
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional Ni4O4-cubane metal–organic framework as a high-performance electrocatalyst for urea oxidation†
Mariam Batool a,
Amir Waseema and
Muhammad Arif Nadeem*ab
a,
Amir Waseema and
Muhammad Arif Nadeem*ab
aCatalysis and Nanomaterials Lab 27, Department of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
bPakistan Academy of Sciences, 3-Constitution Avenue Sector G-5/2, Islamabad, Pakistan. E-mail: manadeem@qau.edu.pk; Tel: +92-51-9064-2062
First published on 5th October 2022
Abstract
The urea oxidation reaction (UOR) is considered to be a replacement of the sluggish anodic oxygen evolution reaction (OER) in overall water-splitting. A three-dimensional (3D) nickel-containing metal–organic framework {[NiII2(pdaa)(OH)2(H2O)]n (MOF 1) (where, H2pdaa = 1,4-phenylene diacetic acid) was investigated as a robust and highly efficient electrocatalyst for the UOR. MOF 1 comprised 1D nickel(II) chains crosslinked through Ni4O4 cubane units to form a 3D extended network. Dangling Ni⋯OH− groups were exposed in the MOF 1 structure, and could act as active catalytic centers for the UOR. MOF 1 required a very small onset potential of 1.18 V for urea oxidation in KOH (1 M) and urea (0.33 M) and had a low Tafel slope of 38.8 mV dec−1 (in contrast to 1.84 V for the oxygen evolution reaction). The overpotential required to attain a catalytic current density of 10 mA cm−2 was 1.24 V, which is much lower than that for many materials. Controlled potential electrolysis, powder X-ray diffraction, and X-ray photoelectron spectroscopy affirmed the physicochemical integrity of the catalyst over a 17 h test reaction. This work not only addresses the problem of urea contamination, it also helps to utilize it in an energy-conversion process.
Introduction
Efficient energy carriers with high energy density, low cost, and ease of availability are needed worldwide.1 Hydrogen is considered to be a promising candidate for replacement of fossil fuels because it has high energy density and is environmentally friendly (the only by-product is H2O).2,3 Electrolyzing water is an old (yet promising) route for hydrogen production because of its high purity. Electrocatalytic water-splitting involves a cathodic hydrogen evolution reaction (HER) and anodic oxygen evolution reaction (OER). However, due to the high kinetic barriers (1.23 V) associated with multiple electron-coupled proton processes in OER, following this route is difficult.4 The actual working potential usually increases to 1.8 V due to the torpid kinetics of OER and HER, resistance from the electrolyte and electrode material, and their contact resistance.5,6 To minimize these factors, several solutions have been suggested, such as using suitable electrocatalysts to amplify slow kinetics, modifying electrolyzer structure, and substituting the OER with other promising anode reactions.7–9Substitution of the OER by readily oxidized chemicals have gained the attention of the scientific community, and use of hydrazine, alcohols, amines, aldehydes and urea has been suggested.10–14 Among these, the urea oxidation reaction (UOR) is preferred due to low thermodynamic voltage (0.37 V vs. reversible hydrogen electrode (RHE)), non-toxicity, stability, abundance, as well as ease of transportation and storage.15,16 In addition to industrial (N-containing fertilizers) and agricultural production, urea is produced as a bio-waste that pollutes the atmosphere and groundwater. It has been estimated that 80% of wastewater is dumped each year.17 The natural decomposition of urea releases nitrates and ammonia, which cause eutrophication and severe hazards to the environment and health.18 Therefore, the UOR is not only a good way to produce hydrogen efficiently, it can also be a remedy for wastewater treatment.19,20 The UOR (like the OER) is a complex process that involves transfer of six electrons with a complex system of gas evolution.13
The oxidation of urea over noble metals (e.g., Ti–Pt, Ti–Pt–Ir and Ru–TiO2) has been studied by several research teams. However, high cost and poisoning of the surface catalyst limits practical application.21–24 Boggs and colleagues documented the electro-oxidation of urea by cost-effective Ni-based catalysts in a basic medium.25 The facile oxidation of Ni(II) to Ni(III) during electrocatalysis makes it a promising candidate for the UOR.26 Moreover, the consistency between the peak of urea oxidation and peak of Ni oxidation makes Ni-based catalysts active for urea oxidation.27,28 This approach led the scientific community in the last decade to search for Ni-based catalysts to understand the reaction mechanisms.29,30 Ni-containing electrocatalysts have been employed for the UOR. For instance, Ni3N nanosheet arrays have been reported to be active catalysts for the UOR, with a small onset potential of 1.33 V vs. RHE.31 Similarly, an onset potential of 1.35 V has been documented for Ni0.9Fe0.1O porous hollow microspheres.32 Also, an onset potential of 1.36 V for Ni(OH)2 leads to an effective UOR.15 Zhang and colleagues demonstrated UOR activity for Ni–Mo nanotubes at 1.36 V vs. RHE.33
Metal–organic frameworks (MOFs) have many metal cations on their surface, electronic structures to attain high oxidation states, unsaturated metal sites, and are versatile. These features make MOFs promising electrocatalysts for the UOR.34 Their three-dimensional (3D) structure provides high specific surface area and facilitates mass transport to enhance electrocatalytic activity.35–39 Recently, NiFe-MIL-53-NH2 was shown to elicit a better UOR with an onset potential of 1.36 V compared with its monometallic counterparts Fe-MIL-53-NH2 (1.61 V) and Ni-MIL-53-NH2 (1.42 V).40 The Ni-MOF@NiO/Ni composite demonstrated electrocatalytic urea oxidation at 1.40 V for 10 mA cm−2.41 Pang and colleagues reported 3D aggregates of Ni-MOF ultrathin nanosheets to display a potential of 1.38 V for the UOR at 10 mA cm−2,42 and ultrathin 2D Ni-MOF has been reported to produce identical current density at a potential of 1.36 V.43 Due to the instability of certain MOFs under electrochemical conditions, MOF-derived Ni/C electrocatalysts have been reported to display the onset of urea oxidation at 1.33 V.44 Rezaee and co-workers discovered that Ni1.6Co0.4P/C@HCNs fabricated from NiCo-MOF could produce 10 mA cm−2 at a potential of 1.33 V.45 Optimizing UOR catalysts via compositional modulation and microstructure design could enhance UOR performance.
We report a 3D MOF, {[NiII2(pdaa)(OH)2(H2O)]n (MOF 1), as a highly efficient electrocatalyst for urea oxidation synthesized via a hydrothermal approach. The exceptionally low onset potential of 1.18 V vs. RHE in contrast to the sluggish OER (onset potential of 1.84 V) and small Tafel slope (38.8 mV dec−1) was displayed by MOF 1. For the UOR, a 3D MOF having such a low overpotential of 1.24 mV to achieve a catalytic current density of 10 mA cm−2 has not been documented. The controlled potential electrolysis (CPE) for 17 h continuously depicts the stability and robustness of MOF 1. We describe a cost-effective electrocatalyst for water-splitting but also propose a hybrid water electrolysis-affiliated urea oxidation to substitute the torpid OER for H2 generation. This strategy would aid energy conversion and treatment of wastewater by electrochemical means.
Experimental
Synthesis of MOF 1
The synthesis of MOF 1 was carried out according to our method, which has been reported previously.46 Briefly, a mixture of 1,4-phenylene diacetic acid (pdaa; 0.5 mmol) and KOH (2 mmol) was stirred for 10 min in distilled water (6 mL). NiCl2·6H2O (1 mmol) was dispersed in water (4 mL) and added slowly to the reaction mixture. This mixture was transferred to a 23 mL autoclave and kept at 170 °C under autogenous pressure for 72 h. Green, diamond-shaped crystals were obtained after cooling to room temperature and used for characterization and electrochemical studies.Preparation of MOF 1-deposited fluorine-dopped tin oxide electrode (FTO)
FTO of area (1 × 2 cm2) was washed successively with deionized water and alcohol by sonication. Then, it was kept in a furnace at 400 °C for 30 min for annealing, and used for catalytic studies. The catalytic suspension was prepared by pouring 5 μL of Nafion (binder) over MOF 1 (2 mg) and ethanol (1 mL). This mixture was sonicated for 1 h to obtain a homogenous ink, drop-casted over annealed FTO, and dried overnight at 50 °C in a desiccator before electrocatalytic studies.Electrochemical measurements
Electrochemical experiments were carried out in a potentiostat (Gamry Interface 5000) electrochemical analyser. A three-electrode system (FTO-coated electrode (working), Hg/HgO (reference), and graphite rod (counter electrode)) was used at room temperature. All potentials were transformed into a reversible hydrogen electrode (RHE) using| ERHE = EHg/HgO + 0.0592 (pH) + 0.098 |
Before electrochemical testing, N2 gas was bubbled through the electrolyte for 30 min KOH (1 M) was used as the electrolyte for the OER. A mixture of KOH (1 M) with urea (0.33 M) was used to carry out studies on urea oxidation. The electrocatalytic performance was evaluated by linear sweep voltammetry (LSV). CPE was employed to check the stability of MOF 1. Cyclic voltammetry (CV) was done in a non-faradaic region to estimate double-layer capacitance (Cdl) in the narrow potential at sweep rates from 2 mV s−1 to 14 mV s−1. Potentiostatic electrochemical impedance spectroscopy (EIS) was carried out to evaluate impedance. All experiments were undertaken under a N2 atmosphere.
Results and discussion
The synthesized MOF 1 was characterized by powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), infrared (IR) spectroscopy, and thermogravimetric analysis (TGA). The phase purity of MOF 1 was evaluated by PXRD. Consistency with the calculated pattern implied a successful synthesis, showing major peaks at 7.7°, 11.5°, 15.5° and 18.2° (Fig. 1d). All peak positions were in accordance with a simulated PXRD pattern from a single crystal. Moreover, the sharpness of peaks showed the crystalline nature of the material.46 When studying electrochemical properties, the shape and morphology of a material have crucial roles. High-magnification SEM revealed a pyramidal morphology (Fig. S1†). Fourier transform (FT)-IR spectroscopy verified the synthesis of MOF 1 because the peaks matched well with reported data (Fig. S2†). A broad peak at 3353 cm−1 indicated the presence of hydroxyl groups (a water molecule on the Ni ion).41 A sharp peak at 1558 cm−1 appeared due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching. Peaks at 1260 and 1386 cm−1 were ascribed to the –COO– group of pdda coordinated to Ni(II).47 Peaks below 1000 cm−1 were assigned to Ni–O bending and stretching vibrations.48 The thermal stability of MOF 1 was assessed through TGA: it showed thermal stability up to 380 °C (Fig. S3†).
O stretching. Peaks at 1260 and 1386 cm−1 were ascribed to the –COO– group of pdda coordinated to Ni(II).47 Peaks below 1000 cm−1 were assigned to Ni–O bending and stretching vibrations.48 The thermal stability of MOF 1 was assessed through TGA: it showed thermal stability up to 380 °C (Fig. S3†).
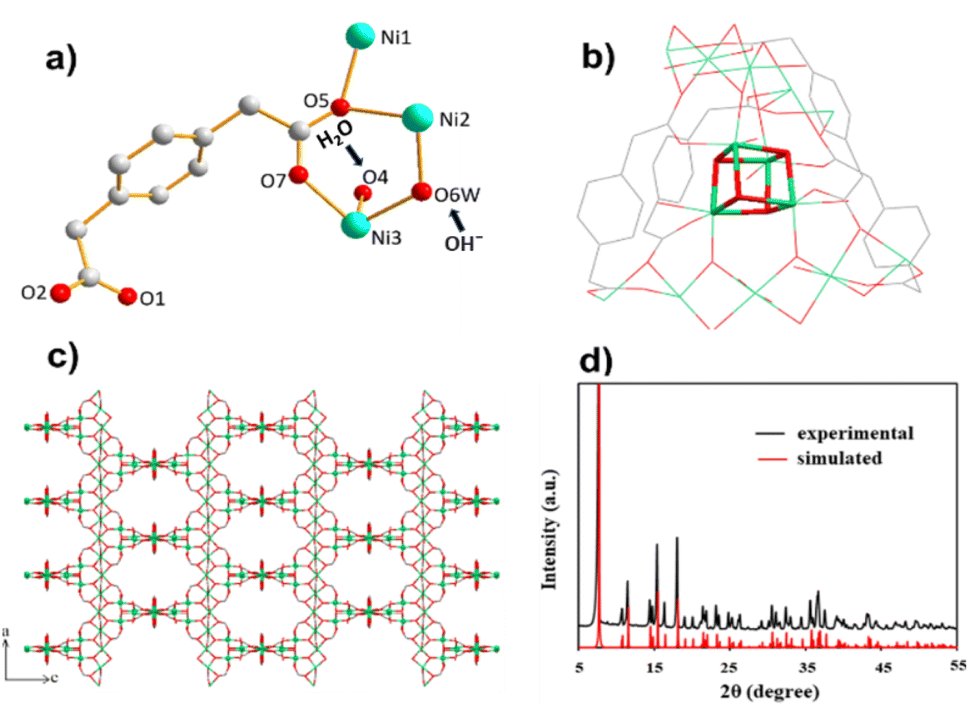 | ||
| Fig. 1 (a) Asymmetric unit of MOF 1. (b) Fragment emphasizing interconnections between Ni chains and Ni4O4 cubane units. (c) Packing diagram of MOF 1 demonstrating the separation of linear Ni chain through Ni4O4 cubane units. Ni: green, O: red C: grey sticks. H atoms have been omitted (d) comparison of PXRD of experimental (black) and calculated (red) patterns. | ||
Crystallographic studies revealed that 3D MOF 1 crystallized in the tetragonal space group I41/a. The basic structural unit of MOF 1 is shown in Fig. 1a. Structurally, MOF 1 showed two features. The first feature was a linear Ni atom bonded octahedrally to four oxygen atoms from four pdaa ligands, and the two O atoms from two non-identical OH− gave a slightly distorted octahedral geometry. The second feature was distorted Ni cubane units bridging linear chains. The nickel center was attached to three O atoms of the bridging –OH− groups of cubane, one O atom of the pdaa ligand, one O atom of the water molecule, and one O atom that linked the nickel center to linear chains (Fig. 1b). The coordination environment of Ni sites with O atoms is shown in Fig. S4.† The 3D network was formed by the bridging of cubanes with linear Ni chains coordinated by flexible pdaa ligands (Fig. 1c). The oxidation state of +II for Ni was confirmed by bond-valence sum analysis (Σbv(Ni(II) = 1.993–2.168)).46
Electrocatalytic studies on urea oxidation
Several electrocatalytic methods were used via a three-electrode arrangement to determine the electrocatalytic performance of MOF 1 for urea oxidation. Fig. 2a depicts the LSV of MOF 1 fabricated on FTO in different electrolyte setups at a sweep rate of 5 mV s−1. The estimated urea concentration in human urine is 0.33 M, so this concentration was used for the UOR.49 Fig. 2a depicts that the OER occurred in KOH (1 M) with an onset potential of 1.84 V (vs. RHE), which was reduced greatly to 1.18 V (vs. RHE) by urea addition. The current density also enhanced markedly. The as-synthesized MOF 1 exhibited better performance for urea oxidation (1.24 V @ 10 mA cm−2) than most of the recently reported Ni-based electrocatalysts given in Table 1. LSV of MOF 1 at various scan rates is shown in Fig. S6.† The extraordinary UOR catalysis of MOF 1 could be studied further by Tafel plots calculated from LSV curves. The lower Tafel slope of 38.8 mV dec−1 (Fig. 2b) compared with the OER (188 mV dec−1) indicated favorable kinetics, which is a preferable feature for commercial applications. This value (in mV dec−1) is lower than that reported previously: Ni(OH)2 (40),50 IrO2 (70),51 Pt/C (187),43 aggregated Ni-MOF ultrathin nanosheets (52)42 and Ni3N NA/CC (57).31 The excellent performance for urea oxidation can be attributed to the 3D porous structure, which facilitates electrolyte diffusion and mass transport.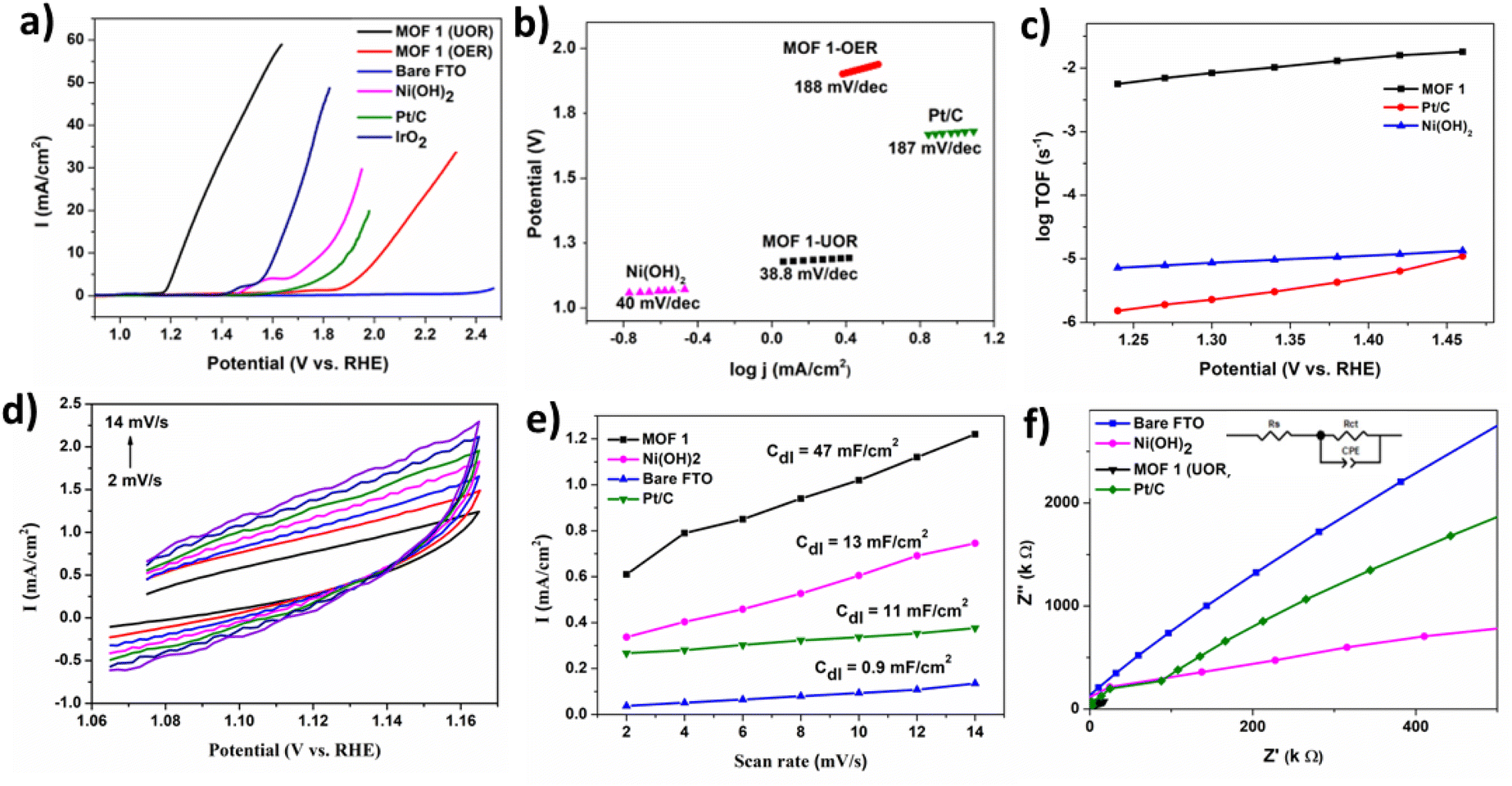 | ||
| Fig. 2 (a) Linear sweep voltammetry for MOF 1, UOR, OER, and comparative materials vs. RHE. (b) Corresponding Tafel plots. (c) Dependence of the TOF in KOH (1 M) with urea (0.33 M). (d) Cyclic voltammetry in a non-faradaic region with an incremental sweep rate of 2 mV s−1. (e) Current density (I) as a function of the sweep rate to determine Cdl. (f) Comparative Nyquist plots and equivalent circuits. | ||
| Material | Electrolyte (1 M KOH) | Onset-potential (V) | Ref. |
|---|---|---|---|
| MOF 1 | +0.33 M urea | 1.18 | Present work |
| NiFe-MIL-53-NH2 | +0.33 M urea | 1.36 | 54 |
| Ni-MOF@NiO/Ni | +0.33 M urea | 1.40 (10 mA cm−2) | 59 |
| Aggregated Ni-MOF nanosheets | +0.5 M urea | 1.38 (10 mA cm−2) | 42 |
| Ni3N nanosheets | +0.33 M urea | 1.33 | 60 |
| Ni0.9Fe0.1O | +0.33 M urea | 1.35 | 32 |
| Ni(OH)2 | +0.33 M urea | 1.36 | 15 |
| Ni-Mo nanotubes | +0.1 M urea | 1.36 | 33 |
| Ni-MOF nanosheets | +0.1 M urea | 1.33 | 50 |
| Ni2P/Fe2P | +0.5 M urea | 1.33 | 61 |
| MnO2/MnCo2O4/Ni | +0.5 M urea | 1.33 | 33 |
| Ni(OH)2@NF | +0.33 M urea | 1.33 | 62 |
| Ni-Bi | +0.33 M urea | 1.34 | 63 |
| Ni-WO2@C/NF | +0.33 M urea | 1.30 | 64 |
The overall performance of a catalyst is dependent upon two major factors: the abundance of active sites and the intrinsic activity of each active site. We used Cdl to measure the electrochemical active-surface area (ECSA) to gain understanding about the intrinsic surface activity. Cdl was calculated by carrying out CV (Fig. 2d and S5†) in a non-faradaic region (1.065–1.165 V). The straight line acquired by plotting current density vs. scan rate (2–14 mV s−1) at a constant potential of 1.11 V gave Cdl. Fig. 2e shows Cdl (mF cm−2) of MOF 1, Ni(OH)2, Pt/C, and bare FTO to be 47, 13, 11, and 0.9, respectively. A Cdl of 47 mF cm−2 indicates a high ECSA, thereby revealing a high density of active sites of MOF 1 responsible for phenomenal UOR activity. Thus, the enhanced electrochemical activity could be credited to the intrinsic high activity of MOF 1. The corresponding ECSA was calculated as ECSA = Cdl/Cs (where specific capacitance (Cs) = 60 μF cm−2)).40 The highest value of ECSA (in cm2) was 783 for MOF 1, followed by 216, 183, and 15 for Ni(OH)2, PT/C, and bare FTO, respectively. A higher ECSA more catalytic sites which, in turn, depicts a higher catalytic performance. The ECSA (in cm2) of MOF 1 was much higher than that for materials reported, such as NiS2–MoS2 (1.45), NiS2 (0.44),26 Ni/Ni(OH)2 (54), β Ni(OH)2 (14),15 Li4Ni3O10 (8.98), Li2NiO4 (5.55), and Li3Ni2O7 (6.33).52 The mass activity was calculated by J m−1 (where J is the current density (10 mA cm−2) at potential (1.24 V) and m is the mass loading (0.32 mg cm−2) on an electrode surface.53 The mass activity of MOF 1 (31.25 A g−1) was estimated to determine the intrinsic activity. The mass activity of MOF 1 was 2.6- and 4.5-times greater than that of Ni-MIL-53-NH2 (12 A g−1) and Fe-MIL-53-NH2 (6.8 A g−1), respectively.54 Thus, the outstanding activity of MOF 1 for the UOR was credited to a high intrinsic activity and large ECSA. Recently, it has been reported that introduction of readily oxidizable metals in catalysts promotes oxidation reactions.55 Thus, the ease of oxidation of Ni2+ to Ni3+ by application of potential during the UOR is correlated to its efficient activity.
Electrochemical impedance spectroscopy (EIS) was undertaken to obtain further insights about kinetics on the electrode/electrolyte interface during the UOR. Nyquist plots along with equivalent circuits are displayed in Fig. 2f in a frequency range of 100 kHz to 0.1 Hz. The smaller semi-circular diameter for MOF 1 revealed the strikingly small resistance to charge transfer and, thus, high performance compared with that of Ni(OH)2, Pt/C and bare FTO. The turnover frequency (TOF) was evaluated from the surface concentration of coated FTO to evaluate catalytic performance further. Assuming that all metal sites were electrocatalytically active, a potential of 1.24 V was required for a TOF of 5 × 10−3 s−1 at 10 mA cm−2 (Fig. 2c), which is greater than the TOF for Ni(OH)2 (1.3 × 10−3 s−1) and 2.9 × 10−4 s−1 for Pt/C. The TOF for MOF 1 was also higher than that reported for Ni-MIL-53-NH2 (1.2 × 10−3 s−1) and Fe-MIL-53-NH2 (6.5 × 10−4 s−1).54
Another critical factor for the efficiency of a catalyst is its stability and durability for commercial applications. CPE was carried out for MOF 1 at a potential of 1.41 V for 17 h (J = 30 mA cm−2). MOF 1 exhibited strong durability with retained UOR activity after 17 h continuously (Fig. 3a). The slight increment in current may be due to conversion of Ni(II) to Ni(III) during pre-oxidation to acquire high-valence, catalytically active Ni(III) species.13 The electrochemical stability of MOF 1 is compared to that of other Ni-based electrocatalysts from the literature. The stability of MOFs such as Ni-MOF-0.5,42 2D Ni-MOF,43 Ni-MOF-100,56 NiO@Ni-MOF,48 NiMn0.14-BDC MOF,57 and 3% CeO2/Ni-MOF has been documented.58 LSV was undertaken after a long-term stability test. There was no noticeable change in the position of the curve for the initial scan and after the stability test. Fig. 3b shows the robustness of the catalyst. Taken together, these results suggest that MOF 1 could be a potent electrocatalyst for the UOR.
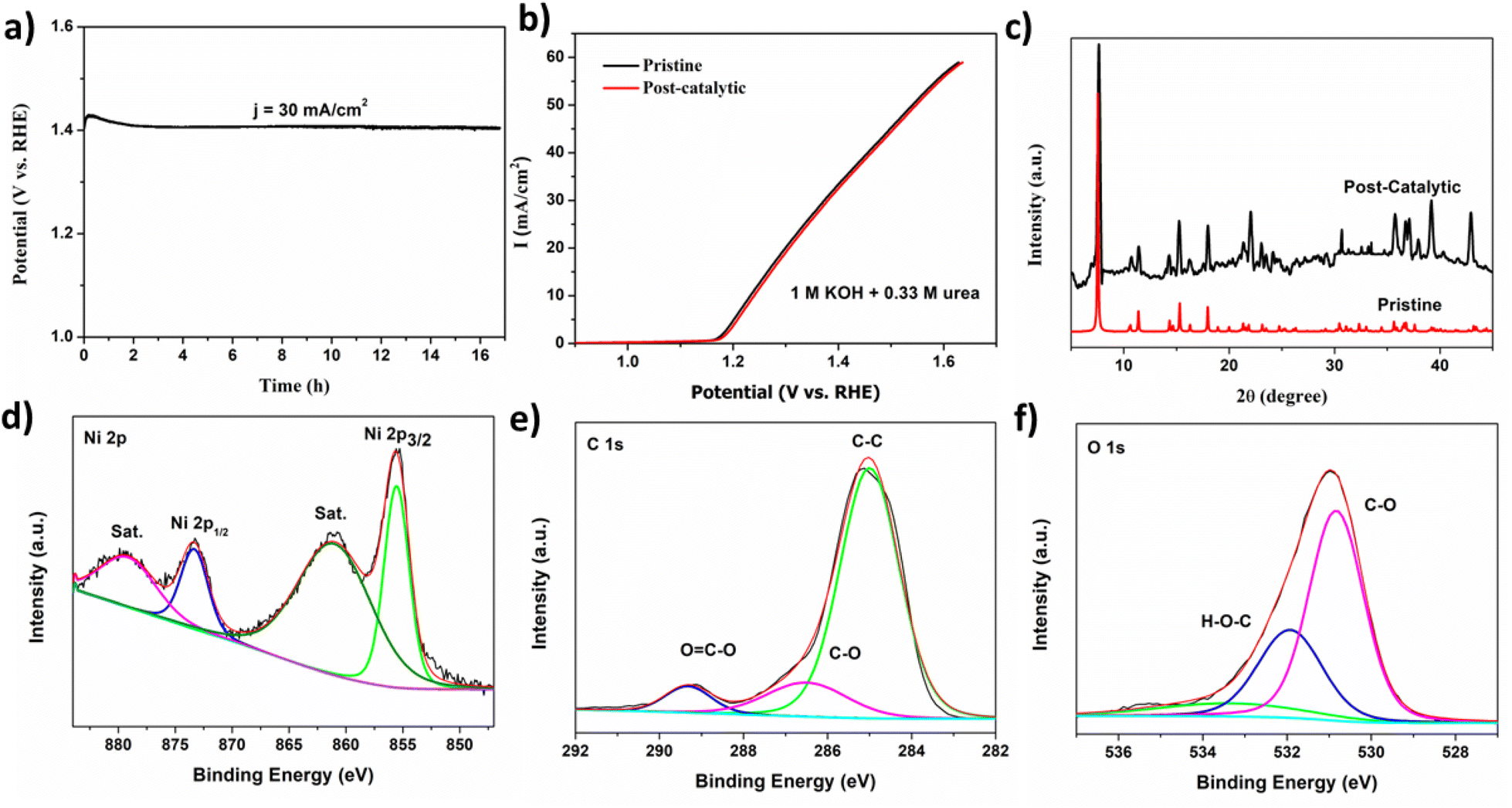 | ||
| Fig. 3 (a) Controlled potential electrolysis at a potential of 1.41 V. (b) LSV curves of pristine and post-catalytic samples. Physiochemical structure of MOF 1 after 17 h of continuous UOR electrolysis. (c) XRD spectrum of pristine and post-catalytic samples. (d) High-resolution spectra for Ni 2p (e) and C 1s (f) O 1s in post-catalytic samples. | ||
The structural integrity of the catalyst was assessed before and after stability tests through PXRD, SEM, and X-ray photoelectron spectroscopy (XPS). A noticeable dissimilarity in the diffraction pattern for MOF 1 before and after catalysis was not observed (Fig. 3c), which indicated the stability of the crystal structure after the UOR. The surface morphology of MOF 1 remained intact after a 17 h stability test (Fig. S7†). To obtain information about the chemical composition and surface electronic state of MOF 1, XPS was employed in a post-catalytic sample. In the high-resolution Ni 2p spectrum, peaks at 855.6 and 873.2 eV were assigned to the core levels of Ni 2p3/2 and Ni 2p1/2, whereas shoulder peaks at 861.8 and 879 eV indicated the Ni2+ oxidation state (Fig. 3d).65 The absence of Ni(0) and NiO peaks suggested that the catalyst was stable and was not converted to metallic nickel or its oxides. The XPS patterns for C 1s and O 1s are in accordance with the literature. The high-resolution C 1s spectrum showed distinctive peaks at 284.81 eV for C–C,66 286.5 eV for C–O, and 289.18 eV for O–CO.67 In the XPS pattern for O 1s, the deconvoluted peak at 530.81 eV corresponded to M–O, the peak at 532.1 eV was ascribed to CO, and unavoidable adsorbed H2O appeared at 534 eV.68 The XPS survey is given in Fig. S8.†
Proposed mechanism
To understand the structure–activity relationship of MOF 1 with abundantly exposed active sites we propose a mechanism involving fast transfer of electrons and low resistance to charge transfer. In an indirect or electrochemical–chemical (EC) mechanism, urea is oxidized to N2, CO2, and H2O with catalyst regeneration:69,70| 6Ni(OH)2 + 6OH− → 6NiOOH + 6H2O + 6e− |
| 6NiOOH + CO(NH2)2 + H2O → CO2 + N2 + 6Ni(OH)2 |
OH− coordinated with Ni(II) in linear chains and dangling OH− groups attached to Ni4O4 cubane makes Ni⋯OH− prone to conversion to NiOOH under an applied potential during the UOR. Due to electrostatic interaction, the urea molecule is adsorbed on the active NiOOH surface via O–C and Ni–O coordination bonds.71 As a result, urea is oxidized and NiOOH is reduced back to Ni⋯OH− to complete the catalytic cycle, and reacts with OH− to repeat the process.
Conclusions
We identified MOF 1 as a unique electrocatalyst that exhibits excellent activity, strong durability, and stability along with favorable kinetics towards the UOR. The low onset potential of 1.18 V and Tafel slope of 38.8 mV dec−1 was due to an intrinsic high TOF of 0.005 s−1. A lowest overpotential of 1.24 V to attain a catalytic current density of 10 mA cm−2 makes MOF 1 a highly efficient electrocatalyst for the UOR. No considerable change in comparative PXRD patterns of a pristine sample and post-catalytic sample signified the structural stability of MOF. MOF 1 could be a competent candidate for urea-associated applications such as hydrogen production, treatment of urea in wastewater, and urea-based fuel cells. Most importantly, our work provides insights for designing heterogenous catalysts at the molecular level. Thus, by incorporating suitable linkers, the electronic environment around metals could be altered to make it conducive to the design of exceptional catalysts having high oxidation states for various oxidation reactions.Author contributions
All authors contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported financially by the Pakistan Academy of Sciences (PAS) and Higher Education Commission (HEC) of Pakistan (8400/Federal/NRPU/R&D/HEC/2017). AW acknowledges the Pakistan Science Foundation for financial assistance under a project code of PSF-NSFC-IV/Chem/C-QAU (27).Notes and references
- M. Dresselhaus and I. Thomas, Nature, 2001, 414, 332 CrossRef CAS PubMed.
- Y. Xing, B. Fang, A. Bonakdarpour, S. Zhang and D. P. Wilkinson, Int. J. Hydrogen Energy, 2014, 39, 7859 CrossRef CAS.
- P. Hao, W. Zhu, L. Li, J. Tian, J. Xie, F. Lei, G. Cui, G. Y. Zhang and B. Tang, Electrochim. Acta, 2020, 338, 135883 CrossRef CAS.
- M. Carmo, D. L Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901 CrossRef CAS.
- X. Sun and R. Ding, Catal. Sci. Technol., 2020, 10, 1567 RSC.
- J. Jiang, L. Chang, W. Zhao, Q. Tian and Q. Xu, ChemComm, 2019, 55, 10174 RSC.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2017, 56, 842 CrossRef CAS PubMed.
- J. Xie, L. Gao, S. Cao, W. Liu, F. Lei, P. Hao, X. Xia and B. Tang, J. Mater. Chem. A, 2019, 7, 13577 RSC.
- X. Yang, L. Kang, Z. Wei, S. Lou, F. Lei, P. Hao, J. Xie and B. Tang, Chem. Eng. J., 2021, 422, 130139 CrossRef CAS.
- J. Zhang, J. H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 7649 CrossRef CAS.
- M. N. Kopylovich, A. P. Ribeiro, E. C. Alegria, N. M. Martins, L. M. Martins and A. J. Pombeiro, Adv. Organomet. Chem., 2015, 63, 91 CrossRef CAS.
- H. G. Cha and K. S. Choi, Nat. Chem., 2015, 7, 328 CrossRef CAS.
- J. Xie, W. Liu, F. Lei, X. Zhang, H. Qu, L. Gao, P. Hao, B. Tang and Y. Xie, Chem.–Eur. J., 2018, 24, 18408 CrossRef CAS PubMed.
- W. Liu, J. Xie, Y. Guo, S. Lou, L. Gao and B. Tang, J. Mater. Chem. A, 2019, 7, 24437 RSC.
- R. K. Singh and A. Schechter, Electrochim. Acta, 2018, 278, 405 CrossRef CAS.
- J. Xie, W. Liu, X. Zhang, Y. Guo, L. Gao, F. Lei, B. Tang and Y. Xie, ACS Mater. Lett., 2019, 1, 103 CrossRef CAS.
- X. Zhu, X. Dou, J. Dai, X. An, Y. Guo, L. Zhang, S. Tao, J. Zhao, W. Chu and X. C. Zeng, Angew. Chem., Int. Ed., 2016, 55, 12465 CrossRef CAS.
- F. Wu, G. Ou, Y. Wang, H. Zhong, L. Zhang, H. Li and Y. Shi, Chem.–Asian J., 2019, 14, 2796 CAS.
- J. Xie, H. Qu, F. Lei, X. Peng, W. Liu, L. Gao, P. Hao, G. Cui and B. Tang, J. Mater. Chem. A, 2018, 6, 16121 RSC.
- Z. Wei, W. Sun, S. Liu, J. Qi, L. Kang, J. Li, S. Lou, J. Xie, B. Tang and Y. Xie, Particuology, 2021, 57, 104 CrossRef CAS.
- W. Simka, J. Piotrowski, A. Robak and G. Nawrat, J. Appl. Electrochem., 2009, 39, 1137 CrossRef CAS.
- J. Wright, A. Michaels and A. Appleby, AIChE J., 1986, 32, 1450 CrossRef CAS.
- W. Simka, J. Piotrowski and G. Nawrat, Electrochim. Acta, 2007, 52, 5696 CrossRef CAS.
- A. Bolzan and T. Iwasita, Electrochim. Acta, 1988, 33, 109 CrossRef CAS.
- B. K. Boggs, R. L. King and G. G. Botte, ChemComm, 2009, 32, 4859 RSC.
- S. Wang, L. Zhao, J. Li, X. Tian, X. Wu and L. Feng, J. Energy Chem., 2022, 66, 483 CrossRef CAS.
- S. Wang, J. Zhu, X. Wu and L. Feng, Chin. Chem. Lett., 2022, 33, 1105 CrossRef CAS.
- P. Hao, H. Wen, Q. Wang, L. Li, Z. Zhao, R. Xu, J. Xie, G. Cui and B. Tang, J. Mater. Chem. A, 2021, 9, 8576 RSC.
- D. Suárez, N. Díaz and K. M. Merz, J. Am. Chem. Soc., 2003, 125, 15324 CrossRef PubMed.
- J. Li, S. Wang, J. Chang and L. Feng, Adv. Powder Mater., 2022, 3, 100030 CrossRef.
- Q. Liu, L. Xie, F. Qu, Z. Liu, G. Du, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2017, 4, 1120 RSC.
- F. Wu, G. Ou, J. Yang, H. Li, Y. Gao, F. Chen, Y. Wang and Y. Shi, ChemComm, 2019, 55, 6555 RSC.
- J. Y. Zhang, T. He, M. Wang, R. Qi, Y. Yan, Z. Dong, H. Liu, H. Wang and B. Y. Xia, Nano Energy, 2019, 60, 894 CrossRef CAS.
- S. Zhao, Y. Wang, J. Dong, C. T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao and L. Zhang, Nat. Energy, 2016, 1, 1 Search PubMed.
- J. H. Kim, B. Fang, M. Y. Song and J. S. Yu, Chem. Mater., 2012, 24, 2256 CrossRef CAS.
- B. Fang, J. H. Kim, M. S. Kim and J. S. Yu, Acc. Chem. Res., 2013, 46, 1397 CrossRef CAS.
- B. Fang, L. Daniel, A. Bonakdarpour, R. Govindarajan, J. Sharman and D. P. Wilkinson, Small, 2021, 17, 2102288 CrossRef CAS PubMed.
- L. Lu, S. Zou and B. Fang, ACS Catal., 2021, 11, 6020 CrossRef CAS.
- B. Fang, J. H. Kim and J. S. Yu, Electrochem. Commun., 2008, 10, 659 CrossRef CAS.
- Z. Gao, Y. Wang, L. Xu, Q. Tao, X. Wang, Z. Zhou, Y. Luo, J. Yu and Y. Huang, Chem. Eng. J., 2022, 433, 133515 CrossRef CAS.
- Q. Li, S. Zheng, M. Du and H. Pang, Chem. Eng. J., 2021, 417, 129201 CrossRef CAS.
- S. Zheng, Y. Zheng, H. Xue and H. Pang, Chem. Eng. J., 2020, 395, 125166 CrossRef CAS.
- D. Zhu, C. Guo, J. Liu, L. Wang, Y. Du and S. Z. Qiao, ChemComm, 2017, 53, 10906 RSC.
- L. Wang, L. Ren, X. Wang, X. Feng, J. Zhou and B. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 4750 CrossRef CAS PubMed.
- S. Rezaee and S. Shahrokhian, Nanoscale, 2020, 12, 16123 RSC.
- M. A. Nadeem, M. C. C. Ng, J. van Leusen, P. Kögerler and J. A. Stride, Chem.–Eur. J., 2020, 26, 7589 CrossRef CAS PubMed.
- M. Radhika, B. Gopalakrishna, K. Chaitra, L. K. G. Bhatta, K. Venkatesh, M. S. Kamath and N. Kathyayini, Mater. Res. Express, 2020, 7, 054003 CrossRef CAS.
- S. Xiong, S. Jiang, J. Wang, H. Lin, M. Lin, S. Weng, S. Liu, Y. Jiao, Y. Xu and J. A. Chen, Electrochim. Acta, 2020, 340, 135956 CrossRef CAS.
- R. P. Forslund, J. T. Mefford, W. G. Hardin, C. T. Alexander, K. P. Johnston and K. J. Stevenson, ACS Catal., 2016, 6, 5044 CrossRef CAS.
- D. Zhu, C. Guo, J. Liu, L. Wang, Y. Du and S.-Z. Qiao, ChemComm, 2017, 53, 10906 RSC.
- S. Hu, C. Feng, S. Wang, J. Liu, H. Wu, L. Zhang and J. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 13168 CrossRef CAS PubMed.
- Y. Tao, L. Chen, Z. Ma, C. Zhang, Y. Zhang, D. Zhang, D. Pan, J. Wu and G. Li, Chem. Eng. J., 2022, 446, 137240 CrossRef CAS.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu and Y. Jiang, Angew. Chem., Int. Ed., 2015, 127, 8846 CrossRef.
- Z. Gao, Y. Wang, L. Xu, Q. Tao, X. Wang, Z. Zhou, Y. Luo, J. Yu and Y. Huang, Chem. Eng. J., 2022, 433, 133515 CrossRef CAS.
- C. Guo, Y. Zheng, J. Ran, F. Xie, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 8539 CrossRef CAS PubMed.
- M. Yuan, R. Wang, Z. Sun, L. Lin, H. Yang, H. Li, C. Nan, G. Sun and S. Ma, Inorg. Chem., 2019, 58, 11449 CrossRef CAS PubMed.
- X. Xu, Q. Deng, H. C. Chen, M. Humayun, D. Duan, X. Zhang, H. Sun, X. Ao and X. Xue, Research, 2022, 2022, 9837109 CrossRef CAS PubMed.
- X. Wu, L. Li, J. Pan, X. Wang, H. Zhang, S. Song and H. Zhang, Mater. Lab., 2022, 1, 220009 Search PubMed.
- Q. Li, S. Zheng, M. Du and H. Pang, Chem. Eng. J., 2021, 417, 129201 CrossRef CAS.
- Q. Liu, L. Xie, F. Qu, Z. Liu, G. Du, A. M. Asiri and X. Sun, Inorg. Chem., 2017, 4, 1120 CAS.
- H. Xu, K. Ye, K. Zhu, Y. Gao, J. Yin, J. Yan, G. Wang and D. Cao, ACS Sustainable Chem. Eng., 2020, 8, 16037 CrossRef CAS.
- L. Xia, Y. Liao, Y. Qing, H. Xu, Z. Gao, W. Li and Y. Wu, ACS Appl. Energy Mater., 2020, 3, 2996 CrossRef CAS.
- J. Ge, Y. Lai, M. Guan, Y. Xiao, J. Kuang and C. Yang, Environ. Sci.: Nano, 2021, 8, 1326 RSC.
- F. Shen, W. Jiang, G. Qian, W. Chen, H. Zhang, L. Luo and S. Yin, J. Power Sources, 2020, 458, 228014 CrossRef CAS.
- L. Qian, Z. Lu, T. Xu, X. Wu, Y. Tian, Y. Li, Z. Huo, X. Sun and X. Duan, Adv. Energy Mater., 2015, 5, 1500245 CrossRef.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti and J. O. Müller, J. Mater. Chem. A, 2008, 18, 4893 RSC.
- H. Cao, X. Wu, G. Yin and J. H. Warner, Inorg. Chem., 2012, 51, 2954 CrossRef CAS PubMed.
- S. Dutta, A. Indra, Y. Feng, H. Han and T. Song, Appl. Catal., B, 2019, 241, 521 CrossRef CAS.
- V. Vedharathinam and G. Botte, Electrochim. Acta, 2012, 81, 292 CrossRef CAS.
- W. Sun, J. Li, W. Gao, L. Kang, F. Lei and J. Xie, ChemComm, 2022, 58, 2430 RSC.
- D. A. Daramola, D. Singh and G. G. Botte, J. Phys. Chem. A, 2010, 114, 11513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM image, FT-IR spectrum, TGA, coordination environment for Ni, cyclic voltammograms for Cdl, LSV at varied scan rates, post-catalytic SEM and XPS survey of MOF 1. See https://doi.org/10.1039/d2ra05145a |
| This journal is © The Royal Society of Chemistry 2022 |