
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecisely controlling the cellular internalization of DNA-decorated semiconductor polymer nanoparticles for drug delivery†
Ying Tan,
Mengyi Xiong*,
Qin Liu,
Yao Yin,
Xia Yin*,
Shiyi Liao,
Youjuan Wang,
Ling Hu and
Xiao-Bing Zhang *
*
Molecular Science and Biomedicine Laboratory, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Collaborative Innovation Center for Chemistry and Molecular Medicine, Hunan University, Changsha 41008, P. R. China. E-mail: xbzhang@hnu.edu.cn; yinxia@hnu.edu.cn; xiongmy@hnu.edu.cn
First published on 1st November 2022
Abstract
Nonspecific adhesivity of nanoparticles to cells is regarded as a significant issue of nanomedicine, which brings about many serious drawbacks in applications, including low detection sensitivity, non-targeted biotoxicity and poor diagnostic accuracy. Here, we propose for the first time, DNA-decorated semiconductor polymer nanoparticles (SPN-DNAs), whose adhesivity can be significantly alleviated by controlling the density and thickness of DNA layers. This property is demonstrated to be independent of external conditions such as temperature, concentration, incubation time, ionic strength and cell lines. The mechanism of this phenomenon is also discussed. Finally, based on minimized nonspecific adhesivity to cells, a triggered nanoswitch can be constructed to control cellular internalization and drug delivery.
1. Introduction
Organic polymeric nanoparticles (OPNs) have shown splendid promise in bioanalysis and biomedical applications.1 For instance, the semiconductor polymer nanoparticles (SPNs) have emerged as attractive imaging nanoagents due to their large absorption coefficients, high photostability and tuneable optical properties.2 Currently, various SPNs-based nanoprobes with multifunctional light-responsive characteristics, such as fluorescence, photoacoustic and photothermal, have been developed for tumor imaging and cancer therapy in vivo.3,4 However, these nanoparticles (NPs) with hydrophobic core and relatively large particle sizes have been reported to show strong nonspecific adhesivity to cells, which leads to many serious issues in applications, including low detection sensitivity, non-targeted biotoxicity and poor diagnostic accuracy, eventually hampering their future clinical applications.5,6In order to reduce the nonspecific adhesivity of NPs to cells, many strategies have been adopted, such as size reduction,7 rapid charge reversal,8 and surface coating.9 Among them, the surface coating strategy has been extensively studied.10 For example, a series of nonionic and hydrophilic polymers are employed in coating NPs, such as polyethylene glycol (PEG),11 poly(glyceryl monomethacrylate) (PGMMA),6 polyphosphoester (PPE),12 and hyperbranched polyglycerol (HPG).13 These polymer coatings can impart the NPs with a steric repulsion and hydration molecular layer, which can reduce the hydrophobic and electrostatic interactions between NPs and biomolecules, thereby alleviating the nonspecific adhesivity of NPs to cells.5 Simultaneously, targeting units are modified on the NPs to enhance the internalization by target cells.11,12 However, due to the heterogeneous expression of receptors on the surface of tumor cells, these NPs are internalized by target cells with different efficiencies, resulting in inaccurate imaging and therapy.14,15 Therefore, it is urgent to develop an alternative strategy to mitigate the nonspecific adhesivity of NPs to cells while maintaining a homogeneous cellular internalization in a controlled way.
In recent years, synthetic nucleic acid strands (DNA), a sequence-manageable biopolymer with high biocompatibility and biodegradability,16,17 has merged as a new category for the decoration of OPNs. For example, Tian proposed that the OPNs with functional nucleic acids could transport a NIR-II emitting nano-fluorophore across the blood-brain barrier (BBB), facilitating non-invasive imaging of brain tumors.18 Zhang reported the drug-loaded OPNs equipped with functional nucleic acids for active tumor targeting and treatment.19 Although the synthetic DNA is also a class of nonionic and hydrophilic polymers with a particularly precise and controllable length, there are no studies and reports that it can be used to alleviate the nonspecific adhesivity of NPs to cell, ultimately achieving controlled cellular internalization and drug delivery.
Herein, we proposed a flexible method for the generation of DNA-decorated SPNs, and systematically studied their capacity in reducing nonspecific adhesivity, while also providing a triggered strategy for controlled cellular internalization and drug delivery. As shown in Scheme 1a, SPN-DNA with various densities and thicknesses of DNA layers could be easily synthesized in one step in the presence of semiconductor polymers (SP, Fig. S1†), cholesteryl-modified DNA (Table S1†), PEG-lipid and lipid. Further, the nonspecific adhesivity of the SPNs to cells was demonstrated to decrease along with the increasing density and thickness of the DNA layers. When the amount of DNA layer was 5 times that of SP core, the formed SPN-DNA40 could significantly alleviate nonspecific adhesivity to cells. Based on the minimized nonspecific adhesivity, a nanoswitch (SPN-DNA40S–S or SPN-DNA40PCL) was constructed, upon triggered, it could be converted to SPN-DNA10 to enhance cellular internalization, thereby achieving controlled drug delivery (Scheme 1b). Overall, this strategy is of great significance for reducing the nonspecific adhesivity of nanomaterials to cells in bioimaging and biomedicine, simultaneously providing a new thought for the controlled internalization and delivery of nanomedicines to cells.
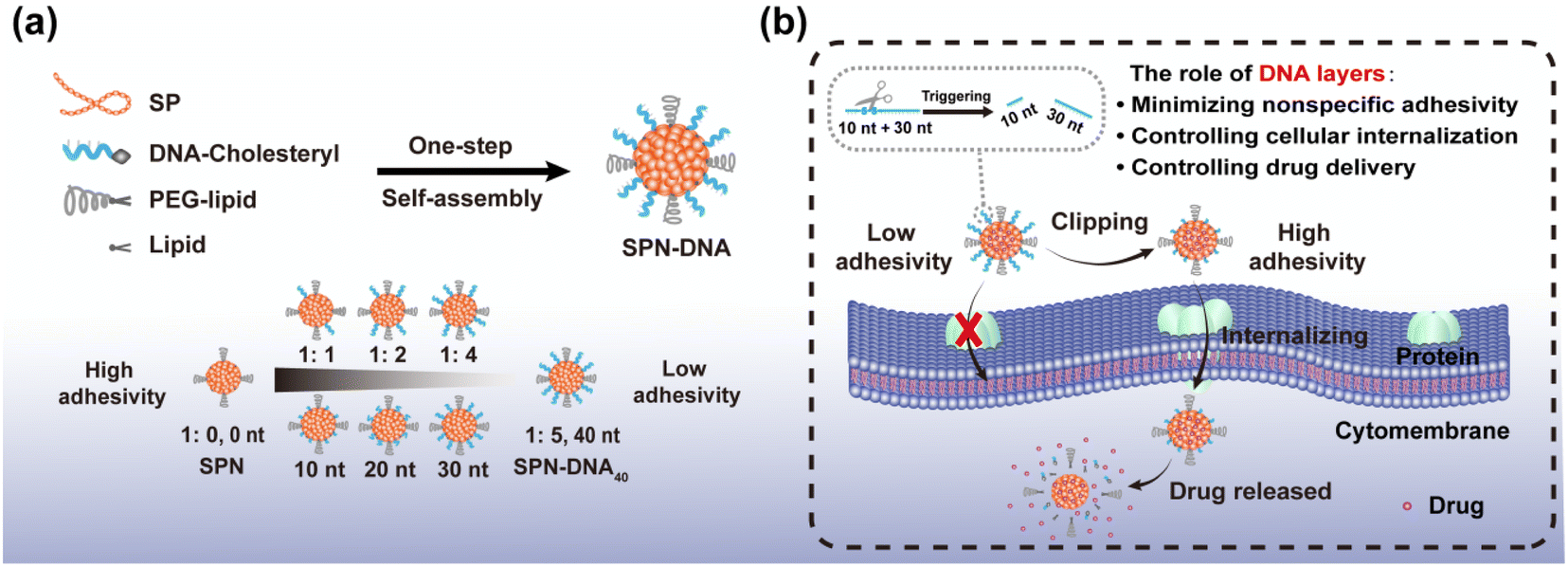 | ||
| Scheme 1 (a) Synthesis of SPN-DNA with tunable DNA layer for reducing nonspecific adhesivity of NPs to cells. The ratio referred to the amount of SP core to DNA layer. (b) A triggered nanoswitch for controlling cellular internalization and drug delivery. | ||
2. Materials and methods
2.1. Chemicals and materials
All the DNA oligonucleotides (Tables S1, S2 and ESI†) were purchased from Sangon Biotech Co., Ltd. (Shanghai, China). All cells used in this research were obtained from ATCC including CCRF-CEM (human acute lymphoblastic leukemia T lymphocyte), Ramos (human B-lymphocytic tumor cells), A549 (human non-small cell lung cancer cells) and HEK293 (human embryonic kidney cells) cells. MEH-PPV, PFO-DBT, streptavidin-modified microbead and Nystatin were purchased from Sigma-Aldrich (Shanghai, China). 1,2-Distearoyl-sn-glycerol-3-phosphoethanolamine-N-maleimide (polyethylene glycol 2000) (PEG-DSPE) was purchased from ToYongBio Tech. Inc. (Shanghai, China). 1,2-Dihexadecanoyl-rac-glycero-3-phosphocholine (DPPC) was purchased from Haohong Biotechnology Co., Ltd. (Shanghai, China). Dynasore and LY 294002 were obtained from Macklin Biochemical Co., Ltd. (Shanghai, China). Chlorpromazine (CPZ), resveratrol (Res) and tris(2-carboxyethyl) phosphine (TCEP) were obtained from Saen Chemical Technology Co., Ltd. (Shanghai, China). All solutions used in the experiments were prepared by ultrapure water (resistance >18 MΩ cm), which was obtained through a Millipore Milli-Q ultrapure water system (Billerica, MA, USA). Unless otherwise specified, all other reagents used in this work were of analytical grade, commercially purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), and used without further treatment.2.2. Preparation of SPN, SPN-DNA and SPN-DNA-Cy5 nanoparticles
All the SPN, SPN-DNA and SPN-DNA-Cy5 nanoparticles were prepared according to a nanoprecipitation method.2–4 Briefly, MEH-PPV or PFO-DBT (20 μg), PEG-DSPE (80 μg) and DPPC (300 μg) were dissolved into THF with a total volume of 100 μL, the solution was rapidly injected into the ultrapure water (900 μL) under vigorous sonication with a sonicator under 110 W. Then, the sonication was continued for 10 min. Next, THF in the obtained solution was removed at 50 °C by the rotary evaporation until it stopped bubbling, and the resulting solution was purified and concentrated for three times by ultrafiltration (5000 rpm, 4 min). The prepared SPN solution was stored at 4 °C for further use.To fabricate the SPN-DNA or SPN-DNA-Cy5, DNA-C (Table S1†) or C-DNA-Cy5 (Table S2†) was added to ultrapure water in advance, with a total volume of 900 μL. The other steps were the same as above.
2.3. Preparation of SPNRes-DNA nanoparticles
SPNRes-DNA nanoparticles were prepared according to a nanoprecipitation method.2–4 Briefly, MEH-PPV (20 μg), Resveratrol (Res, 400 μg), PEG-DSPE (80 μg) and DPPC (300 μg) were dissolved into THF with a total volume of 100 μL, the solution was rapidly injected into the ultrapure water containing the corresponding DNA (100 μg, total volume of 900 μL) under vigorous sonication with a sonicator under 110 W. Then, the sonication was continued for 10 min. Next, THF in the obtained solution was removed at 50 °C by the rotary evaporation until it stopped bubbling, and the resulting solution was purified and concentrated for ten times by ultrafiltration (5000 rpm, 4 min). The prepared SPNRes-DNA solution was stored at 4 °C for further use.The drug loading efficiency of SPNRes-DNA is calculated according to the following equation:
| Loading efficiency = m/m0 × 100% |
In this equation, m and m0 represent the mass of the loaded drug and the total inputted drug, respectively.
2.4. Size, morphology and potential characterization of nanoparticles
After ultrafiltration and concentration, the resulting SPN and SPN-DNA nanoparticles were characterized by JEOL transmission electron microscopy (TEM, Hitachi JEM-2100, Japan) and dynamic light scattering (DLS, Worcestershire, UK). The zeta potential was determined by dynamic light scattering (DLS, Nano-ZS90, Malvern).2.5. UV-vis measurements
UV-vis spectroscopy was achieved with UV-2450 (Shimadzu, Japan). Taking ultrapure water as the baseline, the UV-vis spectra of SPN, DNA-Cy5 and SPN-DNA-Cy5 were collected from 235 nm to 735 nm.2.6. Fluorescent measurements
The fluorescent data were collected at room temperature on a fluorometer (Edinburgh Instruments Ltd, FS5, UK) with both excitation and emission slits of 2 nm. The fluorescence emission spectra of SPN, DNA-Cy5 and SPN-DNA-Cy5 were collected from 510 nm to 800 nm with excitation at 450 nm (Fig. 1b) or collected from 660 nm to 800 nm with excitation at 650 nm (Fig. S2†).2.7. The proof that cholesteryl-modified DNA was decorated on the surface of SPN through hydrophobic interaction rather than adsorption
The experiment was divided into three steps. The first step was the formation of Bead-DNA. The streptavidin-modified bead was mixed with B-DNA (Table S2†) at 37 °C for 40 min, and the final concentration was 1 μM. The obtained resolution was purified by centrifugal washing for 3 times (1000 rpm, 4 min). The second step was the hybridization between SPN-DNA and linker. Both of them were mixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in 1× tris–acetic–EDTA (TAE) buffer (40 mM Tris, 20 mM acetic acid, 2 mM EDTA and 12.5 mM magnesium acetate, pH 8.0) and incubated at 37 °C for 1 h. SPN-DNA–linker was obtained by ultrafiltration for 3 times (5000 rpm, 4 min). Finally, SPN-DNA–linker and Bead-DNA were connected by incubating together at 37 °C for 1 h, followed by centrifuging for 3 times (1000 rpm, 4 min). The results were evaluated by confocal laser scanning microscopy (CLSM).
:1 in 1× tris–acetic–EDTA (TAE) buffer (40 mM Tris, 20 mM acetic acid, 2 mM EDTA and 12.5 mM magnesium acetate, pH 8.0) and incubated at 37 °C for 1 h. SPN-DNA–linker was obtained by ultrafiltration for 3 times (5000 rpm, 4 min). Finally, SPN-DNA–linker and Bead-DNA were connected by incubating together at 37 °C for 1 h, followed by centrifuging for 3 times (1000 rpm, 4 min). The results were evaluated by confocal laser scanning microscopy (CLSM).
2.8. Cell culture
The CCRF-CEM and Ramos cells were cultured in 1640 medium with the addition of 10% FBS (fetal bovine serum, Invitrogen, Carlsbad, CA, USA) and 0.5 mg mL−1 penicillin–streptomycin (KeyGEN Biotech, Nanjing, China) in a 5% CO2 environment at 37 °C, A549 and HEK293 cells were cultured in DMEM medium with the addition of 10% FBS (fetal bovine serum, Invitrogen, Carlsbad, CA, USA) and 0.5 mg mL−1 penicillin–streptomycin (KeyGEN Biotech, Nanjing, China) in a 5% CO2 environment at 37 °C.2.9. Nonspecific adhesivity and cellular internalization analysis
1 × 105 CCRF-CEM (Ramos, A549 or HEK293) cells were incubated with SPN, SPN-DNA or SPNRes-DNA(6 μg mL−1) in a binding buffer at 37 °C for 30 min, followed by washing for 3 times with washing buffer to remove the excess mixture (1000 rpm, 4 min). The efficiency of nonspecific adhesivity and cellular internalization was evaluated by flow cytometry.For experiments to test the inhibitory effect, cells were pretreated with the inhibitors for 20 min, followed by the addition of a solution of SPN, SPN-DNA10 or SPN-DNA40 (Dynasore: 50 μg mL−1, CPZ: 20 μg mL−1, Nystatin: 20 μg mL−1, LY294002: 20 μg mL−1). The inhibition efficiency of cellular internalization was evaluated by flow cytometry and confocal laser scanning microscopy (CLSM).
For experiments to control cellular internalization of SPN-DNA40S–S or SPN-DNA40PCL, the NPs were incubated with different concentrations of TCEP for 1 h or be exposed under UV irradiation for different time at 37 °C. The efficiency of cellular internalization was evaluated by flow cytometry.
The washing buffer was composed of DBPS containing 4.5 g L−1 glucose and 5 mM Mg2+. The binding buffer was composed of the washing buffer containing 0.1 mg mL−1 tRNA.
2.10. Confocal laser scanning microscopy (CLSM) analysis
The samples were analyzed using the New Nikon or Zeiss LSM 880 confocal microscope (Olympus). Correspondingly, images were analyzed with Viewer or ZEN (blue edition) software. The excitation wavelength was 488 nm, and the received emission wavelength ranged from 510 nm to 800 nm.2.11. Flow cytometry
After the incubation of SPN or SPN-DNA, cells were suspended in a washing buffer for measurement. Using a flow cytometer (Beckman), a total of 10000 events were recorded for each sample. The data were analyzed by FlowJo software.
2.12. Gel electrophoresis analysis
Reaction mixtures were finally quantified in a volume of 12 μL for electrophoresis experiments directly with 12% polyacrylamide gel electrophoresis (PAGE). Electrophoresis was carried out in 1× tris–acetic–EDTA (TAE) buffer (40 mM Tris, 20 mM acetic acid, 2 mM EDTA and 12.5 mM magnesium acetate, pH 8.0) at 80 V for 80 min. The gels were imaged and analyzed by a Bio-Rad ChemiDoc XRS System after being stained with Gel green.2.13. Cytotoxicity study of SPN-DNA and SPNRes-DNA for A549 cells
The cytotoxicity of SPN-DNA and SPNRes-DNA were determined by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay. A549 cells were placed in a 96-well plate in 200 μL of culture medium and incubated in 5% CO2 at 37 °C for 24 h. Next, the culture medium was removed and cells were treated with corresponding concentrations of NPs. After incubating for 1 h, NPs were removed and 200 μL of fresh culture medium was added into each well for 24 h. Then the solution in each well was replaced with MTT solution (0.5 mg mL−1). Residual MTT solution was removed after 4 h, and then dimethyl sulfoxide (DMSO, 150 μL) was added to each well to dissolve the formazan crystals. After the plates were shaken for 20 min at 37 °C, the absorbance values of the wells were recorded at 570 nm by a microplate reader. The cytotoxic effects (VR) of these NPs were assessed by the following equation:| VR = A/A0 × 100% |
In this equation, A and A0 represent absorbance of the experimental group and control group, respectively. The assays were performed in three sets for each concentration.
For experiments to control drugs delivery of SPNRes-DNA40S–S to cells, the NPs were incubated certain concentrations of TCEP at 37 °C for 1 h. SPNRes-DNA40 without disulfide bond was used as the control group. The delivery efficiency of drugs to cells was evaluated by MTT assay as above.
3. Results and discussion
For better visualization of the SPN-DNA, a widely used fluorescent SP, MEHPPV was used as an organic core to fabricate SPN-DNA through nanoprecipitation (Scheme 1a). As the UV-vis spectrum displayed in Fig. 1a, the bare SPN showed maximum absorption at about 495 nm, while the SPN-DNA-Cy5 that referred to the SPN modified with C-DNA-Cy5 (Table S2†) showed a new absorption peak of DNA at 260 nm and Cy5 at 650 nm. Simultaneously, with the excitation of 450 nm, the fluorescence resonance energy transfer (FRET) between SPN and Cy5 was observed (Fig. 1b), and when excited at 650 nm, the fluorescence of Cy5 appeared in SPN-DNA-Cy5 (Fig. S2†). Additionally, the dynamic light scattering (DLS) analysis indicated that the size increased from 50.1 nm to 57.5 nm after the SPN was modified with DNA, which was also verified by transmission electron microscopy (TEM) (Fig. S3†). These results demonstrated the successful modification of DNA on the surface of SPN.To verify that cholesteryl-modified DNA was decorated on the surface of SPN through hydrophobic interaction rather than adsorption. A short DNA strand (B-DNA, Table S2†) was immobilized on the surface of the microbead, followed by the connection of SPN-DNA through a linker DNA sequence (Table S2,† Fig. 1c). The confocal laser scanning microscopy (CLSM) images showed that the SPN-DNA could be captured on the surface of microbead in the presence of linker (Fig. 1d and S4†). In contrast, the SPN-DNA without the addition of a linker or the SPN plus linker could not be attached to the microbead (Fig. 1d). These results indicated that cholesteryl-modified DNA was decorated on the surface of SPN through hydrophobic interaction rather than adsorption.
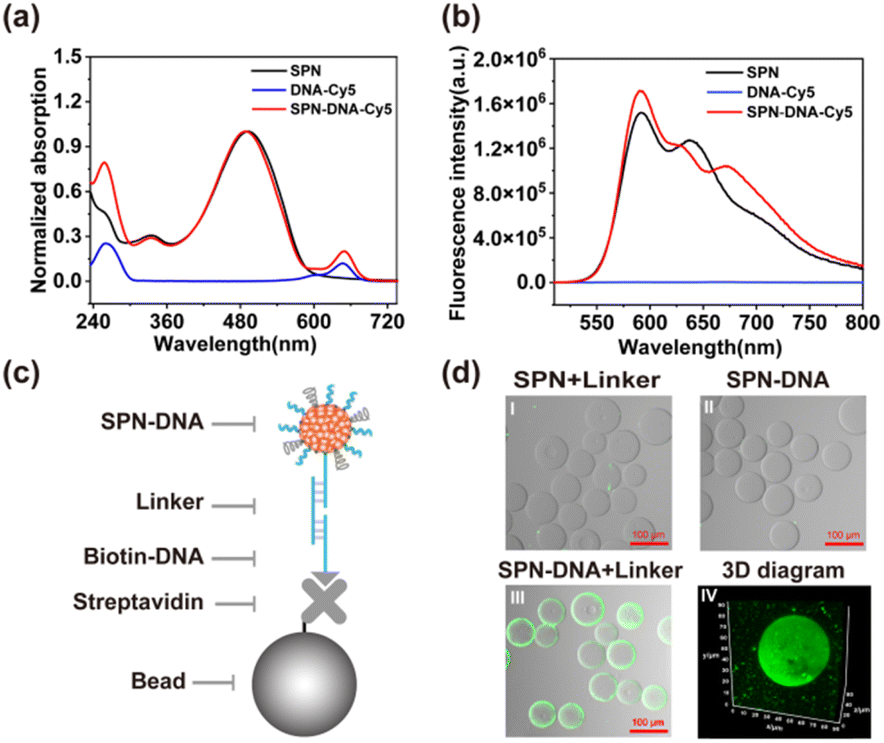 | ||
| Fig. 1 (a) UV absorption and (b) fluorescence emission of SPN, DNA-Cy5 and SPN-DNA-Cy5. (c) Schematic illustration and (d) CLSM imaging of the hybridization of SPN-DNA to microbeads. | ||
Next, the density and thickness of DNA layers on the surface of SPN were modulated in the process of synthesis. As shown in Fig. 2a, when the amount of DNA layer increased relative to the SP core, the UV-vis absorption of DNA at 260 nm rose, demonstrating a higher density of DNA on the surface of SPN. Furthermore, the zeta potential of the synthesized SPN-DNA was observed to decrease continuously with the increasing amount of DNA until the amount of DNA layer was 5 times that of SP core, where the DNA on the surface of SPN reached saturation (Fig. 2b). Similarly, the thickness of the DNA layer could also be modulated by regulating the length of input DNA.
As demonstrated by UV-vis spectrum and zeta potential, the absorption of DNA at 260 nm increased (Fig. 2c) while the potential decreased (Fig. 2d) with the length of DNA extending from 0 nt to 60 nt. These results indicated that the density and thickness of DNA layers on the surface of SPN were flexibly controllable, which was beneficial for studying the role of DNA in reducing nonspecific adhesivity to cells.
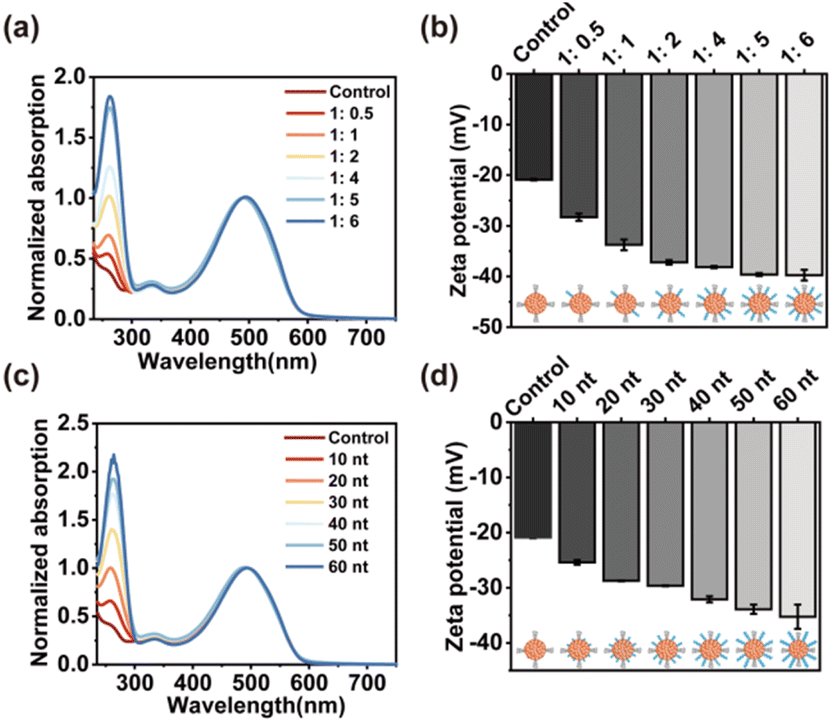 | ||
| Fig. 2 (a) UV absorption and (b) zeta potential of SPN-DNA with different density of DNA. The ratio referred to the amount of SP core to DNA layer. (c) UV absorption and (d) zeta potential of SPN-DNA with different lengths of DNA. | ||
Then, the above SPN-DNAs decorated with different densities and thicknesses of DNA layers were incubated with cells to assess their nonspecific adhesivity and cellular internalization. As the flow cytometry analysis displayed in Fig. 3a and b, the SPN-DNA covered with higher DNA density or longer DNA strand remarkably reduced nonspecific adhesivity against living cells. When the amount of DNA layer was 5 times that of the SP core, the formed SPN-DNA40 could significantly minimize nonspecific adhesivity and cellular internalization to CCRF-CEM cells (human acute lymphoblastic leukemia T lymphocyte). Therefore, it was the DNA layer that could alleviate the nonspecific adhesivity of SPN to cells.
To further explore whether the SPN-DNA40 could maintain low nonspecific adhesivity and escape cellular internalization in different incubation conditions, a series of influence factors were studied. Usually, the temperature matters a lot for the cellular internalization of NPs, in which the endocytosis of NPs by cells would decrease with the dropping of temperature.20,21 Here, the nonspecific cell adhesivity of SPN and SPN-DNA40 at 37 °C and 4 °C was studied. The results revealed that compared to SPN, SPN-DNA40 could keep extremely low nonspecific adhesivity and cellular internalization at both 37 °C and 4 °C (Fig. S5a†). It had been reported that the cellular internalization of NPs was usually concentration-dependent and time-dependent.20,21 The effects of particle concentration and incubation time were investigated. Results showed that SPN-DNA40 could still maintain low nonspecific adhesivity to living cells when the concentration increased to 25 μg mL−1 (Fig. 3c) or the incubation time was prolonged to 90 min (Fig. S5b†). The changes in ionic strength in the incubation medium also demonstrated the low nonspecific adhesivity of SPN-DNA40 (Fig. S5c†). To evaluate whether the proposed strategy could be generalized to other SPNs, another fluorescent polymer, PFODBT was employed as the core to form SPN-DNA40. The result indicated that these NPs also gained the capability of reducing nonspecific adhesivity to cells (Fig. S5d†). In addition, the minimized nonspecific adhesivity of the SPN-DNA40 was observed in other cell lines, such as CCRF-CEM, Ramos, A549 and HEK293 cells (Fig. 3d). Collectively, the DNA layer on the surface of SPN could effectively alleviate nonspecific adhesivity against living cells in most incubation conditions.
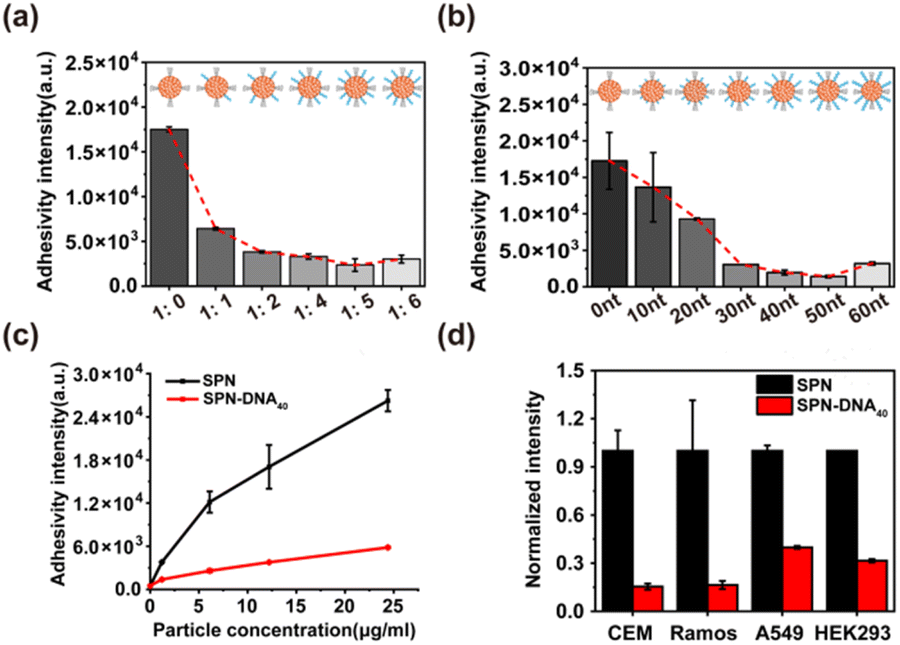 | ||
| Fig. 3 The cellular internalization of SPN-DNA with various (a) DNA density and (b) length on the surface of SPN. The ratio referred to the amount of SP core to DNA layer. (c) The cellular internalization of different concentrations of SPN or SPN-DNA40. (d) The cellular internalization of SPN or SPN-DNA40 by different cell lines. | ||
To explore the mechanism of NPs in evading internalization pathways,22 several endocytosis inhibitors were employed in previous reports, including Dynasore,23 chlorpromazine (CPZ),24 Nystatin,25 LY294002 (Fig. S6†).7 Herein, the cellular internalization behavior of SPN, SPN-DNA10, and SPN-DNA40 was investigated after the pretreatment of these above inhibitors. As the results shown in Fig. 4a, Dynasore and CPZ could significantly inhibit cellular internalization of SPN and SPN-DNA10, demonstrating these NPs were internalized primarily by clathrin-mediated endocytosis (CME)23,24 rather than by caveolae-mediated endocytosis (CvME),25 phagocytosis,26 or macropinocytosis.7 However, the SPN-DNA40 showed negligible change in cellular internalization after the pretreatment of these inhibitors, which was also verified by CLSM (Fig. S7†). Therefore, it could be concluded that the SPN-DNA40 principally evaded energy-dependent clathrin-mediated endocytosis to escape cellular internalization.
Further, based on minimized nonspecific adhesivity to cells, a nanoswitch, SPN-DNA40S–S or SPN-DNA40PCL was constructed to control cellular internalization (Fig. 4b). Specifically, a disulfide bond (S–S) or PC-Linker (PCL) was integrated into the DNA strand (Table S1†) so that the DNA could be cleaved into short fragments after being triggered (Fig. S8†). As shown in Fig. 4b and c, the cellular internalization of the SPN-DNA40S–S and SPN-DNA40PCL could be turn “ON” by corresponding triggers such as the addition of TCEP or UV irradiation, which was demonstrated by flow cytometry. Upon triggered, the cellular internalization of the nanoswitch (SPN-DNA40S–S or SPN-DNA40PCL) would be turned “ON”, and with the increase of the corresponding trigger factor (TCEP concentration or UV irradiation time), cellular internalization would be greatly enhanced (Fig. 4d and e). Therefore, a triggered nanoswitch that could precisely control the cellular internalization had been successfully constructed.
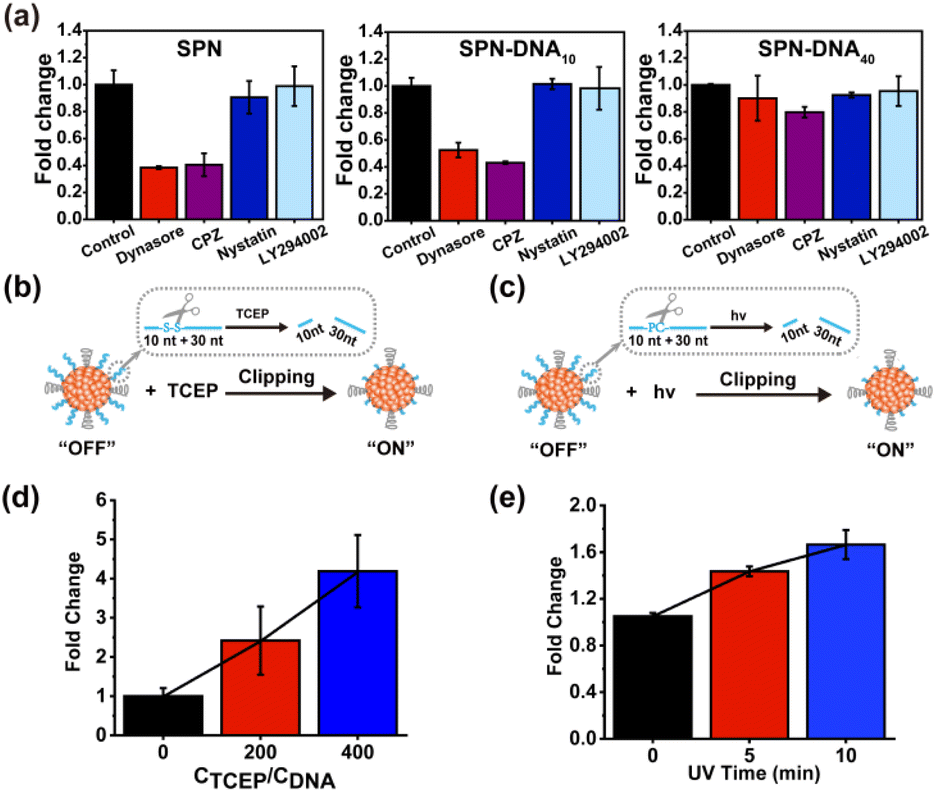 | ||
| Fig. 4 (a) Fold change of SPN, SPN-DNA10 and SPN-DNA40 internalized by cells after the pretreatment of inhibitors. The triggered nanoswitch, SPN-DNA40S–S (b) or SPN-DNA40PCL (c) for controlled cellular internalization. The change in cellular internalization of SPN-DNA40S–S (d) or SPN-DNA40PCL (e) after being triggered by TCEP or UV irradiation. | ||
Finally, the above nanoswitch was employed to encapsulate resveratrol (Res, a hydrophobic chemotherapeutics drug) via hydrophobic interaction for expanding the application of DNA in drug delivery (Fig. S9a†). The absorption peak of Res appeared in SPNRes-DNA (SPN-DNA loading Res), indicating the successful encapsulation of this drug (Fig. S9b†). The DLS analysis showed that the sizes of SPNRes-DNA10 and SPNRes-DNA40 were 52.2 nm and 58.7 nm, respectively, which was also confirmed in TEM (Fig. S9c†). Subsequently, CLSM experiments were performed, and as expected, SPNRes-DNA10 exhibited greater cellular internalization than SPNRes-DNA40 (Fig. S10†). Next, the controlled drug delivery was achieved by triggering the nanoswitch, whose delivery efficiency was evaluated by cytotoxicity (Fig. 5a). As shown in Fig. 5b, with the increase of particle concentration, SPNRes-DNA10 showed increasing cytotoxicity, while SPNRes-DNA40 exhibited inappreciable cytotoxicity. Both exhibited good biocompatibility without drug loading (Fig. S11†). These outcomes were beneficial for triggering the nanoswitch for controlled cellular internalization and drug delivery. As exhibited in Fig. 5c, in the presence of the trigger, SPNRes-DNA40S–S exhibited significant difference in cytotoxicity, while SPNRes-DNA40 as the control showed negligible change, which indicated that the controllable DNA layers played a vital role in controlled cellular internalization and drug delivery.
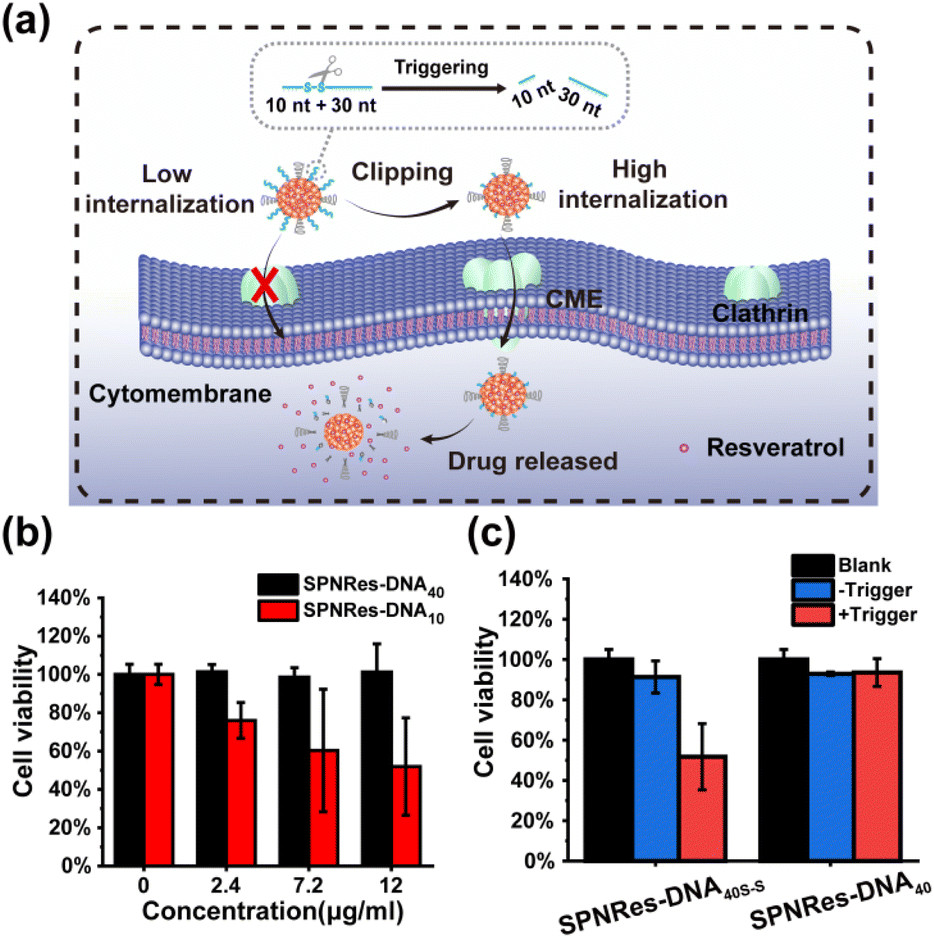 | ||
| Fig. 5 (a) The triggered nanoswitch integrating controlled cellular internalization and drug delivery. (b) The difference in cytotoxicity when cells incubated with SPNRes-DNA10 or SPNRes-DNA40 with the increasing concentration of NPs. (c) The cytotoxicity of SPNRes-DNA40 or SPNRes-DNA40S–S after being triggered. | ||
4. Conclusions
In summary, this work successfully developed a novel and flexible method to minimize nonspecific adhesivity of the SPNs to cells by regulating DNA layers on their surface, ultimately achieving controlled cellular internalization and drug delivery. The nonspecific adhesivity of SPN-DNA to cells was found to decrease with the increasing density and depth of the DNA layer, which was independent of the culture conditions including temperature, concentration, incubation time, ionic strength, and could be generalized to various cell lines. The mechanism of this phenomenon was also investigated, which indicated the SPN-DNA40 could escape cellular internalization by evading an energy-dependent clathrin-dependent endocytosis pathway. Further, via integrating a switch into the DNA strands, the controlled internalization and drug delivery of SPNs to cells was realized by triggers. This study might have an enlightenment significance for the design of controlled delivery systems for nanodrugs with low nonspecific adhesivity to cells and their successful translation in vivo to achieve long blood circulation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21890744, 21974039, 22104032), the National Key R&D Program of China (2019YFA0210100), and the China Postdoctoral Science Foundation (2020M672470), the National Postdoctoral Program for Innovative Talents (BX2020118).Notes and references
- J. Karlsson, H. J. Vaughan and J. J. Green, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 105–127 CrossRef.
- Y. Jiang, J. Huang, X. Zhen, Z. Zeng, J. Li, C. Xie, Q. Miao, J. Chen, P. Chen and K. Pu, Nat. Commun., 2019, 10, 2064 CrossRef.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- J. Li and K. Pu, Acc. Chem. Res., 2020, 53, 752–762 CrossRef.
- Y. Li, X. Qian, X. Tan, X. Liu, Y. Li, D. Ming, L. Zhang and Y. Yang, ACS Appl. Polym. Mater., 2020, 2, 2238–2245 CrossRef.
- C. Vancaeyzeele, O. Ornatsky, V. Baranov, L. Shen, A. Abdelrahman and M. A. Winnik, J. Am. Chem. Soc., 2007, 129, 13653–13660 CrossRef.
- G. Feng, J. Liu, R. Liu, D. Mao, N. Tomczak and B. Liu, Adv. Sci., 2017, 4, 1600407 CrossRef PubMed.
- X. Xu, J. Duan, Q. Lan, Y. Kuang, T. Liao, Y. Liu, Z. Xu, J. Chen, B. Jiang and C. Li, J. Drug Delivery Sci. Technol., 2021, 66, 102817 CrossRef.
- D. Haute, A. Liu and J. Berlin, ACS Nano, 2018, 12, 117–127 CrossRef PubMed.
- S. Fam, C. Chee, C. Yong, K. Ho, A. Mariatulqabtiah and W. Tang, Nanomaterials, 2020, 10, 787 CrossRef.
- J. G. Dancy, A. S. Wadajkar, N. P. Connolly, R. Galisteo, H. M. Ames, S. Peng, N. L. Tran, O. G. Goloubeva, G. F. Woodworth, J. A. Winkles and A. J. Kim, Sci. Adv., 2020, 6, eaax3931 CrossRef.
- J. Wang, S. Shen, D. Li, C. Zhan, Y. Yuan and X. Yang, Adv. Funct. Mater., 2018, 28, 1704806 CrossRef.
- N. K. Y. Wong, R. Misri, R. A. Shenoi, I. Chafeeva, J. N. Kizhakkedathu and M. K. Khan, J. Biomed. Nanotechnol., 2016, 12, 1089–1110 CrossRef.
- P. Bedard, A. Hansen, M. Ratain and L. Siu, Nature, 2013, 501, 355–364 CrossRef PubMed.
- A. Salvati, A. Pitek, M. Monopoli, K. Prapainop, F. Bombelli, D. Hristov, P. Kelly, C. Aberg, E. Mahon and K. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef.
- Y. H. Roh, R. C. H. Ruiz, S. Peng, J. B. Lee and D. Luo, Chem. Soc. Rev., 2011, 40, 5730–5744 RSC.
- D. Wang, P. Liu and D. Luo, Angew. Chem., Int. Ed., 2022, 61, e202110666 Search PubMed.
- F. Xiao, L. Lin, Z. Chao, C. Shao, Z. Chen, Z. Wei, J. Lu, Y. Huang, L. Li, Q. Liu, Y. Liang and L. Tian, Angew. Chem., Int. Ed., 2020, 59, 9702–9710 CrossRef.
- Y. Guo, J. Zhang, F. Ding, G. Pan, J. Li, J. Feng, X. Zhu and C. Zhang, Adv. Mater., 2019, 31, 1807533 CrossRef.
- M. Benfer and T. Kissel, Eur. J. Pharm. Biopharm., 2012, 80, 247–256 CrossRef PubMed.
- L. Wang, Y. Sun, C. Shi, L. Li, J. Guan, X. Zhang, R. Ni, X. Duan, Y. Li and S. Mao, Acta Biomater., 2014, 10, 3675–3685 CrossRef PubMed.
- S. Xiang, H. Tong, Q. Shi, J. C. Fernandes, T. Jin, K. Dai and X. Zhang, J. Controlled Release, 2012, 158, 371–378 CrossRef PubMed.
- G. Preta, J. G. Cronin and I. M. Sheldon, Cell Commun. Signaling, 2015, 13, 24 CrossRef.
- M. Wiranowska, L. O. Colina and J. O. Johnson, Cancer Cell Int., 2011, 11, 27 CrossRef.
- Y. Chen, S. Wang, X. Lu, H. Zhang, Y. Fu and Y. Luo, Blood, 2011, 117, 6392–6403 CrossRef.
- X. Song, S. Tanaka, D. Cox and S. C. Lee, J. Leukocyte Biol., 2004, 75, 1147–1155 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05172a |
| This journal is © The Royal Society of Chemistry 2022 |