered nanoplates can be explained by the photocatalysis process under UV-vis irradiation depending on two mechanisms. Firstly, the physical process in which disruption of the bacterial membrane occurs by rupturing the potential and integrity due to surface interactions between the cell wall and nanoplates. Secondly, the chemical process includes the production of enhanced levels of ROS such as superoxide anion radical (*O−2), hydroxyl radical (*OH), and hydrogen peroxide radicals (*H2O2). The capacity of Sm-incorporated α-MoO3 nanoplates to generate ROS is the primary cause for their improved antibacterial activity. Generally, many transition metal oxides are sportive against micro-organisms due to the interaction of hydrophobic and electrostatic forces.100 The production of enhanced levels of these ROS can be explained as follows:96,97,99 when 2D layered nanoplates of pristine and Sm-incorporated α-MoO3 are exposed to light with the energy of photon equal to or higher than the energy bandgap of the α-MoO3 semiconductor, electrons from the valence band (VB) are transferred to the conduction band (CB), leaving holes in the valence band (eqn (10)). These excited electrons (e−) in the CB can be trapped by the oxygen molecules (O2) present on the surface, producing superoxide anion radicals (*O2−) (eqn (11)). Simultaneously, the holes (h+) in the VB reacts with the water molecules (H2O), emitting hydroxyl ion (OH−) and hydrogen ion (H+) (eqn (12)). The superoxide anion radicals (*O2−) thereafter interacts with H+ to produce hydroperoxyl radical (*HO2) (eqn (13)). The hydroperoxyl radical (*HO2) combines with e− and H+ once more, resulting in hydrogen peroxide radical (*H2O2) (eqn (14)). At the same time, hydrogen peroxide radical (*H2O2) can react with superoxide anion radicals (*O2−) to generate hydroxyl radical (*OH), hydroxyl ion (OH−) and singlet oxygen (1O2) (eqn (15)). The reactions that occur in this mechanism are illustrated in the following eqn (10–15):
| | |
MoO3 + hn(photon) → h+(hole) + e−(excited electron) + Mo6+
| (10) |
| | |
e−(excited electron) + O2(surface oxygen molecule) → *O−2(superoxide anion radical)
| (11) |
| | |
h+(hole) + H2O(water molecule) → OH− + H+
| (12) |
| | |
*O2−(superoxide anion radical) + H+ → *HO2(hydroperoxyl radical)
| (13) |
| | |
*HO2(hydroperoxyl radical) + e−(excited electron) + H+ → *H2O2(hydrogen peroxide radical)
| (14) |
| | |
*H2O2(hydrogen peroxide radical) + *O2−(superoxide anion radical) → *OH(hydroxyl radical) + O2 (singlet oxygen) + OH−(hydroxyl ion)
| (15) |
The electron–hole pairs serve as the primary unit for the generation of ROS. The gradually decreasing optical band gap (from 2.96 to 2.83 eV) accelerates this ROS-producing mechanism by creating more electron–hole pairs as the Sm admixing level increases in the α-MoO3 semiconductor. The negatively charged radicals (hydroxyl radicals and superoxide anions) cannot penetrate the cell membrane of micro-organisms, but they can induce critical damage to the bacteria's outer surface, whereas positively charged hydrogen peroxide radicals (*H2O2) can penetrate easily through the negatively charged bacterial cell membranes and kill the bacteria. Therefore, hydrogen peroxide radical (*H2O2) is highly harmful to damaging bacterial cell membranes. Further, the superoxide dismutase (SOD) mimetic/catalase assay analysis would be helpful to understand the levels of ROS with the incorporation of Sm. The detailed mechanism of SOD and catalase mimic activities of MoO3-based materials like MoO3−x nanodots were explained by Han et al.101 It is worth noting that when the Sm level was increased up to 2.5 M%, in the α-MoO3 semiconductor, the antibacterial efficiency against all three micro-organisms was found to be higher than that of the pristine. In this experiment, the improved antibacterial activity of α-MoO3 nanoplates after Sm-incorporation is due to the following factors: decreased crystallite sizes, formation of defects and oxygen vacancies, two-dimensional plate type layered shape, improved ferromagnetic behavior, sinking optical band gap, increased levels of ROS, and (Mo6+ and Sm3+) ions formation from the α-MoO3 nanoplates.
4 Summary
Here, we have reported the impact of Sm contents on the physical properties and antibacterial activity of hydrothermally synthesized α-MoO3 nanoplates. The single-phase orthorhombic structure with the polycrystalline nature was revealed by XRD patterns, supported by FTIR and Raman results. The overall crystallite size decreased with the increment of Sm contents, resulting in an increment of defects, strain, and dislocation density. The EDX spectra confirmed the Sm incorporation and the plate-type 2D layered shape was identified from the FE-SEM micrograph. The bandgap energy decreased gradually from 2.96 to 2.83 eV as the Sm content increased. The measured magnetic hysteresis loops lead to the ferromagnetic behavior of α-MoO3 nanoplates. The room temperature ferromagnetism of 2.5 M% Sm α-MoO3 increased compared to the pristine sample which is attributed to the existence of a large number of oxygen vacancies, supported by Raman studies as well. In comparison to the pristine one, the antibacterial activity enhanced as Sm contents increased in both Gram-positive (S. aureus) and Gram-negative (E. coli and S. enteritidis) bacteria. The reason might cause the decreased crystallite sizes, formation of defects and oxygen vacancies, 2D plate type layered morphology, enhanced ferromagnetic behavior, lowering optical band gap, enhanced levels of ROS, and (Mo6+ and Sm3+) ions formation in the α-MoO3 nanoplates. The investigated opto-structural and magnetic properties suggested that the synthesized Sm-incorporated 2D layered α-MoO3 nanoplates might be suitable for photocatalysts, magneto-optic, and spintronic nanodevice applications. The enhanced antibacterial activity postulated that Sm-incorporated 2D layered α-MoO3 nanoplates could be an innovative antibacterial approach for eradicating microorganisms in medicinal as well as public health sectors.
Data availability
All data used to evaluate the conclusion of this study are presented in the manuscript. Additional data can be available from the corresponding authors upon reasonable request.
Author's contributions
S. K. Sen: conceptualization, methodology, software, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, supervision, project administration; M. R. Munshi: software, writing – original draft; A. Kumar: investigation, data curation; A. A. Mortuza: writing – review & editing, software; M. S. Manir: resources, investigation, data curation; M. A. Islam: writing – review & editing; M. N. Hussain: data curation; M. K. Hossain: software, visualization; validation, writing – review & editing.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Colloids Surf., B, 2013, 112, 521–524 CrossRef CAS PubMed.
- K. Krishnamoorthy, M. Premanathan, M. Veerapandian and S. Jae Kim, Nanotechnology, 2014, 25, 315101 CrossRef PubMed.
- M. I. Anik, M. K. Hossain, I. Hossain, A. M. U. B. Mahfuz, M. T. Rahman and I. Ahmed, Nano Sel., 2021, 2, 1146–1186 CrossRef CAS.
- M. Moritz and M. Geszke-Moritz, Chem. Eng. J., 2013, 228, 596–613 CrossRef CAS.
- S. Khan and M. K. Hossain, in Nanoparticle-Based Polymer Composites, Elsevier, 2022, pp. 15–54 Search PubMed.
- M. I. Anik, M. K. Hossain, I. Hossain, I. Ahmed and R. M. Doha, in Magnetic Nanoparticle-Based Hybrid Materials, Elsevier, 2021, pp. 463–497 Search PubMed.
- N. Mahmud, M. I. Anik, M. K. Hossain, M. I. Khan, S. Uddin, M. Ashrafuzzaman and M. M. Rahaman, ACS Appl. Bio Mater., 2022, 5, 2431–2460 CrossRef CAS PubMed.
- R. D. Holtz, B. A. Lima, A. G. Souza Filho, M. Brocchi and O. L. Alves, Nanomedicine, 2012, 8, 935–940 CrossRef CAS PubMed.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27, 4020–4028 CrossRef CAS PubMed.
- S. O. Ali Ahmad, M. Ikram, M. Imran, S. Naz, A. Ul-Hamid, A. Haider, A. Shahzadi and J. Haider, RSC Adv., 2021, 11, 23330–23344 RSC.
- U. Qumar, M. Ikram, M. Imran, A. Haider, A. Ul-Hamid, J. Haider, K. N. Riaz and S. Ali, Dalton Trans., 2020, 49, 5362–5377 RSC.
- Y.-N. Chang, M. Zhang, L. Xia, J. Zhang and G. Xing, Materials, 2012, 5, 2850–2871 CrossRef CAS.
- A. M. Allahverdiyev, E. S. Abamor, M. Bagirova and M. Rafailovich, Future Microbiol., 2011, 6, 933–940 CrossRef CAS PubMed.
- R. Kumar, A. Umar, G. Kumar and H. S. Nalwa, Ceram. Int., 2017, 43, 3940–3961 CrossRef CAS.
- A. Sirelkhatim, S. Mahmud, A. Seeni, N. H. M. Kaus, L. C. Ann, S. K. M. Bakhori, H. Hasan and D. Mohamad, Nano-Micro Lett., 2015, 7, 219–242 CrossRef CAS PubMed.
- S. M. Dizaj, F. Lotfipour, M. Barzegar-Jalali, M. H. Zarrintan and K. Adibkia, Mater. Sci. Eng., C, 2014, 44, 278–284 CrossRef CAS PubMed.
- A. Bouafia and S. E. Laouini, Mini-Rev. Org. Chem., 2021, 18, 725–734 CrossRef CAS.
- S. Yadav and G. Jaiswar, J. Chin. Chem. Soc., 2017, 64, 103–116 CrossRef CAS.
- J. He, P. Xu, R. Zhou, H. Li, H. Zu, J. Zhang, Y. Qin, X. Liu and F. Wang, Adv. Electron. Mater., 2022, 8, 2100997 CrossRef CAS.
- H. Zhang, Y. Meng, L. Song, L. Luo, Y. Qin, N. Han, Z. Yang, L. Liu, J. C. Ho and F. Wang, Nano Res., 2018, 11, 1227–1237 CrossRef CAS.
- T. Dietl, Nat. Mater., 2010, 9, 965–974 CrossRef CAS PubMed.
- F. Pulizzi, Nat. Mater., 2010, 9, 955–956 CrossRef CAS PubMed.
- M. K. Hossain, M. F. Pervez, M. N. H. Mia, S. Tayyaba, M. J. Uddin, R. Ahamed, R. A. Khan, M. Hoq, M. A. Khan and F. Ahmed, Mat. Sci., 2017, 35, 868–877 CAS.
- N. H. Mia, S. M. Rana, F. Pervez, M. R. Rahman, K. Hossain, A. Al Mortuza, M. K. Basher and M. Hoq, Mat. Sci., 2017, 35, 501–510 CAS.
- M. T. Rahman, M. A. Hoque, G. T. Rahman, M. M. Azmi, M. A. Gafur, R. A. Khan and M. K. Hossain, Radiat. Eff. Defects Solids, 2019, 174, 480–493 CrossRef CAS.
- M. N. H. Mia, U. Habiba, M. F. Pervez, H. Kabir, S. Nur, M. F. Hossen, S. K. Sen, M. K. Hossain, M. A. Iftekhar and M. M. Rahman, Appl. Phys. A, 2020, 126, 162 CrossRef CAS.
- M. Ali, S. Sharif, S. Anjum, M. Imran, M. Ikram, M. Naz and S. Ali, Mater. Res. Express, 2020, 6, 1250d5 CrossRef.
- B. Choudhury, R. Verma and A. Choudhury, RSC Adv., 2014, 4, 29314 RSC.
- A. Boukhachem, M. Mokhtari, N. Benameur, A. Ziouche, M. Martínez, P. Petkova, M. Ghamnia, A. Cobo, M. Zergoug and M. Amlouk, Sens. Actuators, A, 2017, 253, 198–209 CrossRef CAS.
- O. Kamoun, A. Boukhachem, S. Alleg, B. Jeyadevan and M. Amlouk, J. Alloys Compd., 2018, 741, 847–854 CrossRef CAS.
- M. Imran, M. Ikram, A. Shahzadi, S. Dilpazir, H. Khan, I. Shahzadi, S. A. Yousaf, S. Ali, J. Geng and Y. Huang, RSC Adv., 2018, 8, 18051–18058 RSC.
- J. Hassan, M. Ikram, A. Ul-Hamid, M. Imran, M. Aqeel and S. Ali, Nanoscale Res. Lett., 2020, 15, 75 CrossRef CAS PubMed.
- M. Nafees, M. Ikram and S. Ali, Appl. Nanosci., 2017, 7, 399–406 CrossRef CAS.
- D. Sundeep, A. Gopala Krishna, R. V. S. S. N. Ravikumar, T. Vijaya Kumar, S. Daniel Ephraim and Y. L. Pavan, Int. Nano Lett., 2016, 6, 119–128 CrossRef CAS.
- D. Xiang, C. Han, J. Zhang and W. Chen, Sci. Rep., 2015, 4, 4891 CrossRef PubMed.
- A. L. F. Cauduro, R. dos Reis, G. Chen, A. K. Schmid, C. Méthivier, H.-G. Rubahn, L. Bossard-Giannesini, H. Cruguel, N. Witkowski and M. Madsen, ACS Appl. Mater. Interfaces, 2017, 9, 7717–7724 CrossRef PubMed.
- A. Arfaoui, S. Touihri, A. Mhamdi, A. Labidi and T. Manoubi, Appl. Surf. Sci., 2015, 357, 1089–1096 CrossRef CAS.
- A. Phuruangrat, U. Cheed-Im, T. Thongtem and S. Thongtem, Mater. Lett., 2016, 173, 158–161 CrossRef CAS.
- A. Hojabri, F. Hajakbari and A. E. Meibodi, J. Theor. Appl. Phys., 2015, 9, 67–73 CrossRef.
- S. K. Sen, M. S. Manir, S. Dutta, M. H. Ali, M. N. I. Khan, M. A. Matin and M. A. Hakim, Thin Solid Films, 2020, 693, 137700 CrossRef CAS.
- O. Kamoun, A. Boukhachem, M. Amlouk and S. Ammar, J. Alloys Compd., 2016, 687, 595–603 CrossRef CAS.
- S. K. Sen, U. C. Barman, M. S. Manir, P. Mondal, S. Dutta, M. Paul, M. A. M. Chowdhury and M. A. Hakim, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2020, 11, 025004 CAS.
- A. Phuruangrat, U. Cheed-Im, T. Thongtem and S. Thongtem, Mater. Lett., 2016, 172, 166–170 CrossRef CAS.
- S. Bai, C. Chen, D. Zhang, R. Luo, D. Li, A. Chen and C.-C. Liu, Sens. Actuators, B, 2014, 204, 754–762 CrossRef CAS.
- Z. Li, W. Wang, Z. Zhao, X. Liu and P. Song, RSC Adv., 2017, 7, 28366–28372 RSC.
- A. Phuruangrat, S. Thipkonglas, T. Thongtem and S. Thongtem, Mater. Lett., 2017, 195, 37–40 CrossRef CAS.
- S. Yang, Y. Liu, T. Chen, W. Jin, T. Yang, M. Cao, S. Liu, J. Zhou, G. S. Zakharova and W. Chen, Appl. Surf. Sci., 2017, 393, 377–384 CrossRef CAS.
- G. Sanal Kumar, N. Illyaskutty, S. Suresh, R. S. Sreedharan, V. U. Nayar and V. P. M. Pillai, J. Alloys Compd., 2017, 698, 215–227 CrossRef CAS.
- M. K. Hossain, M. H. Ahmed, M. I. Khan, M. S. Miah and S. Hossain, ACS Appl. Electron. Mater., 2021, 3, 4255–4283 CrossRef CAS.
- M. K. Hossain, M. I. Khan and A. El-Denglawey, Appl. Mater. Today, 2021, 24, 101104 CrossRef.
- M. K. Hossain, G. A. Raihan, M. A. Akbar, M. H. Kabir Rubel, M. H. Ahmed, M. I. Khan, S. Hossain, S. K. Sen, M. I. E. Jalal and A. El-Denglawey, ACS Appl. Electron. Mater., 2022, 4, 3327–3353 CrossRef CAS.
- M. K. Hossain, S. Hossain, M. H. Ahmed, M. I. Khan, N. Haque and G. A. Raihan, ACS Appl. Electron. Mater., 2021, 3, 3715–3746 CrossRef CAS.
- M. K. Hossain, M. H. K. Rubel, M. A. Akbar, M. H. Ahmed, N. Haque, M. F. Rahman, J. Hossain and K. M. Hossain, Ceram. Int., 2022, 48, 32588–32612 CrossRef CAS.
- A. Mehtab, J. Ahmed, S. M. Alshehri, Y. Mao and T. Ahmad, Nanotechnology, 2022, 33, 142001 CrossRef PubMed.
- P. A. Arasu and R. V. Williams, Surf. Rev. Lett., 2015, 22, 1550054 CrossRef CAS.
- A. Chithambararaj and A. C. Bose, J. Alloys Compd., 2011, 509, 8105–8110 CrossRef CAS.
- J. Ortiz-Landeros, C. Gómez-Yáñez, R. López-Juárez, I. Dávalos-Velasco and H. Pfeiffer, J. Adv. Ceram., 2012, 1, 204–220 CrossRef CAS.
- M. K. Basher, M. K. Hossain, M. J. Uddin, M. A. R. Akand and K. M. Shorowordi, Optik, 2018, 172, 801–811 CrossRef CAS.
- M. K. Basher, M. K. Hossain, R. Afaz, S. Tayyaba, M. A. R. Akand, M. T. Rahman and N. M. Eman, Results Phys., 2018, 10, 205–211 CrossRef.
- J. S. Chen, Y. L. Cheah, S. Madhavi and X. W. Lou, J. Phys. Chem. C, 2010, 114, 8675–8678 CrossRef CAS.
- S. K. Sen, S. Dutta, M. R. Khan, M. S. Manir, S. Dutta, A. Al Mortuza, S. Razia and M. A. Hakim, Bionanoscience, 2019, 9, 873–882 CrossRef.
- S. K. Sen, M. A. Jalil, M. Hossain, M. S. Manir, K. Hoque, M. A. Islam and M. N. Hossain, Mater. Today Commun., 2021, 27, 102404 CrossRef CAS.
- S. K. Sen, S. Dutta, L. Paik, T. C. Paul, M. S. Manir, M. Hossain and M. N. Hossain, J. Alloys Compd., 2021, 876, 160070 CrossRef CAS.
- H. Y. He, J. Fei and J. Lu, J. Nanostruct. Chem., 2015, 5, 169–175 CrossRef CAS.
- L. Zheng, Y. Xu, D. Jin, Y. Xie, Z. Lei, X. Yang, J. Dong and X. Yi, Chem. Mater., 2009, 21, 5681–5690 CrossRef CAS.
- K. Gomathi, S. Padmanathan, A. M. Ali and A. T. Rajamanickam, Inorg. Chem. Commun., 2022, 135, 109079 CrossRef CAS.
- Y. Chen, C. Lu, L. Xu, Y. Ma, W. Hou and J.-J. Zhu, CrystEngComm, 2010, 12, 3740 RSC.
- P. Wongkrua, T. Thongtem and S. Thongtem, J. Nanomater., 2013, 2013, 1–8 CrossRef.
- G. S. Zakharova, C. Täschner, V. L. Volkov, I. Hellmann, R. Klingeler, A. Leonhardt and B. Büchner, Solid State Sci., 2007, 9, 1028–1032 CrossRef CAS.
- C. D. A. Lima, J. V. B. Moura, G. S. Pinheiro, J. F. D. F. Araujo, S. B. S. Gusmão, B. C. Viana, P. T. C. Freire and C. Luz-Lima, Ceram. Int., 2021, 47, 27778–27788 CrossRef CAS.
- L. G. Pereira, L. E. B. Soledade, J. M. Ferreira, S. J. G. Lima, V. J. Fernandes, A. S. Araújo, C. A. Paskocimas, E. Longo, M. R. C. Santos, A. G. Souza and I. M. G. Santos, J. Alloys Compd., 2008, 459, 377–385 CrossRef CAS.
- Y. Muraoka, J.-C. Grenier, S. Petit and M. Pouchard, Solid State Sci., 1999, 1, 133–148 CrossRef CAS.
- M. A. Py and K. Maschke, Physica B+C, 1981, 105, 370–374 CrossRef CAS.
- M. Dieterle and G. Mestl, Phys. Chem. Chem. Phys., 2002, 4, 822–826 RSC.
- S. K. S. Patel, K. Dewangan and N. S. Gajbhiye, J. Mater. Sci. Nanotechnol., 2015, 31, 453–457 CrossRef CAS.
- G. Mestl, N. F. D. Verbruggen, E. Bosch and H. Knözinger, Langmuir, 1996, 12, 2961–2968 CrossRef CAS.
- G. M. Ramans, J. V. Gabrusenoks, A. R. Lusis and A.
A. Patmalnieks, J. Non-Cryst. Solids, 1987, 90, 637–640 CrossRef CAS.
- M. Vila, C. Díaz-Guerra, D. Jerez, K. Lorenz, J. Piqueras and E. Alves, J. Phys. D: Appl. Phys., 2014, 47, 355105 CrossRef.
- A. Chithambararaj, N. S. S. S. Sanjini, A. C. C. C. Bose and S. Velmathi, Catal. Sci. Technol., 2013, 3, 1405 RSC.
- A. Chithambararaj, D. Bhagya Mathi, N. Rajeswari Yogamalar and A. Chandra Bose, Mater. Res. Express, 2015, 2, 055004 CrossRef.
- S. K. Sen, M. Noor, M. A. Al Mamun, M. S. Manir, M. A. Matin, M. A. Hakim, S. Nur and S. Dutta, Opt. Quantum Electron., 2019, 51, 82 CrossRef.
- J. Okumu, F. Koerfer, C. Salinga, T. P. Pedersen and M. Wuttig, Thin Solid Films, 2006, 515, 1327–1333 CrossRef CAS.
- V. Madhavi, P. Kondaiah, S. S. Rayudu, O. M. Hussain and S. Uthanna, Mater. Express, 2013, 3, 135–143 CrossRef CAS.
- A. Arunachalam, S. Dhanapandian, C. Manoharan and G. Sivakumar, Spectrochim. Acta, Part A, 2015, 138, 105–112 CrossRef CAS PubMed.
- E. Ghaleghafi and M. B. Rahmani, Solid State Sci., 2019, 94, 85–91 CrossRef CAS.
- M. H. K. Rubel and M. K. Hossain, in Fundamentals of Low Dimensional Magnets, CRC Press, Boca Raton, 2022, pp. 183–205 Search PubMed.
- N. Benameur, A. Boukhachem, M. Ghamnia, M. A. Chakhoum, M. A. Dahamni and C. Fauquet, Int. J. Chem. Eng. Appl., 2019, 10, 33–39 CAS.
- H. Ueda, N. Todo and M. Kurita, J. Less-Common Met., 1974, 36, 387–394 CrossRef CAS.
- A. Agarwal, S. Khasa, V. P. Seth, S. Sanghi and M. Arora, J. Mol. Struct., 2014, 1060, 182–190 CrossRef CAS.
- K. Badreddine, I. Kazah, M. Rekaby and R. Awad, J. Nanomater., 2018, 2018, 1–11 CrossRef.
- K. S. Siddiqi, A. ur Rahman, Tajuddin and A. Husen, Nanoscale Res. Lett., 2018, 13, 141 CrossRef PubMed.
- A. Azam, Int. J. Nanomed., 2012, 3527 CrossRef CAS PubMed.
- G. A. Martínez-Castañón, N. Niño-Martínez, F. Martínez-Gutierrez, J. R. Martínez-Mendoza and F. Ruiz, J. Nanopart. Res., 2008, 10, 1343–1348 CrossRef.
- H. Li, Q. Cui, B. Feng, J. Wang, X. Lu and J. Weng, Appl. Surf. Sci., 2013, 284, 179–183 CrossRef CAS.
- A. N. P. Raj, R. B. Bennie, G. A. I. Xavier, C. Joel, D. A. Chelliah and S. H. Kengaram, J. Cluster Sci., 2022, 33, 2429–2441 CrossRef CAS.
- Z. N. Kayani, M. Sahar, S. Riaz, S. Naseem and Z. Saddiqe, Opt. Mater., 2020, 108, 110457 CrossRef CAS.
- N. Rajiv Chandar, S. Agilan, R. Thangarasu, N. Muthukumarasamy, J. Chandrasekaran, S. Arunachalam and S. R. Akshaya, J. Sol-Gel Sci. Technol., 2021, 100, 451–465 CrossRef CAS.
- M. I. Anik, N. Mahmud, A. Al Masud, M. I. Khan, M. N. Islam, S. Uddin and M. K. Hossain, ACS Appl. Bio Mater., 2022, 5, 4028–4054 CAS.
- A. Robert Xavier, A. T. Ravichandran, K. Ravichandran, S. Mantha and D. Ravinder, J. Mater. Sci.: Mater. Electron., 2016, 27, 11182–11187 CrossRef.
- E. R. Weiner, Applications of Environmental Chemistry, CRC Press, 2010 Search PubMed.
- Q. Han, X. Wang, X. Liu, Y. Zhang, S. Cai, C. Qi, C. Wang and R. Yang, J. Colloid Interface Sci., 2019, 539, 575–584 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
M. Rajib Munshib,
Arup Kumarc,
A. A. Mortuza
*a,
M. Rajib Munshib,
Arup Kumarc,
A. A. Mortuza




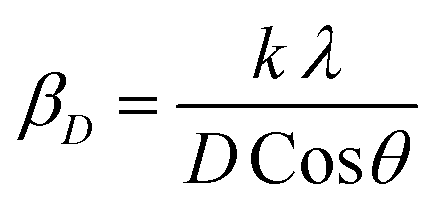 and βS = 4εtanθ, one gets eqn (6).
and βS = 4εtanθ, one gets eqn (6).